
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical one-pot Barbier/Simmons–Smith reaction via sequential zinc activation†
Asma A.
Alharthi
ad,
Benson M.
Kariuki
 a,
Louis C.
Morrill
*ab and
Duncan L.
Browne
*c
a,
Louis C.
Morrill
*ab and
Duncan L.
Browne
*c
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: lcm71@bath.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cDepartment of Pharmaceutical and Biological Chemistry, University College London (UCL), School of Pharmacy, 29-39 Brunswick Square, Bloomsbury, London, WC1N 1AX, UK. E-mail: duncan.browne@ucl.ac.uk
dDepartment of Chemistry, College of Science, University of Jeddah, Jeddah 21589, Saudi Arabia
First published on 23rd December 2025
Abstract
We report a mechanochemical one-pot Barbier/Simmons–Smith reaction enabled by ball-milling-mediated activation of zinc(0). This operationally simple method generates organozinc intermediates in situ and uses them sequentially in allylation and cyclopropanation without intermediate work-up. The protocol tolerates a broad range of ketones, exhibits selectivity over other carbonyl groups, and is compatible with various physical forms of zinc metal. In selected cases, the reaction proceeds with markedly enhanced diastereoselectivity under minimal-solvent milling conditions and was demonstrated on a gram scale using standard ball-milling equipment. Comparative studies show advantages over conventional solution and neat conditions, highlighting how mechanochemistry can uniquely enable tandem organometallic processes as for C–C bond construction.
Organometallic reagents such as Grignard and organozinc compounds are widely used in synthesis, with well-established protocols for their preparation and application across numerous transformations.1 However, their handling often requires rigorous exclusion of air and moisture, low temperatures, and extensive solvent use.2 These practical challenges have prompted interest in alternative approaches that simplify reagent generation and streamline transformations.
Mechanochemistry has emerged as a powerful method for promoting chemical reactivity under solvent-free or solvent-minimised conditions, enabling efficient formation of both carbon–carbon and carbon–heteroatom bonds.3,4 A key feature is the ability to generate reactive organometallic intermediates in situ from zero-valent metals (including Zn, Mn, Mg, Ca, Na, Li), allowing multi-step processes to be carried out in a single operation.5 For example, Browne and co-workers have reported a ball-milling strategy for direct activation of zinc metal to form organozinc reagents, which were applied in one-pot Negishi-type couplings (Scheme 1(A)).6 In related studies, Kubota and Ito demonstrated the generation of Grignard reagents by ball milling in air, followed by nickel-catalysed cross-coupling under solvent-minimised conditions.7
 | ||
| Scheme 1 Key inspiration and outline of the mechanochemical strategy. (A) Some previous examples of ball-milling-enabled generation and in situ use of organometallic reagents. (B) This work: a one-pot mechanochemical Barbier/Simmons–Smith sequence enabled by sequential zinc activation. | ||
This platform has since been extended to manganese chemistry, where reactive arylmanganese intermediates were generated from elemental Mn and subsequently employed in addition and coupling reactions.8
These studies highlight the potential of mechanochemistry to combine multiple synthetic steps in a single jar, minimising handling and solvent use while enabling access to reactive species.9 Building on this concept, we envisaged a one-pot sequence comprising a Barbier-type allylation followed by a Simmons–Smith cyclopropanation, mediated by mechanochemical activation of zinc metal (Scheme 1B). Herein, we report the development of this tandem protocol, which enables in situ generation and sequential use of organozinc intermediates for efficient C–C bond formation. The method is compatible with a wide range of ketones, exhibits selectivity in the presence of other carbonyl groups, and demonstrates a distinct advantage over traditional solution-phase approaches.
Our investigation commenced with 2-tetralone (1) as a model substrate (Table S1). Milling this substrate with zinc dust (10 equivalents), allyl bromide (2, 1.5 equivalents), diiodomethane (5 equivalents), and 2-methyltetrahydrofuran (2-MeTHF, 1.5 equivalents) as a liquid-assisted grinding (LAG) agent at 30 Hz for 3 hours yielded the desired cyclopropane (5) with a 62% NMR yield and a 54% isolated yield (Table S1, entry 1; see SI).10 Reducing the quantity of zinc to 7 equivalents resulted in a diminished, but nonetheless serviceable, NMR yield of 52% (Table S1, entry 2). Variations in the LAG agent or increasing its loading led to further reductions in yield (entries 3, 4, and 5). Notably, omitting 2-MeTHF entirely caused a significant decrease in product yield to 19%. Adjusting the amount of CH2I2 to either 3.5 or 7.5 equivalents adversely affected the reaction, producing yields of 51% and 61%, respectively (Table S1, entry 7). Furthermore, reducing the allyl bromide quantity to 1.1 equivalents resulted in a 10% reduction in NMR yield (Table S1, entry 8). The reaction proved less efficient when grinding auxiliaries, such as sand or magnesium sulfate, were introduced (Table S1, entries 9 and 10). Across most experiments, the intermediate allyl alcohol (4) was detectable. To mitigate the persistence of intermediate 4—challenging to separate from the cyclopropane product (5)—a one-pot, two-step protocol was devised. The initial Barbier allylation proceeded for 1 hour, after which diiodomethane (3) was added, and milling continued for an additional 2 hours. This sequence yielded product 5 at 68% NMR yield (64% isolated), with no residual alkene detected (Table S1, entry 11), thus establishing it as the optimised condition. Considering prior reports indicating that zinc activation is largely independent of its physical form,6,11 we evaluated five additional zinc sources under the optimised conditions. Granular (20–30 mesh), flake, foil, wire, and shot forms all demonstrated comparable efficacy to zinc dust. For instance, zinc foil afforded a 58% NMR yield of product 5.
Next, under the optimised conditions (Table S1, entry 11), the scope and limitations of the one-pot Barbier/Simmons–Smith process were evaluated (Scheme 2).12 Replacing diiodomethane with bromo-iodomethane resulted in a diminished yield, although the reaction still proceeded. The protocol demonstrated efficacy across a diverse range of cyclic ketones, with substrates comprising four- to eight-membered rings yielding the corresponding cyclopropanes (7–11) in moderate to good yields. Notably, 2-indanone was a competent substrate, furnishing compound 6 with a 58% yield. Tetrahydropyranones also participated smoothly, affording products 12 and 13.
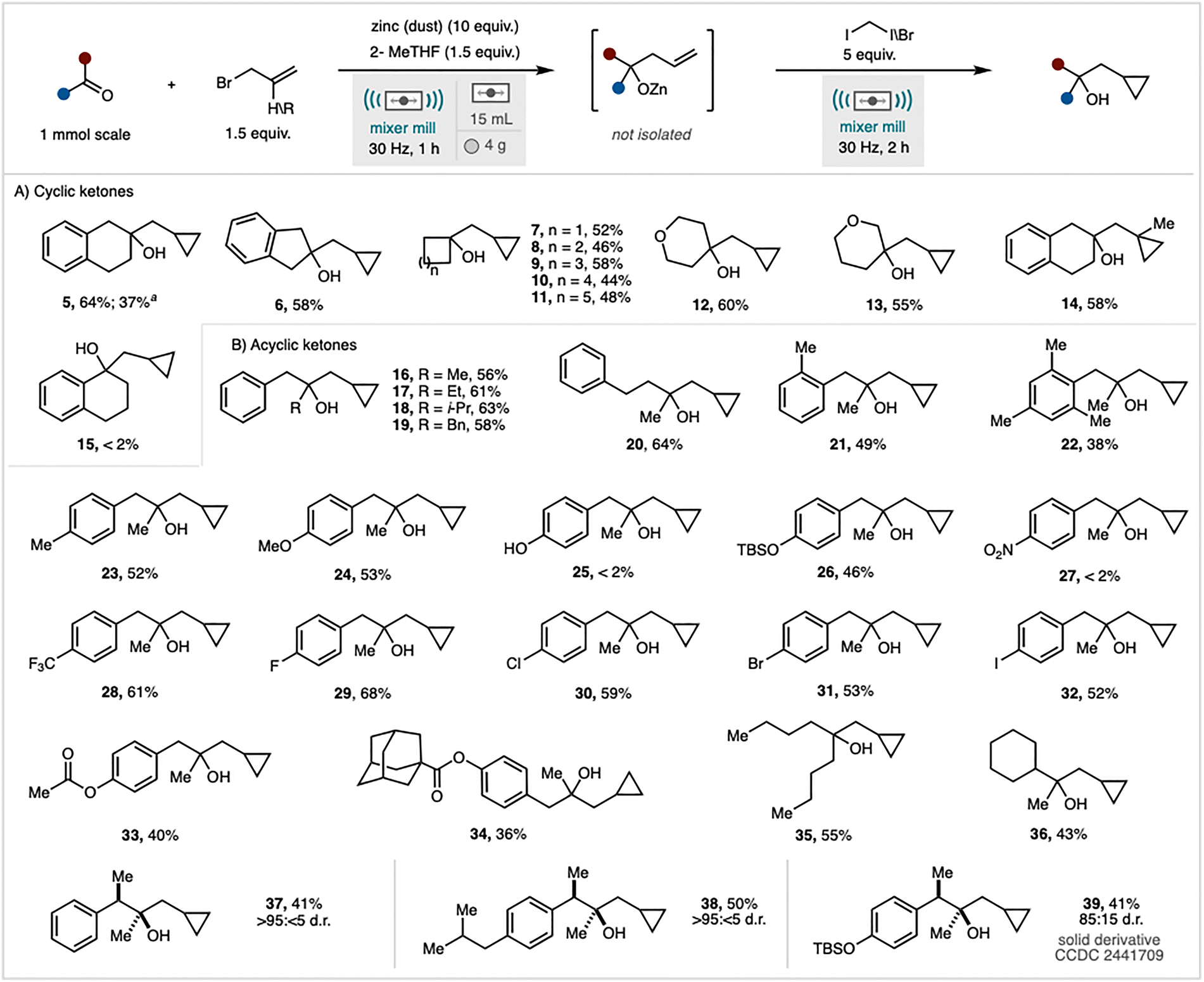 | ||
Scheme 2 Scope and limitations of the sequential process. Reactions performed using 1.00 mmol of starting material. All yields are isolated yields after chromatographic purification. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction performed using bromo-iodomethane instead of diiodomethane. Compound 39 was deprotected, derivatised to a solid ester and a combination of X-ray analysis and NMR spectroscopy used to establish diastreoselectivity. Scope and limitations of the mechanochemical Barbier/Simmons–Smith reaction. (A) Cyclic ketones. (B) Acyclic ketones. Reaction performed using bromo-iodomethane instead of diiodomethane. Compound 39 was deprotected, derivatised to a solid ester and a combination of X-ray analysis and NMR spectroscopy used to establish diastreoselectivity. Scope and limitations of the mechanochemical Barbier/Simmons–Smith reaction. (A) Cyclic ketones. (B) Acyclic ketones. | ||
Under the standard conditions, phenylacetaldehyde afforded low conversion (28% NMR yield), while representative imines, esters, and acids were unreactive (see SI). Alternative allyl sources were investigated. Allyltrimethylammonium chloride, a solid-state reagent, proved ineffective under these conditions, with 93% of the starting material recovered.13 Conversely, employing 3-bromo-2-methylpropene facilitated the formation of product 14 in 58% yield. The reaction did not proceed with conjugated ketones, such as 1-tetralone, indicating incompatibility with enone-like systems under the current conditions. To assess whether this limitation was due to product instability—potentially from benzylic carbocation formation in the presence of a Lewis acid—1-phenylethanol was subjected to the standard reaction protocol. Recovery of only 23% of the starting material indicated the instability of benzylic alcohols under milling conditions. In contrast, compound 5, derived from a non-conjugated ketone, remained unaffected upon re-exposure to the reaction conditions, confirming its stability (see SI). Acyclic ketone substrates with increased steric hindrance or extended side chains (16–22) were well tolerated, showing no significant reduction in efficiency, except for the mesityl product (22), which yielded 38%. The reaction demonstrated broad functional group compatibility. Aryl ketones featuring electron-donating groups (4-OMe, 4-OTBS), electron-withdrawing groups (4-CF3), and halogen substituents efficiently furnished the desired cyclopropanes (23, 24, 26, 28–32) in good to high yields. Conversely, the reaction failed to proceed with aromatic substrates bearing 4-OH (25) or 4-NO2 (27) substituents. The selectivity of the method towards ketones was thoroughly evaluated in the presence of other carbonyl functional groups, including methyl esters and adamantane esters. The results demonstrated high selectivity, with the reaction exclusively targeting the ketone functionalities to yield the corresponding cyclopropane products (33, 34). Furthermore, two aliphatic ketones were subjected to the optimised reaction conditions, both of which reacted efficiently to afford the desired cyclopropane products (35, 36) in good yields.
A direct comparison of stirred neat and solution-phase conditions (2-Me-THF solvent, Scheme 3A) were run using the tetralone model substrate and keeping stoichiometric ratios and reaction times (3 hours) consistent between methods. These control reactions showed no product at room temperature in either a neat reaction mixture or with 2-MeTHF solution and only modest conversion when the latter was heated to reflux (31%). Under identical stoichiometry and reaction time, the mechanochemical protocol delivered a significantly higher yield of 68% highlighting a benefit of ball milling over conventional stirring methods.14 Most notably, the use of a substrate bearing a stereocentre revealed a significant enhancement in diastereoselectivity under mechanochemical conditions.
 | ||
| Scheme 3 Further investigations of the mechanochemical Barbier/Simmons–Smith reaction. (A) Comparison of mechanochemical, neat, and solution-phase conditions. (B) Effect of solvent equivalency on diastereoselectivity under mechanochemical and solution-phase conditions. (C) Reaction scale-up under ball-milling conditions. aAs determined by 1H NMR analysis of the crude reaction mixture. | ||
The analogous solution-phase reactions consistently gave d.r. values of ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, whereas the ball-milling protocol delivered cyclopropanes 37 and 38 as single diastereomers. Cyclopropane 39, obtained as an oil, was deprotected and derivatised to a crystalline ester and its structure confirmed by X-ray analysis (CCDC 2441709, see SI), establishing the anti relationship of the methyl groups. To explore the relationship between solvent loading and stereocontrol, cyclopropane 38 was prepared using 1.5, 6, and 12 equivalents of 2-MeTHF under ball-milling conditions, and 20 and 40 equivalents under reflux. A clear trend emerges whereby increasing solvent content progressively reduced diastereoselectivity (Scheme 3B). The mechanistic basis of this behaviour is not fully established. The observed anti-product is consistent with a Felkin–Anh-type transition state.15 This is potentially facilitated by coordination/chelation of the allyl zinc to the carbonyl electrophile. This latter chelation clearly improves as the solvent loading is lowered since competition for zinc coordination is reduced. The mechanistic rationale as to why such chelation leads to enhanced discrimination between syn and anti addition is not yet understood. It is also noted that in order to elicit any reaction from the solvent based process, the reaction needs to be heated, where upon discrimination between diastereo-transistion states will be poorer. To assess the scalability of the process, the reaction was scaled from 1 mmol to 5 mmol by increasing the grinding ball size from 4 g to 8 g. Under these conditions, 1.21 g of the cyclopropane product was obtained after 3 hours, with no significant reduction in isolated yield compared to the optimised conditions, despite the change in ball size/mass (Scheme 3C). Mechanistically, each stage follows the established solution-phase organozinc pathways: in situ formation of allylzinc species during the Barbier step and ICH2ZnI-mediated cyclopropanation in the Simmons–Smith step. Because the two transformations were developed sequentially and behave analogously to their solution counterparts, we have no evidence to suggest any mechanistic deviation under milling. The novelty therefore lies in the ability to perform both steps in a single vessel, enabled by zinc activation through attrition as an operational advantage of the mechanochemical environment.
:25, whereas the ball-milling protocol delivered cyclopropanes 37 and 38 as single diastereomers. Cyclopropane 39, obtained as an oil, was deprotected and derivatised to a crystalline ester and its structure confirmed by X-ray analysis (CCDC 2441709, see SI), establishing the anti relationship of the methyl groups. To explore the relationship between solvent loading and stereocontrol, cyclopropane 38 was prepared using 1.5, 6, and 12 equivalents of 2-MeTHF under ball-milling conditions, and 20 and 40 equivalents under reflux. A clear trend emerges whereby increasing solvent content progressively reduced diastereoselectivity (Scheme 3B). The mechanistic basis of this behaviour is not fully established. The observed anti-product is consistent with a Felkin–Anh-type transition state.15 This is potentially facilitated by coordination/chelation of the allyl zinc to the carbonyl electrophile. This latter chelation clearly improves as the solvent loading is lowered since competition for zinc coordination is reduced. The mechanistic rationale as to why such chelation leads to enhanced discrimination between syn and anti addition is not yet understood. It is also noted that in order to elicit any reaction from the solvent based process, the reaction needs to be heated, where upon discrimination between diastereo-transistion states will be poorer. To assess the scalability of the process, the reaction was scaled from 1 mmol to 5 mmol by increasing the grinding ball size from 4 g to 8 g. Under these conditions, 1.21 g of the cyclopropane product was obtained after 3 hours, with no significant reduction in isolated yield compared to the optimised conditions, despite the change in ball size/mass (Scheme 3C). Mechanistically, each stage follows the established solution-phase organozinc pathways: in situ formation of allylzinc species during the Barbier step and ICH2ZnI-mediated cyclopropanation in the Simmons–Smith step. Because the two transformations were developed sequentially and behave analogously to their solution counterparts, we have no evidence to suggest any mechanistic deviation under milling. The novelty therefore lies in the ability to perform both steps in a single vessel, enabled by zinc activation through attrition as an operational advantage of the mechanochemical environment.
In conclusion, we have developed a mechanochemical one-pot Barbier/Simmons–Smith reaction. By employing organozinc generated in situ through milling, we established a system that facilitates the allylation of diverse ketones, followed by cyclopropanation, irrespective of the physical form of the zinc metal. The reaction exhibited a broad substrate scope and was successfully scaled up to 5 mmol. The enhanced diastereoselectivity observed under minimal-solvent milling conditions further underscores how solid-state reactivity can modulate selectivity in ways that are difficult to access in solution. These results highlight the potential of mechanochemistry to streamline tandem organometallic processes, opening opportunities for further multi-step solvent-minimised sequences.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). The data supporting the findings of this study, including raw NMR, IR, and mass spectrometry datasets are openly available from the Cardiff University data repository at https://doi.org/10.17035/cardiff.30287158. See DOI: https://doi.org/10.1039/d5cc05732a.CCDC 2441709 contain the supplementary crystallographic data for this paper.16
Acknowledgements
We gratefully acknowledge the School of Chemistry at Cardiff University, the Department of Chemistry at the University of Bath and the School of Pharmacy at University College London for generous support. We thank the Saudi Arabia cultural mission in the UK and the Department of Chemistry at the University of Jeddah (A.A.) for PhD studentship. We also thank Dr Matthew Tredwell for his supervision and support.References
- (a) U. Tilstam and H. Weinmann, Org. Process Res. Dev., 2002, 6, 906–910 CrossRef CAS; (b) P. Knochel and C. Diène, C. R. Chimie, 2011, 14, 842–850 CrossRef CAS; (c) J. Kim, Y. Ko, J. Bouffard and S. Lee, Chem. Soc. Rev., 2015, 44, 2489–2507 RSC; (d) A. Dilman and V. Levin, Tetrahedron Lett., 2016, 57, 3986–3992 CrossRef CAS; (e) R. Peltzer, J. Gauss, O. Eisenstein and M. Cascella, J. Am. Chem. Soc., 2020, 142, 2984–2994 CrossRef CAS; (f) A. Kremsmair, J. Harenberg, K. Schwärzer, S. Schwärzer, A. Hess and P. Knochel, Chem. Sci., 2021, 12, 6011–6019 RSC.
- (a) J. Lee, R. Velarde-Ortiz, A. Guijarro, R. Wurst and R. Rieke, J. Org. Chem., 2000, 65, 5428–5430 CrossRef CAS; (b) T. Rathman and J. Schwindeman, Org. Process Res. Dev., 2014, 18, 1192–1210 CrossRef CAS.
- (a) S. Wada, N. Hayashi and H. Suzuki, Org. Biomol. Chem., 2003, 1, 2160–2163 RSC; (b) Q. Cao, R. Stark, I. Fallis and D. L. Browne, ChemSusChem, 2019, 12, 2554–2557 CrossRef CAS PubMed; (c) J. Yin, R. Stark, I. Fallis and D. L. Browne, J. Org. Chem., 2020, 85, 2347–2354 CrossRef CAS PubMed; (d) H. Chen, J. Fan, Y. Fu, C.-L. Do-Thanh, X. Suo, T. Wang, I. Popovs, D. Jiang, Y. Yuan, Z. Yang and S. Dai, Adv. Mater., 2021, 33, 2008685 CrossRef CAS; (e) A. C. Jones, J. A. Leitch, S. E. Raby-Buck and D. L. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef CAS; (f) R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., Int. Ed., 2022, 61, e202210508 CrossRef CAS; (g) L. Pontini, J. Leitch and D. L. Browne, Green Chem., 2023, 25, 4319–4325 Search PubMed; (h) J. V. Nallaparaju, T. Nikonovich, T. Jarg, D. Merzhyievskyi, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2023, 62, e202305775 Search PubMed; (i) J. F. Reynes, F. Leon and F. García, ACS Org. Inorg. Au., 2024, 4, 432–470 CrossRef CAS PubMed; (j) K. Kubota, S. Kawamura, J. Jiang, S. Maeda and H. Ito, Chem. Sci., 2024, 15, 17453–17459 RSC.
- For selected reviews, see: (a) G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (b) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 Search PubMed; (c) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (d) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS; (e) R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 Search PubMed; (f) M. T. J. Williams, L. C. Morrill and D. L. Browne, ChemSusChem, 2022, 15, e202102157 Search PubMed; (g) F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 Search PubMed; (h) A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 Search PubMed; (i) S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC; (j) N. Fantozzi, J. N. Volle, A. Porcheddu and D. Virieux, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC; (k) S. Harder and J. Langer, Nat. Rev. Chem., 2023, 7, 843–853 Search PubMed; (l) T. Auvray and T. Friščić, Molecules, 2023, 28, 897 CrossRef CAS; (m) I. R. Speight, K. J. Ardila-Fierro, J. G. Hernández, F. Emmerling, A. A. L. Michalchuk, F. García, E. Colacino and J. Mack, Nat. Rev. Methods Primers, 2025, 5, 29 Search PubMed.
- (a) H. W. Liu, H. Xu, G. Shao and G. W. Wang, Org. Lett., 2019, 21, 2625–2628 Search PubMed; (b) I. R. Speight and T. P. Hanusa, Molecules, 2020, 25, 570 CrossRef CAS; (c) W. Nicholson, J. Howard, G. Magri, A. Seastram, A. Khan, R. Bolt, L. C. Morrill, E. Richards and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 23128–23133 CrossRef CAS; (d) P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 Search PubMed; (e) S. Chen, C. Fan, Z. Xu, M. Pei, J. Wang, J. Zhang, Y. Zhang, J. Li, J. Lu, C. Peng and X. Wei, Nat Commun., 2024, 15, 769 CrossRef; (f) C. Wu, J. Lv, H. Fan, W. Su, X. Cai and J. Yu, Chem. Eur. J., 2024, 30, e202304231 CrossRef; (g) P. Gao, J. Jiang, Y. Fukuzawa, S. Maeda, K. Kubota and H. Ito, RSC Mechanochem., 2024, 1, 486–491 RSC; (h) J. V. Nallaparaju, R. Satsi, D. Merzhyievskyi, T. Jarg, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2024, 63, e202319449 CrossRef PubMed; (i) K. Kondo, K. Kubota and H. Ito, Chem. Sci., 2024, 15, 4452–4457 RSC; (j) K. Kondo, K. Kubota and H. Ito, Nat. Synth., 2025, 4, 744–753 CrossRef; (k) T. Čarný, D. Mravcová, B. Steinhüblová and R. Šebesta, Adv. Synth. Catal., 2025, 367, e202401403 Search PubMed; (l) W. Xihong, F. Yamato, P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, RSC Mechanochem., 2025, 2, 256–262 RSC; (m) N. Biedermann and M. Schnürch, Chem. Eur. J., 2025, 31, e202500798l CrossRef PubMed; (n) J. V. Nallaparaju, R. Satsi, D. Merzhyievskyi, T. Jarg, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2024, 63, e202319449 CrossRef PubMed.
- Q. Cao, J. Howard, E. Wheatley and D. L. Browne, Angew. Chem., Int. Ed., 2018, 57, 11339–11343 CrossRef PubMed.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- R. Takahashi, P. Gao, K. Kubota and H. Ito, Chem. Sci., 2023, 14, 499–505 RSC.
- J. L. Howard, W. Nicholson, Y. Sagatov and D. L. Browne, Beilstein J. Org. Chem., 2017, 13, 1950–1956 CrossRef PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef.
- (a) A. C. Jones, W. I. Nicholson, J. A. Leitch and D. L. Browne, Org. Lett., 2021, 23(16), 6337–6341 CrossRef PubMed; (b) M. T. J. Williams, L. C. Morrill and D. L. Browne, Adv. Synth. Catal., 2023, 365, 1477 CrossRef; (c) A. C. Jones, M. T. J. Williams, L. C. Morrill and D. L. Browne, ACS Catal., 2022, 12, 13681–13689 CrossRef PubMed.
- See the SI for full experimental details.
- J. Templ and M. Schnürch, Angew. Chem., Int. Ed., 2024, 63, e202314637 CrossRef.
- (a) Q. Xu, X. Sheng, N. Li, J. Zhang, H. Shi, M. Niu, Q. Ping and N. Li, ACS Sustainable Chem. Eng., 2021, 9, 8232–8237 CrossRef; (b) D. Breilly, S. Fadlallah, V. Froidevaux, F. Lamaty, F. Allais and T. X. Métro, Green Chem., 2022, 24, 7874–7882 RSC.
- (a) C. H. Heathcock, S. Kiyooka and T. A. Blumenkopf, J. Org. Chem., 1984, 49, 4214–4223 CrossRef; (b) C. Felix, A. Laurent and P. Mison, J. Fluor. Chem., 1995, 70, 71–82 CrossRef; (c) V. De Sio, A. Massa and A. Scettri, Org. Biomol. Chem., 2010, 8, 3055–3059 RSC.
- CCDC 2441709: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2n57kn.
Footnote |
| † We dedicate this work to Professor Steven V. Ley FRS on his 80th birthday, with admiration for his pioneering work and his long-standing inspiration to our community. |
| This journal is © The Royal Society of Chemistry 2026 |