
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ generation of Cu- and Ag–Sn alloys from metal sulfides for CO2 reduction†
Sebastian A.
Sanden
a,
Anne
Schmidt
b,
Miłosz
Kożusznik
 c,
Yannik
Haver
a,
Yannick
Weidemannn
a,
Kevinjeorjios
Pellumbi
d,
Sven
Rösler
b,
Kai
junge Puring
d,
Andrzej
Mikuła
c and
Ulf-Peter
Apfel
*ad
c,
Yannik
Haver
a,
Yannick
Weidemannn
a,
Kevinjeorjios
Pellumbi
d,
Sven
Rösler
b,
Kai
junge Puring
d,
Andrzej
Mikuła
c and
Ulf-Peter
Apfel
*ad
aRuhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de
bLeuchtstoffwerke Breitungen, Lange Sömme 17, 98597 Breitungen, Germany
cAGH University of Science and Technology, Adama Mickiewicza 30, 30-059 Kraków, Poland
dFraunhofer UMSICHT, Osterfelder Straße 3, 46047 Oberhausen, Germany
First published on 12th March 2025
Abstract
Ag, Cu and Sn based electrocatalysts promise high CO2 reduction kinetics and efficiencies on gas diffusion electrodes. Ag, Cu, Sn sulfide catalysts in particular may offer altered electronic properties and product selectivity, while still being easy to manufacture in scaleable synthesis routes. Comparing the CO2 reduction (CO2RR) performance of Cu3SnS4, Ag3SnS4, Cu2S, SnS and Ag8SnS6 at 100 mA cm−2, formate is found to be the primary CO2RR product with a faradaic efficiency of 57% for Cu3SnS4 and 81% for Ag3SnS4. Characterization by X-ray photoelectron spectroscopy (XPS) and X-ray diffraction revealed the formation of Ag3Sn and Cu3Sn alloys from the corresponding sulfide species during CO2RR. But while the Cu3Sn based electrode surface decomposed into CuO and SnO after 2 h at −100 mA cm−2, metallic Ag3Sn sites on the corresponding electrode surface could be detected by XPS after removing the surface layer. Using density functional theory, the binding energies of *H, *CO and *OCHO on Cu3Sn and Ag3Sn were computed to identify possible catalytic sites. Thereby, Sn was found to render both Cu and Ag highly oxophilic resulting in strong adsorption of carboxylic functionalities, enabling formate production with a partial current density of up to 162 mA cm−2.
Introduction
CO2 reduction on large scales is imperative for decreasing the impact of greenhouse gases and creating a circular carbon economy. Electrochemical CO2 reduction reaction (CO2RR) offers a selective way to recycle CO2 using renewable energies. To realize this goal by obtaining high CO2 conversion and energy efficiency, gas diffusion electrodes (GDE) need to be employed to avoid limitations concerning CO2 mass transfer to the catalyst while operating at current densities >100 mA cm−2.1 While the catalyst material of the GDE is crucial to CO2RR and a plethora of metal chalcogenides and molecular catalysts have been tested, the application of strong reductive currents can lead to the degradation of the catalyst.2–4In situ X-ray absorption spectroscopy measurements have demonstrated the reduction of CuO catalysts to Cu0 and SnO to Sn0 and therefore, the catalytic properties of plain metal electrodes remain relevant.5,6Nano-structuring of electrocatalysts can significantly enhance their catalytic efficiency,7–9 but up-scaling such materials to industrial scales and the implementation into large electrolyzers that meet the global demand of CO2 reduction, appears impractical. The in situ generation of catalytic sites from bulk materials under electrolytic conditions offers an alternative approach that alleviates any additional synthetic steps and demands. Recently, a Cu2SnS3 and CuS composite was reported to produce formate at a partial current density of up to 240 mA cm−2, with the catalytically active species, a Cu24Sn20 alloy, being generated in situ during electrolysis.10 With Cu-based catalysts being known to be prone to corrosion,11 an AgSn alloy promises a higher corrosion resistance and a lower oxophilicity. Therefore, we prepared silver tin sulfide catalysts for comparison to their respective copper tin sulfides counterparts concerning their electrocatalytic performance and corrosion stability. Previous work on AgSn-alloys explored the ideal stoichiometry for AgSn alloys with a SnO2 surface for CO2 reduction with Ag3Sn obtaining a partial current density for formate production of 25.4 mA cm−2 at −0.8 V vs. RHE using a H-type cell.12 But instead of utilizing a nanostructured core–shell-type catalyst with an Ag3Sn alloy core and a SnO surface, the corresponding Ag3SnS4 sulfide material offers the possibility of generating the alloy in situ under electrocatalytic conditions. Here, we envisioned that transferring the previously optimized noble metal to tin ration of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to CuSn alloys may thus lead to the optimum composition of CuSn alloys for CO2RR and allow for the comparison of their electrocatalytic properties and stabilities with Ag3Sn.
:1 to CuSn alloys may thus lead to the optimum composition of CuSn alloys for CO2RR and allow for the comparison of their electrocatalytic properties and stabilities with Ag3Sn.
In the herein presented work, we prepared Cu3SnS4, Ag3SnS4 and Ag8SnS6 through mechanochemical or high-temperature synthesis to assess the CO2 reduction activity of the corresponding Cu3Sn, Ag3Sn and Ag8Sn alloys. The targeted metal sulfide stoichiometries Cu3SnS4 and Ag3SnS4 were found to consist of Cu2SnS3 and Ag8SnS6 phases respectively with an excess of amorphous SnS, thus being composite materials rather than pure metal sulfide phases. Upon reduction these mixed phases were expected to yield the previously reported ideal Ag3Sn ratio and its Cu3Sn analogue. For AgxSnSy the corresponding alloying processes similar to Cu3SnS4 to Cu3Sn have not been reported yet, although Ag, Sn are among the most prominent metals for CO2RR catalysts.11,13 The compositions of these electrodes were spectroscopically characterized after electrolysis to determine the changes in chemical speciation on the electrode surface and the bulk material. Furthermore, we evaluated the catalytic processes using density functional theory by calculating the binding energies of *H, *CO and *OCHO on Cu3Sn and Ag3Sn. Based on these results, possible catalytic sites and the influence of Sn are discussed, as well as the stability of the alloy surfaces under electrocatalytic conditions up to −300 mA cm−2.
Experimental section
Material synthesis and characterization
For synthesis of Cu3SnS4 and Ag3SnS4 a planetary ball mill (Fritsch, Pulverisette 7 premium line) was employed. The synthesis of Cu3SnS4 was performed by weighing stoichiometric amounts of Cu2S (99%, Tribotecc), SnS (99%, Tribotecc), and elemental sulfur (99.9%, Sigma Aldrich) for a batch size of 4 g. The reagents were added together with 24 g of 2 mm ZrO2 balls into the milling vessel under argon atmosphere. Afterwards, the reaction mixture was milled at 1100 rpm for 2 h.Ag3SnS4 was prepared using stoichiometric amounts of Ag2S (99%, Tribotecc), SnS and S0 and 5 g of the powders were milled with 24 g of 2 mm ZrO2 balls at 1100 rpm for 2 h. Ag8SnS6 was synthesized from the above-described powders using evacuated quartz ampules, which were gradually heated to 900 °C within 24 h, and the final temperature kept constant for 72 h. The resulting Ag8SnS6 ingot was reduced to particulate size by ball milling the ground powder at 350 rpm for 30 min. The milled material consisted of 5 g of 2 mm sized ZrO2 balls and 200 mg of Ag8SnS6 including 10 wt% stearic acid, which was subsequently removed by washing with isopropanol.
The identification of the catalyst materials was carried out using a Bruker Phaser D2 powder diffractometer equipped with a Cu Kα radiation source (λ = 1.5406 Å) at 30 kV and 10 mA. The open crystallography database was used for the identification of the prepared materials.
The prepared electrodes were analyzed by a powder X-ray photoelectron spectroscopy using micro-focused Al Kα X-rays of a Nexsa G2 Surface Analysis System (ThermoFischer). The detector was 128-channel together with a 180°, double-focusing, hemispherical analyzer. The electrodes were analyzed using a band pass energy of 50.0 eV and a 200 μm wide analysis area; and the resulting peaks were fitted using a Shirley-type background and a Lorentzian line shape, as implemented in CasaXPS. Ag0 (99.9%, Abcr) and Ag2S (99.9%, Abcr) powders were used as references for the measurement of the Auger parameters of silver. If indicated, additional Ar sputtering of the samples was performed for 100 s with a set cluster size of 300 and an ion energy of 6000 eV to remove surface layers.
Scanning electron microscopy images and energy dispersive X-ray spectra and maps were recorded with a Dualbeam FIB-SEM SCIO2 (ThermoFischer) equipped with a Ultimax silicon drift detector (170 mm2, Oxford Instrument) for EDX. Electron microscopy images were recorded at an acceleration voltage of 4 kV, while EDX mappings were performed at 20 kV.
Testing electrochemical activity for CO2RR
The GDE were prepared using H23C6 carbon paper (Freudenberg) and drop coated with ink containing 10 mg mL−1 catalyst and 20 wt% binder in respect to the resulting catalyst layer after drying. For the dispersion of the catalyst ink a 2:1 v/v isopropanol and water solution was sonicated for 30 min. To obtain electrodes with a loading of 3 mg cm−2, the resulting ink was drop casted in 0.2 mL steps on carbon paper, which was heated to 75 °C. Cu2S and SnS GDE as reference experiments were prepared using the reagents for the Cu3SnS4 synthesis.
Electrocatalytic CO2 reduction experiments were performed using an in-house built electrolyzer using titanium plates as flow fields and copper current collectors.14 In the assembled electrolyzer, the GDE have an active area of 2 cm−2, encased by PTFE gaskets, and are in contact with a catholyte chamber of a 2 mL volume. 40 mL of 1 M KOH (hyd.) (≥86.0%, Fisher) was supplied as anolyte and catholyte through a Minipuls 2 pump (Gilson) at a 15 mL min−1 flow rate. Mini HydroFlex RHE (Gaskatel) were employed as reference electrode. Nafion™ N117 (Chemours) was used as ion exchange membrane and nickel foam as anode (Goodfellow, 99.5%, porosity 95%, 1.6 mm thickness). The flow field on the cathode was passed over with 20 mL min−1 CO2 (air liquide, >99.95 vol%) and 2 mL min−1 N2 (air liquide, 99.999%) controlled by mass flow controllers (EL-Prestige, Bronkhorst). For each experiment, two linear sweep voltammograms (LSV) were recorded before and after 2 h chronopotentiometries. LSV were measured in the potential range from 0 V to −1.5 V vs. RHE with a scan rate of 50 mV s−1. If not stated otherwise, chronopotentiometries were measured with a current density of 100 mA cm−2, in respect to the geometric area of the electrode. The gaseous products were analyzed with an online gas chromatograph (Agilent Technologies 7820A), equipped with two columns: HP-Molsieve 5 Å 30 m, ID 0.53 mm, 25 μm film and HPPLOTQ 30 m, ID 0.53 mm, 25 μm film, as well as a flame ionization detector (FID) and a thermal conductivity detector (TCD). The gaseous products were analyzed every 30 min by the FID-TCD and 1 mL of catholyte was sampled for liquid analysis.
The liquid samples were analyzed in a GC–MS-QP2020 gas chromatograph equipped with a HS-20 headspace analyzer and a SH-Rtx-200MS column. Since formic acid was difficult to be chromatographically separated from the solvent peaks, the samples were derivatized using 1-propanol (HPLC grade, Sigma-Aldrich) and concentrated sulfuric acid. The samples were prepared by diluting 100 μl sample with 300 μl of 1 M KOH solution and to derivatize formate to propyl-formate, 100 μl of sulfuric acid (95–97%, Sigma Aldrich) and 500 μl of 1-propanol were added. The GC vials were then heated in the GC–MS autosampler for 20 min at 60 °C before injection. Standard dilutions of formic acid (≥96%, Sigma Aldrich) derivatized by this method were used as calibration standards.
Theoretical investigations via DFT
Density functional theory (DFT) calculations were performed within the DFT formalism using VASP code15,16 and the Perdew–Burke–Ernzerhof (PBE)17 potential with DFT-D2 dispersion to correct for van der Waals interactions. The plane-wave energy cut-off was set to 520 eV and the k-point mesh was generated using the Monkhorst–Pack scheme with distances of 0.025 Å. 2 × 2 × 2 M3Sn (M = Ag, Cu) supercells were modelled using VESTA18 and the resulting structures were relaxed using convergence criteria of 1 × 10−5 eV and 2 × 10−2 eV Å−2 for electronic and ionic relaxation, respectively. After relaxation, the energy of the systems was evaluated again, using a stricter electronic convergence criterion of 5 × 10−6 eV and subsequently, slab models of (100), (110) and (010) planes were created with a vacuum level of 15 Å (ESI,† Fig. S20) and were again relaxed with two bottom layers being fixed in the initial positions.The (010) surface was the focus of the investigation due to the presence of multiple non-equivalent adsorption sites, as well as the lowest surface energy. Placing the adsorbate on the surface resulted in surface coverage of 0.125 ML (1 × 1). The same numerical parameters and convergence criteria were used for supercells and pristine slabs, except for systems with *OCHO adsorbate for which the ionic convergence criterium was increased to 5 × 10−2 eV Å−2. Additional details concerning the theoretical investigations are described in the ESI.†
Results and discussion
Three metal sulfides with the stoichiometries Cu3SnS4, Ag3SnS4 and Ag8SnS6 were synthesized to yield upon electrochemical reduction the corresponding alloys of Cu3Sn, Ag3Sn and Ag8Sn. Here, Cu3SnS4 and Ag3SnS4 represent mixtures of crystalline and amorphous metal sulfide phases prepared by mechanochemical synthesis in a planetary ball-mill from Cu2S and elemental Ag, Sn and sulfur, following previously established procedures.19,20 To track the apparent stoichiometries from the initial synthesis throughout the electrode preparation, PXRD and SEM/EDX are measured of the synthesized powder, the prepared electrodes and the resulting electrode material after electrolysis in 1 M KOH and room temperature at up to 300 mA cm−2.
Fig. 1(A) depicts powder X-ray diffraction patterns of Cu3SnS4 which matches the highest intensity reflexes of Cu2SnS3 at 28.4°, 32.8°, 47.2° and 56.0°. EDX mappings of electrodes coated with this material find a stoichiometry of 3.16:1.06:4.0 for Cu, Sn and S (ESI,† Fig. S3), which is close to the nominal stoichiometry albeit with a slight excess of copper. The PXRD pattern is preserved after preparation of the GDE, aside from a broad reflex at 26° originating from carbon paper. (Fig. 1(C)) Post-electrolysis, the PXRD pattern of the Cu3SnS4 GDE shows a new broad reflex at 42.9°, which matches the reported reflexes of a Cu3Sn alloy, akin to the findings of Li et al., who started from a Cu2SnS3 precatalyst and obtained a Cu24Sn20 alloy through electrolysis; as confirmed by in-operando XPS and XRD. Several other reflexes visible <40° in the here prepared GDE post-electrolysis could not be clearly assigned, but as elucidated below by XPS and EDX, metal oxides and hydroxides are the most abundant species aside from Cu3Sn.
 | ||
| Fig. 1 Powder X-ray diffraction patterns of Cu3SnS4 (A) and Ag3SnS4 (D) as synthesized displayed in black, coated on a GDE (red) and of the GDE post-electrolysis in blue. Faradaic efficiencies obtained during CO2RR for Cu3SnS4 (B) and SnS at 100 mA cm−2 (C) and Ag3SnS4 (E) and Ag8SnS6 (F) at 100 mA cm−2 for 1 h and 300 mA cm−2 for 1 h. CO2RR measurements were conducted in triplicates and standard deviations are depicted as ranges at the corresponding FE. The average of the corresponding electrode potentials in V referenced to RHE with IR correction are displayed in red, with the standard errors indicated as shaded areas. | ||
The Cu3SnS4 pre-catalyst has a high activity in respect to CO2RR with an average of 56.8 ± 5.7% formate over 2 h electrolysis, 17.2 ± 0.8% CO and 19.3 ± 2.0% H2 at an electrode potential of −1.0 V vs. RHE and 100 mA cm−2. To ascertain whether a CuSn alloy is responsible for the observed electrochemical activity or rather Cu or Sn oxides, the starting materials Cu2S and Sn used in the synthesis of Cu3SnS4 were also employed as electrocatalysts. Residual sulfur in copper sulfide electrodes for CO2RR has recently been found to reduce hydrogen evolution compared to pristine copper electrodes while producing formate with high FE.21 Similarly, SnS is known to be a highly active formate producing catalyst, but with an average potential of −1.33 V vs. RHE and 67.4 ± 7.1% H2, CO2RR performance is comparatively low (ESI,† Table S5). Together with the gradual drop in FEtotal, SnS and its derivatives such as metallic Sn or SnO appear to not be responsible for the observed activity of Cu3SnS4 and require nano-structuring of the electrode surface for higher FE.22 Cu2S shows an even lower performance with an average potential of −1.72 V vs. RHE and FEHCOO− of 29.7 ± 7.4% and FECO of 8.0 ± 0.7% and a decrease in FEtotal down to <30% after 2 h of electrolysis, likely due to corrosion. (ESI,† Fig. S1).
Under alkaline conditions, Cu based catalysts are often subjected to severe corrosion and oxidation; and we therefore synthesized and tested its more noble counterpart Ag3SnS4.11,23Fig. 1(D) displays the PXRD of the as prepared powder of Ag3SnS4, which contains Ag8SnS6 as a crystalline phase and a stoichiometry of Ag2.56Sn0.97S4 based on EDX, when normalized to the sulfur content. (ESI,† Fig. S5) Post-electrolysis, the highest intensity reflexes at 39.6° and 37.6° match the reported reflexes for an Ag3Sn, which indicates that Ag3SnS4 is converted, similar to Cu3SnS4, into its corresponding alloy.10
Ag3SnS4 shows an even higher FEHCOO− than Cu3SnS4 of initially 81.2 ± 2.9% at 100 mA cm−2 and 7.6 ± 0.7% CO and 10.8 ± 0.4% H2 at −0.54 V vs. RHE (Fig. 1(E)). Based on the higher performance of this catalyst, the current density was increased to 300 mA cm−2 after 1 h. An average potential of −1.0 V vs. RHE was recorded at 300 mA cm−2 but FEHCOO− experienced a gradual drop from 53.5 ± 4.3% to 44.9 ± 11.2%. As the prepared Ag3SnS4 also consists of Ag8SnS6, we synthesized the corresponding mineral phase to test whether the observed CO2RR activity may also be obtained from an alloy with an 8:1 Ag:Sn ratio. The actual stoichiometry of the prepared GDE of Ag8SnS6 was Ag8.9Sn0.9S6 (normalized to sulfur) and deviates from the expected ratio due to partial oxidation of the catalyst, as 1.1 eq. of oxygen was detected as well (ESI,† Fig. S7). During CO2RR the FEtotal of Ag8.9Sn0.9S6 drops from 73.6% with 51.6% FEHCOO− down to 43.9% FEtotal after 1 h at 100 mA cm−2. The CO2RR activity of Ag8.9Sn0.9S6 is significantly lower than Ag3SnS4 and Cu3SnS4 and after 1 h at 300 mA cm−2, only 14 ± 7.2% FEHCOO−, 2.1 ± 1.9% FECO and 6.2 ± 4.6% FEH2 were measured. Compared to Ag3SnS4, and especially at 300 mA cm−2, FEtotal below 100% and a more negative electrode potential of about −1.3 V were recorded. No additional CO2RR products were observed using GC–MS, which may relate the parasitic currents to an inefficient restructuring process of Ag8.9Sn0.9S6 to an AgSn alloy or hydroxylation of the electrode surface.
Post-mortem surface analysis
To assess changes in chemical composition on the electrode surface post electrolysis, XPS and EDX spectra were recorded for Cu3SnS4, Ag3SnS4 and Ag8SnS6. While the as prepared Cu3SnS4 exhibits a strong signal at 932.7 eV with 67% of the entire spectral area, small signals corresponding to Cu2O (932.2 eV), Cu(OH)2 (934.7 eV), CuO (933.7 eV) and CuSO4 (935.8) eV are detected (Fig. 2).24 High intensity S 3/2p and S 1/2p signals for metal sulfide species are measured at 161.7 eV and 162.9 eV. EDX images of the electrode surface before electrolysis similarly show a homogenous distribution of Cu, S and Sn and only traces of oxygen. (ESI,† Fig. S1) After electrolysis, 77.6% of the Sn 3d spectrum is assigned to SnO and sulfidic species only make up 4.2% of spectral intensity. The Sn 3d5/2 transitions observed at 486.3 eV did not exhibit a shift in binding energies after electrolysis, which signifies an isoelectronic change from Sn2+ in Cu3SnS4 to SnO. Blue colored, spherical copper oxide crystals were visible and a phase separation between Cu2O and SnO on the electrode surface was also detected via EDX (Fig. 2(D)). Only traces of sulfide were detected via XPS and EDX. This indicates a near complete loss of sulfur during the electroreduction of Cu3SnS4 to Cu3Sn (Fig. 2(C), inset). Additionally, about two equivalents of Cu are leached from the electrode surface. Normalizing the Cu and O content to Sn in the EDX spectra yields a ratio of 1.18:1.03:1. (ESI,† Fig. S4) While the bulk of the electrode contains an Cu3Sn alloy according to PXRD, the surface is populated with Cu2O and SnO species, which are likely formed after electrolysis by oxidation in air. Li et al. determined through in situ XRD and XPS a metallic Cu24Sn20 alloy at the surface of Cu2SnS3 as catalytically active sites.10 Similarly, Cu3Sn is considered here to be the catalytically active species since the applied electrode potential of −1.2 V exceeds the known stability range of copper tin sulfides, resulting in metallic phases.25
 | ||
| Fig. 2 XPS and EDX Imaging of Cu3SnS4 electrode surfaces before (panel (A), (B)) and after electrolysis (C), (D) at 100 mA cm−2. The XPS spectra depict the Cu 3d transitions with orange peaks corresponding to metal sulfides, green to Cu2O, purple to Cu(OH)2, CuO (blue), and dark green to CuSO4. The corresponding S 2p regions are shown as insets. Overlays of EDX mappings before and after electrolysis (B), (D) are composed of the EDX mappings shown on the right with Cu Kα in blue, sulfur in orange, Sn Lα in red and oxygen Kα in green. | ||
For Ag3SnS4 and Ag8SnS6, the extent of oxidation of the electrodes post electrolysis was expected to be diminished. However, SEM/EDX mappings of the electrodes post electrolysis showed a drastic morphological change of the electrode surface. The initial micrometer sized particles of metal sulfide were replaced by a fibrous structure consisting largely of C, O and Ag. The Ag3Sn electrode consisted of 55.4% O, 34.5% C and 9.7% Ag and Ag8Sn of 46.6% O, 30.2% C and 22.4% Ag (ESI,† Fig. S6 and S8). Only trace amounts of tin were detected for both electrodes. Likewise, 0.2% and 0.7% S were found for Ag3Sn and Ag8Sn respectively and most of the electrode surface is covered by carbonate salts. To remove the carbonate layer and allow an accurate assessment of surface of the suspected Ag3Sn alloy, the electrodes were sonicated in HPLC water.
XPS Sn 3d spectra of the prepared electrodes show two peaks corresponding to Sn4+ at 487.1 eV and Sn2+ at 486.2 eV for both Ag3SnS4 and Ag8SnS6 (Fig. 3(A) and (B)). The mineral Ag8SnS6 consists of Sn with a net oxidation state of +4, but for Ag8SnS6, as well as Ag3SnS4, Sn4+ only makes up 24.5 and 43.7% of the Sn 3d spectrum respectively.26 Sn2+ is likely related to oxidation of the surface in air to SnO, although metastable SnO2 species may persist under electrocatalytic conditions.27 Post electrolysis and after removal of the carbonate layer, the surface of Ag3Sn consists of 63.1% Sn2+, 33.2% Sn4+ and 3.7% metallic Sn0, whereas Ag8Sn consists of 84.7% Sn2+ and 13.8% Sn0 (ESI,† Fig. S12). While metallic Sn is expected for alloys, the tin oxide species present on the surface are proposed to be rapidly formed on air after removal of the surface layer. The surface of Ag3Sn appears to be more prone to oxidation and exhibits a higher oxophilicity compared to Ag8Sn, which may also indicate a higher possible binding strength of carbonates and CO2RR intermediates such as *OCHO producing formate.28 While metallic Sn was present on Ag8Sn, for Cu3Sn no metallic species were detected on the electrode surface by XPS. Together with the extensive morphological changes evidenced by SEM, this shows a higher corrosion stability of silver tin alloys compared to copper tin alloys.
 | ||
| Fig. 3 X-ray photoelectron spectra in the Sn 3d region of Ag3SnS4 and Ag8SnS6 before and after electrolysis, panel (A)–(D) respectively. The peak corresponding to Sn at a 4+ oxidation state is depicted in green, 2+ in blue and metallic tin in orange. Scanning electron microscopy images of Ag3SnS4 and Ag8SnS6 before electrolysis (E), (F) and after electrolysis (G), (H) performed at 100 mA cm−2 and 300 mA cm−2 for 1 h each. The Auger parameters determined from the silver 3d and M4N4,5N4,5 transitions are indicated for the corresponding sample on the right, pre electrolysis and post electrolysis, with and without removal of the surface layers through ultrasonication. | ||
The oxidation state of silver before and after electrolysis was tracked by determining the Auger parameter of silver 3d and M4N4,5N4,5 transitions. The Auger parameters of Ag3SnS4 and Ag8SnS6 are initially closely centered around Ag1+ similar to Ag2S at 724.6 eV and after electrolysis, Ag3Sn is located at 724.9 eV indicating only minor changes due to electrochemical reduction. Ag8Sn however, displays metallic character similar to AgO with 725.5 eV. After electrolysis and removal of the surface oxide layer through sonication in HPLC water for 30 s, a significantly more metallic bulk material was exposed for both silver tin materials. With 725.9 eV, Ag3Sn now has a metallic character similar to Ag0 at 725.9 eV and Ag8Sn to an even greater extent with 726.0 eV. A value exceeding Ag0 is surprising. However, a similar effect was observed in CuHf alloys and analogously, the shift of the Auger parameter is likely related to charge transfer of Sn to Ag, as well as a shift of the typical CO2RR product spectrum of metallic Ag from CO to HCOO−.29 The increased metallic character of silver is also confirmed by Bader charge analysis. In both 2 × 2 × 2 supercells the surface slabs, Ag atoms exhibit a negative residual charge near Sn in comparison to metallic silver. An exception to this observation is the (010) slab, where net positively charged Ag atoms are also present (ESI,† Fig. S24).
PXRD recorded of Ag8SnS6 after electrolysis also showed predominantly an Ag0 phase at 38.3° and only a minor reflection of a possible Ag3Sn alloy at 39.5° (ESI,† Fig. S2). Together with the main CO2RR product of Ag8SnS6 still being formate – rather than CO as expected for metallic silver – an AgSn alloy appears to have formed during electrolysis. SEM analysis of the GDE surface only finds traces of Sn, which suggests a low number of possible, accessible catalytic sites consisting of Sn due to carbonate adsorption and precipitation (ESI,† Fig. S8). The large parasitic currents observed for CO2RR with Ag8Sn6 might therefore be related to accumulation of carbonate layers on the electrode, aside from a possible inefficient restructuring process.
Adsorption energies of the catalytic sites
To investigate potential compositions of the catalytic sites and whether product inhibition could occur, we calculated the adsorption energies of *H, *CO and *OCHO on the surfaces of Cu3Sn and Ag3Sn alloys. Thus, supercells and consequently slab models of M3Sn (M = Ag, Cu), characterized by an orthorhombic Pmmn structure, were considered. To describe the enhanced selectivity towards CO and formate, *CO and *OCHO molecules were chosen as adsorbates. Additionally, proton adsorption was also considered due to hydrogen evolution being a competing process. To assess the stability of the modelled surfaces, the surface energy Esurf of the slabs was evaluated (ESI,† Tables S2 and S3). The (010) surface was the focus of the investigation due to the presence of multiple non-equivalent adsorption sites, as well as the lowest surface energy.The catalytic activity of M3Sn materials towards particular catalytic processes is directly reflected in the energy diagrams. The changes in Gibbs free energy for various adsorbates on (010) planes of M3Sn are depicted in Fig. 4(a)–(c). Generally, ΔG for all processes is more exergonic in Cu-based systems compared to Ag3Sn considering monometallic and trimetallic binding sites. The smaller radius of copper results in shorter intermetallic distances, leading to charge accumulation in smaller spaces. This promotes adsorption processes and facilitates charge transfer from the material surface to the adsorbate. Multi-metallic active sites (MM, MMM), whether in Cu3Sn or Ag3Sn, are overall more effective regardless of the process or adsorbate considered. This configuration likely arises from a local minimum in electron density placed between the atomic spheres of surface atoms, which is particularly significant in HER. Especially the CuCuCu active site exhibits significantly lower binding energy for *H with −0.23 eV, suitable for HER (Fig. 3(a)).30 The adsorption site with the second lowest energy, AgAgAg with 0.05 eV, already partakes in an endergonic adsorption process, followed by energetically unfavorable adsorptions at AgAg and CuCu sites with 0.17 and 0.25 eV respectively. These calculations align with the observation that Cu3Sn produces more H2 than Ag3Sn (19% and 11% FEH2 at 100 mA cm−2 respectively), likely at CuCuCu sites. Multi-metallic sites in the near vicinity of Sn appear unfavorable for HER with >0.25 eV. Monometallic active sites, especially those involving Sn are distinctly disadvantageous for HER, consistent with other studies, where vacancies typically constitute ideal active sites for binding with an individual transition metals.31
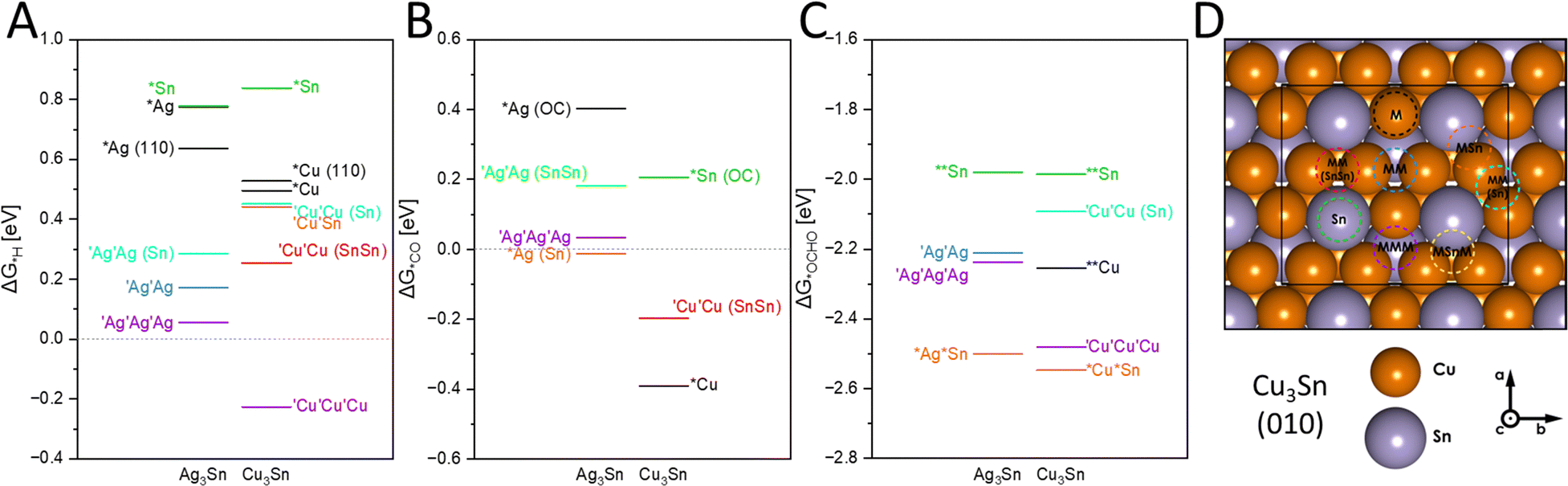 | ||
| Fig. 4 Calculated Gibbs free energy of adsorption on M3Sn (010) surfaces with panel A corresponding to *H, panel B to *CO and C to *OCHO, using PBE-D2. For *CO adsorption rotation of the adsorbent and coordination with O was observed (abbreviated as OC) and for *OCHO, binding through both O occurred. (abbreviated *O*OCH). Panel D displays the investigated binding sites on the example of Cu3Sn, with a surface coverage of 0.125 ML (1 × 1). An asterisk * denotes a single bond (or close-range interaction) between an adsorbate atom/molecule and the element after the asterisk and a quotation mark indicates a more diffuse bonding interaction distributed among atoms marked with single quotation marks. | ||
Adsorption of CO on the (010) surface occurs overall in a more spontaneous reaction than HER (Fig. 4(b)), preferentially on monometallic Cu and Ag or CuCu (−0.39, −0.01 and −0.19 eV respectively). Conversely to the observations for HER, monometallic active sites exhibit lower energies for CO adsorption. In our electrochemical experiments, a higher FECO of 17.2% was obtained with Cu3Sn, whereas Ag3Sn produced only 7.6% at 100 mA cm−2. With Cu and CuCu sites allowing for a stronger binding of *CO, than at a monometallic Ag site, CO2RR to CO appears favored on these Cu sites compared to Ag. It is also worth noting that in the case of *Sn and *Ag sites, the relaxation process resulted in molecule rotation, facilitating binding via oxygen. This initially led to the hypothesis that *OC could be the preferential binding mode. However, subsequent calculations refuted this hypothesis due to the significant distances observed between the molecules and the surface (ESI,† Table S4).
The free energy of adsorption of the CO2RR intermediate *OCHO ranges from −1.9 to −2.9 eV (Fig. 4(c)) and demonstrates the high oxophilicity of M3Sn surfaces. The energetically most favorable adsorption sites are CuSn and AgSn with −2.55 and −2.53 eV, as well as the trimetallic sites CuCuCu and AgAgAg with −2.48 and −2.24 eV respectively. Here, a beneficial influence of tin atoms in the vicinity of Cu and Ag is suggested, as evidenced by the lowest energies of bimetallic AgSn/CuSn sites. Ternary active sites follow in sequence, consistently demonstrating high activity in catalytic processes, albeit strongly favoring *OCHO binding over *H and *CO. Two binding modes for *OCHO were analyzed: *OCHO and *O*OCH, with the latter mode being more prevalent on bimetallic and monometallic sites. Monodentate binding modes of *OCHO on Ag are hypothesized to produce CO and in the here studied surfaces, the reaction pathway from the bidentate *O*OCH binding mode to formate is more energetically favorable after optimization (ESI,† Fig. S4).28
Overall, *OCHO adsorption is the most spontaneous process, followed by *CO, and least favorable for *H, with multi-metallic active sites being highly desirable for all these processes. The presence of tin generally modulates the energetics of the adsorption towards higher energies, rendering the reactions less or even non-spontaneous in some cases, except for *O*OCH adsorption where bimetallic MSn sites are particularly active. The high binding affinity for *OCHO and *O*OCH on Ag3Sn and Cu3Sn may also serve as a proxy for the binding of bicarbonate species, for which we observed extensive coverage on the electrode surfaces by EDX and XPS. The parasitic currents observed at −300 mA cm−2 could be related to increased bicarbonate binding on the catalytic sites. A pulsed application of the applied voltage has been shown to alleviate carbonate formation and could potentially be applied to the AgSn-catalysts.32
Conclusions
Cu3SnS4 and Ag3SnS4 were found to be suitable precursors to the formation of Cu3Sn and Ag3Sn alloys through in situ electrochemical reduction. Both materials show a high selectivity for formate production during CO2RR with 81% FEHCOO at −100 mA cm−2, 7% FECO and 11% H2 at −0.54 V vs. RHE for Ag3Sn. Sn is found to render the alloy surfaces highly oxophilic as evidenced by extensive oxidation in EDX and accumulation of carbonate on the electrode surfaces. High binding energies of up to −2.5 eV for HCOO on Cu3Sn and Ag3Sn steer the expected CO2RR product spectrum from H2 and CO on metallic Ag – and multicarbon compounds in the case of Cu – towards formate.In the case of Cu3Sn, Cu2O and SnO were found as separated phases on the electrode surface post-electrolysis, demonstrating an unstable alloy structure. For Ag3Sn, the expected oxidation states of an alloy structure could be detected by XPS and PXRD after electrolysis at 300 mA cm−2. The gradual decrease in FEHCOO at high current densities is likely caused by strong adsorption of carbonate or formate species on the catalyst surface, as evidenced by post-mortem SEM/EDX analysis and the computed binding energies. This limitation can possibly be overcome by the application of alternating currents to Ag3Sn to reverse the excessive adsorption of carbonate on the electrode surface at current densities above −100 mA cm−2. Future work at our laboratory is aimed at this mitigation strategy for the application of alloys for CO2RR. Considering the high FEHCOO− obtained with Ag3Sn despite its high oxophilicity and exposure to high OH− concentrations, testing of CO2RR activity with O2 diluted gas streams may still yield favorable results, thus showing the applicability for CO2 reduction of industrial flue gases.
Considering the vast experimental space alloying can provide, suitable binding energies can likely be tailored to the desired catalytic process.33,34 The example of Ag3Sn and Cu3Sn demonstrates that a high oxophilicity may cause partial inhibition of the catalytic sites through carbonate adsorption or decomposition of the catalyst into i.e. Cu2O and SnO. Therefore, an assessment of this property through ab initio methods and post-mortem spectroscopy after the application of high current densities appears necessary to select the highest performing alloy CO2RR catalysts.
Author contributions
Conceptualization: SAS, UPA, KjP. Formal analysis: SAS, AS, MK, AM, YH. Funding acquisition: UPA, SR. Investigation SAS, AS, MK, YW methodology SAS, AS, KP, SR. Project administration: UPA, SR, KjP, SAS resources: UPA, SR. Supervision: UPA, SR. Validation: SAS, AS, MK, AM. Visualization: SAS, MK, AM. Writing – original draft: SAS, MK, AM. Writing – review & editing SAS, AS, MK, AM, KP, UPA.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Further information not provided can be obtained upon request by the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of SAS, AS, YH, YW, KP, KjP, SR and UPA was funded by grant number 03EE5104A, “CO2-Syn”, of the German Federal Ministry for Economic Affairs and Climate Action. MK and AM gratefully acknowledge Poland's high-performance computing infrastructure PLGrid (HPC Center: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016825.Notes and references
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12(5), 1442–1453 CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei and A. Zitolo, et al., Electroreduction of CO2 on single-site copper–nitrogen-doped carbon material: selective formation of ethanol and reversible restructuration of the metal sites, Angew. Chem., Int. Ed., 2019, 58(42), 15098–15103 CAS.
- W. Wang, S. Gong, H. Wang, Y. Tan, X. Zhu, X. Wang, J. Liu, W. Yu, G. Zhu and X. Lv, Surface-modified silver aerogels combining interfacial regulation for electrocatalytic CO2 reduction under large current density, Chem. Eng. J., 2024, 490, 151849, DOI:10.1016/j.cej.2024.151849.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Nanoarchitectonics for Transition-Metal-Sulfide-Based Electrocatalysts for Water Splitting, Adv. Mater., 2019, 31(17), e1807134, DOI:10.1002/adma.201807134.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano and A. Mehta, et al., Oxidation state and surface reconstruction of Cu under CO2 reduction conditions from in situ X-ray characterization, J. Am. Chem. Soc., 2020, 143(2), 588–592 CrossRef PubMed.
- K. Ye, Z. Zhou, J. Shao, L. Lin, D. Gao, N. Ta, R. Si, G. Wang and X. Bao, In Situ Reconstruction of a Hierarchical Sn–Cu/SnOx Core/Shell Catalyst for High-Performance CO2 Electroreduction, Angew. Chem., Int. Ed., 2020, 59(12), 4814–4821 CrossRef CAS PubMed.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. Luna, T. de; Burdyny, F. Che, F. Meng, Y. Min and R. Quintero-Bermudez, et al., Steering post-C–C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols, Nat. Catal., 2018, 1(6), 421–428 CrossRef CAS.
- J. He, X. Liu, H. Liu, Z. Zhao, Y. Ding and J. Luo, Highly selective electrocatalytic reduction of CO2 to formate over Tin(IV) sulfide monolayers, J. Catal., 2018, 364, 125–130 CrossRef CAS.
- J. Wang, J. Mao, X. Zheng, Y. Zhou and Q. Xu, Sulfur boosting CO2 reduction activity of bismuth subcarbonate nanosheets via promoting proton-coupled electron transfer, Appl. Surf. Sci., 2021, 562, 150197 CrossRef CAS.
- K. Li, J. Xu, T. Zheng, Y. Yuan, S. Liu, C. Shen, T. Jiang, J. Sun, Z. Liu, Y. Xu, M. Chuai, C. Xia and W. Chen, In Situ Dynamic Construction of a Copper Tin Sulfide Catalyst for High-Performance Electrochemical CO2 Conversion to Formate, ACS Catal., 2022, 12(16), 9922–9932, DOI:10.1021/acscatal.2c02627.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Stability and Degradation Mechanisms of Copper-Based Catalysts for Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2020, 132(35), 14844–14854, DOI:10.1002/ange.202000617.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, Ag–Sn Bimetallic Catalyst with a Core–Shell Structure for CO2 Reduction, J. Am. Chem. Soc., 2017, 139(5), 1885–1893, DOI:10.1021/jacs.6b10435.
- N. S. Shaikh, J. S. Shaikh, V. Márquez, S. C. Pathan, S. S. Mali, J. V. Patil, C. K. Hong, P. Kanjanaboos, O. Fontaine, A. Tiwari, S. Praserthdam and P. Praserthdam, New perspectives, rational designs, and engineering of Tin (Sn)-based materials for electrochemical CO2 reduction, Mater Today Sustain., 2023, 22, 100384, DOI:10.1016/j.mtsust.2023.100384.
- L. Hoof, N. Thissen, K. Pellumbi, K. junge Puring, D. Siegmund, A. K. Mechler and U.-P. Apfel, Hidden parameters for electrochemical carbon dioxide reduction in zero-gap electrolyzers, Cell Rep. Phys. Sci., 2022, 3(4), 100825, DOI:10.1016/j.xcrp.2022.100825.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561, DOI:10.1103/PhysRevB.47.558.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276, DOI:10.1107/S0021889811038970.
- D. Tetzlaff, K. Pellumbi, D. M. Baier, L. Hoof, H. Shastry Barkur, M. Smialkowski, H. M. A. Amin, S. Grätz, D. Siegmund, L. Borchardt and U.-P. Apfel, Sustainable and rapid preparation of nanosized Fe/Ni-pentlandite particles by mechanochemistry, Chem. Sci., 2020, 11(47), 12835–12842, 10.1039/D0SC04525J.
- S. A. Sanden, M. Smialkowski, S. Y. Hu, N. Suvagiya, S. Angel, C. Schulz and U.-P. Apfel, Ternary Pentlandites as Hydrogen Evolution Catalysts in Alkaline Media, Adv. Energy Sustainability Res., 2024, 5(10), 2400128, DOI:10.1002/aesr.202400128.
- S. Stojkovikj, G. A. El-Nagar, S. Gupta, M. Najdoski, V. Koleva, T. Tzanoudakis, F. Firschke, P. Bogdanoff and M. T. Mayer, Facile Synthesis of CuxS Electrocatalysts for CO2 Conversion into Formate and Study of Relations Between Cu and S with the Selectivity, Adv. Funct. Mater., 2024, 2415405, DOI:10.1002/adfm.202415405.
- H. Da Won, C. H. Choi, J. Chung, M. W. Chung, E.-H. Kim and S. I. Woo, Rational design of a hierarchical tin dendrite electrode for efficient electrochemical reduction of CO2, ChemSusChem, 2015, 8(18), 3092–3098 Search PubMed.
- J. Kok, J. Ruiter, W. de van der Stam and T. Burdyny, Interrogation of Oxidative Pulsed Methods for the Stabilization of Copper Electrodes for CO2 Electrolysis, J. Am. Chem. Soc., 2024, 146(28), 19509–19520, DOI:10.1021/jacs.4c06284.
- M. C. Biesinger, Advanced analysis of copper X-ray photoelectron spectra, Surf. Interface Anal., 2017, 49(13), 1325–1334, DOI:10.1002/sia.6239.
- A. Giaccherini, G. Montegrossi and F. Di Benedetto, Stability of naturally relevant ternary phases in the Cu–Sn–S system in contact with an aqueous solution, Minerals, 2016, 6(3), 79 CrossRef.
- B. Li, Y. Xie, J. Huang, H. Su and Y. Qian, Synthesis and Characterization of Ternary Chalcogenides Ag8SnE6 (E = S, Se), J. Solid State Chem., 2000, 149(2), 338–340, DOI:10.1006/jssc.1999.8537.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy, ACS Catal., 2015, 5(5), 3148–3156, DOI:10.1021/acscatal.5b00402.
- O. Christensen, A. Bagger and J. Rossmeisl, The Missing Link for Electrochemical CO2 Reduction: Classification of CO vs. HCOOH Selectivity via PCA, Reaction Pathways, and Coverage Analysis, ACS Catal., 2024, 2151–2161, DOI:10.1021/acscatal.3c04851.
- C. Walker, S. A. Morton, G. Beamson, J. Matthew and F. N. Yousif, Auger parameter studies of amorphous CuHf alloys, J. Electron Spectrosc. Relat. Phenom., 1994, 70(1), 73–81, DOI:10.1016/0368-2048(94)02216-M.
- F. C. Østergaard, A. Bagger and J. Rossmeisl, Predicting catalytic activity in hydrogen evolution reaction, Curr. Opin. Electrochem., 2022, 35, 101037, DOI:10.1016/j.coelec.2022.101037.
- X.-Y. Zhang, J.-Y. Xie, Y. Ma, B. Dong, C.-G. Liu and Y.-M. Chai, An overview of the active sites in transition metal electrocatalysts and their practical activity for hydrogen evolution reaction, Chem. Eng. J., 2022, 430, 132312, DOI:10.1016/j.cej.2021.132312.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, Self-Cleaning CO2 Reduction Systems: Unsteady Electrochemical Forcing Enables Stability, ACS Energy Lett., 2021, 6(2), 809–815, DOI:10.1021/acsenergylett.0c02401.
- J. He, K. E. Dettelbach, A. Huang and C. P. Berlinguette, Brass and Bronze as Effective CO2 Reduction Electrocatalysts, Angew. Chem., Int. Ed., 2017, 129(52), 16806–16809, DOI:10.1002/ange.201709932.
- X.-Q. Duan, G.-Y. Duan, Y.-F. Wang, X.-Q. Li, R. Wang, R. Zhang and B.-H. Xu, Sn–Ag Synergistic Effect Enhances High-Rate Electrocatalytic CO2-to-Formate Conversion on Porous Poly(Ionic Liquid) Support, Small, 2023, 19(18), e2207219, DOI:10.1002/smll.202207219.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00603h |
| This journal is © The Royal Society of Chemistry 2025 |