
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic water oxidation with a tris-chelating bipyridine/bis-pyrimidinethiolate Co(III) complex covalently immobilized onto MoS2 nanosheets
Michail P.
Minadakis
a,
Dimitra K.
Gioftsidou
b,
Panagiotis A.
Angaridis
 b,
Verónica
Pérez-Luna
c,
Mildred
Quintana
c,
Ruben
Canton-Vitoria
de and
Nikos
Tagmatarchis
*a
b,
Verónica
Pérez-Luna
c,
Mildred
Quintana
c,
Ruben
Canton-Vitoria
de and
Nikos
Tagmatarchis
*a
aTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens 11635, Greece. E-mail: tagmatar@eie.gr
bLaboratory of Inorganic Chemistry, Department of Chemistry, Aristotle University of Thessaloniki, Thessaloniki 54124, Greece
cHigh Resolution Microscopy-CICSaB and Faculty of Science, Universidad Autonóma de San Luis Potosi, Av. Sierra Leona 550, Lomas de San Luis Potosi, 78210 SLP, Mexico
dDepartment of Materials Chemistry & Institute of Materials and Systems for Sustainability (IMaSS), Nagoya University, Nagoya 464-8601, Japan
eJoining and Welding Research Institute, The University of Osaka, Osaka 567-0047, Japan
First published on 3rd December 2025
Abstract
The development of an oxygen evolution reaction (OER) electrocatalyst based on 2H-MoS2 nanosheets bearing covalently linked octahedral tris-chelating bipyridine/bis-pyrimidinethiolate Co(III) complexes as catalytic centers is presented. The nanohybrid material exhibits superior OER electroactivity and facilitates electron flow, compared to 2H-MoS2, which is attributed to the dispersed, surface-located molecular Co(III) catalytic centers and supported by a catalytic cycle for water oxidation that proceeds through a single-site water nucleophilic attack mechanism, operating within the Co(III)/Co(IV) redox manifold.
The oxygen evolution reaction (OER) is a key half-reaction in energy devices such as water electrolyzers, metal–air batteries and reversible fuel cells. As a multielectron process, the OER is both thermodynamically and kinetically unfavorable due to complex O–O bond formation and mass transfer limitations.1 Its harsh oxidative and acidic/alkaline conditions demand highly durable electrocatalysts.2 State-of-the-art Ru- and Ir-based electrocatalysts offer high activity but are economically unsustainable because of scarce noble metal content and face dissolution and surface reconstruction issues, thus suffering from long-term degradation.3,4 In contrast, non-noble metal catalysts (Mn, Co, Ni) offer tunable molecular structures and improved stability. Moreover, single-atom electrocatalysts, where activity arises from isolated metal sites, exhibit atom economy.5 Also, understanding metal-reaction mechanisms further enables rational ligand and oxidation state design for optimized catalytic systems.
Layered transition metal dichalcogenides (TMDs) are widely studied as water-electrocatalysis substrates, especially for the hydrogen evolution reaction (HER), with MoS2 being the most prominent example.6,7 Although MoS2 shows minimal activity toward O-based electrocatalysis,8 and thus has limited use in the OER,9 it is often overlooked that it retains activity and conductivity, even when locally oxidized, and that its Mo centres can still bind O species and support O-based reactions. Combined with its highly tunable properties (bandgap, defects, nanosheet dimensions) and the ease of incorporating electroactive units (metal complexes, nanoparticles) through various synthetic routes, MoS2 can yield catalytically promising systems.
MoS2-based materials have been decorated on the basal plane with pyridine moieties to non-covalently attach Co-phthalocyanine for the oxygen reduction reaction (ORR) to H2O2,10 and with imidazolium-end alkyl chains that bear Co-porphyrin units, exhibiting a dual activity in both the OER and ORR.11 Edge-located S-defect capping has been proven as a facile method to introduce electroactive units, without altering conductivity, as it has been done on 2H-WS2, where interfacing a Ni-porphyrin lipoic acid derivative led to a highly active OER electrocatalyst.12 However, such a strategy has as a downside that the inherent TMD HER activity is compromised, since S-edges, which strongly bind H atoms, are deactivated.12 Collectively, to avoid the easily affected by the local environment dynamic equilibrium of a non-covalent system and the S-defect capping strategy that reduces HER activity, controlled basal plane activation through pinning electroactive moieties is preferred.
Here, we prepared a nanohybrid OER electrocatalyst by covalently immobilizing an octahedral tris-chelating bipyridine/bis-pyrimidinethiolate Co(III) complex onto exfoliated 2H-MoS2 nanosheets. Co(III) complexes offer tunable chemistry, high chemical stability, strong electrochemical activity, and good environmental compatibility, making them attractive candidates. To avoid classic acidic diazotization, we used isoamyl nitrite for aprotic diazotization, following methods typical for carbon nanomaterial modification.13 The H2N-phen-fuctionalized precursor [Co(5-NH2-phen)(pymt)2]PF61, where 5-NH2-phen is 5-amino-1,10-phenanthroline and pymt is 2-pyrimidinethiolate, was synthesized and modified using conditions adapted directly from carbon nanomaterials diazotization.14 Co(III) was selected over the more labile Co(II) for its greater stability and easier access to higher oxidation states.15 Multidentate Co(III) complexes are known OER catalysts due to their stabilized high-valence Co centers.16,17 In complex 1, the chelating ligands enhance robustness and help stabilize Co(IV), while the hemilabile, proton-responsive pyrimidinethiolate ligands18 may facilitate proton-coupled electron transfer (PCET), a key step in efficient OER.19,20
Complex 1 was obtained by reacting CoCl2·6H2O with pymtH/MeONa in MeOH, then adding 5-NH2-phen and NH4PF6 to give dark green crystals (SI, Fig. S1). FT-IR confirmed the expected ligand and PF6− bands (Fig. S2), and HR-ESI+ MS verified the cation [Co(5-NH2-phen) (pymt)2]+ (Fig. S3). The 1H-NMR spectrum in CH3CN showed the anticipated diamagnetic Co(III) ligand resonances and remained unchanged over time, indicating solution stability. Its UV-Vis spectrum in DMSO (Fig. S4) displayed a strong π–π* band at 325 nm, weaker charge transfer bands at 350–500 nm, and faint d–d transitions at 600–700 nm. Cyclic voltammetry in CH3CN (Fig. S5 and Table S1) revealed an irreversible Co(III)/Co(IV) oxidation at ca. +1.0 V (vs. Fc+/Fc), a reversible Co(III)/Co(II) reduction at ca. −0.7 V, and an irreversible Co(II)/Co(I) reduction at ca. −1.8 V. Incremental addition of water converted the single-electron oxidation into a growing anodic wave, indicating the complex's ability to catalyze water oxidation (Fig. S6).
2H-MoS2 nanosheets were prepared through standardized liquid phase exfoliation in the presence of chlorosulfonic acid.21 Apart from the drastic increase in dispersibility, the exfoliated nature of 2H-MoS2 is further spectroscopically corroborated. In the UV-Vis spectrum of 2H-MoS2, excitonic bands A and B, located at 681 and 622 nm, respectively, and direct transitions from the valence to the conduction band C and D, located at 448 and 413 nm, respectively, were observed (Fig. S7a) to be more pronounced compared to the bulk material, pointing to a few-layered material.22 Raman spectroscopy (Fig. S7b) also supports the nature of the exfoliated material, since an increase in the intensity of the 2LA(M) mode, reflecting the number of lattice defects (S vacant sites), is observed. Furthermore, the widening of the distance between the E12g and A1g modes by 2–3 cm−1 is characteristic.23,24
Nanohybrid 2 was prepared by reacting the Co(III) complex 1 with exfoliated 2H-MoS2 (Fig. 1a) and fully characterized by spectroscopic, microscopic and thermal methods. UV-Vis spectra showed no major changes or shifts in the 2H-MoS2 excitonic peaks (Fig. S8a), likely because 2H-MoS2 strong absorption masks the Co(III) signals. ATR-IR revealed no distinct new bands except in the 990–1240 cm−1 region, where multi-signal features likely arise from the complex (Fig. S8b), and a weak 723 cm−1 peak consistent with S–C bond formation, confirming covalent attachment, as it has been reported in past works.25 TGA under N2 for 2 indicated a total 7.53% weight loss in the temperature range of 100–600 °C attributable to the Co(III) complex (Fig. 1b), corresponding to roughly one complex per ∼41 MoS2 units and 0.73% Co per mg of material.26 Raman spectroscopy further confirmed the covalent functionalization in 2: the 2LA(M) mode increased (Fig. 1c), in spectra normalized at A1g, and Raman mapping showed a consistent decrease in the A1g/2LA(M) ratio (Fig. 1d and e), across the surface. No peak shifts were observed, indicating that Co(III) conjugation does not significantly alter the MoS2 electronic environment in 2.
 | ||
| Fig. 1 (a) Illustrative preparation of 2. (b) TGA graphs (solid lines) and differential thermograms (dashed lines) for 2H-MoS2 (black), 1 (olive) and 2 (red) under a N2 atmosphere. (c) Raman spectra (633 nm) for 2H-MoS2 (black) and 2 (red), normalized at A1g (402 cm−1), and, (d) and (e) Raman spectral maps of the 30 × 30 µm region for 2H-MoS2 (left panel) and 2 (right panel) depicting the A1g/2LA(M) ratio. | ||
TEM and EDS analyses of nanohybrid 2 confirmed successful functionalization. TEM images (Fig. 2a) show few-layer MoS2 sheets with a transversal size of around 600 nm. HR-TEM reveals clear layered edges (Fig. 2b) and a 0.27 nm lattice spacing (Fig. 2c) consistent with the (100) plane.27 EDS mapping (Fig. 2d) detects Co and N, evidence of the attached Co(III) complex, with their distribution shown in Fig. 2e and f. Mo and S maps (Fig. 2g and h) verify the structural integrity of the MoS2 substrate.
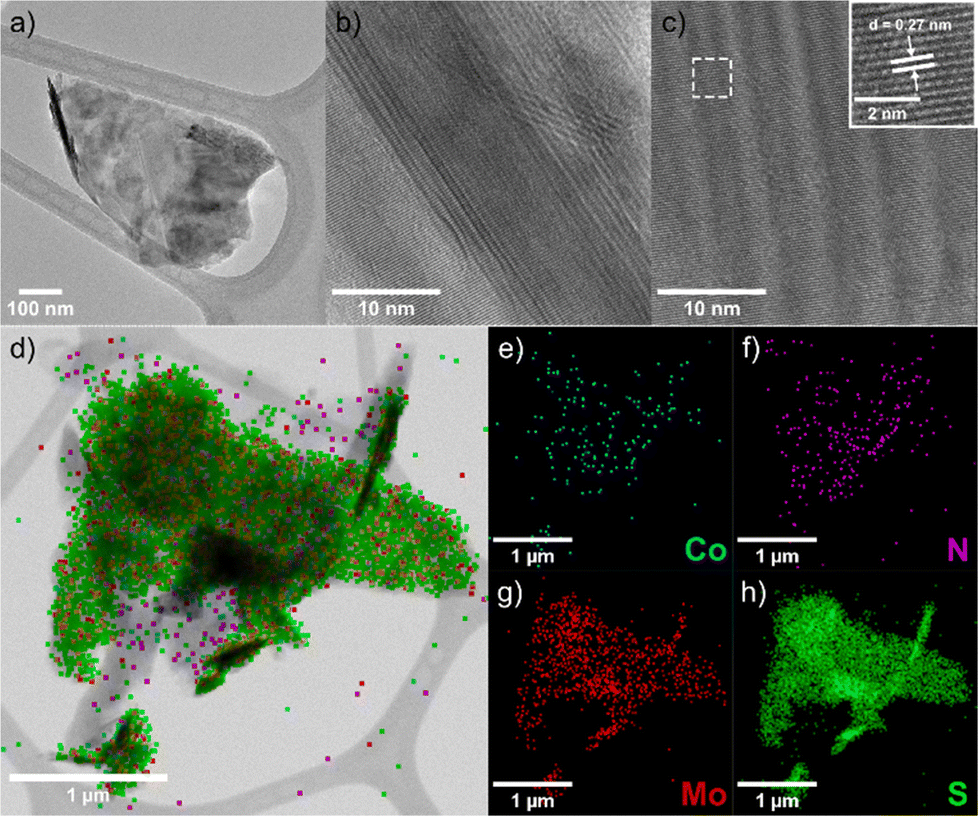 | ||
| Fig. 2 (a)–(c) TEM images, (d) EDS map, and (e)–(h) EDS elemental analysis for Co, N, Mo, and S, on a modified MoS2 flake of 2. | ||
Next, the S 2p XP spectrum displays the characteristic 2p1/2 and 2p3/2 doublet of crystalline MoS2 at 163.71 and 162.55 eV, together with additional  and
and 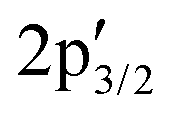 components at 163.43 and 162.54 eV associated with intrinsic defects (Fig. 3a).28 The Mo 3d region shows the expected 3d5/2 and 3d3/2 peaks at 229.73 and 232.91 eV, with only negligible Mo6+ contributions at 232.27 and 236.16 eV (Fig. 3b). In material 2, defect-related S
components at 163.43 and 162.54 eV associated with intrinsic defects (Fig. 3a).28 The Mo 3d region shows the expected 3d5/2 and 3d3/2 peaks at 229.73 and 232.91 eV, with only negligible Mo6+ contributions at 232.27 and 236.16 eV (Fig. 3b). In material 2, defect-related S  and
and 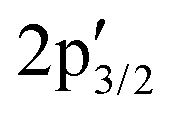 signals increase ∼20-fold vs. pristine MoS2, indicating higher defect density from the covalent attachment of complex 1 on MoS2, while Mo6+ remains unchanged, ruling out oxidation (Fig. 3c and d). The C 1s and N 1s regions overlap but can be reasonably assigned (Fig. 3e): C 1s shows a C
signals increase ∼20-fold vs. pristine MoS2, indicating higher defect density from the covalent attachment of complex 1 on MoS2, while Mo6+ remains unchanged, ruling out oxidation (Fig. 3c and d). The C 1s and N 1s regions overlap but can be reasonably assigned (Fig. 3e): C 1s shows a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C component (284.75 eV), Co-coordinated aromatic N (286.02 eV), non-coordinated phenanthroline N (287.80 eV), and metal–S–C/C–S–Mo bonds (289.26 eV); N 1s shows aromatic and Co-bound N at 398.69 and 400.31 eV, with no –NH2 peak (Fig. 3e). Co 2p1/2 and 2p3/2 peaks at 797.07 and 781.82 eV indicate a Mo
C component (284.75 eV), Co-coordinated aromatic N (286.02 eV), non-coordinated phenanthroline N (287.80 eV), and metal–S–C/C–S–Mo bonds (289.26 eV); N 1s shows aromatic and Co-bound N at 398.69 and 400.31 eV, with no –NH2 peak (Fig. 3e). Co 2p1/2 and 2p3/2 peaks at 797.07 and 781.82 eV indicate a Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co ratio of 1.25%, slightly higher than that extracted from TGA due to signal attenuation but broadly consistent (Fig. 3f).
:Co ratio of 1.25%, slightly higher than that extracted from TGA due to signal attenuation but broadly consistent (Fig. 3f).
 | ||
| Fig. 3 XP spectra of (a) S 2p and (b) Mo 3d for exfoliated 2H-MoS2, and (c) S 2p, (d) Mo 3d, (e) C 1s (right) and N 1s (left), and (f) Co 2p for material 2. | ||
Nanohybrid 2 was evaluated for the OER using advanced electrochemical methods. Initially, linear sweep voltammograms (LSVs) were recorded in N2 saturated 0.1 M aqueous KOH electrolyte and presented with the potential compensated for solution resistance (Fig. 4a). A drastic increase in recorded current was observed, verifying that Co(III) moieties attached to MoS2 have a strong impact on the performance of 2, therefore validating the original proof-of-concept. The overpotential at 10 mA cm−1 (η10) for 2 is 1.79 V, only 90 mV higher than RuO2 (1.7 V). In terms of kinetics, Tafel analysis revealed two regions: 61 mV dec−1 at low overpotential and 129 mV dec−1 at high, likely due to changes in active site coverage (Fig. 4b). Nanohybrid 2 excels in kinetics when compared with RuO2, which also exhibits an equivalent behavior. The charge transfer resistance (Rct) of 2, which is a metric of how easily charge travels through the electrochemical double layer region, is 298 Ω, one order of magnitude lower than pristine MoS2 (3420 Ω) and close to RuO2 (206 Ω), indicating that Co centres improve conductivity despite lattice defects (Fig. 4c). Finally, cyclic voltammetry in the non-Faradaic region gave the double layer capacitance (Cdl) and electrochemically active surface area (ECSA) (Fig. 4d). Overall, Co(III) functionalization endows 2H-MoS2 in nanohybrid 2 with efficient OER activity.
 | ||
| Fig. 4 (a) Polarization curves of 2 (red), 2H-MoS2 (black), and RuO2 (wine) before (solid) and after (dashed) 10000 cycles. (b) Tafel slopes for 2, and RuO2 (black dotted). (c) Nyquist plots (dotted) and fits (solid) for 2 (red), 2H-MoS2 (black), and RuO2 (wine) [inset: blown-up image of Nyquist semicircles of 2 and RuO2]. (d) CVs of 2 in the non-Faradaic region at various scan rates [inset: (janode−jcathode)/2 plotted vs. scan rate for Cdl extrapolation]. | ||
Since the coordination sphere of complex 1 remains intact upon immobilization, it is safe to assume that the mechanism for the oxidation of H2O by the MoS2-bound Co(III) complex moieties in 2 is expected to follow that of the molecular analogue in free form. Based on CV data and established precedents for other systems with Co-oxo intermediates, a plausible single-site water nucleophilic attack pathway within the Co(III)/Co(IV) manifold is proposed (Fig. 5) and described by the series of equations in Fig. S9. The mechanism involves coordination of a H2O molecule to the catalyst [CoIII(κ2-L)2(N^N)]+ (L = pymt and ^ = 5-NH2-phen) followed by intramolecular proton transfer to one of the proton-responsive thioamidate ligands (as κ1-LH) generating a Co(III)–OH intermediate. The initial PCET oxidizes this intermediate to the high-valent oxo species Co(IV)O. The key O–O bond-forming step occurs through nucleophilic attack of a second H2O molecule on the electrophilic Co(IV)O, affording a Co(III)–OOH intermediate and releasing a H+ to the solution. The hydroperoxo fragment is subsequently oxidized and deprotonated in a second PCET, affording a Co(IV)–OO species. The peroxo intermediate undergoes two sequential 1-electron oxidations at the electrode, producing a ligand-centered radical and then liberating O2. In this final step, the H+ that had been stored on the κ1-LH ligand is returned to solution, regenerating the catalyst. Overall, four electrons and four protons are transferred: 2H2O → O2 + 4H+ + 4e−. The key to this mechanism is the synergy between the redox active Co(III) center and proton relaying pyrimidinethiolate ligands, enabling Co(IV)O formation without Co(V) ensuring stepwise, energetically feasible and balanced electron-proton transfer. Nevertheless, we emphasize that this mechanism is supported by a literature precedent, and it is compatible with the electrochemical data and structural features of the Co(III) complex.
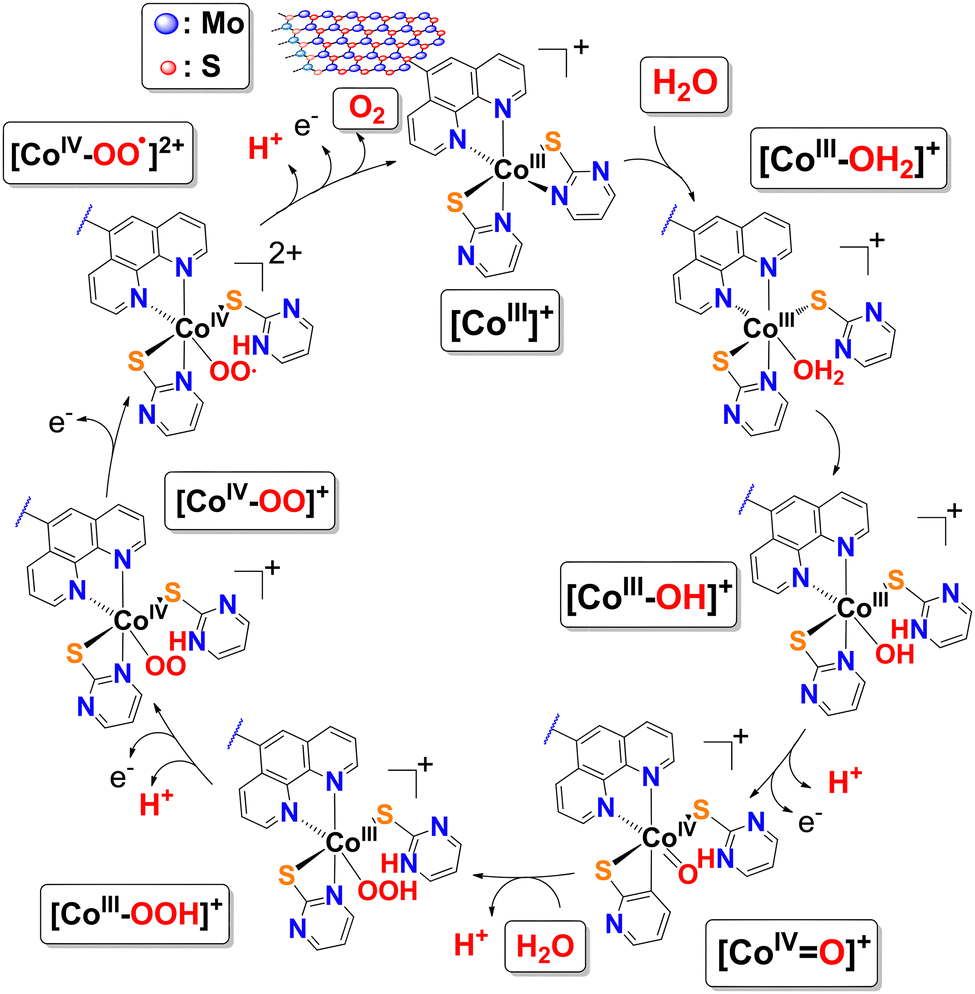 | ||
| Fig. 5 Plausible mechanism for the electrocatalytic oxidation of H2O to O2 by nanohybrid 2. | ||
We conceptualized, prepared, characterized and assessed a nanohybrid material based on semiconducting 2H-MoS2 nanosheets bearing covalently attached Co(III) complexes coordinated by a set of chelating bipyridine and pyrimidinethiolate ligands. The non-precious metal molecular moieties enable efficient water oxidation, making nanohybrid 2 superior to exfoliated MoS2 nanosheets, with a Co content as low as 0.73%. The presented synthetic route and structural innovation may inspire the development of next-generation TMD-based nanohybrid materials for effective electrocatalytic water oxidation, featuring localized non-noble metal molecular centers.
This work was carried out within the framework of the Action “Flagship Research Projects in challenging interdisciplinary sectors with practical applications in Greek Industry”, implemented through the National Recovery and Resilience Plan Greece 2.0 and funded by the European Union-NextGenerationEU (project code: TAEDR-0537347).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc05575j.References
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- K. Zhang and R. Zou, Small, 2021, 17, e2100129 CrossRef PubMed.
- H. Yu and J. Ge, Curr. Opin. Electrochem., 2023, 39, 101296 CrossRef CAS.
- C. Wang, L. Jin, H. Shong, H. Xu, Y. Shiraishi and Y. Du, Chin. Chem. Lett., 2021, 32, 2108–2116 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS PubMed.
- T. Shaker, H. Mehdipour and A. Z. Moshfegh, Int. J. Hydrogen Energy, 2022, 47, 1579–1588 CrossRef CAS.
- J. Tang, J. Huang, D. Ding, S. Zhang and X. Deng, Int. J. Hydrogen Energy, 2022, 47, 39771–39795 CrossRef CAS.
- E. German and R. Gebauer, Appl. Surf. Sci., 2020, 528, 146591 CrossRef CAS.
- M. P. Minadakis and N. Tagmatarchis, Adv. Sustainable Syst., 2023, 7, 2300193 CrossRef CAS.
- H. Meng, X. Liu, X. Chen, Y. Han, C. Zhou, Q. Jiang, T. Tan and R. Zhang, J. Energy Chem., 2022, 71, 528–538 CrossRef CAS.
- D. K. Perivoliotis, C. Stangel, Y. Sato, K. Suenaga and N. Tagmatarchis, 2D Mater., 2023, 10, 014007 CrossRef CAS.
- M. P. Minadakis, R. Canton-Vitoria, C. Stangel, E. Klontzas, R. Arenal, J. Hernandez-Ferrer, A. M. Benito, W. K. Maser and N. Tagmatarchis, ChemSusChem, 2023, 16, e202202322 CrossRef CAS PubMed.
- J. L. Bahr and J. M. Tour, Chem. Mater., 2001, 13, 3823–3824 CrossRef CAS.
- D. Saha, S. Angizi, M. Darestani-Farahani, J. Dalmieda, P. R. Selvaganapathy and P. Kruse, Langmuir, 2022, 38, 3666–3675 CrossRef CAS PubMed.
- S. Biswas, S. Bose, J. Debgupta, P. Das and A. N. Biswas, Dalton Trans., 2020, 49, 7155–7165 RSC.
- P. Lepcha, S. Biswas, S. N. Chowdhury, S. Bose, J. Debgupta, S. Paul and A. N. Biswas, Eur. J. Inorg. Chem., 2023, e202200611 CrossRef CAS.
- Y. Chen, X. Meng, X. Chen, X. Li, H. Ye, S. Liu, Z. Ruan, X. Liang and J. Lin, Sustainable Energy Fuels, 2023, 7, 242–247 RSC.
- D. K. Gioftsidou, M. G. Kallitsakis, K. Kavaratzi, A. G. Hatzidimitriou, M. A. Terzidis, I. N. Lykakis and A. A. Angaridis, Dalton Trans., 2024, 53, 1469–1481 RSC.
- H.-Y. Du, S.-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- S. Dey, T. K. Todorova, M. Fontecave and V. Mougel, Angew. Chem., Int. Ed., 2020, 59, 15726–15733 CrossRef CAS PubMed.
- G. Pagona, C. Bittencourt, R. Arenal and N. Tagmatarchis, Chem. Commun., 2015, 51, 12950–12953 RSC.
- A. K. Mishra, K. V. Lakshmi and L. Huang, Sci. Rep., 2015, 5, 15718 CrossRef CAS PubMed.
- J.-H. Fan, P. Gao, A.-M. Zhang, B.-R. Zhu, H.-L. Zeng, X. D. Cui, R. He and Q.-M. Zhang, J. Appl. Phys., 2014, 115, 053527 CrossRef.
- N. Dong, Y. li, Y. Feng, S. Zhang, X. Zhang, C. Chang, J. Fan, L. Zhang and J. Wang, Sci. Rep., 2015, 5, 14646 CrossRef CAS PubMed.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, ACS Nano, 2015, 9, 6018–6030 CrossRef CAS.
- E. Er, H.-L. Hou, A. Criado, J. Langer, M. Moller, N. Erk, L. M. Liz-Marzan and M. Prato, Chem. Mater., 2019, 31, 5725–5734 CrossRef CAS.
- M. Rajput, S. K. Malik, S. Chatterjee, A. Shukla, S. Hwang, S. Sahoo, G. V. P. Kumar and A. Rahman, Commun. Mater., 2024, 5, 190 CrossRef CAS.
- R. Canton-Vitoria, Y. Sayed-Ahmad-Baraza, M. Pelaez-Fernandez, R. Arenal, C. Bittencourt, C. P. Ewels and N. Tagmatarchis, npj 2D Mater. Appl., 2017, 1, 13 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |