
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixed squaramide thioesters as phosphate group analogues for non-competitive antagonists of the phospholipid-sensing G protein-coupled receptor GPR55
Junpei Abe†
 a,
Xianyue Huang†b,
Nozomi Ishiic,
Itaru Imayoshibde,
Yoshio Hirabayashif,
Ichiro Matsuoc,
Hiroyuki Kamiguchig,
Yukishige Itoa,
Adam T. Guy*bg and
Peter Greimel*g
a,
Xianyue Huang†b,
Nozomi Ishiic,
Itaru Imayoshibde,
Yoshio Hirabayashif,
Ichiro Matsuoc,
Hiroyuki Kamiguchig,
Yukishige Itoa,
Adam T. Guy*bg and
Peter Greimel*g
aGraduate School of Science, The University of Osaka, Toyonaka, 560-0043, Japan
bGraduate School of Biostudies, Kyoto University, Kyoto, 606-8507, Japan. E-mail: guy.adam.3m@kyoto-u.ac.jp
cGraduate School of Science and Technology, Gunma University, Gunma, 376-8515, Japan
dInstitute for Life and Medical Science, Kyoto University, Kyoto, 606-8507, Japan
eCenter for Living Systems Information Science, Kyoto University, Kyoto, 606-8501, Japan
fRIKEN Cluster for Pioneering Research, RIKEN, Wako, Saitama, 351-0198, Japan
gRIKEN Center for Brain Science, RIKEN, Wako, Saitama, 351-0198, Japan. E-mail: petergreimel@riken.jp
First published on 24th April 2026
Abstract
High-affinity inhibitors of specific receptors are a valuable tool to elucidate cellular signaling, control biological systems and develop therapeutic drugs. Here, we report mixed squaramide thioesters as a novel electrophilic handle, demonstrating an optimal balance between nucleophile accessibility and hydrolytic stability in aqueous environments. Density function theory calculations show that the energy of the lowest unoccupied molecular orbital (LUMO) of mixed squaramide thioesters is lower compared to mixed squaramide esters, rendering them more reactive to nucleophilic attack. Synthetic access to various lysophosphatidylglucoside (LysoPtdGlc) analogues incorporating mixed squaramide thioesters as phosphate bioisosteres was readily established by condensation of mixed squaramide esters with the corresponding thiols. Next, we characterised the inhibitory activity of these analogues in biological assays of axon growth cone chemotropism in cultured primary nociceptive neurons. These synthetic analogues induce both acute and sustained (more than 12 h) inhibition of the GPR55/LysoPtdGlc signaling. This inhibition resulted in sustained antagonistic attenuation of GPR55-mediated axon chemotropism while preserving growth cone sensitivity to other GPR55-independent chemotropic signaling molecules. Our findings demonstrate the potential of thiosquaramide-based phosphate bioisosteres as highly specific inhibitors with well controlled reactivity, expanding the repertoire of modulators for lipid-sensing GPCRs.
Introduction
GPR55 is a class A (rhodopsin-like) G protein-coupled receptor that is highly expressed in the nervous system,1 bone marrow and lymphoid tissues, testes and the gastrointestinal tract.2 GPR55 was first designated as cannabinoid receptor3 but has subsequently been reported to be sensitive to lysoglycerophospholipids.4,5 We found that the endogenous lysoglycerophospholipid lysophosphatidyl-β-D-glucoside (LysoPtdGlc, Fig. 1A) acted as the most potent GPR55 ligand, inducing GPR55 dependent chemorepulsion in nociceptive afferents via the LysoPtdGlc/GPR55/Gα13/Rho/ROCK signalling pathway in vitro and in vivo.5 In addition to the role of the LysoPtdGlc/Gpr55 signalling axis in spinal cord sensory circuit development,5 GPR55 has been implicated in modulating inflammatory response,6 neuropathic pain and neurodegeneration.7 | ||
| Fig. 1 Representation of natural GPR55 agonist LysoPtdGlc (A), synthetic analogue LysoSqrlGlc (B) and envisaged Type 1 (C) and Type 2 (D) mixed squaramide thioester analogues. Schematic representation of model substrates (E) and their respective relative HOMO and LUMO energies (F) in aqueous media obtained through DFT calculations with B3LYP/aug-cc-pVTZ basis set. Depiction of LUMO (red/blue) electronic structures at isosurface value of 0.04 (G) of squarate model compounds. | ||
Following extensive screening efforts, a series of small molecule-based agonists8 and antagonists9 for GPR55 were identified. While X-ray crystallography based structural information on GPR55 remains elusive, cryo-EM structures of GPR55-G13 complexes, in the presence and absence of potential ligands, have recently been reported.10,11 The observed heterogeneity in relation to the location of the individual antagonists suggests the existence of a broad variety of distinct agonist binding modes. Further insight into the binding modes of GPR55 agonists and antagonists has been gained by means of homology models and point mutation studies.12,13 These studies have indicated that GPR55 residues K802.60 and E983.29 may potentially contribute to the binding of lysoglycerophospholipid ligands, given their close proximity to the phosphate moiety of the ligand. In addition, the endogenous ligand LysoPtdGlc was used as a lead to develop GPR55 agonists14 and competitive antagonists15 featuring a squaramide as phosphate bioisostere, referred to as LysoSqrlGlc (Fig. 1B).
Beyond lipid-based ligands, squaramide linkages have also been introduced as phosphate diester bioisosteres in oligonucleotides, demonstrating their enzymatic recognition by exonucleases16 as well as their compatibility with DNA duplex formation.17 These observations emphasize the versatility of squarate-based motifs to reproduce essential structural and electronic features of phosphate diesters, supporting their broader implementation in the development of phosphate-mimetic bioactive compounds.
Non-competitive or irreversible antagonists, in contrast to competitive antagonist cause persistent inhibition often over the lifetime of the targeted protein. Non-competitive inhibitors, such as dizocilpine (MK-801), induce a persistent functional blockade through strong, noncovalent interactions.18 In comparison, covalent inhibitors, upon binding to the target protein, an electrophilic moiety is engaged by a nucleophilic residue in close proximity of the binding pocket facilitating the formation of a covalent bond forming an inhibitor-enzyme complex. While high target selectivity requires that covalent bond formation be sufficiently attenuated in solution to minimize nonspecific reactions and ensure aqueous stability, while remaining capable of rapid and efficient engagement upon target binding, where proximity effects and the protein microenvironment can substantially accelerate the reaction. Commonly used electrophilic moieties include acrylamide, vinyl sulphonamide and sulfonium moieties to target cysteine residues, and aryl sulfonyl fluoride, isothiocyanate or thioester moieties to target lysine,19 although reactivity toward cysteine, albeit often reversible, has also been reported for some of these electrophilic moieties.20 Squarate esters have been shown to undergo chemoselective reactions with pKa-depressed amines in neutral aqueous environment, whereas the ε-amino group of lysine required more alkaline conditions (≥ pH 8.5) to undergo reaction.21 This reflects the requirement for amine deprotonation to initiate nucleophilic attack on the electrophilic squarate moiety. A pronounced rate enhancement was observed under neutral conditions in the presence of pKa-depressed cysteine thiols, promoting the formation of transient thioester intermediates that undergo migration to yield corresponding amide products, reminiscent of ligation reactions. Recently, mixed squaramide esters have been demonstrated to retain sufficient reactivity in aqueous environment to covalently modify lysine residues of a target protein,22 presumable rendered sufficiently nucleophlic though local pKa depression. Here we report the development of an non-competitive GPR55 inhibitor based on our LysoSqrlGlc analogue scaffold, and its in vitro evaluation using our previously established primary neuron based activity assay.
Results and discussion
To assess the relative chemical reactivity of mixed squaramide esters and thioesters, density functional theory (DFT) calculations were performed. While earlier studies reported calculations at the B3LYP/6-31G* level,22 the present calculations were carried out at the B3LYP/aug-cc-pVTZ level, whose triple-ζ basis set with diffuse functions enables improved description of delocalized electron density and low-lying virtual orbitals characteristic of extended conjugated systems. We opted to focus on frontier molecular orbitals (FMO), to circumvent the well established basis set dependency23 and inherent neglect of anisotropic charge distribution topography associated with atomic charge assignments. Analogous to acylesters and acylthioesters (Fig. S1A and B), the smaller HOMO–LUMO gap of mixed squaramide thioesters (4.3 eV) compared to mixed squaramide esters (4.9 eV) indicates a higher degree of electrophilicity for mixed squaramide thioesters (Fig. 1E and F). In addition, the lower energy of the mixed squaramide thioester LUMO and hence, smaller gap between the HOMO of the attacking nucleophile and LUMO of the electrophile, renders it more susceptible to a nucleophilic attack compared to a mixed squaramide ester. The LUMO was primarily localised on the vinyl carbon (CV) adjacent to the alkylthiolate and the diagonal carbonyl carbon, rendering a nucleophilic attack on either site possible. This orbital distribution is consistent with resonance structures I and III of the thiosquarate moiety (Fig. S1C), whereas resonance structure II, involving a carbanionic CV adjacent to the alkylthiolate, is expected to be less prominent. In accordance with the documented reduced hydrolytic stability of acylthioesters,24 their relative free energy is elevated in comparison to acylesters (Fig. S1D). In contrast, the hydrolysis rate of vinyl thioethers (vinyl sulfides) has been reported to be approximately 30 times slower than that of vinyl ether homologues,25 due to their lower relative free energy (Fig. S1E). DFT calculations of mixed squaramide thioesters show a lower relative free energy in comparison to their ester analogues (Fig. S1F), which is consistent with the reduced ability of the larger sulphur atom to donate electron density compared to its oxygen counterpart. As a consequence of this thermodynamic stabilisation, hydrolysis of the squaramide thioester becomes markedly less favourable. Taken together, in contrast to their ester analogues, the DFT results suggest an enhanced hydrolytic stability of mixed squaramide thioesters whilst retaining high susceptibility to nucleophilic attack. This observation is consistent with earlier ligation studies proposing transient squaramide thioesters as reactive intermediates that facilitate subsequent amide formation.21 Therefore, we conclude that mixed squaramide thioesters are suitable electrophilic moieties for the development of targeted inhibitors that may engage in nucleophile-driven bond formation at the protein interface.Following the selection of mixed squaramide thioester as electrophilic handle, two types of lead structures were envisaged. In the first type (Type 1 lead), the thioester linkage is positioned between the squaryl and glycerol moieties (Fig. 1C), while in the second type (Type 2 lead), the thioester linkage is located between the carbohydrate head group and the squaryl moiety (Fig. 1D). To investigate the conformational space of the glycosidic linkage of the aforementioned lead structures, DFT calculations at the MP2/6-31g(d) level of model structures were performed (Fig. 2), and compared with a tetrahydropyranephosphatemethyl ester (TPMe) model (Fig. S2). To evaluate the similarity of individual Type 1 model conformations against the lowest energy conformation of the TPMe model (Fig. 2G), the sum of the displacement between the respective terminal methyl groups and between the closest carbonyl oxygen of the squaryl moiety to the non-bonding oxygen of the phosphate moiety was calculated following superposition of their respective tetrahydropyrane moieties. The resulting landscape exhibited a minimum of ∼0.35 nm total displacement that is in close proximity to the lowest energy conformation of the Type 1 model (Fig. 2C, area III, and Fig. 2F) and is associated with an ∼1 kcal mol−1 energy penalty. The lowest energy conformation of Type 1 (Fig. 2B, area I) exhibited a 0.25 nm relative displacement of the terminal methyl groups and 0.16 nm displacement of the closest oxygen atoms compared to the TPMe model (Fig. 2C, area I and Fig. S2C). Together this corroborates the previously reported suitability of squaramides as phosphate diester bioisosters.26
 | ||
| Fig. 2 DFT calculations of model structures (A) at the MP2/6-31g(d) level. Energy landscape of tetrahydropyrane rings featuring an equatorial squaramide (B), Type 1, and an equatorial thiosquarate (D), Type 2. Degree of similarity to the lowest energy conformation of the TPMe model to Type 1 (C) and Type 2 (E) models, expressed as the sum of the displacement between the respective terminal methyl groups and between the closest carbonyl oxygen of the squarate moiety to the non-bonding oxygen of the phosphate moiety. Superposition of the closest matching conformations of Type 1 (F) and Type 2 (H) models against the lowest energy conformation of TMPE (G and gray structure in F and H). Area I, lowest energy conformation; area II, secondary energy minimum; area III, closest matching conformation with TPMe model; carbon, cyan; oxygen, red; hydrogen, white; nitrogen, blue; sulphur, yellow; phosphate, purple. | ||
The energy landscape of the Type 2 differs considerably (Fig. 2D). The lowest energy conformation deviates significantly from the preferred conformation of phosphate diesters (Fig. S2E) as implied by a total displacement of 0.96 nm (Fig. 2E, area I). Conformations associated with the secondary minima (Fig. 2D, area II) carry an energy penalty of ∼0.7 kcal mol−1 and better resemble the TPMe conformation (Fig. 2E, area II and Fig. S2F), as indicated by a 0.46 nm total displacement. Conformations that most closely resemble TPMe (Fig. 2E, area III and Fig. 2H) are associated with a considerable energy penalty of ∼6 kcal mol−1. This suggests that Type 2 compounds exhibit a reduced degree of conformational similarity compared to TPMe and thus are less adequate to act as phosphate diester bioisoster, implying a lower biological activity compared to Type 1 compounds.
Point mutation studies have highlighted the importance of K802.60 for lysolipid-mediated GPR55 activation. Molecular docking simulations of Type 1 and 2 leads using a previously reported GPR55 homology model13,15 with AutoDock have indicated an average lysine Nε to CV distance of 0.44 ± 0.05 nm and 0.40 ± 0.06 nm respectively of the top five conformations, exhibiting a chemically relevant27,28 average distance to facilitate the desired nucleophilic attack. Recent cryo-EM structural information on GPR55 suggests a highly flexible ligand binding pocket. While some ligands exhibit close proximity to K802.60, in other cases the highly flexible extracellular loop 2 (ECL2) partially obstructs access to K802.60. Nevertheless, the extracellular region of GPR55 is decorated with five lysine and two arginine residues that could potentially be targeted.
The synthetic access to Type 1 leads was established from (S)–(−)–glycidol (Scheme 1). Following protection of the primary alcohol as trityl ether, the epoxide was opened with octadecane-1-thiolate under alkaline conditions. Liberation of the primary alcohol and subsequent selective activation yielded the 2,4,6-triisopropylbenzenesulfonate 6. Nucleophilic substitution by S-potassium thioacetate and removal of the acetyl group afforded the glycerol building block 8. Coupling with previously reported14 glucosyl configured squaramide ester 9a gave desired mixed squaramide thioester 1a. Condensation of 8 and 9b readily yielded galactosyl configured 1b. Due to the inherently amphiphilic nature of Type 1 leads, NMR analysis was performed in d6-pyridine. In line with previous reports on squaramides,29 NMR of 1a revealed the presence of a mixture of rotamers which altered its ratio at elevated temperature (Fig. S3).
 | ||
| Scheme 1 Synthetic access to Type 1 lead structures. | ||
To furnish the bridging thioester linking the carbohydrate head group with the squaryl moiety of Type 2 lead structures, we envisaged the condensation of 1-thioglucose (10) with corresponding squaramide ester 11a (Scheme 2). 10 was readily prepared from commercial 1-thio-β-D-glucose sodium salt30 and 11a was accessed by condensation of diethyl squarate14 with the matching aminoglycerol derivative.31 The condensation of 10 and 11a in ethanol as well as DMSO failed to yield the desired product, even at elevated temperature, corroborating previous reports32 on the reduced nucleophilicity of 1-thioglucose (10). To improve the reactivity of 11a, the ethoxy leaving group was replaced with a phenoxy group in 11b, by linkage of the matching aminoglycerol derivative with diphenyl squarate.14 Subsequent condensation with 10 proceeded smoothly in DMSO at room temperature, yielding desired 3. Replacement of 10 with its commercial precursor 1-thio-β-D-glucose sodium salt failed to produce 2 even at elevated temperature.
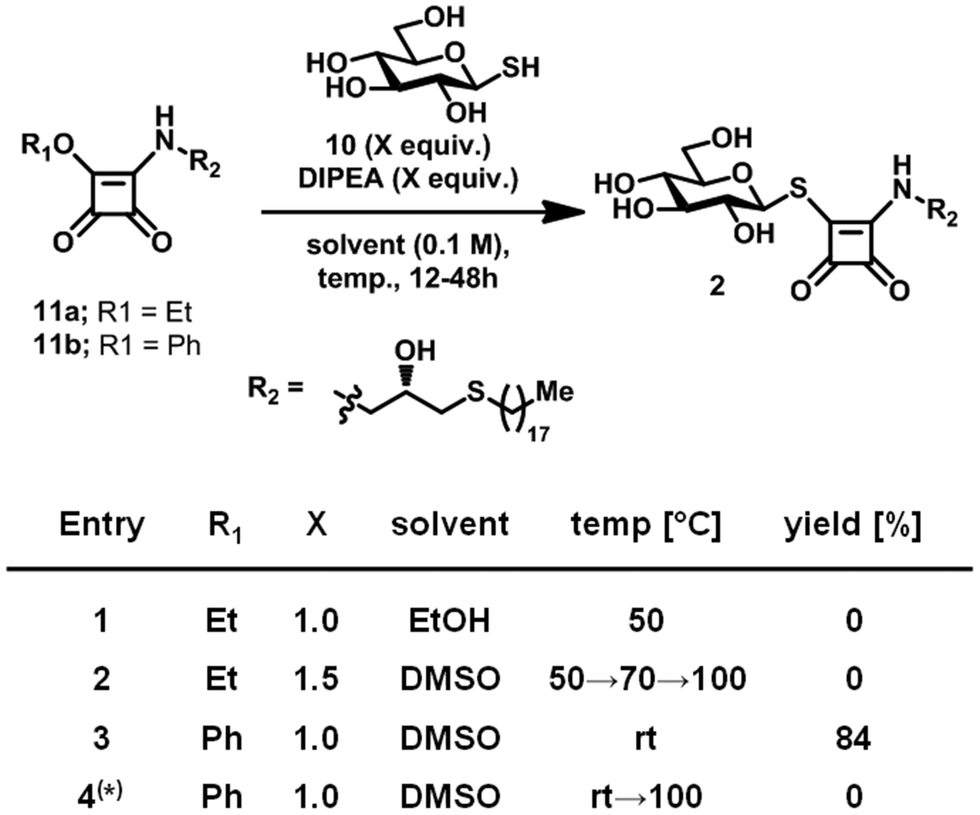 | ||
| Scheme 2 Optimisation of reaction condition towards Type 2 leads. (*) 1-Thio-β-D-glucose sodium salt was used instead of 10. | ||
To evaluate the susceptibility of mixed squaramide thioesters toward nucleophilic attack by lysine Nε and their hydrolytic stability, 1a was incubated with the soluble, capped lysine residue 12 in aqueous solution (Fig. S4A). In agreement with earlier reports on squaramide esters,21 HPLC analysis revealed that after 24 hours of incubation at neutral pH, no detectable formation of the squaramide product 13 occurred, consistent with the low reactivity of the predominantly protonated lysine ε-amino group (pKa ∼10.5). Even after one week, the starting material 1a remained largely intact, with only trace amounts of the squaramide product detected in the presence of protonated Nε (Fig. S4B). In contrast, limited deprotonation of Nε giving rise to a small fraction of neutral primary amino function at a moderate alkaline pH of 9.08 resulted in the formation of minor quantities of the squaramide product within 24 hours, with a significant increase observed after one week. These results suggest that 1a retains substantial hydrolytic stability under neutral and mildly alkaline conditions while maintaining reactivity toward neutral primary amine groups at room temperature.
The biological activity of 1a, 1b and 2 were assessed using an in vitro turning assay with primary cultured sensory neurons endogenously expressing GPR55.5,14 In this assay, axon growth cones exhibit a positive or negative chemotropic response upon exposure to a microscopic concentration gradient of the chemoattractive guidance cue NGF and the chemorepulsive etherLysoPtdGlc (Fig. S5). EtherLysoPtdGlc is a synthetic homologue of LysoPtdGlc (Fig. 1A) that features an ether instead of an ester bond between the glyceryl and acyl moiety, resulting in increased chemical and biological stability but identical signaling activity.13 This assay is highly specific for the LysoPtdGlc/GPR55 signalling axis, as we have demonstrated previously using primary sensory neurons derived from GPR55 knock-out mice.5 The LysoPtdGlc-induced chemotropic responses were abolished in the absence of GPR55, whilst the neurons retained their responsiveness to LysoPtdIns and both attractive and repulsive GPCR-independent chemotropic guidance cues. Bath application of 1a (5 µM) 40 min prior to the challenge with chemorepulsive etherLysoPtdGlc revealed efficient inhibition of the expected chemotropic response (Fig. 3A), consistent with an antagonistic, GPR55-inhibiting activity of 1a. To discriminate between competitive and non-competitive inhibitory activity of 1a, sensory neurons were incubated with 1a for 4 h, washed and subsequently cultured in the absence of 1a for 13 h prior to implementation of the turning assay. Challenge of 1a-pulsed neurons with chemorepulsive etherLysoPtdGlc failed to induce a chemotropic response, suggesting inhibition of the LysoPtdGlc/Gpr55 signalling axis (Fig. 3B). To assess whether incomplete washout of lipophilic 1a might account for the observed lack of response, 1a-pulsed neurons were challenged with a twofold higher concentration of etherLysoPtdGlc. The continued absence of a chemotropic response remained consistent with inhibition of the LysoPtdGlc/GPR55 signalling axis, rendering incomplete washout unlikely. Challenge of 1a-pulsed neurons with chemorepulsive LysoPtdIns elicited the expected negative chemotropic response. This response of 1a-pulsed neurons to LysoPtdIns is consistent with previous observations using GPR55 knock-out neurons5 and GPR55 specific inhibitors13,15 confirming that the chemorepulsive activity of LysoPtdIns acts via a GPR55-independent pathway. Similarly, 1a-pulsed neurons retained chemorepulsive responses to Sema3A, a well-established sensory neuron guidance cue. The maintained chemorepulsive responses to LysoPtdIns and Sema3A indicate that the cellular machinery required for chemotropic axon growth remained functionally intact upon 1a treatment. In all assays, neurons exposed to 1a retained a healthy axon extension rate (Fig. S6). Taken together, these results suggest that treatment with 1a elicits rapid and sustained non-competitive inhibition of GPR55, whilst not affecting other chemotropic signalling mechanisms or overall axon health.
 | ||
| Fig. 3 Chemotropic response in presence of 1a. (A) Axon repulsion was abolished upon 40 min bath application of 1a prior to challenge with etherLysoPtdGlc. (B) 4 h pulse application of 1a followed by a washing step and 13 h incubation in absence of 1a abolished negative chemotropic response to etherLysoPtdGlc, but not to LysoPtdIns and Sema3A. (C) and (D) Representative images at the beginning (left, 0’) and end (right, 45’) of the assay. Graphs: broken line and bars, mean ± standard deviation; grey circles, individual axon responses; images: bar, 20 µm; white arrow, origin of guidance cue gradient. | ||
Bath application of the galacto-configured homologue 1b (5 µM, 40 min) moderately reduced the chemorepulsive response to etherLysoPtdGlc (Fig. 4A). Doubling of the etherLysoPtdGlc concentration restored chemorepulsion, indicating a weak competitive antagonistic activity of 1b against the LysoPtdGlc/GPR55 signalling axis. This low affinity of 1b for GPR55 is in line with previous reports that the galacto-configured homologue of LysoPtdGlc does not exhibit agonistic activity for GPR55.13 Challenge of 1b pulsed neurons with etherLysoPtdGlc elicited a similar chemorepulsive response as observed in vehicle-treated neurons. Extension of the 1b-pulse to 8 h, followed by washing and 13 h of culture in the absence of 1b, likewise failed to restore the moderate inhibitory effect observed during bath application of 1b. Given the highly similar lipophilicity and electrophilic reactivity of 1a and 1b, incomplete washout or unspecific off-target effects are unlikely to account for the persistent inhibition observed for 1a. These findings show that 1b exhibits only weak competitive antagonistic activity toward GPR55 and lacks the lasting non-competitive inhibitory behaviour observed for 1a.
 | ||
| Fig. 4 Chemotropic response of primary cultured sensory neurons endogenously expressing GPR55. (A) Bath and 4 h or 8 h pulse application of 1b (A) and 2 (B) prior to challenge with etherLysoPtdGlc. The datasets used for etherLysoPtdGlc and etherLysoPtdGlc + DMSO (4 h pulse) are the same as used in Fig. 3A and B, respectively. Graphs: broken line and bars, mean ± standard deviation; grey circles, individual axon responses. | ||
Similar to 1b, bath application of Type 2 lead compound 2 (5 µM, 40 min) prior to the challenge with etherLysoPtdGlc exhibited a moderate antagonistic activity against the LysoPtdGlc/GPR55 signalling axis (Fig. 4B). In contrast to 1b, challenge of 2-pulsed neurons with etherLysoPtdGlc retained the moderate antagonistic activity observed in bath application. Extension of the pulse duration to 8 h completely abolished the response to the etherLysoPtdGlc. This enhanced suppression of the chemotropic response upon increased pulse duration of 2 further argues against incomplete washout. In all cases, neurons exposed to 1b or 2 retained a healthy axon extension rate (Fig. S7). Taken together, these results suggest that 2 can establish sustained non-competitive inhibition of GPR55 following extended exposure, but with a significantly lower efficiency compared to 1a.
Summary and conclusions
Highly specific inhibitors capable of extended, non-competitive target engagement represent valuable tools in research, therapeutic intervention and diagnostics by enabling precise control over their biological targets. Here we demonstrate synthetic accessibility and biological applicability of mixed squaramide thioesters as phosphate diester bioisosteres that retain their electrophilic properties in aqueous environment, combining susceptibility to nucleophilic attack by amino and thio functionalities with pronounced hydrolytic stability. Similar to the difference between acylthioesters and acylesters, mixed squaramide thioesters exhibit a smaller HOMO–LUMO gap compared to their mixed squaramide ester analogues, indicating their increased nucleophile accessibility. In contrast to their respective acyl analogues, the relative free energy of mixed squaramide thioesters compared to their ester analogues is significantly lower, mimicking the relationship between vinyl thioethers (vinylsulfides) and their vinyl ether analogues. Collectively, the DFT results indicate that mixed squaramide thioesters compared to their ester analogues achieve enhanced hydrolytic stability through thermodynamic stabilisation, while retaining substantial electrophilic character towards nucleophilic attack.Type 1 and Type 2 lead compounds were readily available via condensation of the squaramide ester with the corresponding thiol moiety. In vitro assays demonstrated substantial hydrolytic stability under both neutral and mildly alkaline conditions, consistent with the DFT predictions. In line with earlier reports21 on mixed squaramide esters and thioesters, nucleophilic attack by the ε-amino group of lysine (pKa ∼10.5) in aqueous solution occurred only under mildly alkaline conditions, where a sufficient fraction of the neutral amine is present. Notably, limited reactivity in solution does not preclude rapid target engagement, as proximity effects and the protein microenvironment, including local pKa depression, can markedly promote nucleophilic attack.
In vitro turning assays using primary sensory neurons demonstrated that Type 1 lead compound 1a exerts strong acute and lasting inhibition of the LysoPtdGlc/GPR55 signalling axis. Even a twofold increase in the concentration of the guidance cue following pulse application failed to overcome 1a-induced inhibition, suggesting either high-affinity binding or a non-competitive inhibitory mechanism.
In contrast, its galacto-configured epimer 1b produced only moderate acute inhibition that was not sustained and easily reversed by raising the guidance cue concentration. This behaviour is consistent with moderate competitive inhibition, in agreement with previous reports describing the inability of the galacto-headgroup featuring LysoPtdGlc analogue to activate the LysoPtdGlc/GPR55 signalling axis.
The Type 2 lead compound 2 displayed moderate acute inhibition of the LysoPtdGlc/GPR55 signalling axis that persisted after pulse application, consistent with moderate non-competitive inhibition. This reduced activity aligns with the DFT analysis, which suggests that Type 2 compounds exhibit lower conformational similarity to phosphate diesters, rendering them less effective phosphate-diester bioisosteres. Interestingly, prolonged exposure to compound 2 resulted in complete and lasting suppression of GPR55 signalling, indicating a time-dependent inhibitory effect, pointing towards a more direct GPR55 engagement.
Neuronal responsiveness to Sema3A and LysoPtdIns remained preserved following pulse treatment with 1a, effectively excluding general toxicity, unspecific impairment of downstream signalling pathways or general disruption of the growth cone turning machinery. Moreover, the differential effects of the two epimers 1a and 1b upon pulse treatment strongly support a stereochemically defined, target-specific mechanism rather than nonspecific off-target cellular effects considering their identical electrophilic reactivity. This divergence closely parallels the GPR55 head group selectivity previously reported13 for the phospholipid analogues LysoPtdGlc (agonist) and LysoPtdGal (inactive).
Incomplete washout of lipophilic 1a seems unlikely, as 1a-pulsed neurons remained unresponsive even when challenged with higher concentrations of etherLysoPtdGlc. By contrast, the equally lipophilic epimer 1b completely lost activity upon pulse treatment, even after prolonged exposure, rendering residual compounds an unlikely explanation for the observed effect.
Collectively, the electrophilic character of the squaramide thioester moiety, the retention of general turning competence, and the strict epimer-dependent and prolonged inhibitory activity point to an amino- or thiol-selective, receptor-mediated mechanism, most plausibly involving direct, potentially covalent, interference with GPR55 signalling. These findings establish mixed squaramide thioesters as potent phosphate-diester bioisosteres exhibiting both hydrolytic stability in aqueous biological environments and with electrophilic reactivity toward amino and thiol functionalities, offering broad potential for the selective targeting of biological macromolecules.
Experimental
The full experimental procedures, equipment used and characterisation data for this article have been included as part of the SI.Author contributions
Conceptualization: PG, ATG and YI; methodology: JA, NI, YI, IM, XH, ATG and PG; investigation: JA, NI, IM, XH, ATG and PG; visualization: JA, XH, ATG and PG; supervision: ATG, JA, YI, II, YH, HK and PG; writing-original draft: PG, ATG, JA and YI. All authors approved the final version of the manuscript.Conflicts of interest
All authors declare no conflict of interest.Data availability
The data supporting the findings of this study are available within the main article and the supplementary information (SI) accompanying this article. All relevant datasets, figures, and tables have been provided as part of the main article and supplementary materials submitted with the manuscript. Supplementary information is available. See DOI: https://doi.org/10.1039/d5cb00231a.Acknowledgements
Funding for this work was provided by Grant-in-Aid for Scientific Research 25K09810 (A.T.G.), 23K27273 (H.K.) and 23K23463 (Y.I.), by the Grant in Aid for Challenging Research (Pioneering) 24K21253 (I.M.), by the Kyoto University Foundation (A.T.G.), by SPRING Fellowship grant JPMJSP2110 (X.H.), by the Program for Technological Innovation of Regenerative Medicine Grant 24bm1123049h0001 (I.I.) and Moonshot Research & Development Program JP22zf0127007 (I.I.) from the Japanese Agency for Medical Research and Development (AMED), by AMED grant JP23wm0625001 (H.K.) and by the Glyco-lipidologue Initiative Program (P.G.) from RIKEN. DFT calculations and docking analysis were performed in part on the RIKEN Hokusai Bigwaterfall 2 supercomputer system.Notes and references
- M. Sawzdargo, T. Nguyen, D. K. Lee, K. R. Lynch, R. Cheng, H. H. Q. Heng, S. R. George and B. F. O’Dowd, Brain Res. Mol. Brain Res., 1999, 64, 193–198, DOI:10.1016/s0169-328x(98)00277-0.
- M. Uhlén, L. Fagerberg, B. M. Hallström, C. Lindskog, P. Oksvold, A. Mardinoglu, Å. Sivertsson, C. Kampf, E. Sjöstedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A.-K. Szigyarto, J. Odeberg, D. Djureinovic and J. O. Takanen, et al., Science, 2015, 347, 1260419, DOI:10.1126/science.1260419.
- E. Ryberg, N. Larsson, S. Sjögren, S. Hjorth, N. Hermansson, J. Leonova, T. Elebring, K. Nilsson, T. Drmota and P. J. Greasley, Br. J. Pharmacol., 2007, 152, 1092–1101, DOI:10.1038/sj.bjp.0707460.
- S. Oka, K. Nakajima, A. Yamashita, S. Kishimoto and T. Sugiura, Biochem. Biophys. Res. Commun., 2007, 362, 928–934, DOI:10.1016/j.bbrc.2007.08.078.
- A. T. Guy, Y. Nagatsuka, N. Ooashi, M. Inoue, A. Nakata, P. Greimel, A. Inoue, T. Nabetani, A. Murayama, K. Ohta, Y. Ito, J. Aoki, Y. Hirabayashi and H. Kamiguchi, Science, 2015, 349, 974–977, DOI:10.1126/science.aab3516.
- P. C. Staton, J. P. Hatcher, D. J. Walker, A. D. Morrison, E. M. Shapland, J. P. Hughes, E. Chong, P. K. Mander, P. J. Green, A. Billinton, M. Fulleylove, H. C. Lancaster, J. C. Smith, L. T. Bailey, A. Wise, A. J. Brown, J. C. Richardson and I. P. Chessell, Pain, 2008, 139, 225–236, DOI:10.1016/j.pain.2008.04.006.
- S. Kallendrusch, S. Kremzow, M. Nowicki, U. Grabiec, R. Winkelmann, A. Benz, R. Kraft, I. Bechmann, F. Dehghani and M. Koch, Glia, 2013, 61, 1822–1831, DOI:10.1002/glia.22560.
- E. Kotsikorou, K. E. Madrigal, D. P. Hurst, H. Sharir, D. L. Lynch, S. Heynen-Genel, L. B. Milan, T. D. Y. Chung, H. H. Seltzman, Y. Bai, M. G. Caron, L. Barak, M. E. Abood and P. H. Reggio, Biochemistry, 2011, 50, 5633–5647, DOI:10.1021/bi200010k.
- S. Heynen-Genel, R. Dahl, S. Shi, L. Milan, S. Hariharan, E. Sergienko, M. Hedrick, S. Dad, D. Stonich, Y. Su, M. Vicchiarelli, A. Mangravita-Novo, L. H. Smith, T. D. Y. Chung, H. Sharir, M. G. Caron, L. S. Barak and M. E. Abood, Probe Reports from the NIH Molecular Libraries Program [Internet], Bethesda (MD): National Center for Biotechnology Information (US), 2011, available from: https://www.ncbi.nlm.nih.gov/books/NBK66153/.
- R. Xia, Q. Yuan, N. Wang, L. Hou, J. Abe, J. Song, Y. Ito, H. E. Xu and Y. He, Cell Res., 2024, 35, 76–79, DOI:10.1038/s41422-024-01044-w.
- H. Chang, X. Li, L. Shen, X. Ge, S. Hao, L. Wu, S. Liu, J. Liu, V. Cherezov and T. Hua, Cell Res., 2024, 35, 80–83, DOI:10.1038/s41422-024-01046-8.
- M. A. Lingerfelt, P. Zhao, H. P. Sharir, D. P. Hurst, P. H. Reggio and M. E. Abood, Biochemistry, 2016, 56, 473–486, DOI:10.1021/acs.biochem.6b01013.
- A. T. Guy, K. Kano, J. Ohyama, H. Kamiguchi, Y. Hirabayashi, Y. Ito, I. Matsuo and P. Greimel, ACS Chem. Neurosci., 2018, 10, 716–727, DOI:10.1021/acschemneuro.8b00505.
- F. Ding, A. T. Guy, P. Greimel, Y. Hirabayashi, H. Kamiguchi and Y. Ito, Chem. Commun., 2018, 54, 8470–8473, 10.1039/c8cc04467h.
- A. T. Guy, F. Ding, J. Abe, M. Inoue, Y. Hirabayashi, Y. Ito, H. Kamiguchi and P. Greimel, ACS Chem. Neurosci., 2020, 11, 3635–3645, DOI:10.1021/acschemneuro.0c00521.
- J. Du, B. Mahcene, V. Martynov, E. Frezza, C. Vasnier, L. Ponchon, D. Coelho, F. Bonhomme, E. Braud, M. Etheve-Quelquejeu and B. Sargueil, Commun. Chem., 2025, 8, 244, DOI:10.1038/s42004-025-01627-7.
- K. Sato, R. Tawarada, K. Seio. and M. Sekine, Eur. J. Org. Chem., 2004, 2142–2150, DOI:10.1002/ejoc.200300682.
- A. Janus, K. Lustyk and K. Pytka, Psychopharmacology, 2023, 240(12), 2435–2457, DOI:10.1007/s00213-023-06454-z.
- L. H. Jones, Chem. Sci., 2025, 16, 10119–10140, 10.1039/D5SC02647D.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62(12), 5673–5724, DOI:10.1021/acs.jmedchem.8b01153.
- P. Sejwal, Y. Han, A. Shah and Y.-Y. Luk, Org. Lett., 2007, 9, 4897–4900, DOI:10.1021/ol702275q.
- K. I. Taylor, J. S. Ho, H. O. Trial, A. W. Carter and L. L. Kiessling, J. Am. Chem. Soc., 2023, 145, 25056–25060, DOI:10.1021/jacs.2c05691.
- J. Rigby and E. I. Izgorodina, Phys. Chem. Chem. Phys., 2012, 15, 1632–1646, 10.1039/c2cp42934a.
- W. P. Jencks, S. Cordes and J. Carriuolo, J. Biol. Chem., 1960, 235, 3608–3614, DOI:10.1016/S0021-9258(18)64517-X.
- R. A. McClelland, Can. J. Chem., 1977, 55, 548–551, DOI:10.1139/v77-076.
- F. Ding, A. T. Guy, P. Greimel, Y. Hirabayashi, H. Kamiguchi and Y. Ito, Chem. Commun., 2018, 54, 8470–8473, 10.1039/c8cc04467h.
- H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067, DOI:10.1021/ja00796a058.
- S. K. Sadiq and P. V. Coveney, J. Chem. Theory Comput., 2014, 11, 316–324, DOI:10.1021/ct5008845.
- K. A. Agnew-Francis and C. M. Williams, Chem. Rev., 2020, 120, 11616–11650, DOI:10.1021/acs.chemrev.0c00416.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802, DOI:10.1002/anie.200903135.
- P. Sejwal, Y. Han, A. Shah and Y.-Y. Luk, Org. Lett., 2007, 9, 4897–4900, DOI:10.1021/ol702275q.
- K. A. F. Banisalman, A. Polykandritou, F. M. Barnieh, G. R. Morais and R. A. Falconer, J. Inorg. Chem., 2022, 87, 14026–14036, DOI:10.1021/acs.joc.2c01651.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |