
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubunit-specific isotope labelling of heteromeric complexes using cell-free protein expression: application to the 760 kDa ClpXP molecular machine
Astrid
Audibert
,
Annelise
Vermot
 ,
Mathieu
Trauchessec
,
Karine
Giandoreggio
,
Daphna
Fenel
,
Aline
Le Roy
,
Chloee
Tymen
,
Basile
Moscatello
,
Lauren
Gandy
,
Caroline
Mas
,
Lionel
Imbert
and
Jerome
Boisbouvier
*
,
Mathieu
Trauchessec
,
Karine
Giandoreggio
,
Daphna
Fenel
,
Aline
Le Roy
,
Chloee
Tymen
,
Basile
Moscatello
,
Lauren
Gandy
,
Caroline
Mas
,
Lionel
Imbert
and
Jerome
Boisbouvier
*
Univ. Grenoble Alpes, CNRS, CEA, Institut de Biologie Structurale (IBS), 71, Avenue des Martyrs, F-38044 Grenoble, France. E-mail: jerome.boisbouvier@ibs.fr
First published on 2nd December 2025
Abstract
The specific insertion of methyl (13CH3) probes into deuterated proteins enables study of the structure, dynamics, and mechanisms of symmetric complexes up to 1 MDa by solution nuclear magnetic resonance (NMR). For asymmetric or heteromeric high molecular weight complexes, subunit-specific labelling is required to simplify spectra, reduce resonance overlap, and facilitate data interpretation. However, the instability of one component may prevent the reconstitution of a functional complex with a single labelled subunit. Here, we employed a simple, multi-step expression protocol that enabled the sequential in vitro synthesis of various subunits, each with a different isotopic labelling scheme. We exploited the open nature of the cell-free synthesis expression system by introducing a stable subunit directly into the synthesis of the unstable or poorly soluble component of the complex. This protocol was used to produce the ClpXP 26-subunit complex, in which the methyl groups were labelled on either ClpP or ClpX. The stabilisation of the newly synthesised ClpX subunits by the ClpP core enabled production of a stable and functional 760 kDa AAA+ proteolytic machine. 13CH3-labelling of the alanine and methionine in ClpXP allowed acquisition of high quality 2D solution NMR spectra, the characterization of the oligomeric size using diffusion-ordered NMR spectroscopy, and observation of structural rearrangements induced by nucleotide binding.
Introduction
Nuclear magnetic resonance (NMR) spectroscopy is a well-established and powerful tool for studying the structure, dynamics, and interactions of biomolecules at the atomic level. While backbone amide proton (HN)-detected NMR coupled with deuteration of non-exchangeable hydrogens is usually limited to proteins with a maximum molecular weight of around 50–100 kDa, the introduction of methyl (13CH3)-labelled probes in fully deuterated proteins1,2 coupled with optimized NMR experiments,3–5 has allowed the study of supramolecular complexes of up to 1 MDa at atomic resolution.6,7 However, these studies of large structures have been mainly restricted to symmetric model protein assemblies to limit the number of overlapping signals in 2D NMR spectra. Investigation of asymmetric hetero-oligomeric functional complexes requires separate labelling of each subunit and reassembly of the complex to acquire well-resolved spectra, a staunch requirement for the site-specific analysis of NMR data.8 However, incorporating separate labelling, reconstituting the complex, and ensuring its functionality remains challenging to generalise. Although an efficient in vivo co-expression method has been proposed for the separate isotope labelling of hetero-oligomers,9 the method is complex to use and is inapplicable to proteins with low expression yield in E. coli.Such is the case with the ClpXP molecular machine, which contains two subunits: the unfoldase ClpX and the protease ClpP. The ClpXP complex participates in cellular homeostasis, as well as proteomic quality control in bacteria and the mitochondria of eukaryotic cells.10–14 The machine targets incomplete proteins resulting from abortive translation as well as damaged proteins.15,16 The ClpXP machine is comprised of 14 ClpP protomers that assemble into a self-compartmentalised protease formed by two layers of heptameric ClpP rings, and one or two ClpX AAA+ ATPase hexameric rings stacked on each side of the ClpP peptidase compartment. The ClpXP complex is particularly difficult to produce in vivo due to the instability and low solubility of ClpX itself, which has prevented the production of a 13CH3 methyl-labelled ClpXP machine, a prerequisite for solution NMR investigation of this 760 kDa molecular machine.
As an open system, in vitro protein production enables the sequential synthesis of proteins, offering the opportunity to label each different subunit of a hetero-oligomer with a different isotope pattern.17,18 The stable individual subunits are produced first, enabling the most challenging component to be stabilised and solubilised. In this article, we present an optimised and economical cell-free protocol for the production of perdeuterated and methyl-labelled large assemblies. This straightforward strategy was successfully applied to the production and characterisation of the ClpXP AAA+ proteolytic machine from Thermus thermophilus, which contains 26 subunits of two different types. The ClpX and ClpP components were separately 13CH3-labelled on alanine and methionine, enabling the acquisition of high-resolution 2D methyl-TROSY on a functional ClpXP machine.
Results and discussion
A simple and cost-effective protocol to produce in vitro perdeuterated and specific 13CH3-labelled proteins
The specific incorporation of 13CH3 probes in a fully deuterated protein background is a powerful method for investigating large protein complexes using solution NMR with a molecular weight of up to 1 MDa.2,7,19 While the 300 kDa tetradecameric ClpP14 assembly can be successfully produced in E. coli and studied by NMR,20 the inherent instability and low solubility of its associated AAA+ unfoldase ClpX make producing the ClpX12P14 complex in a methyl-specific labelling form particularly challenging. To solve this bottleneck, we decided to use an in vitro cell-free protein synthesis approach,21 which has shown great promise as an alternative technology for the production of proteins.22–25 Although cell-free optimised, deuterated amino acid mixtures are commercially available for the in vitro production of uniform isotopically labelled proteins, the specific incorporation of 13CH3 probes is more challenging. Previous strategies have been proposed to express suitably labelled protein on a large scale in E. coli and subsequently used it, after purification and hydrolysis, as a starting material for another protein production in vitro.26 Alternatively, each perdeuterated amino acid can be acquired separately and mixed with a methyl-selectively labelled one,27 or specific keto-acid precursors28,29 to reconstitute the amino acid mix with suitable labelling schemes for cell-free protein production. While efficient, all these strategies are time consuming and expensive.To simplify the process, we decided to specifically label alanine and methionine methyl groups by adding an excess of deuterated, 13CH3-labelled amino acid to a commercial mix of deuterated amino acids. Given the efficiency of isotope incorporation using cell-free technology, each amino acid is present at a concentration of ca. one millimolar in a cell-free reaction.21 The addition of an excess of methyl-labelled amino acid should therefore enable an efficient isotope labelling of these methyl groups. This labelling scheme was used to produce the ClpP14 protease from Thermus thermophilus in vitro in continuous exchange mode (CECF).30 A mix of 20 deuterated cell-free amino acids at 3 mg mL−1 in cell free reaction and dialysis mix was complemented by 0.23 mg mL−1 of [13CH3, 2H5]-L-Met and 0.36 mg mL−1 of [13CH3, 2H1]-L-Ala. After incubating overnight, the protein was purified using a combination of heat shock and anion exchange chromatography. The average yield of the ClpP14 complex (300 kDa) was 2.7 mg mL−1 of cell-free reaction mixture. The 2D methyl TROSY spectra of the 300 kDa ClpP14 complex (Fig. 1(a)) display disperse signals characteristic of a well-folded protein. The well-resolved signals for the 5 methionine and 24 alanine methyl groups indicate that the complex is formed and homogeneous.
 | ||
| Fig. 1 Spectra of cell-free produced, methyl labelled ClpP alone or within ClpXP complex. 2D (1H, 13C) SOFAST-methyl-TROSY spectra of ClpP14 and ClpX12P14 complexes from Thermus thermophilus. ClpP14 was perdeuterated and specifically 13CH3-labelled on alanine and methionine methyl groups. When present, ClpX is protonated. NMR data were acquired at 60 °C in a 90%/10% D2O/glycerol buffer at pH 7.5 (Tris 20 mM, NaCl 100 mM) to stabilize the complex. (a) Spectrum of ClpP14 (300 kDa) at 170 µM of ClpP monomer, acquired on a NMR spectrometer operating at a 1H frequency of 850 MHz (b) spectrum of ClpP within ClpX12P14 (760 kDa) at 38 µM of ClpP monomer (and 32 µM of ClpX monomer) acquired on a NMR spectrometer operating at a 1H frequency of 950 MHz. Star indicate a signal from a contaminant. | ||
To quantify the level of isotope incorporation, we produced in vitro the 17 kDa nucleotide-binding fragment of ATPase HMA8 from Arabidopsis thaliana as a model protein.31 While the deuteration level of exchangeable hydrogens for proteins produced and maintained in a D2O-based buffer is expected to match that of the solvent, we used mass spectrometry to analyse the level of deuteration of non-exchangeable proton sites. Analysis of the protein produced in cell-free with either a mix of natural amino acids or a commercial mix of uniformly deuterated amino acids revealed an average deuterium incorporation level at 96% for all non-exchangeable proton sites (see Fig. S1 and Table S1). Preparations with the addition of alanine and methionine (at concentrations mentioned previously) were sufficient to enable their incorporation at 65% and 76% respectively (see Fig. S1 and Table S1). Similar incorporation yields of 70% were obtained for threonine with its addition at 0.48 mg mL−1.
The methyl-specific labelling protocol described herein is a simple and fast alternative to E. coli-based approaches, as purification can be initiated immediately after overnight synthesis. This enables several preparations to be carried out in the same week. Furthermore, the isotope cost, required for 1 mL of a CECF in vitro reaction, is approximately €30 for 21 mg of deuterated amino acids mix and ca. €2–€15 per methyl-labelled alanine (2.5 mg), methionine (1.6 mg), or threonine (3.4 mg). Doubling the quantities of methyl-labelled amino-acids can increase 13CH3 incorporation yields to 80–90%, at modest additional cost. Besides the cost of labelled materials, the cell-free reaction mix can be prepared in-house using a published protocol21 at less than €40 per mL of reaction, including all the consumables required for cell-free protein synthesis. While the yield of protein expression is protein-dependent, the proposed protocol can be a cost-effective alternative to the production of methyl-labelled proteins using the E. coli expression system. Note that leucine, valine, and isoleucine can also be labelled using such protocol, but at a significantly higher cost, given the difficulty to synthetise these amino acids with deuterated and 13CH3-labelling on a single methyl group.32,33
Production of functional ClpX12P14 machine using a two steps in vitro production protocol
Although the 300 kDa tetradecameric ClpP can be produced successfully in vitro, ClpX is unstable and poorly soluble on its own, even when using the ClpXΔN construct that lacks the 52 flexible residues located on the N-terminal end (for simplicity, herein, this construct is referred to as ClpX). We exploited the open nature of the cell-free production mode by introducing the purified ClpP protein into the cell-free reaction before the ClpX plasmid was introduced (Fig. 2(A)). Consequently, once synthesised, ClpX can be stabilised directly as a hexameric oligomer that stacks on either side of the ClpP complex. Following the purification and cleavage of the His-tag, the ClpX12P14 complex was successfully isolated using size-exclusion chromatography (Fig. 2(B) and (C)). Negative staining imaging of the produced sample allowed observation of the characteristic morphology of the two ClpX hexameric rings separated by the ClpP barrel (Fig. 3(A)). SEC-MALS and analytical ultracentrifugation analyses confirmed the formation of a homogeneous population of the proteolytic machine with the expected molecular weight of 760 kDa (Fig. S2). To verify if this oligomeric state of ClpXP is the most stable one, we used highly resolved, small scale size exclusion chromatography to separate both the single and double ClpX ring oligomeric states of ClpXP complex (Fig. S3). Each fraction was flash frozen and stored at −20 °C, thawed, and injected a second time in the Superose 6 analytical column. In contrast to the double ring fraction that exhibits great stability over time, we observed that most of the ClpXP complex containing a single ring of ClpX dissociates to form the more stable double ring ClpX12P14 complex and an isolated ClpP14 oligomer. | ||
| Fig. 2 Two-step cell-free production and purification protocol of labelled ClpX12P14 complex for NMR analysis. (A) Left: Schematic view of ClpXP production in cell-free dialysis system and purification protocol. Right: Schema of the corresponding ClpXP production step. RM reaction mix, DM dialysis mix. (B) SDS-PAGE gel of soluble fractions at the different ClpXP production steps (ClpP: 21 kDa (blue), ClpX 38 kDa (red), TEV: 28 kDa (green)). (C) Gel filtration chromatogram of ClpXP at final purification stage. Green area: fractions selected for NMR analysis. | ||
 | ||
| Fig. 3 Verification of the integrity and activity of cell-free produced ClpXP (A) Left: Electron microscopy negative staining image of ClpX12P14 complex. Center: Three-fold zoom of the two red rectangles. Right: Two views (top and side) of a ClpXP modified Cryo-EM structure (modified from PDB 6VFS). (B) Degradation assay of FtsZ by ClpXP. SDS-PAGE gel shows the time-resolved degradation of FtsZ-ssrA by ClpX12P14. | ||
To confirm that our in vitro produced ClpX12P14 machine retains ATPase activity, we used a Malachite green colorimetric test to monitor the formation of inorganic phosphate, released as a result of ATP hydrolysis. ClpXP incubated with ATP for 5 minutes produced a deep green color, indicating the release of large quantity of inorganic phosphate, confirming protease function (Fig. S4). Proteolysis activity was also assessed by incubating the complex with a 10-fold excess of substrate protein FtsZ,35,36 and the level of intact FtsZ was monitored using SDS-PAGE (Fig. 3(B)). Proteolysis of FtsZ was clearly observed after 5 minutes of incubation with ClpXP at 55 °C in the presence of a constant concentration of ATP. These biochemical tests confirm that the in vitro produced ClpX12P14 oligomer is the stable form of the functional AAA+ proteolytic machine.
Subunit specific labelling of ClpXP complex
Simultaneous uniform methyl labelling of both subunits of the functional ClpXP machine would lead to significant spectral overlap and impossibility of assignment. The two-step in vitro production protocol enabled the individual isotope enrichment of the ClpP and ClpX subunits. Introducing U-2H, 13C-Ala/Met ClpP14 complex for ClpX synthesis with unlabelled amino acids produced a ClpX12P14 complex with only the ClpP subunits methyl labelled. The corresponding 2D methyl-TROSY spectra is presented in Fig. 1(b). As expected for a 760 kDa particle with a predicted correlation time of τC ∼ 300 ns,37 the NMR signals are broader compared to free ClpP14 (300 kDa, τC ∼ 90 ns; see Fig. 1(a)). The position of the resonance peaks are globally similar, confirming that the overall ClpP structure is preserved. Signs of heterogeneity (peak doubling) are observed for a few peaks, probably due to a mismatch in symmetry between the six interacting ClpX subunits with each heptameric ClpP rings. To verify the oligomeric state of ClpXP in NMR conditions, we acquired diffusion-ordered spectroscopy experiments (DOSY)38 on ClpP and ClpXP samples labelled only on the ClpP subunits. Fitting the DOSY data (Fig. 4) enabled us to derive the translational diffusion coefficient for both oligomers. As expected, faster translation diffusion was observed for ClpP than for ClpXP. The ratio of the two diffusion coefficients was 1.58 ± 0.09, which is in close agreement with the predicted 1.49 ratio using the ClpP14 and ClpX12P14 structures (PDB 6HWN39 and 6VFS40 respectively) for hydrodynamic properties calculation37 (Table S2).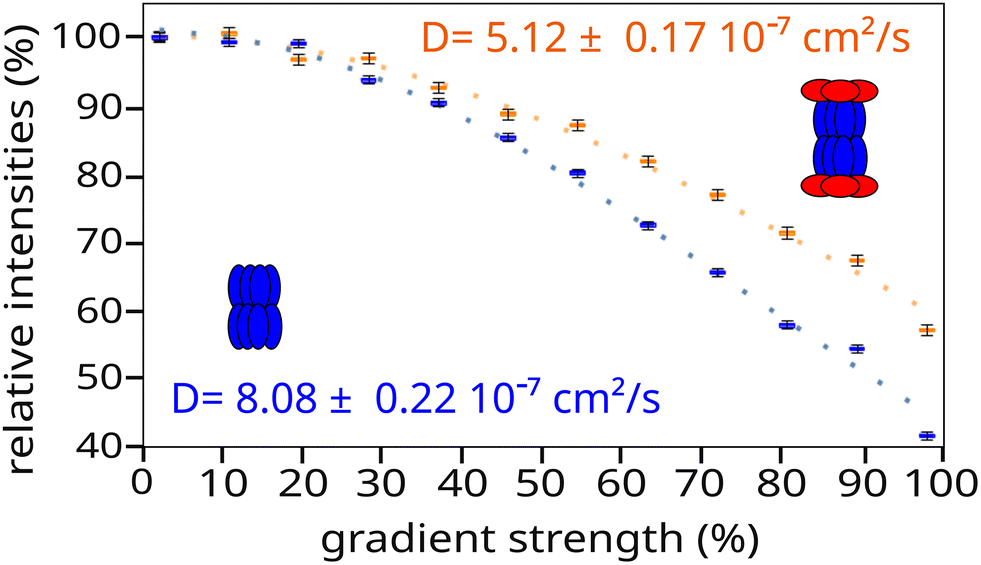 | ||
| Fig. 4 Translational diffusion properties of ClpP alone (blue) and ClpXP complex (orange) characterised by diffusion-ordered NMR spectroscopy. In the ClpXP complex, only ClpP protein was methyl-labelled. Derived diffusion coefficients are shown on the graph. | ||
The same labelling strategy was used to produce deuterated and 13CH3-labelled ClpX on Met/Ala or Met/Thr residues, by providing unlabelled ClpP prior to ClpX synthesis. Fig. 5(A) and Fig. S5 present the 2D methyl TROSY of ClpX within the full ClpXP machine. The well-dispersed signals indicate that the ATPase subunit is well folded, but minor peaks are also visible, confirming the presence of heterogeneity in the nucleotide-free ClpXP machine, as observed for the ClpP14 complex (Fig. 1(B)). Interestingly, upon the addition of ADP, we observed a significant change in the spectra (see Fig. 5(B) and Fig. S6), with the appearance of well-resolved signals corresponding to the 33 alanine and 5 methionine residues in ClpX, despite the mismatch in symmetry at the interface between ClpP and ClpX. This result suggests that ClpX undergoes structural rearrangement upon nucleotide binding, with fast conformational exchange between the different subunits. The conformational exchange coalesces into a single set of NMR signals due to the averaging of different frequencies.
 | ||
| Fig. 5 ClpX spectra within ClpXP complex in absence (A) or in presence (B) of ADP. 2D (13C,1H) SOFAST-methyl-TROSY spectra of ClpX12P14 complexes from Thermus thermophilus. ClpX was perdeuterated and specifically 13CH3-labelled on alanine and methionine methyl groups. ClpP was protonated. NMR data were acquired at 60 °C using a high field NMR spectrometer operating at a 1H frequency of 850 MHz. ClpXP complexes are in an 90%/10% D2O/glycerol buffer at pH 7.5 (Tris 20 mM, NaCl 100 mM) to stabilize the complex. (A) Spectra of ClpXP apo at 140 µM of ClpX monomer. (B) Spectra of ClpXP with 1 mM of ADP at 34 µM of ClpX monomer. | ||
Conclusions
This article presents a straightforward and fast (approx. 1.5 days) protocol for producing a deuterated and methyl-labelled multimeric complex using cell-free technology. The method is a cost-effective way to specifically insert methionine, alanine, and threonine 13C-methyl probes into the desired subunit, and offers an attractive alternative to standard in vivo perdeuteration and methyl-labelling methods which consume milligram to gram quantities of isotopic material with variable control over the material's direct usage in the target protein. By exploiting the open nature of cell-free production, this approach can be used to obtain differential labelling of subunits in hetero-oligomeric complexes, as well as stabilising unstable or insoluble components of a complex.17,18 The direct integration of detergents, chaperones, cofactors, lipids, enzymes, and other stabilizing agents present a multitude of avenues to exploit for complicated but controlled protein production and isotopic integration. Therefore, when in vivo methods fail, cell-free protein production may provide solutions where otherwise research would be stopped.This approach has been successfully applied to separately label the two major subunits of the ClpX12P14 supramolecular complex, proving the viability of this method on a historically challenging target. Much of the structure of the ClpXP molecular machine was deduced from static images of the individual subunits and the ClpXP complex, visualized with cryo-EM.44 Our protein production method led to the acquisition of high-resolution 2D methyl-TROSY spectra of both the protease and the AAA+ unfoldase compartments in the functional machine, opening up the possibility of studying the dynamics, interactions, and mechanisms of this complex in molecular motion.
Materials and methods
Protein plasmids preparation
The wild-type sequences of the Thermus thermophilus ClpP (21.4 kDa), ClpXΔN (38.3 kDa) and FtsZ (37.3 kDa) proteins were cloned separately into pIVEX 2.3d plasmids. The open reading frames of the synthetic genes were optimised for expression in E. coli and ordered from a commercial vendor (GeneCust). In the ClpXΔN constructs, the first 52 amino acids of the ClpX sequence were omitted to stabilise the ClpX protein,34,41 and an N-terminal 6-His tag followed by a TEV protease cleavage site was inserted. An uncleavable 6-His tag was inserted at the N-terminus of FtsZ with a GEGDGEGDGEGDG linker,42 followed by a modified ssrA sequence (SENYALAA)43 at the C-terminus of FtsZ. Each plasmid was transformed into a TOP10 E. coli strain (Thermo Fisher), which was then grown in 500 mL of LB medium containing 100 µg mL−1 ampicillin. The plasmids were then purified using a NucleoBond Xtra Maxi Plus kit (Macherey-Nagel), and the final elution concentration was set at 1 mg mL−1 in RNase-free water.Cell-free protein production
The proteins were produced in vitro in continuous exchange mode (CECF), with a reaction-to-dialysis volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.21,44 All reactions were performed at 27 °C with gentle rotation at 18 rpm in a hybridisation oven (Hybrynogene K38, Ascon Technologic, Techne) using 3 mL dialysis system (Maxi GeBaFlex, 8 kDa MWCO, Gene Bio-Application Ltd) placed in 25 mL Eppendorf tubes containing 18 mL of feeding solution. The reaction mixtures contained the following: target protein pIVEX plasmid (16 µg mL−1); E. coli BL21(DE3) S30 cellular extract; and T7 polymerase. Other standard cell-free components in the reaction and dialysis solutions (e.g. amino acids, NTPs, tRNAs, HEPES, MgOAc2, creatine phosphate, creatine kinase, NH4OAc, spermidine, cAMP, folinic acid) were added as described in a previously published protocol.21 The cell-free reaction was stopped after 16 hours. The reaction mixtures were then centrifuged for 20 minutes at 14000 g prior to purification. To stabilise the synthesised ClpX in the ClpXP complex, 1.2 mg mL−1 of purified ClpP was added to the reaction mixture before adding the ClpX pIVEX plasmid.
:6.21,44 All reactions were performed at 27 °C with gentle rotation at 18 rpm in a hybridisation oven (Hybrynogene K38, Ascon Technologic, Techne) using 3 mL dialysis system (Maxi GeBaFlex, 8 kDa MWCO, Gene Bio-Application Ltd) placed in 25 mL Eppendorf tubes containing 18 mL of feeding solution. The reaction mixtures contained the following: target protein pIVEX plasmid (16 µg mL−1); E. coli BL21(DE3) S30 cellular extract; and T7 polymerase. Other standard cell-free components in the reaction and dialysis solutions (e.g. amino acids, NTPs, tRNAs, HEPES, MgOAc2, creatine phosphate, creatine kinase, NH4OAc, spermidine, cAMP, folinic acid) were added as described in a previously published protocol.21 The cell-free reaction was stopped after 16 hours. The reaction mixtures were then centrifuged for 20 minutes at 14000 g prior to purification. To stabilise the synthesised ClpX in the ClpXP complex, 1.2 mg mL−1 of purified ClpP was added to the reaction mixture before adding the ClpX pIVEX plasmid.
Isotope labelling
To produce deuterated proteins with enriched 13CH3-alanine, methionine, threonine, or combinations thereof, all components of the cell-free reaction and dialysis mixtures were freeze-dried and dissolved in 99.8% D2O (Aldrich). The solvent in the S30 extracts was replaced by D2O via lyophilisation, leading to a reduction of the protein yield by a factor of ca. 2.21 The amino acids in the reaction and dialysis solutions were replaced with a cell-free mixture of deuterated amino acids (Cell Free amino acid mix U-D 98%, Cambridge Isotope Laboratories) at 3 mg mL−1, complemented by an excess of 2H5-13CH3-L-Met (0.23 mg mL−1 – Cambridge Isotope Laboratories) and 2Hα-[13CH3]-L-Ala (0.36 mg mL−1 – NMR-Bio) or 2H2-[13CH3]-L-Thr (0.48 mg mL−1 – Cambridge Isotope Laboratories).To assess the level of isotope incorporation, the nucleotide-binding domain of the 16.7 kDa, 155 amino acid P1B-type ATPase HMA8 from Arabidopsis thaliana31 was produced as a model protein using the aforementioned cell-free protocol. This was done in either protonated or perdeuterated and methyl labelled form using the isotope labelling protocol described above. The protein was then purified by Ni-NTA affinity chromatography and exchanged into a Tris buffer containing 50 mM NaCl and pH 7.5. As partial D-to-H back-exchange was possible during the experiment, the perdeuterated samples were kept in an H2O-based buffer for 72 hours to enable D/H exchange at all exchangeable proton sites prior to the mass spectrometry experiments.
Protein purification
ClpP protein samples were purified from the reaction mixtures using an initial heat shock for 20 minutes at 80 °C. After centrifugation for 20 minutes at 14000g, the ClpP protein was purified from the supernatant using an anion exchange chromatography 6 mL Resource Q column (Cytiva). This was followed by a final 20-minute heat shock at 80 °C and subsequent centrifugation for 20 minutes at 14000g. The ClpP protein purification buffer contains 20 mM Tris base (pH 7.5) and up to 0.5 M NaCl. Following purification, the average yield of the ClpP14 complex (300 kDa) was 2.7 mg per 1 mL of cell-free reaction mixture, as determined by absorbance at 280 nm.
The ClpXP complex was first centrifuged for 20 minutes at 14000g, and then purified using metal affinity chromatography with 5 mL HisTalon Superflow cartridges (TaKaRa Bio). The affinity chromatography buffer contained 20 mM Tris base (pH 7.5), 100 mM NaCl and a 5–500 mM imidazole gradient. The ClpX purification tag was cleaved overnight at 34 °C using 2 mg of His-tagged TEV protease for 5 mg of ClpXP complexes. This was performed in 2 L of dialysis buffer containing 20 mM Tris base (pH 7.5), 100 mM NaCl, and 10% glycerol. Following a 20-minute centrifugation step at 14000g, the His-tagged TEV protease and cleaved tags were removed via reverse Ni-NTA chromatography using HisTrap resin (Qiagen). Finally, size-exclusion chromatography was performed using a Superose 6 Increase 10/300 GL column (Cytiva) to isolate the ClpP tetradecamer, which interacts with two hexameric rings of ClpX. After purification, the average yield of U-[2H], [13CH3]-Alaβ, [13CH3]-Metε ClpX12P14 complex (7602 kDa) was 100 µg for 1 mL of cell-free reaction mix.
The FtsZ-ssrA protein samples were purified from the reaction mixtures by initially subjecting them to a 20-minute heat shock at 60 °C, followed by centrifugation for 20 minutes at 14000g, and then by metal affinity chromatography using 5 mL HisTalon Superflow cartridges (TaKaRa Bio). The affinity chromatography buffer contained 20 mM Tris base (pH 7.5), 100 mM NaCl and a 5–500 mM imidazole gradient. The protein was concentrated using a centrifugal filtration unit (Amicon® Ultra 15–50 kDa MWCO), after which a final heat shock at 60 °C was performed, followed by centrifugation for 20 minutes at 14000g.
FtsZ degradation assay and SDS-page analysis
A solution of ClpXΔN12P14 at 2 mg mL−1 (equivalent to 2.6 µM of complexes) was mixed with a 10-fold excess of FtsZ (equivalent to 26 µM, or 1 mg mL−1), one unit of pyruvate kinase from Bacillus stearothermophilus (obtained from Sigma), 50 mM phosphoenolpyruvate at pH 7.5, 0.1 mM D-ribose 5-phosphate (to activate the pyruvate kinase), 1 mM MgCl2, and 1 mM ATP. The FtsZ degradation assay was performed at 55 °C in a buffer containing 100 mM NaCl, 20 mM Tris base (pH 7.5), and 10% glycerol. Aliquots were taken at specified times, after which the reactions were stopped by direct dilution in SDS and Coomassie Blue solution for gel electrophoresis analysis (migration at 200 V for 40 minutes) using a Chemidoc MP imaging system (Bio-Rad).Malachite green phosphate assay
Malachite green assay was performed according to manufacturer protocol of the Malachite Green Phosphate Assay Kit (Sigma Aldrich). 1,3 µM of ClpXP (15,8 µM of ClpX and 18,4 µM of ClpP) was incubated with 0,1 mM ATP for 5 or 10 minutes. Samples were quenched by the addition of 10 µL of Working Reagent (1 mL of Reagent A and 10 µL of Reagent B) to 40 µL of reaction. Samples were visually confirmed for colorimetric change compared with a phosphate standard curve from 40 µM to 4 µM. The buffer was separately incubated to ensure that the presence of ATP or magnesium did not cause a false positive result.Sedimentation velocity-analytical ultracentrifugation
ClpXP SV-AUC experiments were performed at 36000 rpm at 22 °C on an XLI analytical ultracentrifuge (Beckman Coulter, Palo Alto, USA) with a Ti-50 rotor and double-sector cells with 1.5- and 3-mm optical path centrepieces equipped with sapphire windows (Nanolytics, Potsdam, Germany). ClpXP was analysed at a concentration of 2 mg mL−1. The final ClpXP buffer (20 mM Tris at pH 7.5, 100 mM NaCl, 10% glycerol at pH 7.5) was used in the reference channel. The data were corrected using the REDATE v1.0.1 software. The SV-AUC profiles were analysed in terms of the continuous size distribution (c(s)) of sedimentation coefficients (s) using SEDFIT v16.36. The c(s) analysis was done using the calculated ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) of 0.748 mL g−1. The density (ρ° = 1.026 g mL−1) and viscosity (η° = 1.245 cp) were calculated using SEDNTERP v3.0.2 and the frictional ratio f/f0 (min) was fitted. Non-interacting species analysis was performed to evaluate the molecular weight of the sample. Peak integration and figure creation were performed using GUSSI v1.4.2 software.
of 0.748 mL g−1. The density (ρ° = 1.026 g mL−1) and viscosity (η° = 1.245 cp) were calculated using SEDNTERP v3.0.2 and the frictional ratio f/f0 (min) was fitted. Non-interacting species analysis was performed to evaluate the molecular weight of the sample. Peak integration and figure creation were performed using GUSSI v1.4.2 software.
SEC-MALS
SEC-MALS analyses were performed using Superose 6 Increase 10/300 GL (Cytiva) gel filtration columns connected to an Omnisec analytical system (Malvern Panalytical). The column and system were equilibrated with 10 column volumes of a buffer solution containing 20 mM Tris-base (pH 7.5), 100 mM NaCl and 10% glycerol. 50 µL of protein samples, at concentrations of 0.5, 1 and 2 mg mL−1, were injected at room temperature. The experiment and analysis were performed using OMNISEC software (Malvern Panalytical).Mass spectrometry
Prior to the mass spectrometry (MS), deuterated protein samples were diluted to a concentration of 1 µM using 0.1% formic acid (FA) prepared with protonated Milli-Q water. Experiments were performed using a liquid chromatography (LC) electrospray (ESI) quadrupole-time of flight (Q-TOF) instrument (AdvanceBio 6545 XT, Agilent Technologies). The HPLC system was an Agilent 1260 Infinity II series. A reverse phase chromatography column for biomolecules was used (PLRP-S 100 Å, 1.0 × 50 mm, 3 µm, Agilent Technologies). HPLC mobile phases were prepared using HPLC-grade protonated solvents. HPLC solvents consisted of 0.1% FA in water (solvent A); 99.9% acetonitrile and 0.1% FA (solvent B). We used a gradient of HPLC solvents for a total time of 5.1 min. Gradient started with 100% solvent A, followed by an increase to 20% solvent B over 1 min. Solvent B increased up to 50% over a period of 2 min (total time from the beginning of the gradient: 3 min). Solvent B went up to 70% (total time: 4 min). Solvent B at 50% was maintained for 1 min before switching back to 100% solvent A over a period of 0.1 min (total time: 5.1 min). The HPLC column was re-equilibrated for 1 min using solvent A. The solvent flow rate was 0.25 mL min−1 and the column temperature was 60 °C.MS acquisition was carried out in positive ion mode. “Dual ESI source” capillary needle voltage (Vcap) was set at 5.5 kV and nozzle voltage at 2 kV. Gas temperature was set at 350 °C, nitrogen was used as drying gas (12.0 L min−1) and as nebulizer gas (60 psi). The capillary needle voltage (Vcap) was set at 5500 V. Spectra acquisition rate was of 1 spectra per s. The instrument was calibrated in a mass-to-charge (m/z) range 600–3000 using a standard calibrant (ESI-L, low concentration tuning mix, Agilent Technologies). MS spectra were processed using Bioconfirm workstation software (v. B.3.4.242, Agilent Technologies). Data were further analysed using with GPMAW software (v. 7.00b2, Lighthouse Data, Denmark).
NMR spectroscopy
For the NMR experiments, the sample was fully exchanged into a final buffer solution of D2O containing 20 mM Tris at pH 7.5, 100 mM NaCl and 10% glycerol, using a centrifugal filtration unit (Amicon® Ultra 3–50 kDa MWCO). The 180 µL deuterated and methyl labelled samples were placed in a 4 mm Shigemi NMR tube (SHIGEMI CO., Ltd) at an average final ClpXP complex concentration of 5 µM (i.e. 3.6 mg mL−1), equivalent to 60 µM and 70 µM of ClpX and ClpP subunits respectively.2D SOFAST-methyl-TROSY4 and DOSY methyl-TROSY38 experiments were acquired at 60 °C on Bruker Avance III HD spectrometers equipped with cryogenic probes operating at 1H frequencies of 850 MHz or 950 MHz. For the DOSY experiments, the diffusion time was set to 100 ms and the total gradient duration to 2 ms at a maximum gradient strength of 59.5 G cm−1. The recovery delay was set to 1.1 s, giving a total acquisition time of 12 hours for ClpXP and 6 hours for ClpP samples. The diffusion coefficients were fitted from the experimental data with curve.fit software using the Stejskal–Tanner equation45 and were then compared with the theoretical diffusion coefficients predicted from PDB structures (6HWN39 for ClpP and 6VFS40 for ClpXP) using Hydropro.37
Electron microscopy
Sample (0.1 mg mL−1) were absorbed to the clean side of a carbon film on mica, stained with sodium silico tungstate and transferred to a 400-mesh copper grid. The images were taken with defocus values between 1.2 and 2.5 µm on a Tecnai 12 LaB6 electron microscope at 120 kV accelerating voltage using CCD Camera Gatan Orius 1000.Author contributions
A. A., A. V., J. B., L. I. and M. T. designed the experiments; A. A., A. V., K. G., B. M. and M. T. prepared the samples, A. L. R., C. M., C. T., and D. F. performed and analysed the AUC, SEC-MALS, MS and EM experiments; A. A. and J. B. collected and analysed the NMR experiments; A. A. performed functional tests. A. A., L. G., and J. B. wrote the manuscript. All authors discussed the results, corrected the manuscript and approved the final version of the paper.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the Methods section, as part of the supplementary information (SI) an in Zenodo repository with number 17264974 (DOI: https://doi.org/10.5281/zenodo.17264974). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cb00259a.Acknowledgements
We would like to thank Dr J. Dos Santos Moreira for help and advice, G. Schoehn and E. Boeri Erba for setting up and maintaining the IBS/ISBG EM and MS platforms. This work is supported by the French National Research Agency in the framework of the “Investissements d’avenir” program (ANR-15-IDEX-02), ProteaseInAction project no ANR-19-CE11-0022 and ERC ADG XXL-NMR – project #101097926. AA acknowledges a PhD CDSN fellowship from ENS-Lyon/MESR. This work used the Biophysics, Cell-Free, EM, MS and NMR facilities at the Grenoble Instruct-ERIC Center (ISBG; UAR 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB). Platform access was supported by FRISBI (ANR-10-INBS-05-02) and GRAL, a project of the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). The IBS Electron Microscope facility is supported by the Auvergne Rhône-Alpes Region, the Fonds Feder, the Fondation pour la Recherche Médicale and GIS-IBiSA. IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, CEA).References
- V. Tugarinov, V. Kanelis and L. E. Kay, Nat. Protoc., 2006, 1, 749–754 CrossRef CAS.
- R. Kerfah, M. J. Plevin, R. Sounier, P. Gans and J. Boisbouvier, Curr. Opin. Struct. Biol., 2015, 32, 113–122 CrossRef CAS PubMed.
- V. Tugarinov, P. M. Hwang, J. E. Ollerenshaw and L. E. Kay, J. Am. Chem. Soc., 2003, 125, 10420–10428 CrossRef CAS PubMed.
- C. Amero, P. Schanda, M. A. Durá, I. Ayala, D. Marion, B. Franzetti, B. Brutscher and J. Boisbouvier, J. Am. Chem. Soc., 2009, 131, 3448–3449 CrossRef CAS.
- S. Schütz and R. Sprangers, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 116, 56–84 CrossRef PubMed.
- A. Mainz, T. L. Religa, R. Sprangers, R. Linser, L. E. Kay and B. Reif, Angew. Chem., Int. Ed., 2013, 52, 8746–8751 CrossRef CAS PubMed.
- G. Mas, J.-Y. Guan, E. Crublet, E. C. Debled, C. Moriscot, P. Gans, G. Schoehn, P. Macek, P. Schanda and J. Boisbouvier, Sci. Adv., 2018, 4, eaau4196 CrossRef PubMed.
- J. Liebau, D. Lazzaretti, T. Fürtges, A. Bichler, M. Pilsl, T. Rudack and R. Sprangers, Nat. Commun., 2025, 16, 7896 CrossRef CAS PubMed.
- M. Mund, J. H. Overbeck, J. Ullmann and R. Sprangers, Angew. Chem., Int. Ed., 2013, 52, 11401–11405 CrossRef CAS.
- H.-H. Lo, C.-T. Liao, C.-E. Li, Y.-C. Chiang and Y.-M. Hsiao, Arch. Microbiol., 2020, 202, 597–607 CrossRef CAS.
- M. E. Aljghami, M. M. Barghash, E. Majaesic, V. Bhandari and W. A. Houry, Front. Mol. Biosci., 2022, 9, 1054408 CrossRef CAS.
- K. Nouri, Y. Feng and A. D. Schimmer, Cell Death Dis., 2020, 11, 841 CrossRef CAS PubMed.
- Y. Feng, K. Nouri and A. D. Schimmer, Cancers, 2021, 13, 2020 CrossRef CAS PubMed.
- A. Audibert, J. Boisbouvier and A. Vermot, Biomolecules, 2025, 15(8), 1097 CrossRef CAS.
- R. T. Sauer and T. A. Baker, Annu. Rev. Biochem., 2011, 80, 587–612 CrossRef CAS.
- A. O. Olivares, T. A. Baker and R. T. Sauer, Annu. Rev. Physiol., 2018, 80, 413–429 CrossRef CAS PubMed.
- K. Ozawa, S. Jergic, J. A. Crowther, P. R. Thompson, G. Wijffels, G. Otting and N. A. Dixon, J. Biomol. NMR, 2005, 32, 235–241 CrossRef CAS PubMed.
- K. Ozawa, S. Jergic, A. Y. Park, N. E. Dixon and G. Otting, Nucleic Acids Res., 2008, 36, 5074–5082 CrossRef CAS PubMed.
- R. Sprangers, A. Velyvis and L. E. Kay, Nat. Methods, 2007, 4, 697–703 CrossRef CAS PubMed.
- R. Sprangers, A. Gribun, P. M. Hwang, W. A. Houry and L. E. Kay, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16678–16683 CrossRef CAS PubMed.
- L. Imbert, R. Lenoir-Capello, E. Crublet, A. Vallet, R. Awad, I. Ayala, C. Juillan-Binard, H. Mayerhofer, R. Kerfah, P. Gans, E. Miclet and J. Boisbouvier, Methods Mol. Biol., 2021, 2199, 127–149 CrossRef CAS.
- D. Schwarz, F. Junge, F. Durst, N. Frölich, B. Schneider, S. Reckel, S. Sobhanifar, V. Dötsch and F. Bernhard, Nat. Protoc., 2007, 2, 2945–2957 CrossRef CAS.
- T. Kigawa, Methods Mol. Biol., 2010, 607, 101–111 CrossRef CAS.
- T. Sawasaki, T. Ogasawara, R. Morishita and Y. Endo, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14652–14657 CrossRef CAS PubMed.
- A. S. Spirin, V. I. Baranov, L. A. Ryabova, S. Y. Ovodov and Y. B. Alakhov, Science, 1988, 242, 1162–1164 CrossRef CAS PubMed.
- R. Linser, V. Gelev, F. Hagn, H. Arthanari, S. G. Hyberts and G. Wagner, J. Am. Chem. Soc., 2014, 136, 11308–11310 CrossRef CAS.
- M. Kainosho, T. Torizawa, Y. Iwashita, T. Terauchi, A. Mei Ono and P. Güntert, Nature, 2006, 440, 52–57 CrossRef CAS.
- J. Boisbouvier, E. Crublet, P. Gans, L. Imbert and R. Kerfah, Pat., WO2018157942A1, 2017 Search PubMed.
- M. Lazarova, F. Löhr, R.-B. Rues, R. Kleebach, V. Dötsch and F. Bernhard, ACS Chem. Biol., 2018, 13, 2170–2178 CrossRef CAS PubMed.
- V. A. Shirokov, A. Kommer, V. A. Kolb and A. S. Spirin, Methods Mol. Biol., 2007, 375, 19–55 CAS.
- H. Mayerhofer, E. Sautron, N. Rolland, P. Catty, D. Seigneurin-Berny, E. Pebay-Peyroula and S. Ravaud, PLoS One, 2016, 11, e0165666 CrossRef PubMed.
- Y. Miyanoiri, M. Takeda, K. Okuma, A. M. Ono, T. Terauchi and M. Kainosho, J. Biomol. NMR, 2013, 57, 237–249 CrossRef CAS.
- A. Dubey, N. Stoyanov, T. Viennet, S. Chhabra, S. Elter, J. Borggräfe, A. Viegas, R. P. Nowak, N. Burdzhiev, O. Petrov, E. S. Fischer, M. Etzkorn, V. Gelev and H. Arthanari, Angew. Chem., Int. Ed., 2021, 60, 13783–13787 CrossRef CAS.
- A. Martin, T. A. Baker and R. T. Sauer, Nature, 2005, 437, 1115–1120 CrossRef CAS PubMed.
- J. L. Camberg, J. R. Hoskins and S. Wickner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10614–10619 CrossRef CAS.
- J. L. Camberg, M. G. Viola, L. Rea, J. R. Hoskins and S. Wickner, PLoS One, 2014, 9, e94964 CrossRef PubMed.
- A. Ortega, D. Amorós and J. García de la Torre, Biophys. J., 2011, 101, 892–898 CrossRef CAS.
- T. Didenko, R. Boelens and S. G. D. Rüdiger, Protein Eng., Des. Sel., 2011, 24, 99–103 CrossRef CAS PubMed.
- J. Felix, K. Weinhäupl, C. Chipot, F. Dehez, A. Hessel, D. F. Gauto, C. Morlot, O. Abian, I. Gutsche, A. Velazquez-Campoy, P. Schanda and H. Fraga, Sci. Adv., 2019, 5, eaaw3818 CrossRef CAS PubMed.
- Z. A. Ripstein, S. Vahidi, W. A. Houry, J. L. Rubinstein and L. E. Kay, eLife, 2020, 9, e52158 CrossRef CAS PubMed.
- U. A. Wojtyra, G. Thibault, A. Tuite and W. A. Houry, J. Biol. Chem., 2003, 278, 48981–48990 CrossRef CAS.
- T. A. Bell, T. A. Baker and R. T. Sauer, eLife, 2019, 8, e46808 CrossRef PubMed.
- M. M. Klimecka, A. Antosiewicz, M. A. Izert, P. E. Szybowska, P. K. Twardowski, C. Delaunay and M. W. Górna, Molecules, 2021, 26, 5936 CrossRef CAS PubMed.
- M. A. Apponyi, K. Ozawa, N. E. Dixon and G. Otting, in Structural Proteomics: High-Throughput Methods, ed. B. Kobe, M. Guss and T. Huber, Humana Press, Totowa, NJ, 2008, pp. 257–268 Search PubMed.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |