
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous solution degradation pathways of trimethylsiloxane surfactants†
Maleigh
Mifkovic
 a,
Brian D.
Etz‡
bc,
Manoj K.
Shukla
*c and
Shubham
Vyas
*a
a,
Brian D.
Etz‡
bc,
Manoj K.
Shukla
*c and
Shubham
Vyas
*a
aDepartment of Chemistry, Colorado School of Mines, Golden, CO 80401, USA. E-mail: svyas@mines.edu
bOak Ridge Institute for Science and Education (ORISE), Oak Ridge, TN 37830, USA
cEnvironmental Laboratory, US Army Engineer Research and Development Center, 3909 Halls Ferry Road, Vicksburg, MS 39180, USA. E-mail: Manoj.K.Shukla@usace.army.mil
First published on 23rd September 2024
Abstract
Trimethylsiloxane (TriSil) surfactants are promising alternatives to per- and polyfluoroalkyl substances (PFAS), which are global recalcitrant and persistent environmental contaminants, in aqueous film-forming fire-fighting foams (AFFF). However, much less information is available on the environmental fate and degradation of TriSil surfactants. Thus, it is important to study the degradation chemistry of fluorine-free TriSil surfactants in the solution phase under various conditions to further assess their environmental impact. This computational study reports the prominent hydrolysis, reduction, and oxidation pathways of a truncated TriSil and proposes the major degradation products using density functional theory (DFT) calculations. We have identified the polydimethylsiloxane unit of TriSil to play a prominent role in aqueous solution reactivity initiated via hydrolysis and reduction, while oxidation mainly proceeds through H-atom abstraction along the polyethylene glycol unit. The results of this study aid in establishing the use of the alternative fluorine-free surfactant, TriSil, for fire-fighting foams from an environmental perspective.
Environmental significancePer- and polyfluoroalkyl substances (PFAS) have been a crucial component in certain aqueous film-forming foams (AFFF) due to their fire suppression capabilities. However, PFAS have become worldwide environmental contaminants due to their toxicity, bioaccumulation, and pervasiveness. Thus, safer alternatives are needed to replace PFAS in AFFF which would not pose a hazardous environmental threat. Trimethylsiloxane (TriSil) surfactants are potential fluorine-free PFAS replacements, however, research is needed to understand the effect of its degradation products in aqueous environmental conditions. The hydrolysis and reduction of TriSil targets the polydimethylsiloxane unit while oxidation occurs through H-atom abstraction along the polyethylene glycol unit. Our results support the use of TriSil in AFFF and may lead to significantly less environmental consequences than PFAS in AFFF. |
1 Introduction
Per- and polyfluoroalkyl substances (PFAS) are a key ingredient in aqueous film-forming fire-fighting foams (AFFF) that greatly contribute to the overall effectiveness of the foam due to its ability to suppress fire vapors and lower the surface tension of water.1,2 On the molecular level, fire-suppression activity is largely attributed to the high C–F bond strength (up to ∼130 kcal mol−1)3 and the oleo- and hydrophobic nature of PFAS,4 which macroscopically prevents the fuel from reigniting5 and provides optimal surfactant properties for quick extinction.4,6 As a result, AFFF with PFAS surfactants are the only established formulations that meet high military specifications (MIL-F-24385F) to eliminate fires.7 Additionally, PFAS have been used in numerous industrial and consumer applications such as stain repellants, non-stick cookware, and consumer-goods packaging. However, PFAS have notoriously become persistent global environmental contaminants that cause adverse health impacts, such as cancer, reproductive health issues, immunosuppression, and kidney disease.8–10 In particular, the use of PFAS in fire-fighting foams are a major contributor to environmental contamination at locations employing AFFF, and the robust nature of PFAS leads to significant challenges in remediation. Therefore, the development of fluorine-free alternative surfactants that are both effective for fire suppression formulations and environmentally friendly has become increasingly important.11–17AFFF formulations are typically proprietary information and contain multiple components, which complicates the development of effective PFAS alternatives.18 However, the composition of fluorine-free alternatives has previously included hydrocarbons,19 protein-based surfactants, detergents,1,2,20 and organosilicone surfactants20–22 with ethylene glycol,23 the latter providing stability as a foaming agent.20 In particular, trimethylsiloxanes (TriSil), such as that in Fig. 1, are potential replacements which contain hydrophobic polydimethylsiloxane (PDMS) units1,2,20 and hydrophilic polyethylene glycol (PEG) segments. The presence of TriSil lowers the surface energy of the foam, which reduces the time to coat a flammable surface.20,24 With additional repeating units to tune properties, and the presence of additives, siloxane surfactants have the potential to extinguish fires within the guidelines of military specifications.22 However, more research is needed to understand how these chemicals will degrade in relevant environmental conditions. Previous work has evaluated the degradation chemistry of TriSil at high temperatures;1,25,26 however, its degradation in various solution conditions at the molecular-level, to the best of our knowledge, has not yet been investigated.
 | ||
| Fig. 1 Chemical structures of (i) trimethylsiloxane (TriSil) surfactant used in fire suppression tests in a laboratory setting1 with PDMS and PEG components, where ‘x’ refers to the repeating PDMS component and ‘n’ refers to the repeating PEG component (ii) PDMS unit (iii) PEG-1n and (iv) PEG-3n. Alphabetical labels relate to radical and reduction positions and numerical positions refer to silicon–oxygen bond lengths pertaining to Section 3.2. | ||
The properties and environmental fate and transport of organosilicone materials, including PDMS, have been previously studied.27–29 Laboratory and field studies have shown that PDMS degrades in soil via hydrolysis and leads to volatile and water-soluble products,29–33 including carbon dioxide, siloxanols, and monomeric dimethylsilanediol (DMSD).33–37 Degradation of DMSD via oxidation in water31 or microbiomes further produces CO2,32,38 water, and inorganic silicate,28,37,39 resulting in no bioaccumulation.33,35–37 Moreover, PDMS and DMSD have zero to little toxicity,28,29,36 respectively, and research has shown that PDMS and its degradation products do not damage microbiomes present in soil or negatively impact seedling germination and survival for wheat and soybeans.40 Polyethylene glycol can also completely biodegrade through multiple pathways and does not pose a threat to the environmental cycle.41–43 However, despite extensive research on the degradation of PDMS and PEG components, other factors such as additional siloxanes could influence TriSil's environmental behavior. Thus, further research is needed to study the degradation pathways of TriSil in relevant environmental conditions to understand its fate and transport.
In this study, we have investigated the aqueous solution degradation channels of TriSil, a fluorine-free alternative to PFAS. Density functional theory (DFT) calculations were employed to probe the potential aqueous degradation reactions in environmentally relevant circumstances, such as hydrolysis, oxidation, and reduction. The outcomes of this work provide a thorough analysis in identifying prominent TriSil decomposition pathways and major degradation products, as well as evaluating their potential environmental impact, as safer fluorine-free fire suppressants for PFAS replacement.
2 Methods
All quantum mechanical calculations were performed using Gaussian 16 Rev C.01 software44 at ωB97X-D/6-311+G(2d,2p) with the universal continuum solvation water model (SMD) to simulate aqueous conditions.45,46 Optimized minima geometries and transition state structures were determined by the presence of either zero or one imaginary frequency, respectively. Intrinsic reaction coordinate (IRC) calculations verified the transition states between the correct reactant and product minima. We chose the ωB97X-D functional due to including long-range and dispersion corrections, which is important for longer chain systems,47 and for its increased accuracy in calculating thermochemical values for covalent systems.45,47,48 In addition, ωB97X-D yields some of the lowest errors in vertical attachment energies (VAE) in the reduction of perfluoroalkanes by an aqueous electron, where VAE is equal to the energy of the optimized anion subtracted from the energy of the dianion-radical at the optimized anion geometry.49 Since we compare the reduction potentials of TriSil-1n to that of short-chain PFAS, we applied the same computational method on TriSil-1n for consistency. Previous computational research have also included dispersion corrections to model organosiloxane50–52 and PFAS49 chemistry. Nevertheless, to investigate the structural dependence on the method, M06-2X53 and B3LYP54–56 optimizations and frequencies were calculated for neutral, oxidized, and reduced TriSil-1n. Slight changes were observed in the optimized redox structures across the three DFT functionals, however the underlying results are consistent (see Section 3.2). As such, we employed ωB97-XD for the reasons previously outlined. Natural Bond Orbital (NBO) analysis was performed using NBO Version 3.1 as implemented in Gaussian 16 to compute spin densities and charge analysis. Standard reduction potentials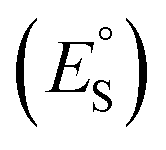 were calculated using the Nernst equation:
were calculated using the Nernst equation:where ΔG is the change in Gibbs free energy between the products and reactants, n is the number of electrons transferred in the reaction, F is Faraday's constant, and SHE is the standard hydrogen electrode (4.44 V). Two methods were used to calculate the reduction potentials of TriSil-1n, PDMS, and PEG-1n, similar to the work by Van Hoomissen et al.49 In method 1, the abstracted hydrogen was included in the same calculation as the reduced molecule, whereas in method 2, the Gibbs free energy of the abstracted hydrogen was considered infinitely separate.

3 Results and discussion
The aqueous solution decomposition of TriSil surfactants via hydrolysis, oxidation, and reduction were investigated using DFT calculations to assess the environmental impact of the parent surfactants and major proposed products. To minimize computational cost and maximize efficiency, and since the exact composition of TriSil surfactants used in commercial fire-fighting formulations is proprietary information,57 a simplified, truncated model, TriSil-1n, system was studied (Fig. 1). This structure was deemed appropriate to study molecular reactions since previous work on the gas-phase reactivity identified that bond dissociation energies (BDEs) and radical intermediates were unchanged with increasing repeating units.26 Thus, the same local chemical structure environment would remain constant, resulting in consistent chemical reactivity for larger TriSil formulations (constructed with more PDMS and PEG monomers). It is important to note that the variation of the TriSil structure (i.e. PDMS and PEG chain lengths) can affect the solution phase macroscopic surfactant properties (such as surface tension and viscosity), which will in turn affect the performance of firefighting formulations.57 However, macroscopic properties cannot be modeled using DFT, whereas molecular-level reactions can be modeled. Therefore, the latter is the focus of this study, and the results of various molecular degradation channels of TriSil-1n in water solution will be discussed in the following sections.3.1 Hydrolysis reactivity
Siloxane-containing surfactants can readily hydrolyze due to the large atomic radius of silicon, and the easy polarizability and strong dipole moment (ionic nature) of silicon–oxygen bonds.58,59 Hydrolysis is especially likely to be the dominant aqueous solution decomposition channel for TriSil-1n when catalyzed by an acid or base.60,61 There has been a significant amount of experimental research on hydrolysis reaction mechanisms for PDMS and PDMS-like chemical systems62 and we have extended these previous studies to TriSil-1n. Many factors such as solvation, pH, temperature, pressure, and humidity influence hydrolysis reactivity,58,60–65 thereby creating potential stability challenges for TriSil surfactants in foam formulations.1 We have investigated the tendency of TriSil-1n to undergo hydrolysis under neutral, acidic, and basic conditions in the present manuscript (Scheme 1). The neutral hydrolysis of TriSil-1n was investigated under two scenarios; firstly, one water molecule nucleophilically attacks the central silicon atom (red line, Fig. 2) via a 4-membered transition state, and secondly, two water molecules (black line, Fig. 2), where one water attacks the silicon and a second water assists in the proton transfer in the transition state, creating a 6-membered transition state (Fig. S1†). This second pathway was investigated because assisting water molecules have been shown to lower the activation energy for hydration/hydrolysis reactions.66,67 | ||
| Scheme 1 Hydrolysis reactions for TriSil-1n (a) under neutral, acidic, and basic conditions. R represents the PEG chain. Energies for these reactions are presented in the potential energy diagram in Fig. 2. | ||
 | ||
| Fig. 2 Potential energy surfaces for hydrolysis reactions at neutral pH (left) and basic and acidic pH (right) of TriSil-1n (a), computed at the ωb97X-D/6-311+G(2d,2p) level of theory. All energies are presented in kcal mol−1 and are normalized to TriSil-1n (a). Transition state energy barriers are in italics and product reaction free energies are in bold. The black pathway corresponds to the reaction facilitated by an additional water molecule. | ||
Regardless of the number of water molecules, both neutral hydrolysis mechanisms lead to the formation of trimethylsilanol (TMS–OH) and PDMS-like intermediates (P1h) with relatively the same stability (4.2 and 5.8 kcal mol−1). The second transition state corresponds to another hydrolysis reaction where the effect of an additional assisting water is observed. When one water performs the hydrolysis, an activation energy of 46.8 kcal mol−1 was determined, like the energetics of the first hydrolysis. However, when a second, assisting water was included, the activation energy becomes 12.3 kcal mol−1 (black line, Fig. 2). This decrease in energy is attributed to the formation of a 6-membered ring transition state structure and the ease of hydrogen atom transfer, where the additional water molecule facilitates hydrogen atom transfer between the attacking water and the PDMS–ether oxygen, forming the alcohol on the leaving group. The results of this second hydrolysis are in stark contrast to the first hydrolysis reaction and are attributed to the decreased steric interactions surrounding the electrophilic silicon atom.
As shown in Fig. 2, the neutral hydrolysis pathway requires either an activation energy of 46.0 kcal mol−1 (TS1h for hydrolysis with only one water attacking the reactant) or an activation energy of 46.7 kcal mol−1 (TS1h for hydrolysis with an additional water facilitating the reaction). Since there is no significant energetic difference between the activation barriers for initial hydrolysis, the number of waters that facilitate the hydrolysis reaction is not a contributing factor. Therefore, the neutral hydrolysis pathway is unlikely regardless of increased solvation. Our results are in agreement with previous computational studies performed regarding the neutral hydrolysis68 of Si(OCH3)4, where in both studies the authors found that the neutral pathway is energetically unfavorable. Interestingly, the addition of a second assisting water molecule does not facilitate the hydrolysis reaction, which has been previously debated in literature. Cypryk et al. found that extra solvation lowers the activation barriers from 34 to 24.5 kcal mol−1 for a water monomer and dimer, respectively, in disiloxane and disiloxanol using DFT methods.61 More recently, DFT results obtained by Cheng et al. indicate that hydrolysis of Si(OCH3)4 is not facilitated by an additional water molecule.68 In our TriSil-1n system, the lack of effect from more water is likely a consequence of the steric hindrance surrounding the central silicon atom, which prevents the extra water molecule from lowering the activation energy. Despite the contradictions in literature, the neutral hydrolysis pathway is not likely to occur.
Acidic and basic aqueous hydrolysis reactions were investigated by either protonating TriSil-1n at different oxygen sites (acidic conditions) or including a hydroxide ion (basic conditions). To understand the sites within the molecule susceptible to nucleophilic or electrophilic attack by these ions, we performed a NBO analysis on TriSil-1n to assess the partial atomic charges and visualize the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) (Fig. 3).
 | ||
| Fig. 3 (a) Partial atomic charges of neutral TriSil-1n, where green represents carbon, red represents oxygen, and blue represents silicon. All values are presented in a.u. Isosurfaces of the (b) highest occupied molecular orbital (HOMO) and (c) lowest unoccupied molecular orbital (LUMO) for TriSil-1n (isovalue = 0.1 a.u.). NBO analysis was computed at the ωb97X-D/6-311+G(2d,2p) level of theory with SMD implicit water solvation. | ||
In the basic hydrolysis pathway, there are two hydroxide anion attack sites, occurring at the central or terminal silicon of the PDMS component forming a 5-coordinate trigonal bipyramidal intermediate. Hydroxide attack of the central silicon position is more favorable by 3.3 kcal mol−1 (Fig. S2†) when compared to the terminal silicon. This result is unexpected as the LUMO is localized on the terminal silicon group. This reactivity is likely due to the higher electropositive nature of the central silicon (2.18 a.u.) compared to the terminal position (1.91 a.u.) (Fig. 3(a)). Furthermore, analysis of the transition state structure and charge indicates that the 5-coordinate intermediate, formed from attack of the central silicon, is stabilized to a greater degree by the two neighboring electronegative ether oxygen compared to the intermediate formed from attack of the terminal silicon (Table S1†). Although we believe the trend in reactivity is due to electrostatic interactions, the reactivity and stabilization could be affected by the silicon hyperconjugation effect.69 Thus, hydroxide prefers to attack the central silicon rather than the terminal silicon of TriSil-1n (Fig. 3(c)) since the terminal silicon is sterically hindered surrounded by three methyl groups. Hydroxide attack at the central silicon occurs with an energy barrier of 8.1 kcal mol−1, indicating that the rate limiting step is the rearrangement of the tetrahedral geometry to trigonal bipyramidal intermediate. Overcoming this energy barrier will subsequently result in the formation of anionic and neutral trimethylsilanols with an activation energy of 3.5 kcal mol−1 (P2b in Fig. 2). The anionic-TMS produced in basic conditions will likely become protonated and form an alcohol. Product P2b is the same as the intermediate produced (P1h) in the neutral hydrolysis pathway, and thus would no longer require basic conditions to further hydrolyze, but could subsequently undergo a second base catalyzed hydrolysis. In the latter, the central silicon would likely be attacked by hydroxide again since it is more electropositive (2.13 a.u.) than the terminal silicon (1.90 a.u.), forming another anionic-TMS and methylsiloxane-diol product.
In acidic conditions, the preferred protonation site will be the siloxane (Si–O–Si) oxygen along the PDMS chain. The siloxane oxygen will exhibit greater nucleophilic behavior due to its significant negative charge compared to the oxygen along the PEG chain (Fig. 3(a)) and since the HOMO of PDMS lies on the siloxane oxygen (Fig. S3(a)†). This agrees with the findings of Cypryk et al. that the environmental degradation of the PDMS unit via acidic hydrolysis will likely begin with protonation at the siloxane oxygen, as this is an important factor in lowering the activation energy to these bonds.61 This protonation occurs with a reaction energy of 9.7 kcal mol−1 (Fig. 2), followed by nucleophilic attack by water at the terminal silicon of the protonated TriSil-1n (Fig. S3(c)†). This is 21.4 kcal mol−1 kinetically more favorable (Fig. S4†), and 2.6 kcal mol−1 thermodynamically more favorable (Fig. S5†), than the pathway stemming from attack at the central Si–O bond, with both pathways leading to the same products, P1a (Fig. S5†). Previous research by Okui et al. regarding the acidic hydrolysis of tetramethyl-p-silphenylene siloxane (TMPS) and dimethyl siloxane (DMS) determined that hydrofluoric acid prefers to attack the electrophilic Si–O bonds, especially in the PDMS unit of DMS.70 Thus, nucleophiles in an acidic environment will likely degrade TriSil-1n via the silicon–oxygen bonds comprising the PDMS unit.71 Further degradation of product P1a involves a second hydrolysis step resulting in the cleavage of the remaining Si–O bond, and producing the methylsiloxane-diol product and TMS–OH (Fig. S6†).
The reaction energies and transition state energy barriers of the basic and acidic hydrolysis in our study are significantly lower compared to neutral hydrolysis. Thus, hydrolysis in the acidic and basic conditions are expected to play a major role in the aqueous degradation of TriSil-1n. Our basic hydrolysis mechanism agrees with that found by Zerda et al., in which the intermediate structure of the hydrolysis of Si(OCH3)4 is a pentavalent coordination complex.62 Although the authors didn't report energy values, they did report rate constants at different pH, which qualitatively match our results. At 1 bar in acidic conditions (pH = 4.9), the rate constant is smaller (6.2 × 10−4 mol−1 s−1) compared to mild basic conditions (pH = 7.5, 8.0 × 10−4 mol−1 s−1), indicating that hydrolysis occurs faster in basic conditions than in acidic conditions.62 Our results also agree with the experimental study of Ducom et al.72 and the computational study performed by Cheng et al.68 The authors found that the basic and acidic hydrolysis of Si(OCH3)4 is more favorable (8.84 kcal mol−1 and 12.7 kcal mol−1 free energy barriers, respectively) than neutral hydrolysis (28.4 kcal mol−1) using the B3LYP/6-311++G(d,p) method. The high energetic barriers associated with the neutral hydrolysis, along with steric interactions associated with the PDMS backbone, demonstrate that decomposition via neutral hydrolysis is not a feasible pathway. This supports the use of TriSil surfactants in firefighting formulations as these surfactants are typically used under 3% or 6% concentrates in water and held at a neutral pH of 6–8.57 Previous research has shown that double-tail trisiloxane surfactants are stable in firefighting formulations over 270 days at a pH of 7.0.73 Furthermore, the number of PDMS and PEG segments in TriSil impacts their resistance to hydrolysis in acidic and basic conditions,73 which may inhibit the degradation of these surfactants in these conditions or possibly result in the formation of harmful cyclic siloxane compounds, the latter of which was not studied here due to computational restraints. Peng et al. found that trimethylsiloxanes become resistant to hydrolysis when the number of carbon atoms in the PDMS unit is increased or the number of ethylene oxide units is decreased.73 Therefore, the degradation of TriSil surfactants will vary according to the specific chemical composition as well as the solution and soil conditions environmentally present, potentially allowing the surfactants to persist long enough to encounter water treatment facilities or evaporate into the atmosphere.74,75
3.2 Redox reactivity
Although basic and acidic hydrolysis is likely the dominant degradation pathway for TriSil in aqueous solutions, it is important to investigate the applicability of redox approaches towards siloxane surfactants given the possibility of these surfactants to persist in the environment73 and because common water treatment approaches use chemical redox methods.76 Like hydrolysis, oxidation depends on several important factors, however, the oxidant dictates much of the oxidation chemistry. For simplicity, we have only investigated oxidation using hydroxyl radicals, which are the most common oxidants employed in advanced oxidation processes (AOPs) for water treatment.77–79 Hydroxyl radicals have been previously shown to undergo three main types of oxidation: H-atom abstraction, single electron transfer (SET), and OH-addition and adduct formation.77 However, for OH-addition and adduct formation to occur, the presence of sp2-hybridized bonds are necessary, as this pathway undergoes electrophilic addition to systems containing π bonds, which TriSil-1n does not contain.78,79 Therefore, this oxidation pathway is assumed to not be relevant for TriSil-1n and similar siloxane surfactants, and we investigated the H-atom abstraction and SET oxidation channels for hydroxyl radical and TriSil-1n with comparisons to the individual polymer constituents. Furthermore, the results of this analysis can be extended to other oxidants.Bond dissociation energies (BDEs), transition state energy barriers, and reaction free energies using implicit water solvation were computed for seven possible H-abstraction positions (a–g) of TriSil-1n (see atom labels in Fig. 1, Table S2†). The BDEs range from 85.8 kcal mol−1 (position d) to 96.3 kcal mol−1 (position f) in the aqueous solution and increase from positions d < c < e < b < a < g < f, indicating that the C–H bonds along the PEG chain are generally weaker than those within the PDMS unit. The transitions state energy barriers for H-abstraction by hydroxyl radicals range from 4.84–9.23 kcal mol−1, indicating that kinetically all H-abstraction reactions are energetically similar and feasible (Table S2†). Furthermore, the reaction free energies for H-abstraction forming TriSil radicals and water show this pathway is thermodynamically favorable, ranging from −27.6 kcal mol−1 (position d, most favorable) to −17.1 kcal mol−1 (position f, least favorable). These results match the trend in the bond dissociation energies (BDEs) in both the solution and gas phase. Positions d, c, and e are the most energetically favorable H-abstraction sites due to the proximity to neighboring oxygen atoms. The electronegative oxygen helps to delocalize and stabilize the resulting TriSil carbon radicals which lowers the energy for the reaction.
The subsequent reactivity of TriSil-1n radicals involves the reaction with oxygen or further radical recombination with hydroxyl radicals in solution, that are typically barrier-less. For the five most favorable H-atom abstraction sites (a–e), the reaction free energies were computed for O2 addition (Table S3†). These values range from −27.7 kcal mol−1 (position c) to −20.5 kcal mol−1 (position a), indicating the anticipated outcome that O2 addition is energetically favorable at all positions forming peroxyl radical intermediates (R–OO˙). Further reaction with aqueous reactive species is highly variable and dependent on solutions conditions. These reactions include peroxyl radical disproportionation reactions through the formation of a short-lived tetroxide intermediate, bimolecular peroxyl radical decay through self-reactions and reactions with 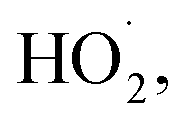 or unimolecular decay via H-shift and elimination of
or unimolecular decay via H-shift and elimination of  80–83 Moreover, propagation of the peroxyl TriSil-1n–O2 radical will likely lead to fragmentation of the PEG chain and products containing aldehyde and alcohol functional groups as well as small oxygen-containing organics (Fig. 4). A proposed reaction diagram for the addition of O2 to the TriSil-1n radical at position d is shown in Scheme S1.†
80–83 Moreover, propagation of the peroxyl TriSil-1n–O2 radical will likely lead to fragmentation of the PEG chain and products containing aldehyde and alcohol functional groups as well as small oxygen-containing organics (Fig. 4). A proposed reaction diagram for the addition of O2 to the TriSil-1n radical at position d is shown in Scheme S1.†
 | ||
| Fig. 4 TriSil-1n degradation reaction channel for hydroxyl radical induced degradation via H-abstraction. See Scheme S1† for a proposed reaction diagram for TriSil-1n–d–O2 peroxyl radical. | ||
Although H-atom abstraction via hydroxyl radical is likely the more favorable oxidation pathway, single electron transfer (SET) could still occur depending on the oxidant or reductant that is encountered in the environment or employed in water treatment facilities.77,79 We investigated the geometric parameters (spin densities and charges) and reduction and oxidation reaction free energies to aid in understanding how SET will affect the chemical structure and degradation of TriSil-1n.
When an electron is removed in a neutral molecule to create the oxidized state (Scheme S2(i)†), it will likely be removed from the highest occupied molecular orbital (HOMO). The HOMO plot of TriSil-1n (Fig. 3(b)) aligns with the computed spin densities of oxidized TriSil-1n (Fig. 5(a)). The spin is mostly located on the PEG ether oxygen (0.27 a.u.) and first three carbons of the PEG chain (0.16–0.23 a.u.), indicating that the electron is primarily removed from the carbons along the PEG unit nearest the central silicon, with stabilization on the neighboring oxygen atom. This also parallels the charge analysis post oxidation, in that the ether oxygen along the PEG unit becomes more electropositive by 0.21 a.u. and the carbon at position a is also more electropositive by 0.15 a.u. (Fig. 5(a)).
 | ||
| Fig. 5 Change in partial atomic charges (left) and spin density isosurfaces (right) for (a) oxidized and (b) reduced TriSil-1n. Green values correspond to carbon, red correspond to oxygen, blue correspond to silicon, and grey corresponds to hydrogen. Positive values indicate a decrease in charge. In the isosurfaces, oxygen atoms are red, silicon atoms are pale blue, carbon atoms are gray, and hydrogen atoms are white. All values are presented in a.u. and only spin densities greater than 0.15 a.u. and changes in partial atomic charge greater than 0.05 a.u. are reported. The isovalues for (a) and (b) are 0.01 and 0.0035, respectively. NBO analysis was computed at the ωb97X-D/6-311+G(2d,2p) level of theory with SMD water solvation. | ||
Furthermore, the geometry of the TriSil-1n upon oxidation significantly changes (Fig. S7†) where the PEG unit twists to stabilize the decrease in electron density. Therefore, geometric and NBO spin density analyses of TriSil-1n were performed to determine the effect of oxidation. It is important to note that the oxidized geometry of TriSil-1n is method dependent, potentially leading to different fragmentation along the PEG chain. Shown in Table S4,† the geometries of oxidized TriSil-1n at the ωB97X-D and B3LYP levels of theory indicate that bond fission would likely occur at either the Cb–Cc or Si1–Ca bonds, since these bonds increased by 0.18 Å and 0.13 Å, respectively (Table S4†). However, using M06-2X, the Cd–Ce bond fission is most probable since this bond lengthens by 0.27 Å. Despite differences in these pathways, bond fission due to oxidation is expected to occur along the PEG chain, likely leading to the formation of small organic products such as formaldehyde and ethylene.
Along with the bond lengthening observed in oxidized TriSil-1n (at the ωB97X-D level), we expect Cb–Cc bond fission to occur more readily due to the lower relative C–C bond strength (84–86 kcal mol−1) compared to Si–C bond strength (90–94 kcal mol−1).26 Furthermore, the charges on the central silicon and carbon at position a are 2.18 a.u. and −0.94 a.u., respectively (Fig. 1 and 3(a)), leading to stronger interactions that decrease the likelihood of Si–C bond fission. Thus, fragmentation from the Cb–Cc bond leads to a shorter PEG chain fragment in addition to a PDMS fragment with an ethyl group hosting the radical.
Analyzing the oxidized geometry of PDMS and PEG provides insight into further reactivity of smaller product fragments from TriSil-1n and TriSil surfactants containing different number of repeating units. The oxidation of PEG-3n will likely lead to internal C–C bond fission and two symmetrical fragments (Fig. S8(a), (b) and S9(a)†). This is due to the removal of charge from the inner oxygen atoms, the increase in the C–C bond length by 0.26 Å, and the slight decrease in the C–O bond length (Table S4†). Therefore, potential oxidized induced C–C bond breakage along the PEG component of TriSil-1n is possible when multiple PEG units (n > 1) are present. In terms of PDMS, oxidation resulted in the Si–C bond length increasing by 0.31 Å (Table S4†) and the increase in electropositivity of the carbon at position a (+0.60) (Fig. S8(d) and (e)†). The change in geometry and charge analysis imply the dissociation of the Si–C bond with the formation of a methyl radical and cationic PDMS product.
No trend is observed in oxidation reaction energies between TriSil-1n and its constituents and a hydroxyl radical, which range from 24.6 to 38.9 kcal mol−1 (Table S5†). These high energy values indicate it may be more difficult to oxidize TriSil-1n, PEG, and PDMS through SET when compared to H-abstraction, however this could change based on the nature of the oxidant. It may be more difficult to oxidize PEG chains containing an odd number of oxygens, such as in PEG-4n and PEG-6n, due to localization of spin radical density around the central oxygen, which stabilizes these chains compared to PEG-5n (Fig. S9(e)–(g)†). However, conformational energies for polyethylene oxide (PEO) and PEG chains are heavily dependent on the solvation environment and usually require explicit solvation,84 which is outside the scope of our work. Regardless, PEG chains are typically resistant to oxidation.85
In addition to oxidative conditions, TriSil surfactants may encounter reductive environments, which are common in wastewater remediation treatments, such as advanced reduction processes.86,87 Thus, it is important to study how the reduction of TriSil-1n (Scheme S2(ii)†) affects its structure and degradation, to aid in determining if this surfactant is a less environmentally toxic replacement to PFAS in fire-fighting formulations. We studied the effect of electron addition on the structure of TriSil-1n, as well as PDMS and increasing PEG units, to understand the locality of the extra charge. The reduction of TriSil-1n, PDMS, and PEG-3n does not lead to any dramatic geometrical changes (Fig. 5(b), S9(b) and (d)†). In PEG-3n, the electron adds to positions Ca and Cb (Fig. 1(iv)) as shown by NBO charge (−0.33 a.u. each) and spin density (0.36 a.u. each) (Fig. S8(b), (c) and S9(b)†). In PDMS, the effect of reduction is observed in localization of the charge (−0.09 to −0.17 a.u.) (Fig. S8(e) and (f)†) and extra spin density (0.10–0.18 a.u.) (Fig. S9(d)†) on the central and terminal silicon atoms, with the remaining charge distributed among the methyl carbons. In TriSil-1n, the change in atomic charge (−0.41 a.u.) and spin (0.42) are primarily located on the central silicon (Fig. 5(b)). This is the opposite of the oxidation pathway, in which the oxygen and carbons along the PEG chain contain most of the charge and spin (Fig. 5(a)). Thus, depending on whether the conditions are oxidative or reductive, TriSil may degrade through different pathways. This has implication regarding the tunability of degradation strategies. By proceeding through the oxidation route, TriSil-1n may break apart and form products along the PEG chain. By proceeding through the reduction route, the PDMS unit of TriSil-1n may initiate degradation and lead to effective elimination of the surfactant.
Reaction free energies and reduction potentials of various electron attachment positions were computed for TriSil-1n, PDMS, and a range of PEG polymer lengths (n = 1–8) (Table S6†). Similar to the calculation of reduction potentials of PFAS by Van Hoomissen et al.,49 we employed two methods to calculate the reduction potentials of these molecules. In method 1  the abstracted hydrogen was included in the same calculation as the reduced molecule. In method 2
the abstracted hydrogen was included in the same calculation as the reduced molecule. In method 2  the energy of the abstracted hydrogen was separately calculated from the TriSil radical. The reduction potentials of TriSil-1n, PDMS, and PEG (1n–8n) do not significantly change by either method, ranging from −4.82 to −3.89 V (Table S6†). Given that the reduction potential of an aqueous electron, a very strong reducing agent, is −2.9 V,88 the reduction of TriSil-1n, PDMS, and PEG are very unlikely. Therefore, oxidation and hydrolysis are much more likely degradation channels for these materials.
the energy of the abstracted hydrogen was separately calculated from the TriSil radical. The reduction potentials of TriSil-1n, PDMS, and PEG (1n–8n) do not significantly change by either method, ranging from −4.82 to −3.89 V (Table S6†). Given that the reduction potential of an aqueous electron, a very strong reducing agent, is −2.9 V,88 the reduction of TriSil-1n, PDMS, and PEG are very unlikely. Therefore, oxidation and hydrolysis are much more likely degradation channels for these materials.
4 Conclusion
Trimethylsiloxane surfactants are potential fluorine-free alternatives to PFAS as fire retardants; therefore, it is important to understand the susceptibility of TriSil to reduction, oxidation, or hydrolysis in relevant environmental conditions or water treatment processes. This work aids in understanding if these fluorine-free surfactants are stable in aqueous fire-fighting formulations and if their aqueous solution degradation reactions and products are environmentally friendly. The primary products identified from neutral, acidic, and basic hydrolysis are neutral or anionic hydroxylated trimethylsiloxanes (TMS–OH or TMS–O−) and stable PDMS–diol products. The TriSil-1n hydrolysis reactions were determined to be more energetically favorable in acidic and basic conditions, as the neutral hydrolysis reaction required large energy barriers due to sterics. Oxidation of TriSil-1n was investigated via H-abstraction and SET reactions using hydroxyl radicals. All radicals produced via the H-abstraction pathway are kinetically and thermodynamically feasible, with the prominent TriSil radical occurring at position d. Subsequent radical propagation via O2 addition leads to the formation of aldehyde and alcohol containing products and other small oxygen-containing organics. Moreover, H-atom abstractions are likely the most favorable oxidation-induced degradation route; however, aqueous solution oxidation reactions are complex and not all oxidants favor H-abstraction. Oxidation via SET determined that electron removal is primarily localized along the PEG chain, facilitating C–C and Si–C bond dissociation and leading to elimination of the PEG unit and formation of PDMS-like products, which are known to completely degrade and not pose an environmental threat. Finally, the reduction of TriSil-1n was investigated and found to be unlikely to occur in aqueous solution degradation. However, although TriSil is not likely to be reduced, TriSil surfactants are a potential safer alternative component to PFAS in fire-fighting foams.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The use of trade, product or firm names in this report is for descriptive purposes only and does not imply endorsement by the U.S. Government. The tests described and the resulting data presented herein were obtained from research conducted under the Installations and Operational Environments Program of the United States Army Corps of Engineers by the USAERDC under the work supported by (OUSD(R&E)) through the Applied Research for the Advancement of Science and Technology Priorities (ARAP) program. Permission was granted by the Chief of Engineers to publish this information. The findings of this report are not to be construed as an official Department of the Army position unless so designated by other authorized documents. Authors also acknowledge grant of computer time from the DOD High Performance Computing Modernization Program at ERDC, Vicksburg MS. This research was supported in part by an appointment to the Department of Defense (DOD) Research Participation Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and the DOD. ORISE is managed by ORAU under DOE contract number DE-SC0014664. All opinions expressed in this paper are the author's and do not necessarily reflect the policies and views of DOD, DOE, or ORAU/ORISE. The work performed by MM and SV was supported by funding provided by Army Corp of Engineers (Cooperative agreement # W912HZ-20-2-0067). MM and SV also gratefully acknowledge allocated computational resources from High Performance Computing facility at Colorado School of Mines.References
- R. Ananth, K. M. Hinnant, S. L. Giles, J. P. Farley and A. W. Snow, Environmentally-Friendly Surfactants for Foams with Low Fuel Permeability Needed for Effective Pool Fire Suppression, NRL Memorandum Report NRL/MR/6180-20-10,145, Naval Research Laboratory: Naval Technology Center for Safety and Survivability Branch Chemistry Division, 2020, pp. 1–139 Search PubMed.
- R. Hetzer, F. Kümmerlen, K. Wirz and D. Blunk, Fire Testing a New Fluorine-Free AFFF Based on a Novel Class of Environmentally Sound High Performance Siloxane Surfactants, Fire Saf. Sci., 2014, 11, 1261–1270, DOI:10.3801/IAFSS.FSS.11-1261.
- U. Mazurek and H. Schwarz, Carbon–Fluorine Bond Activation—Looking at and Learning from Unsolvated Systems, Chem. Commun., 2003,(12), 1321–1326, 10.1039/B211850E.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. van Leeuwen, Perfluoroalkyl and Polyfluoroalkyl Substances in the Environment: Terminology, Classification, and Origins, Integr. Environ. Assess. Manage., 2011, 7(4), 513–541, DOI:10.1002/ieam.258.
- S. Oresky, D. DeYoung and T. Coffey, The Little Book of Big Achievements, Naval Research Laboratory, Washington, DC, 2000 Search PubMed.
- R. L. Tuve and E. J. Jablonski, Method of Extinguishing Liquid Hydrocarbon Fires, US Pat., US3258423, 1966 Search PubMed.
- Military Specification: Fire Extinguishing Agent, Aqueous Film Forming Foam (AFFF) Liquid Concentrate for Fresh and Sea Water; Report No. MIL-F-24385f, U.S. Department of the Navy, Washington, DC, 1992, pp. 1–23 Search PubMed.
- J. N. Meegoda, J. A. Kewalramani, B. Li and R. W. Marsh, A Review of the Applications, Environmental Release, and Remediation Technologies of Per- and Polyfluoroalkyl Substances, Int. J. Environ. Res. Public Health, 2020, 17(21), 8117, DOI:10.3390/ijerph17218117.
- Z. Zeng, B. Song, R. Xiao, G. Zeng, J. Gong, M. Chen, P. Xu, P. Zhang, M. Shen and H. Yi, Assessing the Human Health Risks of Perfluorooctane Sulfonate by In Vivo and In Vitro Studies, Environ. Int., 2019, 126, 598–610, DOI:10.1016/j.envint.2019.03.002.
- E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen, A Review of the Pathways of Human Exposure to Poly- and Perfluoroalkyl Substances (PFASs) and Present Understanding of Health Effects, J. Exposure Sci. Environ. Epidemiol., 2019, 29(2), 131–147, DOI:10.1038/s41370-018-0094-1.
- Å. Høisæter, A. Pfaff and G. D. Breedveld, Leaching and Transport of PFAS from Aqueous Film-Forming Foam (AFFF) in the Unsaturated Soil at a Firefighting Training Facility under Cold Climatic Conditions, J. Contam. Hydrol., 2019, 222, 112–122, DOI:10.1016/j.jconhyd.2019.02.010.
- J. Horst, J. Quinnan, J. McDonough, J. Lang, P. Storch, J. Burdick and C. Theriault, Transitioning Per‐ and Polyfluoroalkyl Substance Containing Fire Fighting Foams to New Alternatives: Evolving Methods and Best Practices to Protect the Environment, Groundwater Monit. Rem., 2021, 41(2), 19–26, DOI:10.1111/gwmr.12444.
- A. R. Sontake and S. M. Wagh, The Phase-out of Perfluorooctane Sulfonate (PFOS) and the Global Future of Aqueous Film Forming Foam (AFFF), Innovations in Fire Fighting Foam, Chem. Eng. Sci., 2014, 2(1), 11–14, DOI:10.12691/ces-2-1-3.
- J. Conder, J. Zodrow, J. Arblaster, B. Kelly, F. Gobas, J. Suski, E. Osborn, M. Frenchmeyer, C. Divine and A. Leeson, Strategic Resources for Assessing PFAS Ecological Risks at AFFF Sites, Integr. Environ. Assess. Manage., 2021, 17(4), 746–752, DOI:10.1002/ieam.4405.
- A. East, R. H. Anderson and C. J. Salice, Per‐ and Polyfluoroalkyl Substances (PFAS) in Surface Water Near US Air Force Bases: Prioritizing Individual Chemicals and Mixtures for Toxicity Testing and Risk Assessment, Environ. Toxicol. Chem., 2021, 40(3), 859–870, DOI:10.1002/etc.4893.
- A. Nickerson, A. E. Rodowa, D. T. Adamson, J. A. Field, P. R. Kulkarni, J. J. Kornuc and C. P. Higgins, Spatial Trends of Anionic, Zwitterionic, and Cationic PFASs at an AFFF-Impacted Site, Environ. Sci. Technol., 2021, 55(1), 313–323, DOI:10.1021/acs.est.0c04473.
- R. Tenorio, J. Liu, X. Xiao, A. Maizel, C. P. Higgins, C. E. Schaefer and T. J. Strathmann, Destruction of Per- and Polyfluoroalkyl Substances (PFASs) in Aqueous Film-Forming Foam (AFFF) with UV-Sulfite Photoreductive Treatment, Environ. Sci. Technol., 2020, 54(11), 6957–6967, DOI:10.1021/acs.est.0c00961.
- K. M. Hinnant, S. L. Giles, A. W. Snow, J. P. Farley, J. W. Fleming and R. Ananth, An Analytically Defined Fire-Suppressing Foam Formulation for Evaluation of Fluorosurfactant Replacement, J. Surfactants Deterg., 2018, 21(5), 711–722, DOI:10.1002/jsde.12166.
- X. Fenghua, L. Xinqiang, D. Jianguo and K. Yan, A Kind of Fluorine-Free Flame Retardant Polyurethane Rigid Foam Material and Preparation Method Thereof, CN102504170, 2012, https://patentscope.wipo.int/search/en/detail.jsf?docId=CN85136542%26_cid=P22-L609OG-16439-1.
- P. Wang, Application of Green Surfactants Developing Environment Friendly Foam Extinguishing Agent, Fire Technol., 2015, 51(3), 503–511, DOI:10.1007/s10694-014-0422-5.
- G. Xiao, L. Lei, C. Chen, Y. Li and W. Hu, Optimization and Performance Evaluation of an Environmentally Friendly Fluorine-Free Foam Extinguishing Agent, J. Nanoelectron. Optoelectron., 2020, 15(7), 884–893, DOI:10.1166/jno.2020.2813.
- K. Rangan, Fluorine Free Aqueous Film Forming Foams Based on Functional Siloxanes; SERDP Final Report WP18-1638, Strategic Environmental Research and Development Program, Fairfax, VA, 2019, pp. 1–16 Search PubMed.
- Y. Sheng, N. Jiang, S. Lu and C. Li, Fluorinated and Fluorine-Free Firefighting Foams Spread on Heptane Surface, Colloids Surf., A, 2018, 552, 1–8, DOI:10.1016/j.colsurfa.2018.05.004.
- J. Liu, F. F. Zhang, Y. H. Song, K. Lv, N. Zhang and Y. C. Li, The Synthesis of Nonionic Hyperbranched Organosilicone Surfactant and Characterization of Its Wetting Ability, Coatings, 2020, 11(1), 32, DOI:10.3390/coatings11010032.
- K. M. Hinnant, M. W. Conroy and R. Ananth, Influence of Fuel on Foam Degradation for Fluorinated and Fluorine-Free Foams, Colloids Surf., A, 2017, 522, 1–17, DOI:10.1016/j.colsurfa.2017.02.082.
- B. D. Etz, M. Mifkovic, S. Vyas and M. K. Shukla, High-Temperature Decomposition Chemistry of Trimethylsiloxane Surfactants, a Potential Fluorine–Free Replacement for Fire Suppression, Chemosphere, 2022, 308(pt 2), 136351, DOI:10.1016/j.chemosphere.2022.136351.
- E. L. DiFilippo and R. P. Eganhouse, Assessment of PDMS-Water Partition Coefficients: Implications for Passive Environmental Sampling of Hydrophobic Organic Compounds, Environ. Sci. Technol., 2010, 44(18), 6917–6925, DOI:10.1021/es101103x.
- N. Carmichael, European Centre for Ecotoxicology and Toxicology of Chemicals, in Encyclopedia of Toxicology, Elsevier, 2014, pp. 547–548, DOI:10.1016/B978-0-12-386454-3.00505-4.
- Organosilicon Materials, ed. G. Chandra and O. Hutzinger, Series: The Handbook of Environmental Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1997, vol. 3/3H, DOI:10.1007/978-3-540-68331-5.
- G. Ducom, B. Laubie, A. Ohannessian, C. Chottier, P. Germain and V. Chatain, Hydrolysis of Polydimethylsiloxane Fluids in Controlled Aqueous Solutions, Water Sci. Technol., 2013, 68(4), 813–820, DOI:10.2166/wst.2013.308.
- R. R. Buch, T. H. Lane, R. B. Annelin and C. L. Frye, Photolytic Oxidative Demethylation of Aqueous Dimethylsiloxanols, Environ. Toxicol. Chem., 1984, 3(2), 215–222, DOI:10.1002/etc.5620030204.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Fate of Silicone Degradation Products (Silanols) in Soil, Environ. Toxicol. Chem., 1994, 13(11), 1753–1759, DOI:10.1002/etc.5620131106.
- J. C. Carpenter, J. A. Cella and S. B. Dorn, Study of the Degradation of Polydimethylsiloxanes on Soil, Environ. Sci. Technol., 1995, 29(4), 864–868, DOI:10.1021/es00004a005.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Degradation of Silicone Polymers in Soil, Environ. Toxicol. Chem., 1994, 13(7), 1061–1064, DOI:10.1002/etc.5620130707.
- R. G. Lehmann, J. R. Miller and G. E. Kozerski, Degradation of Silicone Polymer in a Field Soil under Natural Conditions, Chemosphere, 2000, 41(5), 743–749, DOI:10.1016/s0045-6535(99)00430-0.
- R. G. Lehmann, J. R. Miller and G. E. Kozerski, Fate of Dimethylsilanediol in a Grass and Soil System, Appl. Soil Ecol., 2002, 19(2), 103–111, DOI:10.1016/S0929-1393(01)00186-X.
- C. L. Sabourin, J. C. Carpenter, T. K. Leib and J. L. Spivack, Biodegradation of Dimethylsilanediol in Soils, Appl. Environ. Microbiol., 1996, 62(12), 4352–4360, DOI:10.1128/aem.62.12.4352-4360.1996.
- R. G. Lehmann, J. R. Miller and H. P. Collins, Microbial Degradation of Dimethylsilanediol in Soil, Water, Air, Soil Pollut., 1998, 106(1), 111–122, DOI:10.1023/A:1004933107104.
- Degradation of Silicone Polymers in Nature, Environmental Information – Update, Dow Corning, 01-1113-01, 1998, pp. 1–4 Search PubMed.
- D. Tolle, C. Frye, R. Lehmann and T. Zwick, Ecological Effects of PDMS-Augmented Sludge Amended to Agricultural Microcosms, Sci. Total Environ., 1995, 162(2–3), 193–207, DOI:10.1016/0048-9697(95)04459-E.
- C. A. Staples, J. B. Williams, G. R. Craig and K. M. Roberts, Fate, Effects and Potential Environmental Risks of Ethylene Glycol: A Review, Chemosphere, 2001, 43(3), 377–383, DOI:10.1016/s0045-6535(00)00148-x.
- F. Kawai, Biodegradation of Polyethers (Polyethylene Glycol, Polypropylene Glycol, Polytetramethylene Glycol, and Others): Part 9. Miscellaneous Biopolymers and Biodegradation of Polymers, Biopolym. Online, 2001 Search PubMed.
- E. Beran, S. Hull and M. Steininger, The Relationship Between the Chemical Structure of Poly(Alkylene Glycol)s and Their Aerobic Biodegradability in an Aqueous Environment, J. Polym. Environ., 2013, 21(1), 172–180, DOI:10.1007/s10924-012-0445-2.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. v. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. v. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615, 10.1039/b810189b.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396, DOI:10.1021/jp810292n.
- M. Cypryk and B. Gostyński, Computational Benchmark for Calculation of Silane and Siloxane Thermochemistry, J. Mol. Model., 2016, 22(1), 35, DOI:10.1007/s00894-015-2900-1.
- L. Goerigk and S. Grimme, A Thorough Benchmark of Density Functional Methods for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions, Phys. Chem. Chem. Phys., 2011, 13(14), 6670, 10.1039/c0cp02984j.
- D. J. Van Hoomissen and S. Vyas, Early Events in the Reductive Dehalogenation of Linear Perfluoroalkyl Substances, Environ. Sci. Technol. Lett., 2019, 6(6), 365–371, DOI:10.1021/acs.estlett.9b00116.
- R. S. Alvim and C. R. Miranda, First Principles Characterization of Silicate Sites in Clay Surfaces, Phys. Chem. Chem. Phys., 2015, 17(7), 4952–4960, 10.1039/C4CP05447D.
- A. M. Rodríguez, J. Pérez-Ruíz, F. Molina, A. Poveda, R. Pérez-Soto, F. Maseras, M. M. Díaz-Requejo and P. J. Pérez, Introducing the Catalytic Amination of Silanes via Nitrene Insertion, J. Am. Chem. Soc., 2022, 144(23), 10608–10614, DOI:10.1021/jacs.2c03739.
- K. Garcés, F. J. Fernández-Alvarez, V. Polo, R. Lalrempuia, J. J. Pérez-Torrente and L. A. Oro, Iridium-Catalyzed Hydrogen Production from Hydrosilanes and Water, ChemCatChem, 2014, 6(6), 1691–1697, DOI:10.1002/cctc.201301107.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120(1–3), 215–241, DOI:10.1007/s00214-007-0310-x.
- A. D. Becke, Density‐functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785–789, DOI:10.1103/PhysRevB.37.785.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98(45), 11623–11627, DOI:10.1021/j100096a001.
- R. Ananth, Synergisms between Siloxane-Polyoxyethylene and Alkyl Polyglycoside Surfactants in Foam Stability and Pool Fire Extinction, Colloids Surf., A, 2019, 579, 123686 CrossRef.
- M. Zhang, B. Ning, Y. Bai, X. Tai and G. Wang, Solution Properties of Mixed System Containing Butynediol-Ethoxylate Polysiloxanes and Polyether Trisiloxane Surfactant, Colloid Interface Sci. Commun., 2021, 41, 100367, DOI:10.1016/j.colcom.2021.100367.
- Silicone Surface Science, ed. M. J. Owen and P. R. Dvornic, Advances in Silicon Science, Springer Netherlands, Dordrecht, 2012, vol. 4, DOI:10.1007/978-94-007-3876-8.
- B. Laubie, E. Bonnafous, V. Desjardin, P. Germain and E. Fleury, Silicone-Based Surfactant Degradation in Aqueous Media, Sci. Total Environ., 2013, 454–455, 199–205, DOI:10.1016/j.scitotenv.2013.02.022.
- M. Cypryk and Y. Apeloig, Mechanism of the Acid-Catalyzed Si–O Bond Cleavage in Siloxanes and Siloxanols. A Theoretical Study, Organometallics, 2002, 21(11), 2165–2175, DOI:10.1021/om011055s.
- T. W. Zerda and G. Hoang, High-Pressure Raman Study of the Hydrolysis Reaction in Tetramethylorthosilicate (TMOS), J. Non-Cryst. Solids, 1989, 109(1), 9–17, DOI:10.1016/0022-3093(89)90435-3.
- M. Knoche, H. Tamura and M. J. Bukovac, Performance and Stability of the Organosilicon Surfactant L-77: Effect of PH, Concentration, and Temperature, J. Agric. Food Chem., 1991, 39(1), 202–206, DOI:10.1021/jf00001a041.
- S. K. Lai, A. Batra and C. Cohen, Characterization of Polydimethylsiloxane Elastomer Degradation via Cross-Linker Hydrolysis, Polymer, 2005, 46(12), 4204–4211, DOI:10.1016/j.polymer.2005.02.051.
- A. Michel, H.-J. Brauch, E. Worch and F. T. Lange, Homologue Specific Analysis of a Polyether Trisiloxane Surfactant in German Surface Waters and Study on Its Hydrolysis, Environ. Pollut., 2014, 186, 126–135, DOI:10.1016/j.envpol.2013.11.020.
- D. J. Van Hoomissen and S. Vyas, 1,2-H Atom Rearrangements in Benzyloxyl Radicals, J. Phys. Chem. A, 2019, 123(2), 492–504, DOI:10.1021/acs.jpca.8b10286.
- J. T. Bingham, B. D. Etz, J. M. DuClos and S. Vyas, Structure and Reactivity of Alloxan Monohydrate in the Liquid Phase, J. Org. Chem., 2021, 86(21), 14553–14562, DOI:10.1021/acs.joc.1c01389.
- X. Cheng, D. Chen and Y. Liu, Mechanisms of Silicon Alkoxide Hydrolysis-Oligomerization Reactions: A DFT Investigation, ChemPhysChem, 2012, 13(9), 2392–2404, DOI:10.1002/cphc.201200115.
- J. B. Lambert, Y. Zhao, R. W. Emblidge, L. A. Salvador, X. Liu, J.-H. So and E. C. Chelius, The β Effect of Silicon and Related Manifestations of σ Conjugation, Acc. Chem. Res., 1999, 32(2), 183–190, DOI:10.1021/ar970296m.
- N. Okui and J. H. Magill, Studies of the Chemical Degradation of Polysiloxanes by Hydrofluoric Acid: (2). Poly(Tetramethyl-p-Silphenylene Siloxane-Dimethyl Siloxane) Block Copolymers, Polymer, 1977, 18(8), 845–850, DOI:10.1016/0032-3861(77)90193-8.
- B. Rupasinghe and J. C. Furgal, Degradation of Silicone‐based Materials as a Driving Force for Recyclability, Polym. Int., 2022, 71(5), 521–531, DOI:10.1002/pi.6340.
- G. Ducom, B. Laubie, A. Ohannessian, C. Chottier, P. Germain and V. Chatain, Hydrolysis of polydimethylsiloxane fluids in controlled aqueous solutions, Water Sci. Technol., 2013, 68(4), 813–820, DOI:10.2166/wst.2013.308.
- Z. Peng, C. Lu and M. Xu, Influence of Substructures on the Spreading Ability and Hydrolysis Resistance of Double-Tail Trisiloxane Surfactants, J. Surfactants Deterg., 2010, 13(1), 75–81, DOI:10.1007/s11743-009-1144-4.
- D. Graiver, K. W. Farminer and R. Narayan, A Review of the Fate and Effects of Silicones in the Environment, J. Polym. Environ., 2003, 11, 129–136, DOI:10.1023/A:1026056129717.
- J. C. Furgal and C. U. Lenora, Green routes to silicon-based materials and their environmental implications, Phys. Sci. Rev., 2020, 5(1), 20190024, DOI:10.1515/psr-2019-0024.
- E. B. Esfahani, F. A. Zeidabadi, S. Zhang and M. Mohseni, Photo-Chemical/Catalytic Oxidative/Reductive Decomposition of per- and Poly-Fluoroalkyl Substances (PFAS), Decomposition Mechanisms and Effects of Key Factors: A Review, Environ. Sci.: Water Res. Technol., 2022, 8(4), 698–728 RSC.
- Y. Deng and R. Zhao, Advanced Oxidation Processes (AOPs) in Wastewater Treatment, Curr. Pollut. Rep., 2015, 1(3), 167–176, DOI:10.1007/s40726-015-0015-z.
- T. An, Y. Gao, G. Li, P. V. Kamat, J. Peller and M. V. Joyce, Kinetics and Mechanism of ·OH Mediated Degradation of Dimethyl Phthalate in Aqueous Solution: Experimental and Theoretical Studies, Environ. Sci. Technol., 2014, 48(1), 641–648, DOI:10.1021/es404453v.
- O. Legrini, E. Oliveros and A. M. Braun, Photochemical Processes for Water Treatment, Chem. Rev., 1993, 93(2), 671–698, DOI:10.1021/cr00018a003.
- M. Schuchmann, H. Zegota and C. von Sonntag, Acetate Peroxyl Radicals, O2CH2CO2−: A Study on the y-Radiolysis and Pulse Radiolysis of Acetate in Oxygenated Aqueous Solutions, Z. Naturforsch., 1985, 40b, 215–221, DOI:10.1515/znb-1985-0212.
- D. Kamath, S. P. Mezyk and D. Minakata, Elucidating the Elementary Reaction Pathways and Kinetics of Hydroxyl Radical-Induced Acetone Degradation in Aqueous Phase Advanced Oxidation Processes, Environ. Sci. Technol., 2018, 52(14), 7763–7774, DOI:10.1021/acs.est.8b00582.
- V.-T. Salo, R. Valiev, S. Lehtola and T. Kurtén, Gas-Phase Peroxyl Radical Recombination Reactions: A Computational Study of Formation and Decomposition of Tetroxides, J. Phys. Chem. A, 2022, 126(25), 4046–4056, DOI:10.1021/acs.jpca.2c01321.
- K. Li and J. Crittenden, Computerized Pathway Elucidation for Hydroxyl Radical-Induced Chain Reaction Mechanisms in Aqueous Phase Advanced Oxidation Processes, Environ. Sci. Technol., 2009, 43(8), 2831–2837, DOI:10.1021/es802039y.
- U. R. Dahal and E. E. Dormidontova, The Dynamics of Solvation Dictates the Conformation of Polyethylene Oxide in Aqueous, Isobutyric Acid and Binary Solutions, Phys. Chem. Chem. Phys., 2017, 19(15), 9823–9832, 10.1039/C7CP00526A.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Polyethylene Glycol and Solutions of Polyethylene Glycol as Green Reaction Media, Green Chem., 2005, 7(2), 64, 10.1039/b413546f.
- B. P. Vellanki, B. Batchelor and A. Abdel-Wahab, Advanced Reduction Processes: A New Class of Treatment Processes, Environ. Eng. Sci., 2013, 30(5), 264–271 CrossRef CAS PubMed.
- C. K. Amador, D. J. Van Hoomissen, J. Liu, T. J. Strathmann and S. Vyas, Ultra-Short Chain Fluorocarboxylates Exhibit Wide Ranging Reactivity with Hydrated Electrons, Chemosphere, 2023, 311, 136918, DOI:10.1016/j.chemosphere.2022.136918.
- P. Wardman, Reduction Potentials of One‐Electron Couples Involving Free Radicals in Aqueous Solution, J. Phys. Chem. Ref. Data, 1989, 18(4), 1637–1755, DOI:10.1063/1.555843.
Footnotes |
| † Electronic supplementary information (ESI) available: NBO spin density and charge analyses, alternative pathways, and optimized coordinates for all species. See DOI: https://doi.org/10.1039/d4va00256c |
| ‡ Current affiliation: Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. |
| This journal is © The Royal Society of Chemistry 2025 |