
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA folded mechanochromic organic fluorophore based on thianthrene-fused coumarin
Masafumi
Ueda
 *a,
Asuka
Uehara
a,
Kazuteru
Usui
b and
Masashi
Hasegawa
a
*a,
Asuka
Uehara
a,
Kazuteru
Usui
b and
Masashi
Hasegawa
a
aDepartment of Chemistry, Graduate School of Science, Kitasato University, 1-15-1 Kitazato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan. E-mail: msfmueda@kitasato-u.ac.jp
bFaculty of Pharmaceutical Sciences, Showa Pharmaceutical University, 3-3165 Higashi-tamagawagakuen, Machida, Tokyo 194-8543, Japan
First published on 10th December 2025
Abstract
The powder samples of a novel thianthrene-fused coumarin dye (6,7-BDTC) demonstrated pronounced mechanochromic luminescence, shifting from light-blue to green emission upon mechanical stimulation. X-ray crystallography of polymorphs with different emission colors revealed that this responsiveness originates from variations in the packing mode induced by the D–A type folded structure. These results demonstrate that introducing a folded π-conjugated framework into the coumarin core enables control over the packing mode and emission properties in the solid state.
Coumarin (2H-chromen-2-one) is a bioorganic aromatic lactone that has garnered considerable attention in pharmacology and physiology.1 Its versatile structure allows facile chemical modification, leading to the development of highly efficient luminescent dyes, such as donor–acceptor-type coumarin derivatives, which have a strong presence as key components in organic optical devices.2 To tune the photophysical properties and improve stability, numerous π-expanded coumarins with unique molecular skeletons have been developed. Most of these derivatives exhibit photophysical properties stemming from their rigid and planar π-conjugated structures.3 Conversely, helical π-expansion of the coumarin dyes imparts chiroptical activity,4 while curved or polycyclic π-systems incorporating coumarin often produce bright orange to red emissions in both solution and the solid state.5 Thus, the molecular geometry of π-expanded coumarins critically determines their photophysical properties, and the construction of novel π-frameworks remains essential for expanding the design diversity of coumarin-based luminophores.
Among the possible π-extension strategies, double sulfur bridging (exemplified by the thianthrene skeleton) is a powerful method to enhance conjugation through sulfur orbitals. However, the C–S–C bond introduces structural bending due to the lone pairs on sulfur, resulting in folded π-conjugated architectures. Such a folding often leads to dynamic conformational changes between the ground (S0) and the lowest excited (S1) states, where nonradiative decay dominates and emission efficiency decreases. Indeed, thianthrene itself exhibits low fluorescence efficiency (ΦF = 2%) in THF solution but shows room-temperature phosphorescence in the crystalline state with enhanced efficiency (ΦP = 25%) owing to restricted molecular motion.6 These findings highlight that molecular immobilization in the solid state can suppress non-radiative decay, a key in AIEgens7 and in the design of stimuli-responsive luminescent materials.8 In addition, the π-systems possessing stereospecific frameworks impose steric effects on molecular packing and can modulate π–π interactions, thereby influencing luminescence efficiency and solid-state color tuning.9 Control of such intermolecular arrangements—self-aggregation, hydrogen bonding, encapsulated solvents, or mechanical stress—offers a route to mechanoresponsive materials.10 However, it remains difficult to clearly understand the excited states of the aggregate, and there is little evidence to resolve the question of how applicable π-expanded coumarin dyes incorporating a folding skeleton are to these phenomena.
To address this, we newly designed a novel coumarin dye, 6,7-BDTC, by fusing a folded thianthrene core with a planar coumarin framework (Fig. 1). This structure combines π-extension with a defined folding geometry (dihedral angle: 129°), enabling the study of how molecular shape governs luminescence behavior in both dilute solutions and solid assemblies. Also, enhanced emission efficiency based on the intramolecular donor–acceptor structure of the coumarin core can be expected through electron donation from the thianthrene core.11,12 This asymmetric folding structure, in contrast to conventional planar coumarin dyes, has the potential to induce changes in the stacking pattern within molecular assemblies and control the overlap of conjugated π-planes. This paper describes the synthesis of 6,7-BDTC and its emission behavior in both single-molecule dispersions and solid states.
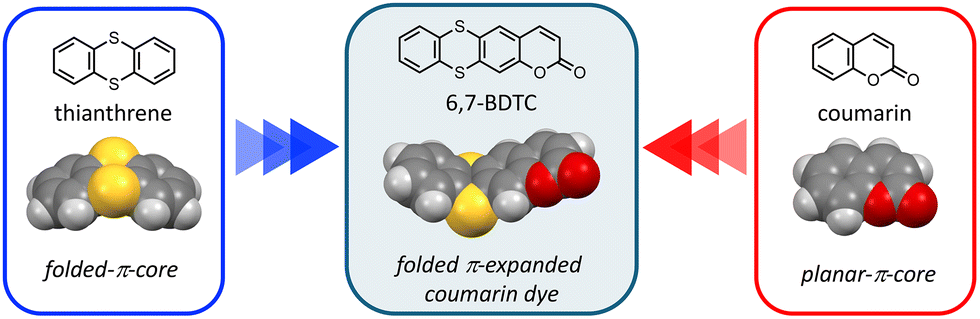 | ||
| Fig. 1 Chemical and optimized structures of 6,7-BDTC, thianthrene, and coumarin. Optimizations were estimated by the CAMB3LYP/6-31G(d,p) level. | ||
The synthetic route to 6,7-BDTC is shown in Scheme 1. 3,4-Difluoroanisole (1) was treated with TiCl4 and dichloromethyl methyl ether (Rieche formylation) to afford 4,5-difluoro-2-methoxybenzaldehyde (2). Compound 3 was prepared from the Wittig reaction of 2 with ethyl(triphenylphosphoranylidene)acetate. 6,7-Difluorocoumarin (4) was formed by intramolecular annulation of 3 in the presence of Lewis acid. Ultimately, the target compound 6,7-BDTC was obtained via aromatic nucleophilic substitution of 1,2-benzenedithiol with 4 in a good yield. Two crystalline polymorphs (crystals I and II) obtained by slow evaporation of acetone and EtOAc revealed that 6,7-BDTC folds at the dihedral angles of 132 and 126° along the S⋯S axis (Fig. S30).
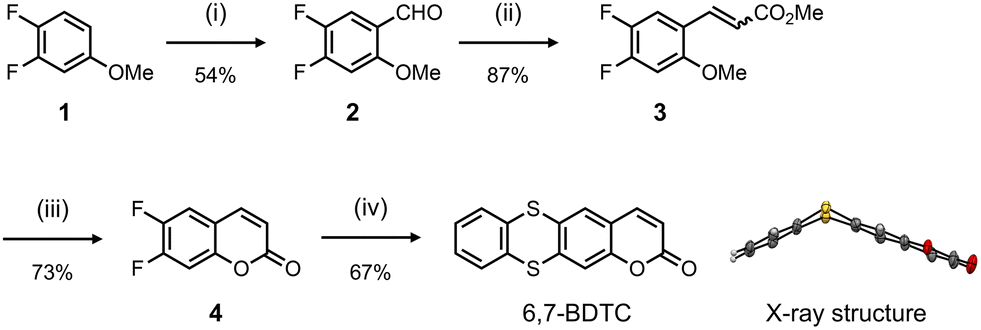 | ||
Scheme 1 Synthetic route for 6,7-BDTC: (i) TiCl4, dichloromethyl methyl ether, CH2Cl2, 0 °C → ambient temperature; (ii) ethyl(triphenylphosphoranylidene)acetate, CH2Cl2, 40 °C, (E/Z = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); (iii) BBr3, CH2Cl2, 0 → 50 °C; (iv) 1,2-benzenedithiol, Cs2CO3, DMF, 120 °C, and X-ray structure of 6,7-BDTC (crystal I). The ellipsoids indicate a 50% probability of formation. :1); (iii) BBr3, CH2Cl2, 0 → 50 °C; (iv) 1,2-benzenedithiol, Cs2CO3, DMF, 120 °C, and X-ray structure of 6,7-BDTC (crystal I). The ellipsoids indicate a 50% probability of formation. | ||
The absorption and emission spectra of 6,7-BDTC in various solvents were recorded to uncover the photophysical properties (Fig. 2a and Table 1). Three main absorption bands at 350, 285, and 255 nm were observed for various solutions of 6,7-BDTC (c = 0.01 mM). The longest wavelength absorption band at 350 nm was attributed to the transition (oscillator strength (f) = 0.2027) from HOMO to LUMO estimated by quantum chemical calculations (Fig. S35 and Table S3). The second and third absorption bands were attributed to mixed configurations with S0 → S3,4 (f = 0.1979 and 0.1871) and S0 → S11 (f = 0.2007) as the main transitions. Focusing on the frontier molecular orbitals, the HOMO of 6,7-BDTC was distributed throughout the entire molecule, while the LUMO was mainly localized on the coumarin core (Fig. 2b). This suggests that 6,7-BDTC adopts a donor–acceptor-type intramolecular charge-separated structure from the thianthrene to the coumarin core during the transition process.12
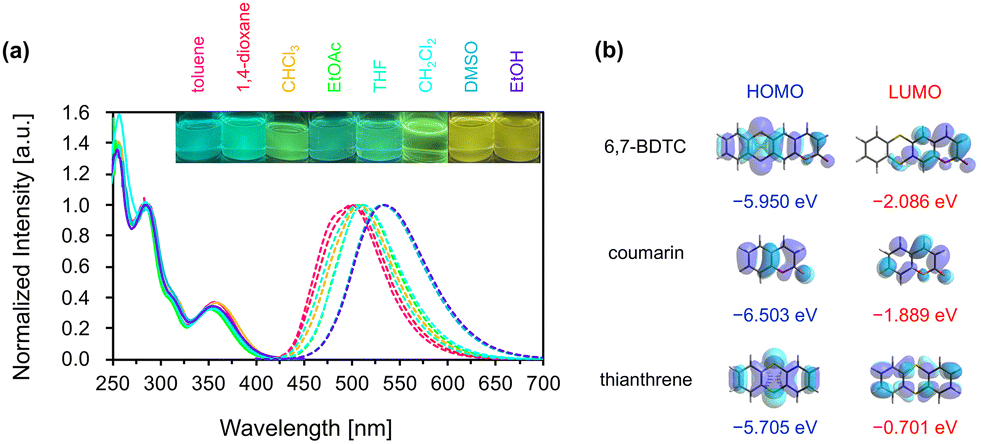 | ||
| Fig. 2 (a) Normalized absorption (solid line) and emission (dashed line) spectra of 6,7-BDTC in various tested solvents, and their photographic images were recorded under the irradiation of a UV lamp (365 nm). (b) Frontier molecular orbitals of 6,7-BDTC, coumarin, and thianthrene. | ||
