
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional Eu(III) and Sm(III) coordination polymers built with silane-bridged dicarboxylate ligand: structure, luminescence and magnetism
Ana
Arauzo
 *a,
Mirela-Fernanda
Zaltariov
b,
Elena
Bartolomé
c,
Sara
Fuertes
d,
Ionut-Radu
Tigoianu
b,
Sergiu
Shova
b and
Maria
Cazacu
b
*a,
Mirela-Fernanda
Zaltariov
b,
Elena
Bartolomé
c,
Sara
Fuertes
d,
Ionut-Radu
Tigoianu
b,
Sergiu
Shova
b and
Maria
Cazacu
b
aInstituto de Nanociencia y Materiales de Aragón (INMA), Departamento de Física de la Materia Condensada, CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: aarauzo@unizar.es
bDepartment of Inorganic Polymers, “Petru Poni” Institute of Macromolecular Chemistry, Aleea Gr. Ghica Voda 41A, 700487, Iasi, Romania
cInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Spain
dDepartamento de Química Inorgánica, Facultad de Ciencias, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC – Universidad de Zaragoza, Pedro Cerbuna 12, 50009, Zaragoza, Spain
First published on 8th August 2025
Abstract
Three novel coordination polymers – [LnLNO3(o-Phen)] (Ln: Eu(III) (1), Sm(III) (2)) and [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O (3) – were synthesized by reacting the respective lanthanide nitrates with the organosilane dicarboxylic acid bis(p-carboxyphenyl)diphenylsilane (H2L) with or without o-phenanthroline (o-Phen) as coligand. The o-Phen-assisted route yielded isostructural one-dimensional coordination polymers (CP) 1 and 2, incorporating both ligands and the nitrate anion. These ribbon-like chains assemble into hydrogen-bonded 2D networks that further stack into 3D architectures through non-covalent interactions. In contrast the o-Phen-free conditions produced the 3D CP 3, based solely on H2L, with ethanol and water molecules retained in the lattice. All three architectures are highly dense, with free volumes of only 2.7% (1, 2) and 11.0% (3). Photophysical studies reveal an exceptional quantum yield of 67% for Eu-based complex 1 attributed to complete ligand-to-Eu energy transfer. In contrast, the Sm-based compound 2 achieves a modest 2% quantum yield, while compound 3 reaches 8%. Magnetic measurements confirm typical Van Vleck paramagnetism for Eu3+ in 1 and 3, with spin–orbit coupling constants of Λ = 379 and 352 cm−1, respectively. The magnetism of the Sm-based CP 2 is governed by its ground 6H5/2 and first excited 6H7/2 multiplets (Λ = 310 cm−1), and notably displays field-induced slow magnetic relaxation. Remarkably, Eu-based CP 1 affords exfoliation by sonication into nanometric flakes that retain a high quantum yield of 57% upon deposition, opening up exciting opportunities for surface-integrated sensing, photonic, and quantum applications.
1. Introduction
The electronic configuration of lanthanide ions underpins their remarkable chemical and photophysical properties.1 Their characteristic luminescence arises from intraconfigurational 4f–4f transitions, as well as interconfigurational 4fn → 4fn5d1 transitions, and charge-transfer processes, such as ligand-to-metal (LMCT) and metal-to-ligand charge transfer (MLCT) mechanisms.2 Among lanthanide-based materials, coordination compounds have attracted significant interest due to their advantageous properties, such as high luminescence efficiency, low excitation energy, and chemical versatility in both solution and solid state.3 In such complexes, luminescence is primarily driven by the “antenna effect” – an energy transfer process, in which the ligand absorbs energy and transfers it to the excited state of the lanthanide ion (Ln3+), enhancing emission efficiency.4Lanthanide ions show flexibility in coordination of ligands, especially with hard-donors (containing oxygen atoms in their structure), and variable coordination number, leading to a variety of original architectural topologies, including coordination polymers (CP) of different dimensionalities and porous metal–organic frameworks (MOFs). In addition, owing to the intrinsic characteristics of 4f electrons, Ln-based complexes exhibit exceptional optical features, including narrow-band emission, long luminescence lifetimes, large Stokes shifts, and excellent color purity.4 Their luminescence may stem from multiple processes, including ligand-centered transitions, metal-centered emissions, photoinduced electron transfer (PET), LMCT, MLCT, ligand-to-ligand (LLCT), or guest-molecule interactions. Nonetheless, ligand-based sensitization via the “antenna effect” remains the most efficient and widely exploited mechanism. Despite their advantages, the luminescence efficiency of Ln-coordination compounds can be compromised by non-radiative decay pathways such as internal conversion and vibrational relaxation. In the solid state or at high concentrations, aggregation-caused quenching can further limit their photoluminescence performance.5 Several strategies combining tailored ligand design with the selective incorporation of homo- or heteronuclear lanthanide centers (e.g., Eu, Sm, Y, Tb) have been developed to enable efficient energy transfer and tunable emission spanning a broad color spectrum – from red and blue to green and white.
The diversity of luminescence mechanisms in Ln-CPs and Ln-MOFs makes them exceptionally versatile platforms for a wide range of applications, including sensing of metal cations, anions, small molecules, gases, vapors, and biomarkers.6 Notably, Ln-MOFs incorporating Eu3+, Tb3+, and Sm3+ have demonstrated significant potential in optical devices (e.g., OLEDs), MRI contrast agents, catalysis/electrocatalysis, and chemical sensing.7 In addition, Ln-MOFs can exhibit single molecule magnet (SMM) behavior when the ligand orbitals effectively overlap with the 4f orbitals of the lanthanide ions, opening opportunities for multifunctional materials.8,9 Lanthanide ions with sharp, line-like emissions – especially Eu(III) – are also considered promising candidates for optically driven qubit operations in quantum information processing.10
To fully exploit the photophysical potential of lanthanide complexes, rational ligand design and coordination are essential. The quantum yield (QY) – the ratio of emitted to absorbed photons – is often limited by Ln3+–Ln3+ interaction (causing concentration quenching) and the presence of C–H and O–H vibrations in the first coordination sphere, which facilitate non-radiative decay. Ideal ligands for high-performance luminescent materials should (i) be sterically bulky to spatially isolate metal centers and solvent coordination, (ii) be at least bifunctional to facilitate extended coordination networks, and (iii) incorporate an extended conjugated system to enhance the antenna efficiency.11 Another approach to design efficient emissive lanthanide systems is to identify the dominant energy transfer mechanism, arising from Förster resonant energy transfer (FRET) involving Coulombic interactions (occurring from ligand singlet state or intraligand charge transfer state) and Dexter electron exchange (occurring from the ligand triplet state).12
Among known sensitizers, β-diketonates are the most recognized and used antenna ligands due to their high sensitization efficiency, which can be controlled via ligand substituents, combined ligands and extended conjugation.13 Aromatic N-donor ligands, such as 1,10-phenanthroline (phen), are also effective sensitizers of lanthanides, as they efficiently absorb and transfer energy to the excited states of Ln3+ ions.14,15 Additionally, phen has been shown to inhibit solvent coordination to lanthanide ions, leading to high quantum yields and remarkable emission in the near-infrared (NIR) region for lanthanides such as Nd, Sm, and Ho.11 In parallel, to construct extended coordination networks, polycarboxylic acids are ideal due to the strong affinity of lanthanides for oxygen donor atoms. This interaction enables various coordination modes, facilitating the formation of diverse multidimensional architectures. As a result, more and more polycarboxylic acids, generally organic, have been used to build such networks. Elemental-organic acids, in contrast, remain less explored. One notable example is tetrakis(4-carboxyphenyl)silane, a rigid tetrahedral carboxylate ligand centered on silicon, which has enabled the construction of Ln (Tb, Er, Dy, Tm, Y, and Pr) 3D MOFs with potential applications as fluorescent probes and antiferromagnetic materials.16 In the present study, we extend this concept by employing a structurally similar dicarboxylated ligand – bis(p-carboxyphenyl)diphenylsilane – a relatively bulky molecule with two carboxyl groups. While this ligand and its coordination with transition metals has been previously reported,17,18 its utility in constructing lanthanide-based frameworks remains unexplored.
In addition to 3D networks, coordination polymers with other dimensionalities (2D and 1D) also present particularities that may be of interest for certain applications. Two-dimensional metal–organic frameworks (2D MOFs), referring to 2D CPs that form network-like sheet-like structures, with weaker interactions between layers, represent a more versatile metal–organic alternative to the established inorganic 2D materials. Due to surface, interface, and edge effects19–21 such complexes can exhibit fascinating properties, including photoluminescence, electrical conductivity, spin-crossover, and magnetism induced by chemical structure and potentiated by the atomic-scale lattice effect.21 Besides, coordination polymers formed by unidirectional extension of coordination structures (1D CP), can assemble into 2D or 3D structures through dynamic supramolecular interactions that can allow the reversible access and orientation of guest molecules larger than common gaseous species, of interest in separation and sensing.22
Within this context, we report here the synthesis, structural, optical, and magnetic properties of novel europium(III) and samarium(III) CPs with different dimensionalities built from diphenylsilane-containing dicarboxylic ligands with phenanthroline as co-ligand. The presence of phenanthroline gives 1D CPs without solvent in the coordination sphere, while its absence leads to 3D CPs that incorporate solvent and water molecules. Notably, such 1D CPs form ribbon-like chains that organize into 2D supramolecular networks through hydrogen bonding, and are further stacked in 3D space. This affords their exfoliation without significant decrease of the luminescent properties. Our results provide insights into the role of ligand design in enhancing lanthanide luminescence, and open new possibilities for designing advanced materials based on lanthanides coordination polymers with multifunctional properties relevant to photonics, sensing, and quantum technologies.
2. Experimental section
2.1. Materials and synthesis
Bis(p-carboxyphenyl)diphenylsilane, (C26H20O4Si) or 4,4′-(diphenylsilanediyl)dibenzoic acid, according to IUPAC nomenclature, H2L, was prepared and characterized as reported in ref. 17. 1,10-Phenanthroline, o-Phen, (≥99%) was purchased from Aldrich, Eu(III) nitrate hexahydrate and Sm(III) nitrate hexahydrate 99.9%, (trace metal basis), were purchased from Thermo Scientific Chemicals, ethanol of gradient grade purity for liquid chromatography was provided from Merck.2.2. Synthesis of [EuLNO3(o-Phen)] (1)
0.05 g (0.12 mmol) of bis(p-carboxyphenyl)diphenylsilane (H2L) was dissolved in ethanol (6 mL). A solution of 1,10-phenathroline (0.02 g, 0.12 mmol) in ethanol (2 mL) was added to the H2L ligand solution and the mixture was ultrasonicated for 2 min. Next, a solution of Eu(NO3)3·6H2O (0.08 g, 0.24 mmol) in distilled water (2 mL) was added and the mixture was ultrasonicated for 2 min and transferred to a glass tube. The tube was placed in the solvothermal oven for 72 h at 90 °C. Cooling to room temperature was done at 1 °C min−1. The single crystals obtained were filtered, washed with water and dried in the oven at 80 °C for 24 h. Yield: 0.048 g (56%). Calcd for C38H26EuN3O7Si·2.5H2O (M = 861.727 g mol−1), %: C, 52.97; H, 3.63; N, 4.88. Found, %: C, 52.49; H, 3.13; N, 4.71.IR νmax(KBr), cm−1: 3420m, 3071w, 3011vw, 2963vw, 2924vw, 1597vs, 1539s, 1518m, 1479s, 1404vs, 1346m, 1292s, 1188w, 1146w, 1105s, 1047vw, 1024m, 993w, 862m, 847m, 812w, 775m, 723s, 636w, 555w, 530w, 507m, 469m, 413w, 390w.
2.3. Synthesis of [SmLNO3(o-Phen)] (2)
The same synthetic procedure was used to prepare the compound 2, with Sm(NO3)3·6H2O used as the metal precursor. Yield: 0.04 g (27%). Calcd. for C38H26N3O7SiSm (M = 815.085 g mol−1), %: C, 56.00; H, 3.22; N, 5.16. Found, %: C, 56.03; H, 3.06; N, 5.45.IR νmax(KBr), cm−1: 3429m, 3071w, 3011vw, 2964vw, 2922vw, 2853vw, 1599s, 1541m, 1518m, 1479s, 1404vs, 1387vs, 1294s, 1188w, 1146w, 1105s, 1047w, 1028m, 993w, 862m, 847m, 812w, 775m, 723s, 700m, 636w, 555w, 528w, 507m, 469m, 413w, 388w.
2.4. Synthesis of [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O (3)
The compound 3 was prepared under the same solvothermal conditions only reacting H2L and Eu(NO3)3. Yield: 0.033 g (25%). Calc. for C83H77Eu2O18.5Si3 (Mr = 1758.694 g mol−1), %: C, 56.68%, H, 4.41%. Found, %: C, 56.24%, H, 4.43%.IR νmax(KBr), cm−1: 3416m, 3068w, 3046w, 2960w, 2924m, 2854w, 1695m, 1601s, 1577s, 1520s, 1500s, 1409, 1308m, 1190w, 1154w, 1106s, 1041m, 1018m, 997w, 858s, 776s, 729vs, 712s, 699s, 634w, 533m, 506m, 474m, 397m.
2.5. Characterization
IR spectra were recorded at room temperature in transmittance mode in the 400–4000 cm−1 spectral range, with a resolution of 4 cm−1 by accumulation of 32 scans, by using a FT-IR spectrometer Bruker Vertex 70. Elemental analysis (C, H and N) was evaluated on a PerkinElmer CHNS 2400 – series II elemental analyzer.X-ray crystallography
Single-crystal X-ray diffraction (SCXRD) data for 1 and 3 were collected on an Oxford-Diffraction XCALIBUR Eos CCD diffractometer with graphite-monochromated Mo Kα radiation, while for 2 on XtaLAB Synergy, Dualflex, HyPix diffractometer using Cu Kα radiation. The unit cell determination and data integration were carried out using the CrysAlisPro package from Oxford Diffraction.23 Multi-scan correction for absorption was applied. The structure was solved with SHELXT program using the intrinsic phasing method and refined by the full-matrix least-squares method on F2 with SHELXL.24,25 Olex2 was used as an interface to the SHELX programs.26 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added in idealized positions and refined using a riding model. The refinement of the 3 structure was performed imposing the necessary restraints on geometry and displacement parameters of disordered molecules. Besides, the Mask option of Olex2 was used to model the contribution to structural factors of disordered solvent molecules that could not be reliably located. Selected crystallographic data and structure refinement details are provided in Table 1 and the corresponding CIF-files. Powder X-ray diffraction analysis (PXRD) was performed on a RigakuMiniflex 600 Diffractometer using CuKα-emission, in the 5–50° (2θ). A scanning step of 0.01° and a registration rate of 2° min−1 were applied.| Compound | [EuLNO3(o-Phen)] (1) | [SmLNO3(o-Phen)] (2) | [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O (3) |
|---|---|---|---|
| Emp. formula | C38H26EuN3O7Si | C38H26N3O7SiSm | C83H77Eu2O18.5Si3 |
| Fw | 816.67 | 815.06 | 1758.63 |
| T [K] | 200 | 156 | 200 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a [Å] | 9.8257(10) | 9.8085(3) | 9.7022(4) |
| b [Å] | 12.6452(9) | 12.5707(3) | 17.9981(7) |
| c [Å] | 13.8342(15) | 13.8397(4) | 25.0737(9) |
| α [°] | 96.921(7) | 97.137(2) | 94.462(3) |
| β [°] | 91.584(9) | 91.692(2) | 96.341(3) |
| γ [°] | 101.544(7) | 101.203(2) | 105.149(4) |
| V [Å3] | 1669.5(3) | 1658.43(8) | 4174.4(3) |
| Z | 2 | 2 | 2 |
| ρ calcd [g cm−3] | 1.625 | 1.632 | 1.399 |
| μ [mm−1] | 1.971 | 14.116 | 1.597 |
| Crystal size [mm] | 0.15 × 0.05 × 0.03 | 0.10 × 0.05 × 0.05 | 0.25 × 0.05 × 0.03 |
| 2θ range | 4.156 to 50.052 | 6.446 to 133.194 | 3.022 to 50.052 |
| Refls. collected | 10379 | 5786 | 30944 |
| Indep. Refls., Rint | 10379, 0.0439 | 5786, 0.0676 | 14728, 0.0427 |
| Data/rests./params. | 10379/0/452 | 5786/0/452 | 14728/21/720 |
| GOF | 1.007 | 1.042 | 1.027 |
| R 1, wR2(all data) | 0.0408, 0.0651 | 0.0719, 0.2013 | 0.0526, 0.1211 |
| CCDC no. | 2314852 | 2314853 | 2314854 |
2.6. Electron microscopy
Field-emission scanning electron microscopy (FESEM) images and energy-dispersive X-ray spectroscopy (EDS) analysis were acquired using a Carl Zeiss MERLIN microscope and a JEOL JXA-IHP200F Field Emission Electron Probe Microanalyzer (FE-EPMA). Transmission electron microscopy (TEM) was performed on a Tecnai F30 microscope operating at an accelerating voltage of 200 kV. Prior to imaging, nanoflake suspensions of compound 1 were prepared by ultrasonic dispersion in tetrahydrofuran (THF). For SEM analysis, the suspension was drop-cast onto quartz substrates, while for TEM, it was deposited onto carbon-coated copper grids. All samples were allowed to dry under ambient conditions before characterization.2.7. Photophysical measurements
Steady-state spectra of polycrystalline solid samples and their exfoliated dispersions were recorded at room temperature (RT) in a FluoTime 300 spectrofluorometer (PicoQuant GmbH) equipped with a NIR-PMT detector (Hamamatsu H10330C-75) using a 300W Xenon lamp. Excitation spectra were recorded at the energy (λem) at which the emission spectra presented the most intense peak for each lanthanide.Time-resolved measurements were recorded with a 300 Xenon lamp 450 nm as excitation source and using the EasyTau II software package (PicoQuant GmbH). Data analysis was performed using the built-in software FluoFit (PicoQuant GmbH).
Quantum yields (QY) at room temperature of polycrystalline solid samples, their exfoliated dispersions and deposited flakes on quartz, were measured under excitation wavelengths ranging from λex = 275 to 415 nm using a Hamamatsu Absolute PL Quantum Yield spectrometer C11347 (Hamamatsu Quantaurus QY). The absolute QY (ratio of the number of photons emitted by photoluminescence to the number of photons absorbed by the light-emitting material) was measured using an integrating sphere within the maximum spectral range from 300 nm to 950 nm. Powdered samples were placed in a capillary with an internal diameter of 4 mm. The absorption and emission spectra of the sample container (the blank) were separately recorded. The QY was calculated as: QY = (Ec − Ea)/(La − Lc), with Ec being the integrated emission spectrum of the sample, Ea the integrated blank emission spectrum, La the blank absorption, and Lc the sample absorption at the excitation wavelength. Emission spectra acquired using this equipment were also used in the analysis, particularly for samples in the form of flakes on quartz.
Transient absorption spectra of the samples dispersed in ethanol were recorded using an LP980 spectrometer (Edinburgh Instruments) with excitation provided by an Nd YAG laser (maximum output 500 mJ, pulse duration 4–6 ns at excitation wavelength 355 nm). LP980 is a fully integrated and sophisticated transient absorption spectrometer that employs the pump–probe technique to measure transient kinetics, and spectra. In kinetic mode, a single point detector is used to measure the transient kinetics at a specific wavelength, and spectra can be built up by automated scanning over a wavelength range and slicing the data.
2.8. Magnetic measurements
The field-dependence of the magnetization at 1.8 K and temperature-dependence of the dc susceptibility in the 1.8 K to 300 K range were measured using a Quantum Design superconducting quantum interference device (SQUID) magnetometer. Powdered sample measurements were performed using a gelatin capsule housed within a plastic straw serving as the sample holder. To immobilize the powder within the capsule, either Daphne oil or cotton was employed. Data were corrected from all diamagnetic contributions used in sample preparation (−1.5 × 10−8 emu Oe−1, −2.4 × 10−8 emu Oe−1 and −1.0 × 10−8 emu Oe−1 for 1, 2 and 3 respectively). Ac measurements in the frequency range between 10 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz, under an excitation field of Hac = 4 Oe, were carried out at temperatures between 1.8–5.0 K, under dc fields between 0–10 kOe, in a Quantum Design PPMS susceptometer.
000 Hz, under an excitation field of Hac = 4 Oe, were carried out at temperatures between 1.8–5.0 K, under dc fields between 0–10 kOe, in a Quantum Design PPMS susceptometer.
3. Results and discussion
3.1. Synthesis and structural characterization
The dicarboxylate ligand, H2L, containing diphenylsilane units, was synthesized according to a previously reported procedure,17 starting from lithium-treated p-bromotoluene in anhydrous diethyl ether to yield p-tolyllithium. This intermediate was then reacted with diphenyldichlorosilane to form bis(p-tolyl)diphenylsilane, which was subsequently oxidized with chromium trioxide in a mixture of glacial acetic acid–acetic anhydride–sulphuric acid to give the corresponding carboxylic acid, having similar characteristics to those presented in ref. 17. The Eu(III)- and Sm(III)-based CPs (Ln-CPs) were successfully prepared in solvothermal conditions, in a 4:1 (v/v) mixture of ethanol and distilled water heated at 90 °C for 72 h by using 1,10-phenanthroline as co-ligand for compounds 1 and 2 (Scheme 1a). Compound 3 (Scheme 1b) was prepared under the same conditions, but in the absence of 1,10-phenanthroline. The structures of the three Ln-CPs were determined by SCXRD analysis, which revealed that compounds 1 and 2 are isostructural. The findings were also supported by elemental analysis, IR spectroscopy (Fig. S1–S3), and PXRD (Fig. S4–S6).
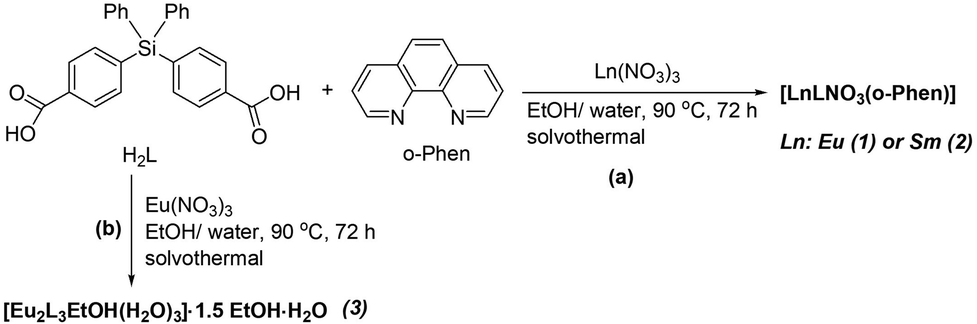 | ||
| Scheme 1 The synthetic route for compounds 1–3. | ||
The most prominent absorption bands in the IR spectra are those attributed to the asymmetric (νas) and symmetric (νs) stretches of the carboxylate groups, identified at 1597 and 1404 cm−1 for compound 1, 1599 and 1404 cm−1 for compound 2 and 1577 and 1409 cm−1 for compound 3 (Fig. S1–S3). The presence of the diphenylsilane units in the structure was confirmed by the bands at 1146, 1105 and 1047 cm−1 for compounds 1 and 2, and 1190, 1106 and 1041 cm−1 for compound 3.27 The phenanthroline fragments in the structure of the compounds 1 and 2 are highlighted by the bands at 1539 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and C–H stretches in aromatic rings at 3071–3011 cm−1. For compound 3, the IR spectrum also reveal the characteristic C–H stretches for coordinated ethanol (2924–2854 cm−1 – νas and νs of C–H groups) and aromatic rings in diphenylsilane units (3068 and 3046 cm−1), as well as O–H deformation vibrations from water and ethanol molecules at 1695 cm−1. In all compounds, the vibration of metal–oxygen bonds are observed at 505 and 555 cm−1 (1 and 2) and 506 and 533 cm−1 (3), while the M–N bond vibrations in compounds 1 and 2 are confirmed by the bands at 469 and 413 cm−1.28
N) and C–H stretches in aromatic rings at 3071–3011 cm−1. For compound 3, the IR spectrum also reveal the characteristic C–H stretches for coordinated ethanol (2924–2854 cm−1 – νas and νs of C–H groups) and aromatic rings in diphenylsilane units (3068 and 3046 cm−1), as well as O–H deformation vibrations from water and ethanol molecules at 1695 cm−1. In all compounds, the vibration of metal–oxygen bonds are observed at 505 and 555 cm−1 (1 and 2) and 506 and 533 cm−1 (3), while the M–N bond vibrations in compounds 1 and 2 are confirmed by the bands at 469 and 413 cm−1.28
X-ray diffraction analysis reveals that compounds 1 and 2 are isostructural. They crystallize in the P space group with closely similar unit cell parameters. Therefore, as an example, only the structure of [SmLNO3(o-Phen)] (2) is described in detail. A view of the asymmetric unit in the crystal structure is shown in Fig. 1a. It consists of one Sm(III) atom, one deprotonated L2− linker, NO3− anion, and o-Phen molecule coordinated as bidentate ligand. There are no co-crystallized solvent molecules in the crystal. The Sm(III) atom is nine-coordinated with seven oxygen atoms from nitrate anions and carboxylate groups, and two nitrogen atoms from o-Phen ligand. The coordination polyhedron is described as a distorted three-capped trigonal prism, as depicted in Fig. 1b. It is to be mentioned that Sm atoms are bridged by two bidentate and two tridentate carboxylate groups, forming centrosymmetrical dinuclear units with a Sm1⋯Sm′ (1 – x, 2 − y, 2 – z) distance of 3.9506(8) Å. The asymmetric units in the crystal structure are assembled into a one-dimensional array, forming a ribbon-like coordination polymer, as shown in Fig. 1c. Further analysis of the crystal packing reveals a 2D supramolecular network (Fig. 1d), reinforced by weak C–H⋯O hydrogen bonding. In turn, these parallel-oriented 2D networks are interconnected by C–H⋯O H-bonds and π–π stacking interactions, resulting in a densely packed supramolecular architecture (Fig. 1e). This arrangement features a reduced volume of solvent accessible voids, which, according to the solvent mask routine of the Olex2 program, constitutes only 2.7%.
 | ||
| Fig. 1 The crystal structure of [SmLNO3(o-Phen)] (2). (a) View of the asymmetric unit showing the metal coordination with atom labeling scheme and thermal ellipsoids at 40% level. Symmetry generated atoms are shown with faded colours. Symmetry codes: i = 1 − x,1 − y,2 − z, ii = x, y, 1 + z. (b) Coordination polyhedron of Sm. (c) 1D coordination polymer (CP) along c direction. (d) View of 2D supramolecular layer in the ac-plane. (e) partial view of crystal packing showing role of H-bonds and π–π stacking interactions. | ||
On the other hand, SCXRD reveals that compound [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O (3) forms a 3D coordination polymer composed by dinuclear {Eu2} nodes and bis(p-carboxyphenyl)diphenylsilane linkers (L2−) in a 1:3 stoichiometric ratio. The asymmetric unit (Fig. 2a) is built up from two Eu atoms, three deprotonated L2− linkers, and three coordinated water molecules. Additionally, there are 1.5 ethanol and one water co-crystallized molecules in the asymmetric unit. The Eu1 atom is coordinated by nine oxygen atoms provided by two bidentate, two tridentate-bridging carboxylate groups, and two water molecules as monodentate ligands. The Eu1–O distances range from 2.348(6) to 2.667(4) Å. As shown in Fig. 2b, the coordination polyhedron of Eu1 is described as a strongly distorted three-capped trigonal prism. In contrast, the Eu2 atom exhibits a coordination number of eight, being surrounded by six carboxylate oxygen atoms and two oxygen atoms from coordinated water and ethanol molecules, with the shortest and largest Eu2–O bond distances of 2.307(4) and 2.529(4) Å, respectively. The next nearest Eu2–O4 distance of 3.280(4) Å is significant longer, and consequently, the coordination polyhedron of the Eu2 atom corresponds to a distorted double-capped trigonal prism, as shown in Fig. 2c. Each of three crystallographically independent bis(p-carboxyphenyl)diphenylsilane linkers are coordinated in a way such as to interconnect two dinuclear {Eu2} fragments, although, the carboxylate groups feature different coordination functions (Fig. 2a). One of the carboxylate groups belonging to linkers denoted by Si1 and Si2 atoms is coordinated in bidentate-chelating, while the second one as bidentate-bridging modes. In contrast, both carboxylate groups of the linker denoted by Si3 atom adopt a bidentate-bridging coordination mode. The crystal packing is described as a compact and complex three-dimensional coordination polymer (3D CP) with lack of solvent accessible voids, representing only 11% of the total unit cell volume.
 | ||
| Fig. 2 Crystal structure of [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O (3). (a) View of the asymmetric unit with selected atom labeling and thermal ellipsoids at 40% level. Symmetry generated atoms are shown with faded colours. Symmetry codes: (1) −x, 2 − y, 1 − z; (2) x, y, z − 2; (3) 1 − x, 2 − y, 2 − z; (4) x, y − 1, z; (5) −x, 1 − y, 1 − z. (b) Coordination polyhedron of Eu1 atom. (c) Coordination polyhedron of Eu2 atom (d) view of 3D coordination polymer (3D CP). | ||
3.2. Microstructural characterization
Crystallographic analysis revealed that the isostructural compounds 1 and 2 form 1D CPs, which are further assembled into parallel-oriented 2D supramolecular networks through directional hydrogen bonding interactions. These 2D layers are subsequently stacked to form a 3D supramolecular framework. This hierarchical arrangement suggested the potential for exfoliation of the bulk material. Upon ultrasonication in an appropriate dispersion medium, the 3D supramolecular framework is presumed to cleave along its weakest intermolecular interactions, resulting in the disassembly of the bulk material into thin flakes composed of the 2D supramolecular layers.To confirm this hypothesis, a comprehensive microstructural analysis was carried out on compound 1 in both its bulk powder and exfoliated forms. Scanning electron microscopy (SEM) of the crystalline powder reveals that the polycrystalline sample primarily consists of plate-like microcrystals, with average lateral dimensions of several micrometers (Fig. 3a and Fig. S9). Energy-dispersive X-ray spectroscopy (EDS) confirms the presence of europium in the sample (Fig. S10).
 | ||
| Fig. 3 (a) SEM image of compound 1 showing lamellar crystals assembled into rose-like clusters. The powder sample was mounted on carbon tape and coated with platinum. (b) Quartz cuvette containing a THF suspension of exfoliated 1, exhibiting the Tyndall effect when illuminated with a laser. (c) TEM image of an individual nanometric flake of 1 deposited on a copper grid (scale bar: 100 nm). | ||
Exfoliation of 1 into nanosheets was achieved by sonicating the powder in tetrahydrofuran (THF) at room temperature. Two colloidal dispersions of exfoliated nanosheets, designated Exf1 and Exf2, were prepared under different conditions. Exf1 was obtained by sonicating 2 mg of compound 1 in 10 mL of THF for 15 minutes. Exf2 was prepared by sonicating 2 mg in 20 mL of THF for 60 minutes, followed by centrifugation at 4500 rpm for 1 minute to remove larger microcrystals. The resulting THF suspensions exhibit a visible Tyndall effect, indicative of colloidal 2D nanosheets (Fig. 3b). The more rigorous treatment in Exf2 is expected to yield a lower concentration of large crystalline particles, favoring thinner nanosheets.
Transmission electron microscopy (TEM) analysis of samples deposited onto copper grids reveals the presence of both individual and aggregated nanosheets. EDS performed on these regions confirms the expected elemental composition, including Eu, C, and O (Fig. S11). TEM images show that the flakes tend to cluster; however, isolated flakes are also observed, typically ranging from 200–300 nm in lateral size (Fig. 3c). Given the electron beam transparency threshold (∼100 nm), most of the flakes are estimated to be only a few nanometers thick. Occasionally, larger flakes (∼1 μm) are observed as darker regions, suggesting a thickness exceeding 100 nm. Electron diffraction patterns exhibit distinct spots, confirming the crystalline nature of the nanosheets, while EDS further verifies the presence of europium (Fig. S12).
Drop-casting of Exf1 and Exf2 dispersions onto quartz substrates enabled the visualization of exfoliated flakes on the surface. SEM images revealed a wide distribution of flake morphologies and sizes, ranging from sub-micrometer thin flakes to larger aggregates and compact lumps (Fig. S13 and S14). The samples contained both small, irregular, ultrathin “petal-like” flakes and larger, micron-sized bulk crystals. Notably, the larger crystals displayed discernible edge-layer stacking characteristic of a ‘mille-feuille’ structure. In sparsely populated regions, isolated, nearly transparent nanometric flakes with lateral dimensions between 100 and 400 nm were also observed.
3.3. Photophysical properties
 | ||
| Fig. 4 Fluorescence spectra of Ln(III)-CPs, Ln = Eu (1, 3) and Ln = Sm (2). Left: Excitation spectra for 1–3 recorded in the 240–350 nm UV range by monitoring two different emission peaks. Right: Emission spectra for Eu(III)-based CPs (1 and 3) in the visible 550–750 nm range, and for Sm(III)-CP (2) in the visible and near infra-red (NIR) ranges. Excitation wavelength: λex = 260 nm for 1, λex = 345, 330 nm for 2, and λex = 270 nm for 3. | ||
The photoluminescence (PL) spectra of 1 (λex = 260 nm) and 3 (λex = 270 nm) show bands associated with the main Eu3+ center transitions: 5D0 →7F0–4. The spectra are dominated by the 5D0 →7F2 transition, centered at 615 nm for 1 and 614 nm for 3. Slight differences in the spectra of the two Eu complexes are clearly seen in the position and width of the peaks (Fig. S7), which can be attributed to the different coordination environment and the presence of two non-equivalent crystallographic sites, Eu1 and Eu2, in compound 3. Crystal field splitting reveals at least three components at 613, 615 and 618 nm for the 5D0 → 7F2 transition in complex 1. The emission at 591 nm for 1 and 3 is assigned to 5D0 → 7F1, which corresponds to the magnetic dipole transition, while the emission at 700 nm is characteristic for the transitions of 5D0 → 7F4.30
The PL spectrum of 2 displays peaks in the visible region, corresponding to the Sm3+ transitions at 561 nm (4G5/2 → 6H5/2), 596 nm (4G5/2 → 6H7/2), and 642 nm (4G5/2 → 6H9/2).31 In the NIR region, the Sm3+ transitions at 1014 nm and 1157 nm are observed, assigned to 4G5/2 → 6F7/2 and 4G5/2 → 6F9/2, respectively. The emission in the 900–950 nm range for Sm3+could not be reliably recorded due to equipment limitations, as this range lies between the visible and the NIR detectors. The observed bands for 1–3 are summarized in Table 2. It is important to note that the excitation wavelength used for recording the emission spectra with the fluorimeter does not correspond to the optimal wavelength derived from the excitation spectra, in order to avoid second harmonic effects from the Xenon lamp. Nonetheless, the quantum yield is still high in this spectral region, as demonstrated below.
| Compound | λ ex (nm) | λ em (nm) | Λ opt (cm−1) |
|---|---|---|---|
| 1 | 260 | 579, 591.7, 613, 615, 618, 649, 687, 694, 697, 701 | 379 ± 2 |
| 2 | 345 | 561, 596, 642 | 310 ± 5 |
| 330 | 1014, 1157 | ||
| 3 | 270 | 578, 590, 594, 614, 651, 693, 698 | 352 ± 2 |
The average spin–orbit parameter can be deduced from the luminescence determination of the energy levels. For lanthanides, the energy of a ground J multiplet is given by:32EJ = 1/2Λ[J(J + 1) − L(L + 1) − S(S + 1)], where Λ is the spin–orbit coupling parameter. The energy difference between the center of mass of the ground state 7F0 and the excited 7F1 multiplets for Eu3+, yields values of Λopt = 379 ± 2 cm−1 for 1 and Λopt = 352 ± 2 cm−1 for 3. In the case of Sm3+, the energy difference between the ground 6H5/2 and the excited 6H7/2 multiplets results in a value of Λopt = 310 ± 5 cm−1.
Fig. 5 illustrates the measured absolute quantum yield (QY) of the three samples as a function of the excitation wavelength. The Eu 1D CP (1) exhibits an exceptionally high QY, reaching a broad maximum of 67% around 300 nm (Fig. 5a, right axis), which is approximately eight times greater than that of the Eu 3D CP (3). For Sm 1D CP (2), the reported QY values consider only the visible portion of the spectrum, providing a lower threshold for the total QY of this compound.
 | ||
| Fig. 5 (a) Quantum yield (%) as a function of excitation wavelength for the three complexes. The compound 1 data (black squares) are referenced to the right-hand scale (up to 100%), while Sm (2) and Eu (3) (blue circles and red stars) are referenced to the left-hand scale, ranging up to 20%. (b) Luminescence decay curves for compounds 1–3. The excitation wavelengths are λex = 330 nm, 345 nm and 290 nm for 1, 2 and 3 respectively, while the monitored emission peaks are at λmon = 615 nm, 640 nm and 614 nm. | ||
Luminescent lifetimes were determined by monitoring the decay of the most intense emission peak for each compound (5D0 → 7F2 for Eu3+, and 1G4 → 6H3 for Sm3+). Table 3 summarizes the QY values, obtained from the emission spectra under ligand excitation at λex, along with the observed lifetimes measured at the monitoring wavelength (λmon) for all characterized complexes.
| Complex | λ ex (nm) | λ mon (nm) | τ 1 (μs) | τ 1, τ2 (μs) | λ ex (nm) | Q LigandLn (%) | τ rad (ms) | Q LnLn (%) | η LnSens (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 330 | 615 | 1104 ± 25 | — | 300 | 67 ± 1 | 1.6 | 67 | 100 |
| 2 | 345 | 640 | 7.9 ± 0.1 | 6 ± 1, 46 ± 2 | 275 | 2 ± 0.5 | — | ||
| 3 | 290 | 614 | 317 ± 2 | 337 ± 1, 65 ± 4 | 285 | 8 ± 0.5 | 1.1 | 15 | 53 |
For Eu3+ compounds, the intrinsic quantum yield of the lanthanide ion under direct excitation can be determined indirectly. This intrinsic quantum yield is calculated as the ratio of the observed to the radiative lifetime (QLnLn = τLnobs/τLnrad). The radiative lifetime for Eu3+ was estimated using the established relationship:33
 | (1) |
 | (2) |
The calculated energy transfer efficiency from the ligand to Eu3+ is 100% in complex 1, but decreases to 53% in complex 3. This result highlights the crucial role of the phenanthroline co-ligand in enhancing the quantum yield, and underscores the higher intrinsic quantum yield of Eu3+ in the Eu 1D CP.
Time-resolved photoluminescence measurements were performed on 1–3 in the solid-state under UV excitation of the ligand, monitoring the main emission bands in the visible range at RT. The resulting luminescence decay curves (Fig. 5) were analyzed to determine the lifetimes, with the results summarized in Table 3. Compound 1 displays a mono-exponential decay with a lifetime of 1104 μs. In contrast, the other two compounds exhibit biexponential decay profiles, with characteristic lifetimes of 6/46 μs (2) and 337/65 μs (3). The presence of a second relaxation time is an indication of the occurrence of non-radiative processes. For comparative purposes between the three compounds, only the fits to a single-exponential model are presented in Fig. 5b.
Photophysical characterization included measurements of excitation and emission spectra, QY and mean luminescence lifetimes. These results were compared to those of the bulk powder of compound 1. The results are summarized in Fig. 6. Interestingly, exfoliation induces a clear shift in the excitation spectra toward higher energies (i.e., shorter wavelengths). This effect is more pronounced in Exf2, which contains fewer residual bulk crystallites. The observed spectral changes are attributed to modifications in the ligand electronic structure, specifically an increase in the singlet excited-state energy levels. Indeed, π–π stacking interactions are known to play a critical role in modulating the optical properties of conjugated systems by reducing the HOMO–LUMO energy gap, frequently resulting in a bathochromic (red) shift in the absorption spectrum.36 The observed blueshift is therefore consistent with a disruption of π–π stacking interactions between phenanthroline ligands in the exfoliated 2D architectures. Similar cases have been reported in the literature, where blue-shifting is associated with decreasing stacking of supramolecular structures.37,38
 | ||
| Fig. 6 Luminescence properties of the compound 1 nanoflakes. (a) Excitation spectra of two exfoliated suspensions in THF (Exf1 and Exf2) compared with the bulk powder sample. (b) Emission spectra of the Exf2 colloidal suspension, flakes1 and the solid-state powder; inset: photo of Exf2 cuvette under UV light. (c) QY (%) as a function of excitation wavelength for different sample preparations: powder (black solid squares), Exf1 (red open squares), Exf2 (orange open squares), and drop-cast flakes on quartz from Exf1 (blue half-filled squares). (d) Luminescence decay profiles and exponential fits for Exf2 in solution (orange) and powder (black), monitored at λmon = 615 nm, corresponding to the 5D0 → 7F2 transition. | ||
Despite the structural differences, the emission spectra of the exfoliated samples remain essentially identical to that of the bulk material, with no notable variation in the relative intensities of the Eu3+ transitions. As a result, the intrinsic radiative lifetime of Eu3+ remains unchanged across all morphologies. Hence the radiative lifetime (τrad ≈ 1.6 ms), calculated viaeqn (1), is preserved in this morphology.
The observed lifetimes for the principal Eu3+ transition (5D0 → 7F2) also exhibit minimal variation: τobs = 1.04 ± 0.01 ms for Exf1, 1.11 ± 0.01 ms for Exf2, and 1.10 ± 0.02 ms for the powder. These small differences fall within experimental error, confirming that the local coordination environment around Eu3+ is largely preserved after exfoliation. Consequently, the intrinsic QY remains ∼67% across all forms. However, differences in the total QY among the two samples point to variations in ligand-to-metal ET efficiency. While both colloidal suspensions maintain high QY values, Exf2 exhibits a slightly reduced QY of 57%, compared to 67% for both Exf1 and the bulk. This reduction is attributed to a diminished “antenna effect” from the phenanthroline ligands, likely caused by reduced π–π stacking and dimensional confinement.39 Specifically, the energy transfer efficiency, as calculated viaeqn (2), drops from near unity in the bulk and Exf1 to ∼85% in Exf2.
In summary, exfoliation of compound 1 leads to only a modest (∼15%) reduction in QY, due to a slightly diminished “antenna effect”, while preserving the fundamental photophysical properties of the Eu3+ emitter. The excitation spectrum undergoes a notable blue shift—up to ∼0.6 eV (5000 cm−1) between Exf1 and Exf2 – reflecting significant changes in the ligand excited states. Nonetheless, the exfoliated nanosheets retain excellent luminescent properties, highlighting their potential for applications in two-dimensional photonic or optoelectronic systems.
 | ||
| Fig. 7 Transient absorption spectra of samples dispersed in ethanol: (a) compound 1, (b) compound 2, and (c) compound 3. | ||
These measurements must be done in solution. The resulting spectral features, which can appear as either positive or negative signals, correspond to transitions from excited states undergoing radiative or non-radiative decay processes. The positive ones are new absorbing species generated by the pump pulse: excited chromophore states, excited state photoproducts, geometric re-organization, solvation effects, while the negative bands originate from ground-state bleach and stimulated emissions resulting from excited-state species stimulated by the sample pulse.40
The transient absorption map of compound 1 reveals negative ΔOD signals (change of optical density) at 218, 260 and 265 nm originating from ground state bleaching (GSB). These bands are attributed to the S0 → S1 transitions, with energy level from 5.68 to 4.67 eV. At shorter wavenumbers, excited state absorptions (ESA) can be seen at 210, 240, 255, 275 and 290 nm, indicating more than one excited state (Sn > 1, S0 → S2 transitions, with energy level from 5.90 to 4.27 eV). The evolution over time of these bands indicates the population of many vibrational states following the initial excitation. At longer wavenumbers (400 and 450 nm), the stimulated emissions from the complex are observed (S1 → S0 transitions), with energy level from 3.09 to 2.75 eV (Fig. 7a and Fig. S8).41
For complex 2 (Fig. 7b), the GSB is evident at 220 and 245 nm assigned to the S0 → S1 transitions, the energy level from 5.63 to 5.06 eV. At shorter wavelengths (235, 250 and 270 nm), the absorptions in excited state are present suggesting more than one excited state (Sn > 1, S0 → S2 transitions, energy level from 5.27 to 4.59 eV). Similar to compound 1, at longer wavelengths 395 and 420 nm, stimulated emission appeared (S1 → S0 transitions, energy level from 3.13 to 2.95 eV) (Fig. 7b and Fig. S8). Strong bleaching in 2, but weak ESA might indicate an efficient excitation but a short-lived excited state.
In the case of compound 3 (Fig. 7c), GSB bands appear at 215 and 260 nm (S0 → S1 transitions, energy level from 5.76 to 4.76 eV). ESA bands at 210, 225, 235, 245, 270, and 285 nm provide additional evidence for multiple excited states (Sn > 1, S0 → S2 transitions, energy level from 5.90 to 4.42 eV). At longer wavelength (400 and 420 nm), the stimulated emission from the complex is also observed (S1 → S0 transitions, energy level from 3.09 to 2.95 eV). Compound 3 shows higher ESA and a larger density of excited states. The bleaching bands are similar to 1 in amplitude. This is indicative of a lower energy transfer from the chromophore to the Eu3+ levels. The ground state is quickly replenished, via non-radiative relaxation (Fig. S8).42 These findings are consistent with our previous results in solid state, confirming a lower ligand-to-Eu3+ energy transfer efficiency in complex 3 (ηEuSens = 53%) compared to the complete transfer observed in complex 1 (see Table 3).
3.4. Magnetic properties
The spin orbit coupling for Eu3+ and Sm3+ is relatively weak, leading to mixing between the ground state and the next higher energy term as a function of magnetic field and temperature. For both lanthanide ions, the energy separation between the J multiplets is about kBT ∼ 200 cm−1, which governs the magnetic properties of these ions. For Sm3+ (4f5, S = 5/2 and L = 5), the 6H5/2 ground state mixes with the higher-lying 6H7/2, term, resulting in an increase in the effective moment from μeff = 0.85μB (S = 5/2, J = 5/2) to μeff = 3.32μB (S = 5/2, J = 7/2). On the other hand, Eu3+(4f6, S = 3, L = 3), has 7F0 (J = 0) ground state, which would give null effective moment, and a first excited multiplet 7F1.The temperature dependence of the susceptibility, χ(T), measured at 1 kOe between 1.8 and 300 K for the two studied Eu-CPs (1, 3) is presented in Fig. 8. For both complexes, the χ(T) increases smoothly as the temperature decreases from room temperature, reaching a plateau in the range of 150–50 K. The low-temperature asymptotic susceptibility value is approximately 0.006 emu mol−1 for compound 1, significantly lower than the 0.010 emu mol−1 for compound 3. Below 50 K, a rapid increase in χ(T) is observed, most likely due to minor spurious magnetic impurities. These features are consistent with previously reported behavior in Eu(III) complexes.43–45 In Eu(III), the 7F ground term is split by the spin–orbit coupling into seven states, 7FJ, where J takes integer values, from 0 to 6, and the energy of each state is given by: EJ = ΛJ (J + 1)/2, where the energy of the 7F0 ground state is taken as the reference. Due to the relatively small spin–orbit coupling, Λ, the crystal field (CF) components of the first and second excited states can be thermally populated, leading to a paramagnetic response upon application of an external magnetic field. At low temperatures, only the non-magnetic ground state J = 0 is populated and the susceptibility becomes temperature-independent. The experimental χ(T) data for the Eu complexes could be well fitted using the theoretical isotropic equilibrium magnetic susceptibility as a function of temperature at zero field within the Van Vleck approximation:46
 | (3) |
 | ||
| Fig. 8 Magnetic susceptibility as a function of temperature, data for the studied Eu compounds, (a) compound 1 and (b) compound 3. Red dashed lines are the corresponding fits to eqn (3). Insets show magnetization in Bohr magneton units per Eu3+ ion, as a function of applied magnetic field at T = 1.8 K. | ||
Isothermal field-dependent magnetization measurements were conducted at 1.8 K for both Eu complexes (see Fig. 8, insets). In both cases, the magnetization increases linearly with the applied field, with small deviations from linearity, likely caused by trace magnetic impurities. At 50 kOe, the magnetization reaches 0.05μB for compound 1 and 0.09μB for compound 3. The larger magnetic susceptibility and magnetization observed for the Eu 3D CP (3) can be attributed to its lower spin–orbit coupling and the closer proximity of the 7F1 levels, which enhance thermal population and paramagnetic response.
Variable temperature (300–1.8 K) magnetic susceptibility data for the Sm-CP (2) was collected under an external magnetic field of 1 kOe. Fig. 9a displays the χT product as a function of temperature, showcasing the typical magnetic behavior of Sm3+. The room temperature χT value 0.37 emu Kmol−1 exceeds the theoretical value for a free Sm3+ ion (6H5/2, 0.089 emu Kmol−1), and aligns well with previously reported values for Sm3+ complexes.47–52 This deviation arises from the partial thermal population of the low-lying first excited state 6H7/2 at room temperature. The χT products decreases almost linearly with cooling, reaching the theoretical value for the 6H5/2 ground state at 35 K. Below this temperature, a further decrease is observed, with χT reaching 0.044 emu Kmol−1 at 1.8 K. This relatively small low-temperature χT value indicates significant crystal field (CF) splitting due to the anisotropic coordination environment.
 | ||
| Fig. 9 Static magnetic properties for Sm-based CP, 2. (a) Magnetic susceptibility-temperature product at 1kO as a function of temperature, χT(T). In red is depicted the fit according to eqn (4) in the T > 125 K region. Red square marks the χT theoretical value for the 6H5/2 ground state multiplet, 0.089 emu Kmol−1. (b) Field-dependent of the isothermal magnetization, M(H) at 1.8 K compared to calculated curve for an effective spin S* = 1/2 and average gyromagnetic value g* = 0.61 for Sm3+. | ||
Sm3+, along with Eu3+, is one of the lanthanides for which Curie law does not apply due to multiplet energy intervals comparable to kBT and significant J-mixing. For Sm3+, the energy splitting between the ground state multiplet 6H5/2 and the first excited 6H7/2 is approximately 3.5 Λ. In the 100–300 K temperature range, the influence of the excited levels manifests as a second-order effect and can be modeled as a temperature-independent contribution to the magnetic susceptibility:46
 | (4) |
Fitting the magnetic susceptibility to the expression above in the 125–300 K range yields the spin–orbit coupling parameter for Sm3+, with a value of Λmag = 315 ± 5 cm−1, which is in excellent agreement with the optically-determined value, Λopt = 310 ± 5 cm−1 (see Table 2).
The isothermal field-dependent magnetization of compound 2 measured at 1.8 K is shown in Fig. 9b. The magnetization increases gradually without reaching saturation, even under a magnetic field of 70 kOe, attaining a maximum of 0.2μB. The M(H) curve can be reasonably modeled with an effective spin S* = 1/2 and effective gyromagnetic factor g* = 0.61, which significantly deviates from the expected value for a free Sm3+ ion in its ground state (J = 5/2, gJ = 2/7). This behavior indicates a significant splitting of the 6H7/2 ground multiplet due to the crystal field.
Sm(III) is an uncommon candidate for single-molecule magnet (SMM) studies, due to its small magnetic moment, with few reporting dynamic properties.47–52 To explore the dynamic behavior of Sm3+ in compound 2, ac magnetic susceptibility measurements were carried out as a function of frequency (10 Hz–10 kHz) and temperature (2–4 K), under various applied static magnetic fields ranging from 0 to 8 kOe. A sizeable non-zero out-of-phase component of the susceptibility (χ′′) was observed between 2.0 K and 4.0 K under the application of a magnetic field, with the highest amplitude at 4 kOe (Fig. S15). Despite a weak signal, and high noise at the lowest frequencies, both in-phase, χ′(f, T) and out-of-phase, χ′′(f, T), curves reveal the presence of a slow relaxation process (Fig. 10a and b). These data were fitted using a generalized Debye model, and the obtained relaxation times as a function of the inverse temperature (1/T) are shown in Fig. 10c. An Arrhenius fit to the data (not shown) using τ = τ0exp(Ea/kBT) yields an activation energy Ea/kB = 3.5 ± 0.3K and a pre-exponential factor τ0 = 2.2 ± 0.3 10−5 s. However, a better fit is achieved when including a Raman relaxation contribution, as shown in Fig. 10c. Fitting the data with the expression: τ−1 = τ0−1exp(−Ea/kBT) + CTn, provides the parameters Ea/kB = 2.4 ± 0.4 K, τ0 = 3.7 ± 0.7 10−5 s, constant C = 1 ± 4 K−n s−1 and n = 6 ± 2.53
 | ||
| Fig. 10 Dynamic magnetic properties for Sm-based CP, 2. (a) and (b) show χ′(f, T) and χ′′(f, T) at H = 4 kOe, where the filled circles represent the experimental data, and the lines correspond to the fit curves using a generalized Debye model; (c) Relaxation time vs. inverse of the temperature on a semilogarithmic scale. The dashed line represents the fit incorporating both Orbach and Raman processes. | ||
Therefore, the Sm3+ compound exhibits field-induced slow magnetic relaxation, with thermally activated relaxation times of the order of 10−4 s. This behavior represents a rare example of a two-dimensional Sm-based compound displaying single-molecule magnet (SMM) characteristics.
4. Conclusions
Our work demonstrates the remarkable versatility of a novel dicarboxylic ligand containing diphenylsilane units to form lanthanide-based coordination polymers (Ln-CPs) with diverse dimensionalities. Through modulation of reaction conditions and the use of o-Phenanthroline as a co-ligand, we obtained two isostructural one-dimensional CPs (Ln = Eu (1), Sm (2)), that assemble into 2D supramolecular layers stacked in 3D architectures, as well as a Eu-based 3D CP (3), and thoroughly characterized their structures and magneto-optical properties. The photophysical performance of these complexes was evaluated through excitation spectra, luminescence, and quantum yield measurements in both visible and NIR regions. Among the complexes, the Eu-based CP (1) stands out with an exceptional 67% quantum yield in the visible region, underscoring the key role of phenanthroline co-ligand as an effective “antenna ligand” for Eu3+ emission. In contrast, the other compounds exhibited moderate quantum yields in visible spectral range (2% for 2, and 8% for 3). Notably, the Sm3+- based 1D CP(2) displayed significant NIR emission and field-induced slow magnetic relaxation – an uncommon feature in Sm3+-based compounds – extending the potential applications of this multifunctional material.The spin–orbit parameters, derived from both optical and magnetic data, were found in close agreement, with values of Λopt (Λmag) = 379 (382) cm−1, 310 (315) cm−1, and 352 (342) cm−1 for complexes 1, 2, and 3, respectively. Furthermore, transient absorption studies revealed the involvement of both fundamental and excited absorption and emission states, providing deeper insight into the excited-state dynamics of these complexes.
The study highlights the profound influence of the coordination sphere and structural topology on the luminescent behavior of Eu3+ complexes. The incorporation of phenanthroline not only significantly enhances the energy transfer to the lanthanide ion but also boosts the intrinsic quantum yield of Eu3, resulting in the creation of an efficient material with 67% quantum yield for red fluorescence. Importantly, the exfoliation of compound 1 retains the essential photophysical characteristics, with only a minor decrease in the energy transfer efficiency from phenanthroline. These findings demonstrate the potential of this compound for integration into two-dimensional and surface-based photonic and optoelectronic applications.
Author contributions
AA: conceptualization, data curation, formal analysis, investigation, methodology, supervision, validation, visualization, writing – original draft, review & editing; MFZ: conceptualization, methodology, resources, formal analysis, investigation, writing – original draft, review & editing; EB: conceptualization, funding acquisition, writing, review & editing; SF: resources, investigation, writing – review; IRT: investigation, writing – review; SS: investigation, formal analysis, writing – review; MC: conceptualization, funding acquisition, supervision; writing – review & editing; All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Further data supporting this article have been included as part of the SI. Additional data that support the findings of this study are available from the corresponding author upon request. Crystallographic data for [EuLNO3(o-Phen)], [SmLNO3(o-Phen)] and [Eu2L3EtOH(H2O)3]·1.5EtOH·H2O are deposited at the CCDC repository under 2314852, 2314853 and 2314854, respectively.Supplementary information available: S1, IR spectroscopy; S2, PXRD powder X-ray diffraction; S3, additional luminescence figures; S4, additional SEM and TEM electron microscopy images; and S5, additional figure of magnetic properties. See DOI: https://doi.org/10.1039/d5tc02436f
CCDC 2314852–2314854 contain the supplementary crystallographic data for this paper.54–56
Acknowledgements
This work was financially supported by Spanish “Ministerio de Ciencia, Innovacion y Universidades” (PID2022-138492NB-I00 and PID2021-122869NB-I00), the Gobierno de Aragón (E12-23R and E17-23R) and the State Investigation Agency, through the Severo Ochoa Program for Centres of Excellence in R&D (CEX2023-001263-S and CEX2023-001286-S). Authors would like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza. Co-authors, M.F.Z. and M.C. thank for the support by a grant of the Ministry of Research, Innovation and Digitization, CCCDI – UEFISCDI, project number PN-IV-P8-8.3-PM-RO-TR-2024-0046, within PNCDI IV, Contract ctr.8 BMTR/2025 (SynTioPOSS).References
- P. Gawryszewska, L. W. Miller and A. J. M. Valente, Front. Chem., 2023, 11, 1258607 CrossRef PubMed.
- Y. Ning, M. Zhu and J. L. Zhang, Coord. Chem. Rev., 2019, 399, 213028 CrossRef.
- H. Iwanaga, J. Lumin., 2018, 200, 233–239 CrossRef.
- E. A. Ivanova, K. S. Smirnova, I. P. Pozdnyakov, A. S. Potapov and E. V. Lider, Inorganics, 2023, 11, 317 CrossRef.
- H. Xu and P. Cheng, Aggregate, 2024, 5, e518 CrossRef.
- M. Yu, Y. Xie, X. Wang, Y. Li and G. Li, ACS Appl. Mater. Interfaces, 2019, 11, 21201–21210 CrossRef PubMed.
- X. Peng, Y. He, K. Tan, R. Yuan and S. Chen, Sens. Actuators, B, 2025, 430, 137343 CrossRef.
- E. Bartolomé, J. Bartolomé, A. Arauzo, J. Luzón, R. Cases, S. Fuertes, V. Sicilia, A. I. Sánchez-Cano, J. Aporta, S. Melnic, D. Prodius and S. Shova, J. Mater. Chem. C, 2018, 6, 5286–5299 RSC.
- E. Bartolomé, X. B. Li, A. Arauzo, J. Luzón, I. García-Rubio and J. G. Planas, ACS Appl. Mater. Interfaces, 2025, 17, 42082–42095 CrossRef PubMed.
- K. S. Kumar, D. Serrano, A. M. Nonat, B. Heinrich, L. Karmazin, L. J. Charbonnière, P. Goldner and M. Ruben, Nat. Commun., 2021, 12, 2152 CrossRef PubMed.
- R. Maouche, S. Belaid, B. Benmerad, S. Bouacida, S. Freslon, C. Daiguebonne, Y. Suffren, G. Calvez, K. Bernot, C. Roiland, L. Le Pollès and O. Guillou, Inorg. Chim. Acta, 2020, 501, 119309 CrossRef.
- M. W. Mara, D. S. Tatum, A. M. March, G. Doumy, E. G. Moore and K. N. Raymond, J. Am. Chem. Soc., 2019, 141, 11071–11081 CrossRef PubMed.
- S. Miyazaki, M. Gotanda, Y. Kitagawa, Y. Hasegawa, K. Miyata and K. Onda, J. Phys. Chem. Lett., 2024, 15, 10718–10724 CrossRef PubMed.
- H. L. Wang, X. F. Ma, H. H. Zou, K. Wang, B. Li, Z. L. Chen and F. P. Liang, Dalton Trans., 2018, 47, 15929–15940 RSC.
- E. Bartolomé, A. Arauzo, J. Luzón and L. Gasque, J. Mater. Chem. C, 2025, 13, 831–841 RSC.
- Y. X. Li, M. Xue, L. J. Guo, L. Huang, S. R. Chen and S. L. Qiu, Chem. Res. Chin. Univ., 2013, 29, 196–200 CrossRef.
- M. Cazacu, A. Vlad, M. F. Zaltariov, S. Shova, G. Novitchi and C. Train, J. Organomet. Chem., 2014, 774, 70–78 CrossRef.
- G. O. Turcan-Trofin, M. Avadanei, S. Shova, A. Vlad, M. Cazacu and M. F. Zaltariov, Inorg. Chim. Acta, 2018, 483, 454–463 CrossRef.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabrés, I. Xamena and J. Gascon, Nat. Mater., 2014, 14, 48–55 CrossRef.
- M. M. Velázquez, T. Alejo, D. López-Díaz, B. Martín-García and M. D. Merchán, Two-Dimensional Materials – Synthesis, Characterization and Potential Applications, InTech, 2015 Search PubMed.
- M. Lippi and M. Cametti, Coord. Chem. Rev., 2021, 430, 213661 CrossRef.
- Rigaku Oxford Difraction. CrysAlisPro Software system (2015).
- G. M. Sheldrick, Acta Crystallogr., A, 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- A. Vlad, M. F. Zaltariov, S. Shova, G. Novitchi, C. Train and M. Cazacu, RSC Adv., 2016, 6, 37412–37423 RSC.
- K. Nakamoto, Handbook of Vibrational Spectroscopy, ed. P. R. Griffiths, John Wiley & Sons, Chichester, UK, 2006 Search PubMed.
- A. Arauzo, L. Gasque, S. Fuertes, C. Tenorio, S. Bernès and E. Bartolomé, Dalton Trans., 2020, 49, 13671–13684 RSC.
- D. Yang, Y. Tian, W. Xu, X. Cao, S. Zheng, Q. Ju, W. Huang and Z. Fang, Inorg. Chem., 2017, 56, 2345–2353 CrossRef PubMed.
- S. S. Mortensen, M. A. M. Nielsen, P. Nawrocki and T. J. Sørensen, J. Phys. Chem. A, 2022, 126, 8596–8605 CrossRef PubMed.
- J. H. Van Vlek, The Theory of Electric and Magnetic Susceptibilities, Oxford University Press, 1965 Search PubMed.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542–1548 RSC.
- N. M. Shavaleev, S. V. Eliseeva, R. Scopelliti and J. C. G. Bünzli, Inorg. Chem., 2015, 54, 9166–9173 CrossRef PubMed.
- A. Aebischer, F. Gumy and J. C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346–1353 RSC.
- R. Zhao and R. Q. Zhang, Phys. Chem. Chem. Phys., 2016, 18, 25452 RSC.
- W. Chai, X. Zhang, L. Song, M. Hong, H. Shi, C. Wang, J. Guo, X. Zheng, G. Chen and K. Shu, Mater. Lett., 2015, 145, 4–6 CrossRef.
- S. Wang, L. Wang, X. Ma, Y. Guo, R. Wang, S. Zhang, Z. Zhang, L. Du and Q. H. Zhao, Inorg. Chem. Front., 2023, 10, 5258 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- R. Hamburger, C. Rumble and E. R. Young, J. Vis. Exp., 2024, 2024, e65519 Search PubMed.
- Y. Tamai, Y. Murata, S. I. Natsuda and Y. Sakamoto, Adv. Energy Mater., 2024, 14, 2301890 CrossRef.
- S. S. Stanimirov, A. A. Trifonov and I. C. Buchvarov, Spectrochim. Acta A: Mol. Biomol. Spectrosc., 2021, 258, 119832 CrossRef PubMed.
- M. Andruh and P. Porchers, Inorg. Chem., 1993, 32, 1616–1622 CrossRef.
- A. Arauzo, A. Lazarescu, S. Shova, E. Bartolomé, R. Cases, J. Luzón, J. Bartolomé and C. Turta, Dalton Trans., 2014, 43, 12342–12356 RSC.
- E. Bartolomé, A. Arauzo, S. Fuertes, L. Navarro-Spreafica, P. Sevilla, H. Fernández Cortés, N. Settineri, S. J. Teat and E. C. Sañudo, Dalton Trans., 2023, 52, 7258–7270 RSC.
- A. Frank, Phys. Rev., 1932, 39, 119–129 CrossRef.
- Y. Z. Pan, Q. Y. Hua, L. S. Lin, Y. B. Qiu, J. L. Liu, A. J. Zhou, W. Q. Lin and J. D. Leng, Inorg. Chem. Front., 2020, 7, 2335–2342 RSC.
- F. A. Mautner, F. Bierbaumer, R. Vicente, S. Speed, Á. Tubau, R. C. Fischer and S. S. Massoud, Magnetochemistry, 2022, 8, 1–16 CrossRef.
- A. Q. Wu, F. K. Zheng, X. Liu, G. C. Guo, L. Z. Cai, Z. C. Dong, Y. Takano and J. S. Huang, Inorg. Chem. Commun., 2006, 9, 347–350 CrossRef.
- M. Andruh, E. Bakalbassis, O. Kahn, J. C. Trombe and P. Porcher, Inorg. Chem., 1993, 32, 1616–1622 CrossRef.
- P. Wytrych, J. Utko, M. Stefanski, J. Kłak, T. Lis and Ł. John, Inorg. Chem., 2023, 62, 2913–2923 CrossRef PubMed.
- K. Kumar, G. Li, O. Stefanczyk, S. Chorazy, K. Nakabayashi and S. I. Ohkoshi, J. Mater. Chem. C, 2022, 11, 1008–1020 RSC.
- K. N. Shrivastava, Phys. Status Solidi (B), 1983, 117, 437–458 CrossRef.
- A. Arauzo, M.-F. Zaltariov, E. Bartolomé, S. Fuertes, I.-R. Tigoianu, S. Shova and M. Cazacu, 2314852: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2hpsnn.
- A. Arauzo, M.-F. Zaltariov, E. Bartolomé, S. Fuertes, I.-R. Tigoianu, S. Shova and M. Cazacu, 2314853: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2hpspp.
- A. Arauzo, M.-F. Zaltariov, E. Bartolomé, S. Fuertes, I.-R. Tigoianu, S. Shova and M. Cazacu, 2314854: Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2hpsqq.
| This journal is © The Royal Society of Chemistry 2025 |