
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Red-fluorescence under UV and green-SHG under NIR dual-mode emission in a yellow crystal of a 1,2,5-thiadiazole derivative
Ryo
Nakamura
a,
Yuta
Sawamura
b,
Kazushi
Nakada
a,
Ryusuke
Mizoguchi
c,
Ayumi
Ishii
 c,
Tomohiro
Yamashita
d,
Hiroko
Yokota
d,
Gary J.
Richards
a and
Akiko
Hori
*ab
c,
Tomohiro
Yamashita
d,
Hiroko
Yokota
d,
Gary J.
Richards
a and
Akiko
Hori
*ab
aGraduate School of Engineering and Science, Shibaura Institute of Technology, Fukasaku 307, Minuma-ku, Saitama 337-8570, Japan. E-mail: ahori@shibaura-it.ac.jp
bUndergraduate School of Engineering, Shibaura Institute of Technology, Fukasaku 307, Minuma-ku, Saitama 337-8570, Japan
cSchool of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
dSchool of Materials and Chemical Technology, Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8501, Japan
First published on 9th December 2025
Abstract
A yellow chiral crystal of a 1,2,5-thiadiazole-substituted pyrazine exhibited red excimer-like emission above 600 nm with a Stokes shift exceeding 200 nm under UV, which also enabled green second harmonic generation (SHG) under near-infrared irradiation.
Invisible light that lies beyond the visible region pervades our surroundings and plays a crucial role in technologies such as optical communication and photochemical reactions. Visualizing such light quantitatively is vital for both applied and fundamental photophysical research. Organic luminescent materials have attracted considerable interest in organic electronics,1–5 particularly in applications such as organic light-emitting diodes (OLEDs)6,7 and bio-probes.8,9 Their lightweight, flexible, and responsive nature, inherent to organic compounds, makes them highly suitable for advanced photonic and optoelectronic devices.1–3 However, nonradiative decay pathways, especially those caused by vibrational relaxation and bond rotations, often reduce their luminescence efficiency.10,11 Addressing these losses requires rigid molecular frameworks with well-defined π-conjugated systems. Among such frameworks, pyrazinacene derivatives—heteroaromatic compounds composed of linearly fused pyrazine units—have emerged as promising candidates due to their electron-accepting properties, structural rigidity, and long-wavelength emission.12–14 Heteroaromatic compounds, particularly those incorporating electron-donating and electron-accepting groups, have attracted considerable interest due to their diverse photophysical properties. Especially when equipped with donor and acceptor substituents, these molecules exhibit tunable photophysical properties and pronounced intramolecular charge-transfer (ICT) behavior. We previously reported TPA-functionalized pyrazinacene derivatives 1 that exhibited efficient ICT emission based on a donor–acceptor system.15 Building on this strategy, we introduced a 1,2,5-thiadiazole unit into the pyrazinacene scaffold, which was expected to enhance the emission efficiency and was anticipated to promote nonlinear optical (NLO) phenomena. This heterocyclic moiety offers a planar, rigid geometry and electron-accepting properties,16–19 making it a promising structural element for new photophysical functions. In this study, we synthesized yellow chiral crystals of thiadiazole-based derivatives that were found to exhibit excimer emission with a Stokes shift exceeding 200 nm, originating from the anisotropic molecular design. Moreover, these crystals were observed to generate second harmonic (SH) signals under near-infrared (NIR) excitation, suggesting a dual-mode emission pathway arising from distinct excitation mechanisms.20–22 Herein, we report the synthesis, crystal structures, and photophysical properties of two 1,2,5-thiadiazole-substituted pyrazine derivatives, 2a and 2b (Scheme 1), bearing phenyl and triphenylamine (TPA) groups, respectively. Single crystals of both compounds were obtained, and their structural features were characterized. Compound 2a not only exhibited excimer emission at 636 nm but also demonstrated pronounced SHG activity in its chiral crystal form, providing a rare solid-state example and offering a potential design strategy for luminescent materials combining fluorescence and nonlinear optical responses.
 | ||
| Scheme 1 Molecular structures of pyrazinacene 1 and thiadiazole derivatives 2; TPA: triphenylamine substituent. | ||
Compound 2 was prepared via a typical dehydration-condensation reaction (Fig. S1–S3).13,23 Specifically, a dicarbonyl intermediate was prepared by coupling oxalyl chloride with an appropriate aromatic substituent. Subsequently, 3,4-diamino-1,2,5-thiadiazole was added, and the mixture was refluxed in the presence of p-toluenesulfonic acid (PTSA), during which a color change indicative of aromatic ring formation was observed. The products were purified by column chromatography (silica gel, CHCl3) to afford single products of 2: 2a as a yellow powder and 2b as a red powder. Single crystals of each compound were obtained by dissolving the products in a good solvent such as CHCl3, followed by addition of a poor solvent such as MeOH and slow concentration of the solution at room temperature to yield single crystals suitable for single-crystal X-ray analysis (Table S1 and Fig. S4–S7).
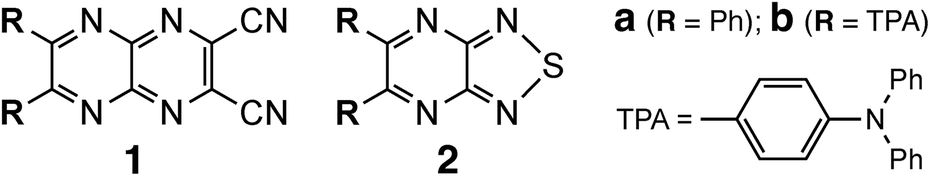
Phenyl-substituted compound 2a crystallized as yellow prismatic crystals in a chiral space group, monoclinic, Cc (No. 9), without inversion symmetry (Fig. 1). The six-membered pyrazine ring (ring-α) and five-membered thiadiazole-NSN ring (ring-β) showed high planarity, and the fused nine-membered ring (αβ) showed minimal deviation from the mean plane (τ = 3.4). The two phenyl rings attached to the pyrazine moiety adopted twisted conformations due to steric hindrance, with relevant dihedral angles of 25.5(3)° and 55.2(3)°, respectively. The resulting planar pyrazinacene core is stacked along the c-axis while avoiding the bulky twisted phenyl groups; the centroid-to-centroid distance between adjacent ab planes is 3.542(4) Å, and the vertical π⋯π overlap ranged from 3.34 to 3.41 Å, indicating strong π-stacking interactions. Notably, the orthogonal packing arrangement was stabilized by intermolecular C–H⋯π interactions between phenyl groups, leading to an alternating stacking along the c-axis without inversion centers. Hirshfeld surface (HS) analysis supports this packing, with moderate H⋯H (29.8%) and notable H⋯N/N⋯H interactions (22.5%) complementing the π⋯π stacking (Fig. S8). This columnar stacking preserved directional anisotropy along the a-axis without cancellation of dipole moments, likely assisted by pseudo-hydrogen bonding between the lone pair of the thiadiazole nitrogen and the adjacent phenyl C–H moiety.
 | ||
| Fig. 1 (a) Appearance of crystal morphology, (b) molecular structure with the selected numbering schemes, and (c) packing structure of 2a (CCDC: 2483803): ORTEP view with 50% thermal ellipsoids; color scheme: C, gray; N, blue; S, yellow. | ||
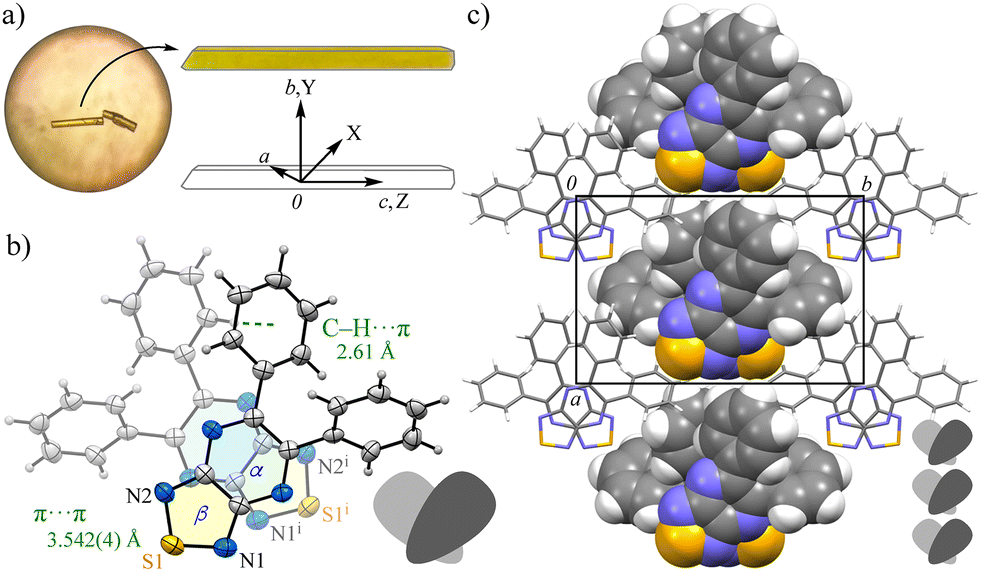
In contrast, compound 2b, bearing a TPA group, formed red needle-like crystals that were resolved in the monoclinic space group P21/c (No. 14) (Fig. 2). The αβ plane, comprising the pyrazine and thiadiazole rings, retained a high degree of planarity (τ = 2.8); however, the bulky TPA group hindered strong intermolecular associations. Only weak π⋯π interactions between the αβ plane and the peripheral phenyl ring (ring-E) were observed; the intermolecular distance of Cg(αβ)⋯Cg(E) is 3.712(3) Å, along with modest C–H⋯π interactions between the TPA moieties of C28–H28⋯Cg(F), C19–H19⋯Cg(B), and C14–H14⋯Cg(B). These findings indicate significantly diminished supramolecular stacking in 2b compared to 2a, as also supported by HS analysis showing dominant H⋯H contacts (48.4%) and fewer H⋯N/N⋯H interactions (15.0%) (Fig. S9).
 | ||
| Fig. 2 (a) Molecular structure with the selected numbering schemes and (b) packing structure of 2b (CCDC: 2483804): ORTEP view with 50% thermal ellipsoids; color scheme: C, gray; N, blue; O, red. | ||
Compound 2a exhibited intriguing photophysical behavior in the crystalline state (Table 1, Fig. 3 and Fig. S10–S13), and the visual appearance together with the absorption spectra of 2 in CCl4 are shown in Fig. 3a. 2a appeared pale yellow with a maximum absorption at 373 nm and showed no detectable emission. In contrast, 2b displayed a red solution with a pronounced absorption band at 494 nm, suggestive of ICT from the TPA moiety to the nine-membered ring (αβ), as supported by theoretical calculations (Fig. 4 and Fig. S14, S15).15,24 It also exhibited an intense emission peak at 592 nm with a photoluminescence quantum yield (PLQY) of ΦPL = 30% and a large Stokes shift of 98 nm. Under the same conditions, the PLQY of 2b was 7% higher than that of its structural analogue 1b (ΦPL = 23%),15 clearly demonstrating that incorporation of the thiadiazole-NSN ring effectively suppresses thermal deactivation. In CHCl3 solution, both 2a and 2b showed similar absorption spectra to those in CCl4, but neither exhibited measurable emission. This absence of emission is likely because the excimer-type excited state that dominates in the solid state cannot form in solution, where the excited energy is efficiently dissipated through nonradiative pathways. The solid-state absorption spectra (measured by drop-casting on filter paper) closely matched the results of the solution-state (Fig. 3b). Solid-state PL spectra of powder samples revealed that 2a emitted strongly at 613 nm with ΦPL = 11%, while 2b showed an emission maximum at 634 nm with ΦPL = 7.9%. As compound 1 exhibited negligible solid-state emission, these results clearly highlight the exceptional luminescence properties of 2, especially 2a, whose Stokes shift exceeds 230 nm, an extraordinary value. Typically, organic molecules display Stokes shifts in the range of 10–50 nm and compounds undergoing excited-state intramolecular proton transfer (ESIPT) may exhibit shifts of 100–200 nm due to substantial structural reorganization in the excited state.25–27 Since excimer or exciplex formation can lead to even larger shifts, often reaching 100–300 nm, the crystal structure of 2a suggests that the supramolecular association of the nine-membered core facilitates excimer emission in the present system,1–3,28,29 accounting for its unique photophysical properties. Although 2b also exhibited a red-shifted emission of 42 nm in the solid state compared to its solution-state emission (with a Stokes shift exceeding 100 nm), the spectral similarity indicates that the emission originates primarily from the monomeric species. Despite the similar emission wavelengths of 2, the corresponding lifetimes were drastically different (Fig. 3c): 2a exhibited long-lived room-temperature emission arising from excimer formation, with an average lifetime of 25.1 µs (τrise = 0.69 µs; τmajor = 14.0 µs, 80%), whereas 2b exhibited a nanosecond-scale lifetime (τavg = 4.3 ns; τmajor = 3.9 ns, 61%) characteristic of fluorescence.
 | ||
| Fig. 3 UV-Vis and emission spectra of 2 in (a) CCl4 solution and (b) solid states with the corresponding appearances. (c) Emission lifetimes of 2a (λem = 636 nm) and 2b (λem = 648 nm) upon excitation at λex = 365 and 470 nm, respectively. | ||
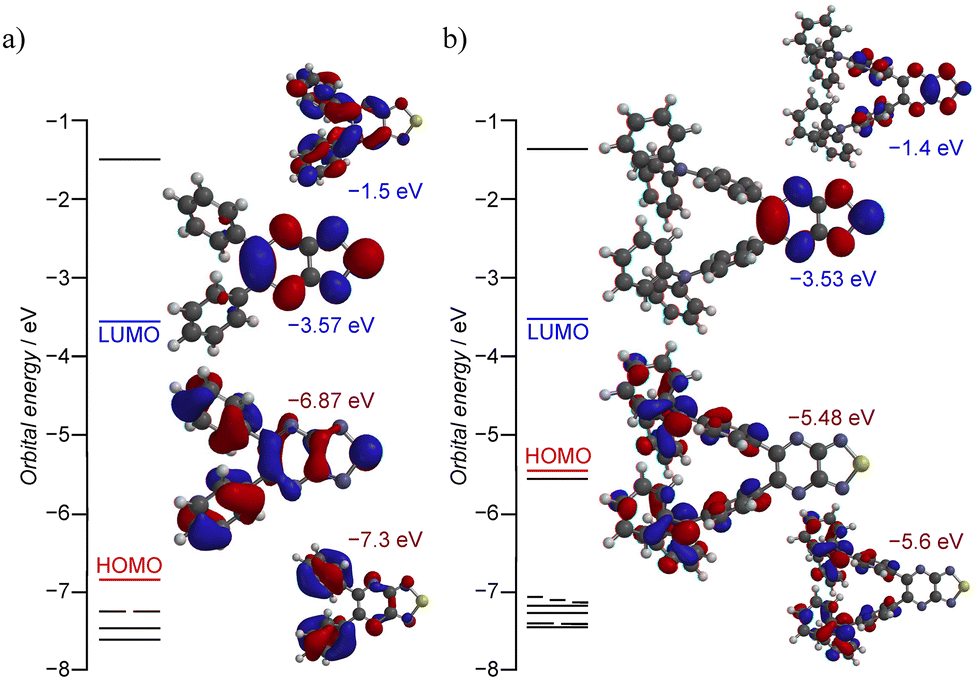 | ||
| Fig. 4 The energy levels near the frontier molecular orbitals, and the contour plots of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of (a) 2a and (b) 2b as optimized isolated molecules [B3LYP/6-311+G(2DF,2P)].25 | ||
In addition to their intriguing emission properties, the crystal of 2a also exhibited SHG activity, as demonstrated by polarization-dependent SHG measurements (Fig. 5a and Fig. S16).20–23,30 When the crystal was irradiated with NIR laser light at 1050 nm, green emission at 525 nm, corresponding to the second harmonic, was observed. Fig. 5b shows a side view of the crystal, where only the irradiated region exhibits weak green emission. The fact that this light originates from the crystal was confirmed by the observation of SH signals on the surface (Fig. 5c). In addition, the relationship between the SHG intensity (photon counts) and the square of the incident laser intensity demonstrated the quadratic nonlinear effect characteristic of SHG (Fig. 5d). Polarization experiments further confirmed the assignment of point group m, in agreement with crystallographic analysis.22,31 The SHG tensor components determined from these measurements exhibited notable anisotropy (d11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :d13:d15:d31:d33:d35 = 57.0:17.2:5.75:4.66:1:1.02), clearly indicating a non-centrosymmetric response consistent with the monoclinic Cc. Among these, the dominant component d11 contributed significantly to the observed SHG signal, especially when the incident polarization is aligned along the c-axis or within the αβ plane. This anisotropy demonstrates local non-centrosymmetry despite the absence of a global inversion center. The SHG intensity reached its maximum along the X-direction, perpendicular to the crystal's elongation axis (c-axis), consistent with the anisotropic nonlinear optical response.32 This behavior offers a unique opportunity to visualize invisible light beyond the visible region, which plays a critical role in optoelectronic applications and photonic signal processing. In this context, 2a serves not only as a model system for studying supramolecular excimer emission but also as a rare π-conjugated molecular crystal combining strong solid-state photoluminescence and measurable SHG activity. Further investigations of phase-matching behavior, SHG intensity versus incident angle, and excitation wavelength dependence are currently underway to evaluate its potential as a functional SHG-active material.
:d13:d15:d31:d33:d35 = 57.0:17.2:5.75:4.66:1:1.02), clearly indicating a non-centrosymmetric response consistent with the monoclinic Cc. Among these, the dominant component d11 contributed significantly to the observed SHG signal, especially when the incident polarization is aligned along the c-axis or within the αβ plane. This anisotropy demonstrates local non-centrosymmetry despite the absence of a global inversion center. The SHG intensity reached its maximum along the X-direction, perpendicular to the crystal's elongation axis (c-axis), consistent with the anisotropic nonlinear optical response.32 This behavior offers a unique opportunity to visualize invisible light beyond the visible region, which plays a critical role in optoelectronic applications and photonic signal processing. In this context, 2a serves not only as a model system for studying supramolecular excimer emission but also as a rare π-conjugated molecular crystal combining strong solid-state photoluminescence and measurable SHG activity. Further investigations of phase-matching behavior, SHG intensity versus incident angle, and excitation wavelength dependence are currently underway to evaluate its potential as a functional SHG-active material.
 | ||
| Fig. 5 SHG activity of 2a: (a) concept of the emission characteristics under UV and NIR excitation, (b) appearance of green emission upon irradiation with NIR (1050 nm), (c) the corresponding photon counts (SH intensity) from the crystal, and (d) the relationship between the incident (1050 nm) and SHG emission (525 nm) light intensities. | ||
This work describes thiadiazole-based π-conjugated compounds featuring a fused pyrazine-NSN ring, 2, which exhibits distinct photophysical properties depending on the substitution pattern. Compound 2a with phenyl groups showed long-lived excimer-like emission in the solid state with an exceptionally large Stokes shift exceeding 230 nm, along with pronounced SHG activity in the crystalline state. In contrast, the TPA-substituted analogue 2b showed efficient ICT and the corresponding emission in both solution and solid states. In particular, 2a demonstrated red emission under UV excitation and green emission under NIR light via SHG, thereby providing a molecular-level strategy to visualize light beyond the visible region. These results highlight a new approach to designing wide-Stokes-shift emitters and nonlinear optical materials by combining supramolecular interactions and local non-centrosymmetry in molecular crystals.
R. N. and Y. S. prepared the samples, performed crystallographic and photophysical investigations, and drafted the manuscript. K. N. and G. J. R. prepared the samples and supported the whole investigation. R. M. and A. I. studied solid state emission with lifetime. T. Y. and H. Y. studied SHG characteristics. A. H. supervised the project and finalized the manuscript. All authors contributed to and approved the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available in the main article and its supplementary information (SI). Supplementary information: the preparation, crystal structures, and photophysical properties of 1,2,5-thiadiazole derivative 2. See DOI: https://doi.org/10.1039/d5cc05735c.Further inquiries regarding specific experimental details can be directed to the corresponding author.
CCDC 2483803 and 2483804 contain the supplementary crystallographic data for this paper.33a,b
Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research B, 23K21122, of JSPS KAKENHI and S-SPIRE project of Shibaura Institute of Technology (A. H.). H. Y. acknowledges financial support for JPMJFR213Z of JST FOREST and Grant-in-Aid for Scientific Research B, 24K00554, of JSPS KAKENHI.References
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388, 10.1039/C1CS15113D.
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53, DOI:10.1038/s41427-021-00318-8.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623, DOI:10.1002/adfm.201807623.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353, DOI:10.1002/chem.201403811.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238, DOI:10.1038/nature11687.
- C. Maeda, S. Nomoto, K. Takaishi and T. Ema, Chem. – Eur. J., 2020, 26, 13016–13021, DOI:10.1002/chem.202001463.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/B105159H.
- J. Yin, L. Huang, L. Wu, J. Li, T. D. James and W. Lin, Chem. Soc. Rev., 2021, 50, 12098–12150, 10.1039/D1CS00645B.
- P. Liu, X. Mu, X.-D. Zhang and D. Ming, Bioconjugate Chem., 2020, 31, 260–275, DOI:10.1021/acs.bioconjchem.9b00610.
- S. Liu, Y. Li, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2021, 12, 3427–3436, 10.1039/d0sc02911d.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680, 10.1039/D1CS00198A.
- G. J. Richards and J. P. Hill, Acc. Chem. Res., 2021, 54, 3228–3240, DOI:10.1021/acs.accounts.1c00315.
- G. J. Richards, K. Nakada, K. Aoki, T. Jitsukata, K. Hashimoto, T. Tajima, R. Mizoguchi, A. Ishii, J. P. Hill and A. Hori, Angew. Chem., Int. Ed., 2025, 64, e202504564, DOI:10.1002/anie.202504564; G. J. Richards, A. Cador, S. Yamada, A. Middleton, W. A. Webre, J. Labuta, P. A. Karr, K. Ariga, F. D’Souza, S. Kahlal, J.-F. Halet and J. P. Hill, J. Am. Chem. Soc., 2019, 141, 19570–19574, DOI:10.1021/jacs.9b10952.
- Y. Shi, Y. Zhang and X. Cai, Chem. Phys. Lett., 2021, 772, 138595, DOI:10.1016/j.cplett.2021.138595.
- K. Nakada, G. J. Richards and A. Hori, Chem. – Eur. J., 2025, 31, e202404487, DOI:10.1002/chem.202404487.
- S. K. M. Nalluri, J. Zhou, T. Cheng, Z. Liu, M. T. Nguyen, T. Chen, H. A. Patel, M. D. Krzyaniak, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1290–1303, DOI:10.1021/jacs.8b11201.
- Y. Duan, Q. Zhao, Y. Yang, J. Zhang, X. Tao and Y. Shen, J. Heterocycl. Chem., 2019, 56, 1464–1471, DOI:10.1002/jhet.3343.
- D. Taylor, T. Malcomson, A. Zhakeyev, S. Cheng, G. M. Rosair, J. Marques-Hueso, Z. Xu, M. J. Paterson, S. J. Dalgarno and F. Vilela, Org. Chem. Front., 2022, 9, 5473–5484, 10.1039/D2QO01316A.
- J. Lei, C.-W. Chang, Y.-K. Chen, P.-Y. Chou, L.-Y. Hsu, T.-L. Wu and C.-H. Cheng, J. Phys. Chem. C, 2024, 128(38), 16189–16198, DOI:10.1021/acs.jpcc.4c04475.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55, DOI:10.1021/cr9000429.
- X. Liu, Z. Yang, D. Wang and H. Cao, Crystals, 2016, 6, 158, DOI:10.3390/cryst6120158.
- H. Yokota, H. Usami, R. Haumont, P. Hicher, J. Kaneshiro, E. K. H. Salje and Y. Uesu, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 144109, DOI:10.1103/PhysRevB.89.144109; H. Yokota and Y. Uesu, J. Appl. Phys., 2021, 129, 014101, DOI:10.1063/5.0032881.
- N. Sato and H. Mizuno, J. Chem. Res. Synopses, 1997, 250–251, 10.1039/A701579H.
- Q. Peng, Y. Niu, C. Deng and Z. Shuai, Chem. Phys., 2010, 370, 215–222, DOI:10.1016/j.chemphys.2010.03.004.
- A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842–8880, 10.1039/C8CS00185E.
- A. Kedzia, A. Kudelko, M. Swiatkowski and R. Kruszynski, Dyes Pigm., 2020, 172, 107865, DOI:10.1016/j.dyepig.2019.107865.
- Y. Wang, J. Ren and Z. Shuai, Nat. Commun., 2023, 14, 5056, DOI:10.1038/s41467-023-40716-w.
- R. E. Cook, B. T. Phelan, R. J. Kamire, M. B. Majewski, R. M. Young and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 1607–1615, DOI:10.1021/acs.jpca.6b12644.
- A. Hori, S. Takatani, T. K. Miyamoto and M. Hasegawa, CrystEngComm, 2009, 11, 567–569, 10.1039/b822007g.
- A. Miniewicz, S. Bartkiewicz, E. Wojaczynska, T. Galica, R. Zalesny and R. Jakubas, J. Mater. Chem. C, 2019, 7, 1255–1262, 10.1039/C8TC05268A.
- S. Cherifi-Hertel, C. Voulot, U. Acevedo-Salas, Y. Zhang, O. Crégut, K. D. Dorkenoo and R. Hertel, J. Appl. Phys., 2021, 129, 081101, DOI:10.1063/5.0037286.
- H. Yokota, T. Hayashida, D. Kitahara and T. Kimura, npj Quantum Mater., 2022, 7, 106, DOI:10.1038/s41535-022-00515-w.
- (a) CCDC 2483803: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pclpc; (b) CCDC 2483804: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pclqd.
| This journal is © The Royal Society of Chemistry 2026 |