| Solvent | λ abs [nm] | λ em [nm] | Φ F [%] | τ [ns] | K r [ns−1] | K nr [ns−1] |
|---|---|---|---|---|---|---|
| Toluene | 356 | 500 | 32 | 5.70 | 0.056 | 0.177 |
| 1,4-Dioxane | 355 | 502 | 34 | 6.97 | 0.048 | 0.144 |
| CHCl3 | 359 | 511 | 38 | 7.14 | 0.053 | 0.141 |
| EtOAc | 353 | 506 | 32 | 6.48 | 0.049 | 0.156 |
| THF | 354 | 508 | 30 | 7.07 | 0.042 | 0.142 |
| CH2Cl2 | 353 | 516 | 47 | 8.08 | 0.058 | 0.125 |
| DMSO | 355 | 531 | 40 | 9.12 | 0.043 | 0.110 |
| EtOH | 356 | 533 | 29 | 7.00 | 0.041 | 0.144 |
The solutions of 6,7-BDTC exhibited a slight solvatochromic effect in the emission spectra with a redshift of ca. 0.15 eV from green to yellow. The Lippert–Mataga plots showed a positive correlation between the Stokes shift (1.0 to 1.2 eV) and solvent parameters (ET(30))13 (Fig. S20); the polarity and hydrogen bonding effect appeared to influence the emission wavelength (Fig. S21). The structural optimization at the CAMB3LYP/6-31G(d,p) level for the S0 and S1 states suggested a dynamic conformational change from the folded to flattened state, with the dipole moment increasing slightly from 3.997 to 4.017 D (Fig. S36). These results indicate that the contribution of the resonance effect based on intramolecular charge-transfer in the flat S1 state lowered the energy level in highly polar solvents (Fig. S37).12 Additionally, it was found that 6,7-BDTC exhibits concentration quenching (Fig. S22). Fluorescence quantum yields (ΦF) were 29–47% regardless of the solvent polarity or viscosity. These were lower than that of 4-methyl-7-methylthio-coumarin (81%)14 but higher than that of planar coumarinacene (24%).15 Conversely, relatively long lifetimes (τ) of 5.7 to 9.1 ns with a single-exponential fluorescence decay were observed (Fig. S24 and Table S1). In all solutions, the nonradiative rate constant (Knr) was higher than the radiative rate constant (Kr), suggesting that the thermal deactivation process induced by dynamic conformational changes dominates during the decay process from flat S1 to folded S0.
The powder sample of 6,7-BDTC exhibited light blue fluorescence, and when this powder was ground in a mortar, the luminescence color changed to green (Fig. 3 and SI, Movie file). The difference between the two states was observed as a slight change involving a slight redshift and spectral broadening. This suggests that 6,7-BDTC can serve as a promising mechanochromic fluorophore. After vapor exposure to CH2Cl2, the greenish emission color returned to its original light blue (Fig. S28). This process was reversible over multiple cycles. The ΦF values (normal powder: 16%; grinding powder: 14%) were slightly lower than those in solution. Despite exhibiting concentration quenching, the emission might occur because the folded 6,7-BDTC was tightly immobilized within the assemblies, suppressing the conformational changes from S1 to S0. Fluorescence lifetime measurements revealed that both normal and ground powder samples exhibit a double-exponential decay process. For the normal powder, the decay curve showed that the short-lived τ1 (1.45 ns) component accounted for 74%, while the long-lived τ2 component accounted for 26% (5.49 ns, average: 2.51 ns). For the ground powder, τ1 (2.67 ns) accounted for 26% and τ2 (6.86 ns) accounted for 74% (average: 5.75 ns, Fig. S25 and Table S1). The excitation spectra of both powders showed slight changes in relative intensity (Fig. S26). These results suggest the presence of two types of luminescent species in each powder based on the stacking order of the aggregates. The ratio of short τ1 to long τ2 is reversed before and after grinding, indicating that mechanical stimulation relatively increased the long-lived luminescent species (Fig. S29). Since its lifetimes resemble those in solution, this suggests that the electronic properties of the excited state in the grinding powder locally approach those of a single-molecule state.
 | ||
| Fig. 3 (a) Emission spectra of 6,7-BDTC in the solid state: powder (light blue dashed line); grinding powder (green dashed line); crystal I (light blue solid line); and crystal II (green solid line). (b) Photographic images of 6,7-BDTC in the solid state were recorded under irradiation by a 365 nm UV lamp. | ||
Fortunately, the obtained crystal polymorphs I and II exhibited light blue and green emissions, respectively (Fig. 3b, bottom). Therefore, we compared them using X-ray crystallography to gain further insights into the intermolecular packing order (Fig. 4). The light blue emissive crystal I had a space group of I2/a, and 6,7-BDTC aligned along the b-axis, maintaining the folded skeleton (the dihedral angle: 132°). The intermolecular stacking distance along the b axis was 3.860 Å. On the long side of 6,7-BDTC, a hydrogen bonding network was formed with neighboring molecules by C–H⋯O and C–H⋯S interactions (Fig. S31). On the short side, at the lactone moiety, 6,7-BDTC formed strong interactions, hydrogen bonds between the oxygen atom of the carbonyl group and the hydrogen atom at the 3-position of the coumarin. Conversely, green emissive crystal II belonged to the P21/n space group. Two molecules of 6,7-BDTC (the dihedral angle: 126°) were dimerized while inverting from each other by partial π–π interactions at the coumarin moiety. The π–π distances between the coumarin cores in the dimer were 3.364 and 3.367 Å. Hydrogen bonding networks consisting of C–H⋯O and C–H⋯S bonds were formed along the molecular short and long sides of 6,7-BDTC, but no significant interactions were observed in the b-axis, except between dimers (Fig. S32).
 | ||
| Fig. 4 Crystal structures of crystals I and II of 6,7-BDTC. Hydrogen atoms are not shown for clarity. | ||
The emission spectra of crystals I and II closely resembled those of the powder samples (Fig. 3, λem: 504 (I); 511 (II) nm), and their quantum yields increased due to the dense immobilization of 6,7-BDTC by crystallization (ΦF: 19 (I); 29 (II)%). However, the excitation spectrum of crystal II differed from those of crystal I and the powder samples (Fig. S26), suggesting that the origin of the luminescence process differed between crystal II and the other solid samples. Furthermore, the decay curve of crystal I was a double-exponential decay, as with that of ordinary powder, where the short-lived τ1 is dominant (τ1 = 1.50 ns (71%), τ2 = 5.33 ns (29%)). Intriguingly, crystal II exhibited a single-exponential decay curve with only a long lifetime component (τ1 = 6.60 ns, Table S1). The differences in lifetime components and stacking patterns between crystals I and II suggest that the origin of the fluorescent species in the solid state of 6,7-BDTC can be explained as follows. In crystal I, 6,7-BDTC adopts a face-to-face arrangement with strong π–π overlap, extending intermolecular interactions into 3-dimensions. Therefore, the light-blue luminescent species observed as a short-lived component is thought to originate from the delocalization of excitons in the aggregate state. The normal powder is also thought to follow this face-to-face stacking (Fig. S33). When this layered structure is disrupted by grinding, the effective intermolecular overlap collapses. As a result, the proportion of long-lived green luminescent species, where transitions to localized excitons dominate similar to single molecules, is expected to increase within the ground powder. In contrast, in crystal II, 6,7-BDTC forms partially overlapping head-to-tail dimers, with intermolecular interactions extending along the a and c axes. However, no interactions are observed along the b axis, indicating the formation of a layer-separated stacking arrangement. Intermolecular interactions are reduced compared to crystal I. It is presumed that the long-lived green luminescent species was obtained as a pure component because the generated excitons prioritized localization in the layer over spatial delocalization.
In conclusion, 6,7-BDTC has been successfully synthesized and characterized. 6,7-BDTC exhibited the solvatofluorochromic effect from green to yellow in dilute solution. Also, the powder samples of 6,7-BDTC changed their emission color from light blue to green in response to mechanical stimulation. Two crystal polymorphs (crystals I and II) with different emission colors and stacking patterns were obtained. Based on the donor–acceptor structure derived from the folded skeleton, we found that suppressing the overlap of intermolecular conjugated planes through face-to-face and head-to-tail stacking formed within the molecular assembly enables the elicitation of different emission properties. Such folded dyes hold the potential to provide insights into the luminescence behavior of molecular assemblies and design guidelines for controlling the excited state.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information; experimental procedures, 1H, 13C and 19F NMR charts, HRMS spectrometry data, optical properties, crystal data and crystal structures, theoretical study, video of emission color change triggered by mechanical stimulation. See DOI: https://doi.org/10.1039/d5cc05320j.CCDC 2487912 (crystal I) and 2487913 (crystal II) contain the supplementary crystallographic data for this paper.16a,b
Acknowledgements
We thank Dr K. Takimoto (Kitasato University) for providing support with X-ray diffraction analysis. This research was funded by JSPS KAKENHI (grant numbers JP22K05070 and JP24K08400). The quantum calculations were performed at the Research Center for Computational Science, Okazaki, Japan (21-IMS-C188).References
- (a) T. O. Soine, J. Pharm. Sci., 1964, 53, 231 CrossRef CAS PubMed; (b) L. Wu, X. Wang, W. Xu, F. Farzaneh and R. Xu, Curr. Med. Chem., 2009, 16, 4236 CrossRef CAS PubMed; P. K. Jain and H. Joshi, J. Appl. Pharm. Sci., 2012, 2, 236 Search PubMed; (c) A. M. Saleh, M. M. Y. Madany and L. González, J. Plant Growth Regul., 2015, 34, 233 CrossRef CAS; (d) J. Sharifi-Rad, N. Cruz-Martins, P. López, E. P.-F. Lopez, N. Harun, B. Yeskaliyeva, A. Beyatli, O. Sytar, S. Shaheen, F. Sharopov, Y. Taheri, A. O. Docea, D. Calina and W. C. Cho, Oxid. Med. Cell Longevity, 2021, 2021, 6492346 CrossRef PubMed; (e) D. Gupta, E. Guliani and K. Bajaj, Top. Curr. Chem., 2024, 382, 16 CrossRef CAS.
- (a) T. Yu, P. Zhang, Y. Zhao, H. Zhang, J. Meng and D. Fan, Org. Electron., 2009, 10, 653 CrossRef CAS; (b) G. Signore, R. Nifosì, L. Albertazzi, B. Storti and R. Bizzarri, J. Am. Chem. Soc., 2010, 132, 1276 CrossRef CAS; (c) R. Qian, H. Tong, C. Huang, J. Li, Y. Tang, R. Wang, K. Lou and W. Wang, Org. Biomol. Chem., 2016, 14, 5007 RSC; (d) W. Xue, D. Wang, C. Li, Z. Zhai, T. Wang, Y. Liang and Z. Zhang, J. Org. Chem., 2020, 85, 3689 CrossRef CAS; (e) K. Górski, I. Deperasińska, G. V. Baryshnikov, S. Ozaki, K. Kamada, H. Ågren and D. T. Gyko, Org. Lett., 2021, 23, 6770 CrossRef; (f) Ł. Kielesiński, I. Deperasińska, O. M. Kateryna, V. Vygranenko, E. T. Ouellette and D. T. Gryko, J. Org. Chem., 2022, 87, 5961 CrossRef PubMed.
- (a) D. Kim, Q. P. Xuan, H. Moon, Y. W. Jun and K. H. Ahn, Asian J. Org. Chem., 2014, 3, 1089 CrossRef CAS; (b) M. Tasior, D. Kim, S. Singha, M. Krzeszewski, K. H. Ahn and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 1421 RSC; (c) Nitisha and P. Venkatakrishnan, J. Org. Chem., 2019, 84, 10679 CrossRef CAS; (d) Nitisha and P. Venkatakrishnan, Tetrahedron Lett., 2020, 61, 15148 CrossRef; (e) A. Kumar, A. Rajpoot, F. Imroze, S. Maddala, S. Dutta and P. Venkatakrishnan, Eur. J. Org. Chem., 2020, 6976 CrossRef CAS; (f) A. Kumar and P. Venkatakrishnan, Asian J. Org. Chem., 2021, 10, 1390 CrossRef CAS.
- (a) A. Mukhopadhyay, T. Hossen, I. Ghosh, A. L. Koner, W. M. Nau, K. Sahu and J. N. Moorthy, Chem. – Eur. J., 2017, 23, 14797 CrossRef CAS PubMed; (b) K. Usui, K. Yamamoto, Y. Ueno, K. Igawa, R. Hagihara, T. Masuda, A. Ojida, S. Karasawa, K. Tomooka, G. Hirai and H. Suemune, Chem. – Eur. J., 2018, 24, 14617 CrossRef CAS PubMed; (c) A. Mukhopadhyay, K. Jana, T. Hossen, K. Sahu and J. N. Moorthy, J. Org. Chem., 2019, 84, 10658 CrossRef CAS PubMed.
- Nitisha, P. Chetti and V. Parthasarathy, Chem. Commun., 2022, 58, 431 RSC.
- H. Liu, Y. Gao, J. Cao, T. Li, Y. Wen, Y. Ge, L. Zhang, G. Pan, T. Zhou and B. Yang, Efficient room-temperature phosphorescence based on a pure organic sulfur-containing heterocycle: folding-induced spin-orbit coupling enhancement, Mater. Chem. Front., 2018, 2, 1853 RSC.
- Z. Zhao, X. Zheng, L. Du, Y. Xiong, W. He, X. Gao, C. Li, Y. Liu, B. Xu, J. Zhang, F. Song, Y. Yu, X. Zhao, Y. Cai, X. He, R. T. K. Kwok, J. W. Y. Lam, X. Huang, D. L. Phillips, H. Wang and B. Z. Tang, Nat. Commun., 2019, 10, 2952 CrossRef PubMed.
- (a) R. Kotani, H. Sotome, H. Okajima, S. Yokoyama, Y. Nakaike, A. Kashiwagi, C. Mori, Y. Nakada, S. Yamaguchi, A. Osuka, A. Sakamoto, H. Miyasaka and S. Saito, J. Mater. Chem. C, 2017, 5, 5248 RSC; (b) S. Amishiro, M. Ueda and Y. Mazaki, Bull. Chem. Soc. Jpn., 2022, 95, 1723 CrossRef CAS.
- W. Liu, J. Wang, Y. Gong, Q. Liao, Q. Dang, Z. Li and Z. Bo, Angew. Chem., Int. Ed., 2020, 59, 20161 CrossRef CAS PubMed.
- (a) Y. Shan, J. Sheng, Q. Zhang, M. C. A. Stuart, D.-H. Qu and B. L. Feringa, Aggregate, 2024, 5, e584 CrossRef CAS; (b) X. Niu, S. Ren, K. Wang, F. Song, X. Dong, W.-J. Guo, H.-Q. Peng, Z. Zhao, J. W. Y. Lam, Y. S. Zhao, F. Li, S.-Y. Yu and B. Z. Tang, Aggregate, 2025, 6, e70003 CrossRef CAS; (c) X. Long, Z. Ma, H. Dai, Y. Wang, H. Xie, X. Ge, Z. Yang, J. Zhao, W. Hong and Z. Chi, Aggregate, 2025, 6, e70006 CrossRef CAS.
- (a) W. Hinrich, P. Berges, G. Klar, E. Sánchez-Martínez and W. Gunsser, Synth. Met., 1987, 20, 357 CrossRef; (b) S. Söderholm, J. Noreland, G. Olovsson, I. Olovsson, J. Helleberg and L. Engman, Mol. Cryst. Liq. Cryst., 1989, 167, 259 CrossRef; (c) J. Noreland, G. Olovsson and I. Olovsson, Synth. Met., 1991, 42, 1961 CrossRef.
- (a) K. H. Drexhage, Dye Laser, ed., F. P. Schäfer, Springer-Verlag, Berlin, Heidelberg, 1973, ch. 4, p. 144 Search PubMed; (b) G. Jones II, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294 CrossRef.
- J. P. Cerón-Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, J. Phys. Org. Chem., 2014, 27, 4199 CrossRef.
- A. E. Lanterna, M. González-Béjar, M. Frenette and J. C. Scaiano, Photochem. Photobiol. Sci., 2017, 16, 1284 CrossRef CAS PubMed.
- A. Kumar, A. Rajpoot, F. Imroze, S. Maddala, S. Dutta and P. Venkatakrishnan, Eur. J. Org. Chem., 2020, 6976 CrossRef CAS.
- (a) CCDC 2487912: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2phw7b; (b) CCDC 2487913: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2phw8c.
| This journal is © The Royal Society of Chemistry 2026 |