
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg(I)-mediated mono-selective C(sp2)–H chalcogenation of α-aminotropones and their peptides at room temperature
Malobika
Kar
ab and
Nagendra K.
Sharma
 *ab
*ab
aSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), Bhubaneswar, 752050, Odisha, India. E-mail: nagendra@niser.ac.in; Tel: +91-674-249-4141
bHomi Bhabha National Institute (HBNI), Training School Complex, Anushaktinagar, Mumbai, 400094, India
First published on 1st December 2025
Abstract
This report describes an Ag(I)-mediated strategy for the mono-selective chalcogenation of α-aminotropones and their peptides with a range of disulfides and diselenides at ambient temperature. The method efficiently furnishes mono-chalcogenated aminotropone derivatives in moderate to good yields, while demonstrating compatibility with diverse substrates and functional groups, offering synthetic versatility in downstream applications.
Tropone and tropolone are constituents of various natural products, troponoids, possessing valuable bioactivities such as antimicrobial, antifungal, anti-protozoal, antibacterial, and anticancer.1 Structurally, tropone has a non-benzenoid aromatic scaffold comprising six conjugated π-electrons in the seven-membered ring. Its distinctive electronic structure and inherent ring strain render it a valuable scaffold in meticulous synthesis design and a topic of particular interest in medicinal chemistry.2,3 In particular, sulfur-containing tropone derivatives such as thiotropocin, tropodithietic acid (TDA), troposulfenin, and roseobacticides highlight their structural diversity (Fig. 1a).4 An unusual aromaticity and strong biological relevance make these molecules attractive precursors for deliberate synthetic design and complicated architecture development. α-Aminotropone is a synthetic analogue of tropolone possessing unique structural and functional properties, including therapeutic value.5 Notably, tropones and their analogues participate in diverse chemical reactions, including cycloaddition,6 nucleophilic substitution,7 α-alkylation,8,9 metal-catalyzed transformations10etc. Despite the growing interest in tropolone chemistry, the majority of previous studies have primarily focused on cycloaddition reactions as a method for structural modification.11 While these transformations have proven effective for generating complex frameworks from the core ring, they often lack precision in functionalizing specific positions on the ring. In contrast, site-selective C–H functionalization of the tropolone core remains significantly underexplored, specifically in the presence of a directing group.3 The development of methodologies for precise and predictable C–H bond functionalization within this non-benzenoid aromatic framework would significantly broaden the synthetic utility of tropolones and enhance their application in medicinal chemistry. In the literature, Janik and co-workers have shown the Pd(0)-catalyzed synthesis of 5-aryl tropolone from 5-iodotropolone, which is derived from tropolone in multiple steps.12 In 2022, our group reported a Pd(II)-catalyzed C7-olefination of α-aminotropones to synthesize various non-benzenoid cinnamate analogs.13 Additionally, a series of alkylaminotroponyl sulfone (ATS) derivatives have been synthesized via a Cu(II)-mediated C(sp2)–H functionalization of α-aminotropones with aryl sulfonyl hydrazides.14 A couple of ATS derivatives exhibit promising anti-quorum-sensing (anti-QS) activity against pathogenic bacteria Pseudomonas aeruginosa. Recently, Bandini and colleagues have reported an Au(I)-catalyzed direct C–H alkylation of α-substituted tropones at the C5 position using allenes as alkylating agents [Fig. 1b(i)].15 Despite the well-established biological activity of sulfur-containing frameworks,16 site-selective C–H chalcogenation of tropones remains largely unexplored. In 2014, Yamamoto and co-workers successfully synthesized a few sulfur-based tropones from 5-bromo-2-methoxytropone [Fig. 1b(ii)].7 However, synthesizing the bromotropolone requires intricate reaction design and multiple steps. This underscores the need for a mild, efficient, and selective method for the C–H chalcogenation of tropolones. Previously, we have developed a site-selective chalcogenation strategy on tryptophan-containing peptides via an Ag(I)-mediated C(sp2)–H functionalisation protocol.17 Herein, we report the mono-selective C5-chalcogenation of α-aminotropones and their peptide derivatives at room temperature, employing various dichalcogenides through Ag(I)-mediated C(sp2)–H functionalisation (Fig. 1c).
 | ||
| Fig. 1 (a) Troponyl natural products; (b) previous works-(i) Au(I)-catalyzed C–H alkylation; (ii) chalcogenation from bromotropolone; (c) this work-C5-chalcogenation of aminotropone derivatives. | ||
Initially, we synthesized various types of 2-aminotropone derivatives (1) from the commercially available Tropolone molecule by following the previous report.13 One of the simple aminotropone derivatives (1a) was treated with diaryl disulfide (2a) in the presence of various Ag(I)-salts under different solvent conditions at room temperature. (Table S1, SI). Pleasingly, we optimized the reaction conditions for the substrate (1a) with the reagent (2a), using metal salt AgCO2CF3 (1.0 equiv.) in solvent toluene at room temperature, which produced the desired sulfenyl product 3a in 76% yield (Table 1, entry 1). Increasing the amount of AgCO2CF3 (2 equiv.) failed to increase the yield. Also, decreasing the amount of AgCO2CF3 (0.5 equiv.) led to a drop in the product yield (entries 2 and 3). Furthermore, in the absence of AgCO2CF3, the formation of the product was not observed, while the use of AgOTf instead of AgCO2CF3 resulted in only a trace amount of the product while with AgOAc, reaction did not proceed (entries 4–6). Also, solvents like MeCN and xylene failed to increase the product yield (entries 7 and 8).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol), AgCO2CF3 (1 equiv.) in toluene (2 mL) at rt for 2 h under air. b Isolated yield. c n.r. = no reaction. | ||
| 1 | None | 76 |
| 2 | 2 equiv. AgCO2CF3 | 74 |
| 3 | 0.5 equiv. AgCO2CF3 | 59 |
| 4 | Without AgCO2CF3 | n.r.c |
| 5 | AgOTf instead of AgCO2CF3 | Trace |
| 6 | AgOAc instead of AgCO2CF3 | n.r.c |
| 7 | MeCN instead of toluene | 41 |
| 8 | Xylene instead of toluene | 53 |
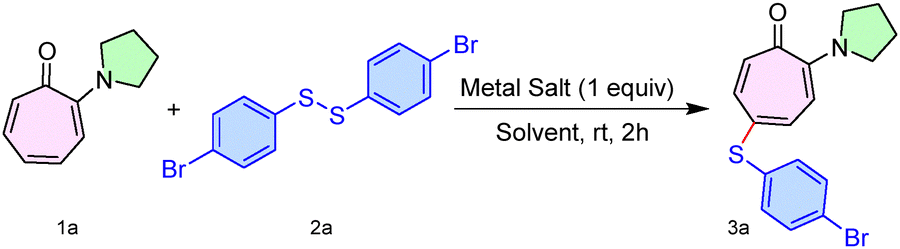
Having the optimal reaction conditions in hand, we next studied the substrate scope of the procedure using various disulfides (2b–l) with N,N-di-alkylaminotropone (1a) as a standard substrate (Scheme 1). To our delight, the simplest disulfide tested, namely diphenyl disulfide (2b), delivered the sulfenyl product (3b) in 73% yield. In contrast, aliphatic disulfides (2j–k) and cysteine-derived disulfides (2l) failed to provide a sulfenylated aminotropone under the optimized reaction conditions. Various aryl disulfides having electron-donating/withdrawing functionalities on the phenyl ring, such as 2-amino (2c), 2-chloro (2d), 3-chloro (2e), 3-fluoro (2f), 3-methoxy (2g), and 4-methyl (2h), produced the desired sulfenyl derivative 3c–h in 76–72% yield. In addition, dinapthyl disulfide (2i) furnished 3i in 75% yield. We also performed a gram-scale reaction of 1a (5.71 mmol, 1.0 g) with 2a (6.85 mmol, 2.58 g), furnishing the desired product 3a (1.32 g) in 56% yield.
 | ||
Scheme 1 Variation of the disulfide partners. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Gram scale yield. Gram scale yield. | ||
The scope of the chalcogenation procedure was next extended for various N,N-dialkylaminotropones (1b–f) with disulfide 2a (Scheme 2). Pleasingly, various N,N-dialkylaminotropones, such as piperidinyl tropone (1b), morpholinyl tropone (1c), N,N-dimethylaminotropone (1d), and N-ethyl-N-methylaminotropone (1e), were found to be effective in delivering the desired sulfenylated compounds (3m–p) in 67–78% yields. Subsequently, the troponyl amino acid derivative, proline-based aminotropone (1f), also produced the sulfenylated prolinyl tropone derivative (3q) in a better yield of 69% under the optimised reaction conditions, having the solvent system DCE/TFE (1:1, 2 mL) instead of toluene.
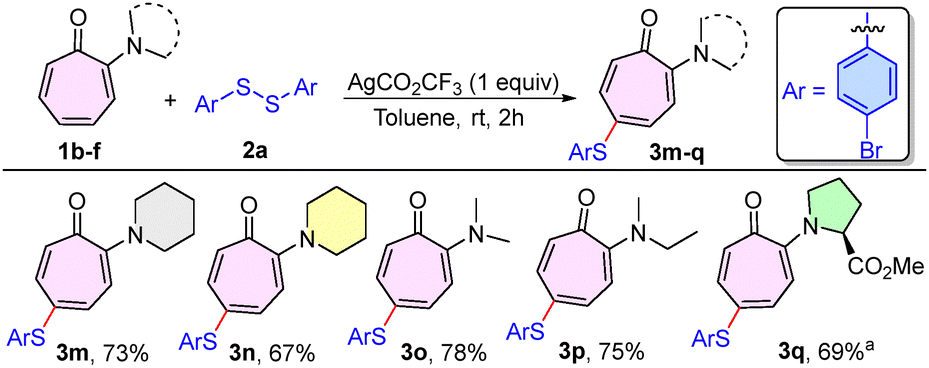 | ||
| Scheme 2 Scope of aminotropone for sulfenylation. aReaction conditions: DCE/TFE (1:1). | ||
Encouraged by the aforementioned successful outcomes, we later tested the workability of various substituted diselenides with 1a (Scheme 3). Interestingly, aryl diselenides bearing various functionalities on the aryl ring, like 2-methoxy (2m), 3-chloro (2n), 3-fluoro (2o), 3-trifluoromethyl (2p), 4-bromo (2q), 4-methoxy (2r), and 4-methyl (2s), afforded the selenylated products in good to moderate yields. Each one formed mono-substituted arylselenides (3r–x) as the major product with a good yield of 52–65%, while di-substituted products (3r′–x′) as minor ones with a yield of 12–20% under the optimized reaction conditions within 5 min. These results reveal a greater reactivity of diselenides towards N,N-dialkylaminotropone as compared to disulfides.
 | ||
| Scheme 3 Variation of diselenides. | ||
Next, the scope of the methodology was investigated for a series of N,N-di-alkylaminotropone (1b–e) with diselenide (2q) (Scheme 4). Several N,N-di-alkylaminotropones, such as piperidinyl tropone (1b), morpholinyl tropone (1c), N,N-dimethylaminotropone (1d), and N-ethyl-N-methylaminotropone (1e) produced the selenylated compounds (3) under the optimized reaction conditions within 5 min. In all the above cases, we observed the formation of mono-substituted 3y-ab as a major product with yields of 59–63%, while the di-substituted product (3y′-ab′) as a minor one with yields of 12–25%. Also, N-troponyl alkylglycinate esters (1g–h) provided 3ac–ad with 71–73% yields under the optimised reaction conditions having solvent system DCE/TFE (1:1, 2 mL) instead of toluene. Furthermore, proline-based aminotropone N-troponyl-4′-OH-proline ester 1i furnished the major mono-substituted selenylated derivative 3ae in a good yield of 57% and minor di-substituted product 3ae′ with 17% yield.
 | ||
| Scheme 4 Scope of aminotropones for selenylation. aReaction conditions: DCE/TFE (1:1). | ||
With these exciting results, late-stage selenylation of structurally more complex aminotropone derivatives such as N-troponyl dipeptides (1j–1o) was treated with diselenides (2q) under the optimised reaction conditions in solvent system DCE/TFE (1:1, 2 mL) for 10 min (Scheme 5). Various troponyl-containing dipeptides such as N-troponyl-hexyl-Gly-OMe (1j), N-troponyl- Pro-Ala-OMe (1k), N-troponyl-Pro-Val-OMe (1l), N-troponyl-Pro-Ile-OMe (1m), BocNH-troponyl-ethyl-Gly-Phe-OMe (1n), and N-troponyl-Pro-Tyr-OMe (1o) were compatible to provide the respective selenylated products (3af–3ak) in good yields of 49–56%. In the case of 1j and 1n, minor di-substituted products 3af′ and 3aj′ were obtained in 19% and 17% yields, respectively. Notably, troponyl peptides (3aj–3ak) produced the aryl selenylation only at the C5 position of the troponyl-ring, though two phenyl/phenol rings were present. Pleasingly, we also noticed that the selenylation of troponyl-containing tripeptide, N-troponyl-octyl-Phe-Phe-OMe (1p) at the C5-position of the troponyl ring afforded the target peptides 3al in a good yield of 47% using diselenide (2t), even in the presence of two phenyl rings of phenylalanine. These results strongly support that site-selective aryl selenylation occurs only at the C5-position of the tropone ring. Furthermore, we also tried the reaction conditions with tripeptides having aminotropones in the middle, Boc-Val-N-troponyl-Pro-Phe-OMe (1q) with diselenide (2t), which provided the desired product 3am in a good yield of 53%.
 | ||
| Scheme 5 Scope of N-troponyl peptide partners. | ||
To understand the reaction mechanism, we conducted radical trapping experiments in the presence of radical scavengers, TEMPO and BHT [Fig. (2a)]. The reaction of aminotropone (1a) with disulfide (2a) under the optimized conditions in the presence of TEMPO and BHT delivered the respective C5-sulfenylated peptide (3a) in a good yield of 54% and 72%. Hence, these results exclude the involvement of a single-electron-transfer (SET) process in the mono-selective chalcogenation of the troponyl ring. Previously, we have reported the chalcogenation of tryptophan at the C2-position through electrophilic substitution reactions at the positively charged S/Se centre of the disulfides/diselenides-Ag(I) complex.17 Also, the role of the Ag(I) ion is purely activating the electrophile, not to coordinate with the amine group of the aminotropones. The amine groups of aminotropones are tertiary and bulky in nature, which may not be feasible for coordination with Ag(I). In the literature, similar observations are noticed with Au(I) catalyzed alkylation at the C-5 position of the tropone ring in aminotropone derivatives.15 Herein, we propose a plausible mechanism. Initially, dichalcogenides (2) give an active electrophilic cationic intermediate (I) with Ag(I) salt (AgCO2CF3). Next, nucleophile aminotropones (1) participate in the electrophilic substitution reaction at the C5 position of tropone with electrophilic, positively charged S/Se intermediate (I), which facilitates the formation of the target chalcogenated aminotropone product (3) along with organosulfide byproduct formation [Fig. 2(b)].
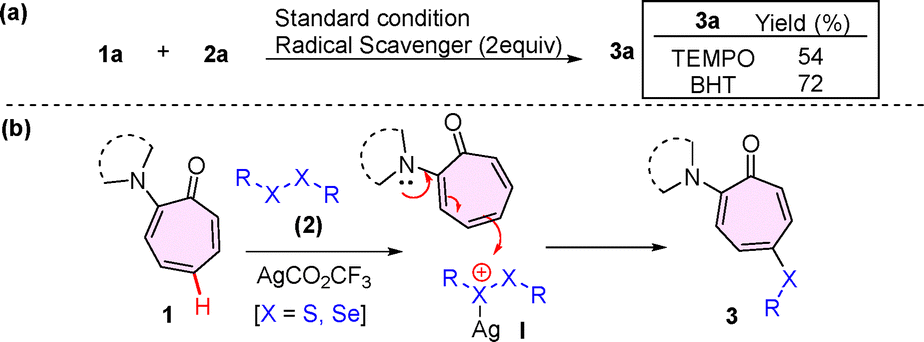 | ||
| Fig. 2 (a) Radical scavenger experiments. (b) Plausible reaction mechanism. | ||
To demonstrate the practical utility of the protocol, various post-synthetic transformations were conducted through reported procedures, such as oxidation of aryl sulfide18 with oxone, borylation of aryl bromide,19 and olefination of the tropone ring.13 For example, the oxidation of 3a with oxone afforded the desired sulfone 4 in excellent yield (Scheme 6a). Also, borylation of a bromo-substituted thiolated compound 3a using bis(pinacolato)diboron and a Pd catalyst was investigated to obtain the corresponding borylated derivative 5 in 53% yield (Scheme 6b). Moreover, Pd catalyzed C(sp2)–H olefination at the C7-position of the aminotropone derivative in the presence of ethyl acrylate starting from 3a was effectively accomplished to give the expected product 6 without affecting the sulfenyl moiety (Scheme 6c). Finally, the removal of the amino group of the sulfenyl aminotropone 3a product was successfully achieved by hydrolysis under alkaline conditions15 in 86% yield (Scheme 6d). The chalcogenated troponyl compounds (4–6) could serve as a synthon for a variety of further modifications (Scheme 6).
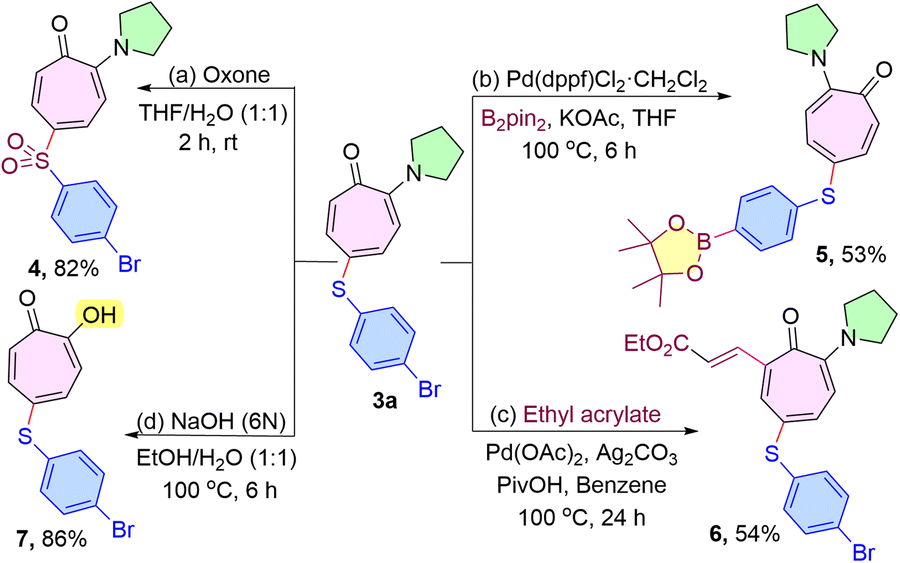 | ||
| Scheme 6 Post synthetic utilities. | ||
In conclusion, we have established an efficient synthetic protocol for site-selective silver-mediated chalcogenation of α-aminotropones with dichalcogenides (disulfides/diselenides) at ambient temperature. The method features broad substrate applicability and tolerance to diverse functionalities, and enables valuable post-synthetic transformations, thereby offering a practical route for the synthesis of chalcogenated aminotropones.
M. K. thanks NISER for the fellowship. We also thank NISER's planned project (RIN4002), the DAE-Govt. of India for the funding. We also acknowledge the SERB (ANRF)-New Delhi CRG research grant (CRG/2020/001028).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general experimental procedures, NMR data/spectra (1H, 13C, 11B, 19F) and HRMS of newly synthesized substrates (1e, 1i, 1k–m, 1o, 1q) and products (3a-am, 4, 5, 6, 7) and crystal data (3a, 3b, 3v, and 3v′). See DOI: https://doi.org/10.1039/d5cc05556c.CCDC 2491071–2491074 contain the supplementary crystallographic data for this paper.20a–d
Notes and references
- H. Guo, D. Roman and C. Beemelmanns, Nat. Prod. Rep., 2019, 36, 1137–1155 RSC.
- N. Liu, W. Song, C. M. Schienebeck, M. Zhang and W. Tang, Tetrahedron, 2014, 70, 9281–9305 CrossRef CAS PubMed.
- P. Pradhan, I. Das, S. Debnath, S. Parveen and T. Das, ChemistrySelect, 2022, 7, e202200440 CrossRef CAS.
- Y. Duan, M. Petzold, R. Saleem-Batcha and R. Teufel, ChemBioChem, 2020, 21, 2384–2407 CrossRef CAS PubMed.
- J. Nagai, M. Imamura, H. Sakagami and Y. Uesawa, Medicines, 2019, 6, 45 CrossRef CAS PubMed.
- C. L. Barløse, J. Faghtmann, M. Kaasik, R. Mastroddi and K. A. Jørgensen, Org. Lett., 2024, 26, 1539–1543 CrossRef PubMed.
- O. Sato, A. Nitta and A. Yamamoto, Heteroat. Chem., 2014, 25, 644–650 CrossRef CAS.
- Q.-X. Zeng, C.-Y. Zheng, Z.-P. Ge, J.-X. Zhao and J.-M. Yue, Chem. Sci., 2025, 16, 8836–8844 RSC.
- A. Brunetti, M. Garbini, N. Gino Kub, M. Monari, R. Pedrazzani, C. Zanardi, G. Bertuzzi and M. Bandini, Adv. Synth. Catal., 2024, 366, 1965–1971 CrossRef CAS.
- B. Seo, W. H. Jeon, C.-E. Kim, S. Kim, S. H. Kim and P. H. Lee, Adv. Synth. Catal., 2016, 358, 1078–1087 CrossRef CAS.
- S. Frankowski, M. Romaniszyn, A. Skrzyńska and Ł. Albrecht, Chem. – Eur. J., 2020, 26, 2120–2132 CrossRef CAS PubMed.
- J. Potenziano, R. Spitale and M. E. Janik, Synth. Commun., 2005, 35, 2005–2016 CrossRef CAS.
- C. K. Jena and N. K. Sharma, Chem. Commun., 2022, 58, 8077–8080 RSC.
- S. Meher, S. Kumari, M. Dixit and N. K. Sharma, Chem. – Asian J., 2022, 17, e202200866 CrossRef CAS PubMed.
- G. Gallorini, S. Kiriakidi, S. Bellini, C. S. López, G. Bertuzzi and M. Bandini, Org. Lett., 2024, 26, 9251–9256 CrossRef CAS PubMed.
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed.
- R. Bag, M. Kar and N. K. Sharma, J. Org. Chem., 2024, 89, 14981–15002 CrossRef CAS PubMed.
- R. Bag and N. K. Sharma, Org. Chem. Front., 2023, 10, 1252–1262 RSC.
- R. Bag and N. K. Sharma, J. Org. Chem., 2023, 88, 15666–15686 CrossRef CAS PubMed.
- (a) CCDC 2491071: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pm54n; (b) CCDC 2491072: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pm55p; (c) CCDC 2491073: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pm56q; (d) CCDC 2491074: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pm57r.
| This journal is © The Royal Society of Chemistry 2026 |