
Paramagnetic discotic liquid crystals based on planar benzo[e][1,2,4]triazin-4-yls: synthesis and properties
Hemant K.
Singh
 a,
Georgia A.
Zissimou
a,
Damian
Pociecha
b,
Martin
Cigl
a,
Anna
Pietrzak
c and
Piotr
Kaszyński
*ad
a,
Georgia A.
Zissimou
a,
Damian
Pociecha
b,
Martin
Cigl
a,
Anna
Pietrzak
c and
Piotr
Kaszyński
*ad
aCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland. E-mail: piotr.kaszynski@cbmm.lodz.pl
bFaculty of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
cFaculty of Chemistry, Łódź University of Technology, 90-924 Łódź, Poland
dFaculty of Chemistry, University of Łódź, 91-403 Łódź, Poland
First published on 11th August 2025
Abstract
Synthetic access to the discotic liquid crystalline derivatives of a planar-Blatter radical has been developed and the properties of several such derivatives and their direct precursors were investigated using optical (POM), thermal (DSC), structural (powder XRD) and magnetic (VT-EPR) methods. The only effective, albeit low-yielding, method for the synthesis of the final radicals was photocyclization of appropriate precursors. These precursors were obtained from a common intermediate already containing two wedge-shaped groups by nucleophilic substitution of the fluorine atom with an aryloxy anion. In an alternative strategy, the photocyclization precursors were obtained by substitution followed by introduction of one or two wedge-shaped groups via Suzuki coupling reactions. Experimental (XRD) and computational (DFT) mechanistic investigation of a model phenoxy derivative demonstrated that the cyclization takes place without Smiles rearrangement, observed in naphthyloxy analogues. Powder XRD analysis revealed that only two radicals form mesophases (Colh), while their precursors exhibit Colh, Colh and 3D ordered Colx phases or only the Colx phase. Results suggest that planarization of the central core in mesogenic Blatter radical derivatives and cyclization of the precursors improve molecular packing and increase the phase stability. VT-EPR results show a significant increase of antiferromagnetic interactions at melting to a Colh phase and then at the clearing temperature for one of the discotic radicals.
Introduction
Functional materials1 derived from stable organic radicals2,3 are of increasing importance for technological advances in areas such as energy storage,4 information processing,5 molecular electronics6 and photonics.7 A subclass of such materials is paramagnetic liquid crystals,8,9 which, due to their self-organizing and fluid nature, offer a viable alternative to solids in formation of functional molecular films.10–12 In this context, benzo[e][1,2,4]triazin-4-yl is a particularly attractive paramagnetic building block for modern open-shell materials.The prototypical Blatter radical13 (Blatter, Fig. 1) and its derivatives14 are characterized by high stability,15 extensive spin π-delocalization,16 favourable electrochemical behavior17,18 and low energy bandgaps,19,20 which have been explored in organic batteries,21–23 molecular electronics,24,25 spintronics,26 and photodetectors,27 among other applications.28 Several years ago, we demonstrated that the Blatter radical substituted with long “arms” at positions 3 and 6 forms lamellar phases,29,30 while substitution with two or three wedge-shaped groups in positions 1, 3 and 6 leads to discotic mesogens (e.g. derivatives 1a–1d)31–34 exhibiting photoconductive and structure-dependent magnetic behavior.31,32 The high torsion angle (∼50°) of the N(1) aryl group in Blatter impacts the molecular packing and consequently the transition temperatures.33,34 Planarization of the core could improve not only the mesophase stability but also could enhance photoconductivity and cooperative magnetic interactions.
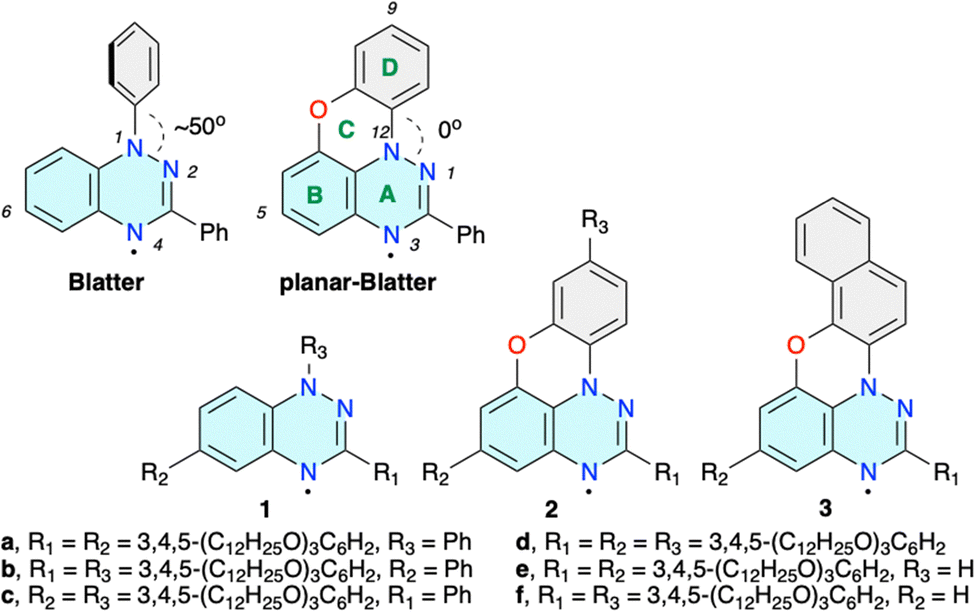 | ||
| Fig. 1 The structure of Blatter and planar-Blatter radicals with a partial numbering system. Disc-shaped derivatives 1–3. | ||
To address this issue, we recently developed an efficient access to the parent planar-Blatter radical35 (Fig. 1) and its derivatives36 using the aza-Pschorr reaction,37 Bu3SnH-assisted cyclization,38 and photocyclization.39 While the last method was used to obtain π-expanded planar Blatter radical 3 (R1 = Ph; R2 = H), aza-Pschorr cyclization gave access to the first bent-core mesogens containing planar-Blatter as the central angular element.40,41 A comparison of these mesogens with those derived from Blatter demonstrated that planarization of the radical indeed increases phase organization and thermal stability by increasing the intermolecular contact of the paramagnetic cores. It is expected that the improved intermolecular contact in discotic derivatives 1 by planarization of the central Blatter radical in closely related discotic derivatives 2 and 3 should also have a positive effect on the mesogenic properties of such materials.
Herein, we explore the methods developed for the preparation of the parent planar-Blatter radicals in the formation of their discotic derivatives 2d–2f and 3e. The effect of planarization of the paramagnetic core on mesogenic behaviour is investigated by comparison between closely related series 1 and 2 and also between precursors before and radicals after cyclization. The regioselectivity of photochemical formation of 2 is investigated by experimental and DFT methods using a model compound. Finally, radicals 2d and 2f are investigated by DSC, POM, powder XRD, and VT EPR methods.
Results and discussion
Synthetic strategy
Preparation of mesogenic radicals 2 and 3 was envisioned using benzo[e][1,2,4]triazine 4 as the key intermediate and strategies shown in Fig. 2. The bromine atom at the C(6) position serves as a handle to introduce the second wedge-shaped 3,4,5-tridodecyloxyphenyl substituent R, while the C(8)–F group enables the construction of ring C of the planar-Blatter radical skeleton (Fig. 1). In strategy A the C(6)–Br atom is transformed first leading to intermediate 5, and ring C is constructed in the second step. This sequence is reversed in strategy B and the C(8)–F atom is replaced first with an appropriate aryloxy fragment followed by Pd-catalysed coupling of the C(6)–Br atom with the R group (Fig. 2). Alternatively, after initial substitution of the C(8)–F atom, the R group is introduced at the C(6) position prior to cyclization and formation of the final radical (strategy BA). All three strategies were explored.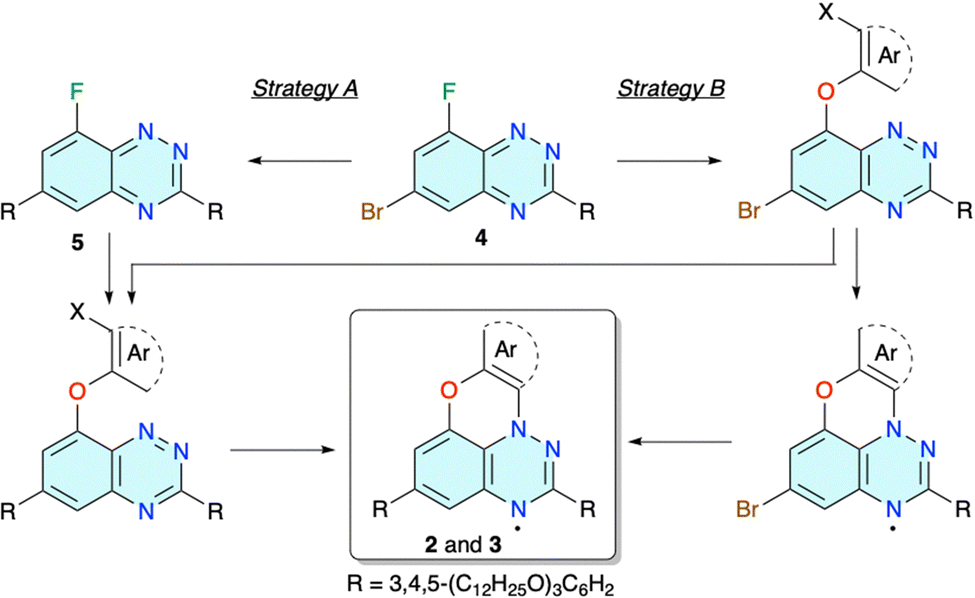 | ||
| Fig. 2 Three strategies for the preparation of liquid crystalline radicals 2 and 3. | ||
The cyclization and formation of the radical can be accomplished using one of the recently developed methods. Thus, intermediates with X = NH2 are designed for aza-Pschorr cyclization.37 Derivatives with X = I could undergo a free radical cyclization process.38 Finally, photocyclization could be performed on several types of derivatives, although experimental data indicate that the effectiveness of the process (conversion and the overall yield) increases in the order X = H < Br < NO2 and Ar = Ph < naphthalene.39,42 Although photocyclization of larger aryloxy derivatives proceeds with a Smiles rearrangement,43,44 this method is particularly attractive since light more easily penetrates to the reaction centre than reagents, such as t-BuONO in the aza-Pschorr or Bu3SnH in the radical cyclization process.
Synthesis
 | ||
Scheme 1 Synthesis of intermediate 4. Reagents and conditions: (i) NBS, TFA/H2SO4 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), rt, 12 h, 73% yield, ref. 41; (ii) 3,4,5-(C12H25O)3C6H2CONHNH2 (7), DMSO, N2, 85 °C, 12 h, 81% yield; (iii) (1) Sn, AcOH/THF (5:1), rt, 8 h, 130 °C, 0.5 h. (2) NaIO4, CH2Cl2/MeOH (1:1), 0.5 h, 75% yield; (iv) 3,4,5-(C12H25O)3C6H2B(OH)2 (10), Na2CO3, Pd-PEPPSI-iPr, THF/H2O (5:2), N2, reflux, 24 h, 78% yield. :4), rt, 12 h, 73% yield, ref. 41; (ii) 3,4,5-(C12H25O)3C6H2CONHNH2 (7), DMSO, N2, 85 °C, 12 h, 81% yield; (iii) (1) Sn, AcOH/THF (5:1), rt, 8 h, 130 °C, 0.5 h. (2) NaIO4, CH2Cl2/MeOH (1:1), 0.5 h, 75% yield; (iv) 3,4,5-(C12H25O)3C6H2B(OH)2 (10), Na2CO3, Pd-PEPPSI-iPr, THF/H2O (5:2), N2, reflux, 24 h, 78% yield. | ||
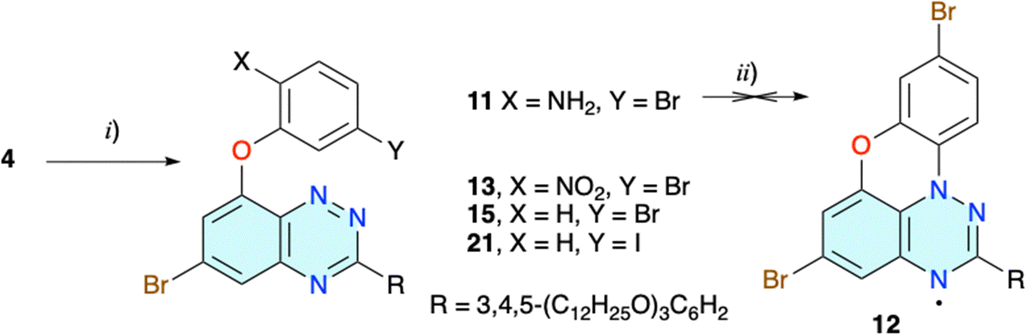 | ||
| Scheme 2 Synthesis of intermediates and attempted preparation of radical 12. Reagents and conditions: (i) for 11: (1) 2-amino-5-bromophenol, NaOH, MeOH, rt, 30 min; (2) DMSO/THF (1:1), rt, 12 h, 85% yield. For 13: (1) 5-bromo-2-nitrophenol, [Me4N]OH × 5H2O, MeOH, rt, 30 min; (2) DMSO, 100 °C, 24 h, 10% yield. For 15 and 21: (1) 3-bromophenol or 3-iodophenol, [Me4N]OH × 5H2O, dry i-PrOH, rt, 30 min; (2) dry i-PrOH, 70 °C, 24 h, 60–90% yield. (ii) t-BuONO, C6H6, rt, 30 min then 70 °C, 2 h, 0% yield. | ||
The strategy was modified and the amino group on the phenoxy substituent, needed for the aza-Pschorr cyclization, was to be generated from the nitro group, after replacement of the two Br atoms in derivative 13 with the wedge-shaped substituents R (strategy BA in Fig. 2 and Scheme 2). The preparation of 13 by a reaction of 5-bromo-2-nitrophenol with 4 under basic conditions was problematic and plagued with low conversion and consequently low yields not exceeding 10%. Standard conditions, NaH/DMSO and 100 °C, gave 13 in 7% yield. Using the [Me4N]+ salt of phenol improved the yield of the isolated product 13 to 10%. Other solvents, such as dry DMA and MeCN, performed worse. A similar decrease of reactivity for less nucleophilic nitrophenols was observed before.37
The obtained dibromo derivative 13 was reacted with boronic acid 10 in the presence of Pd(Ph3P)4, giving the double substituted product 14 (Scheme 3). Purification of this product turned out to be difficult due to its photosensitivity and apparent spontaneous photocyclization in natural light. Therefore, a solution of crude 14 in CH2Cl2 was exposed to sunlight for several days and a black-green solid was isolated in about 20% overall yield after chromatography and repeated recrystallization/precipitation from several combinations of solvents. Although the compound exhibited a columnar discotic phase and MALDI analysis showed the correct mass with little fragmentation, the product never passed the combustion analysis. Three independent samples had low carbon and high nitrogen content, consistent with the presence of an NO2 group in the structure.
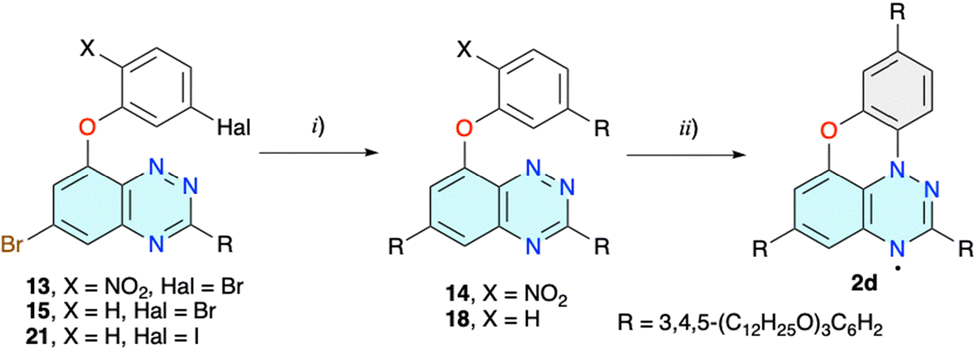 | ||
| Scheme 3 Preparation of discotic mesogen 2d. Reactions and conditions: (i) 3,4,5-(C12H25O)3C6H2B(OH)2 (10), Na2CO3, Pd-PEPPSI-iPr, THF/H2O (5:2), N2, reflux, 24 h, 80% yield. (ii) CH2Cl2, sunlight, 7 d, X = NO2: 20% yield, X = H: 5% yield. | ||
To avoid the complications of the possible insertion of the NO2 group into the molecular structure of 2d, dibromo derivative 15 lacking the nitro group was prepared (Scheme 2). The initial reaction of 4 with 3-bromophenol in DMSO in the presence of NaH gave disubstituted derivative 16 as the sole product (Chart 1). The same reaction conducted in MeCN at reflux in the presence of [Me4N]OH × 5H2O as a base gave no reaction. Changing the solvent to MeOH gave the desired product 15 in 50% yield and 8-methoxy derivative 17(OMe) (Chart 1). To minimize the formation of the alkoxy byproduct, MeOH was replaced by i-PrOH and the desired product 15 was obtained in 75% isolated yield and minimal quantities of byproduct 17(OPr) (Chart 1).
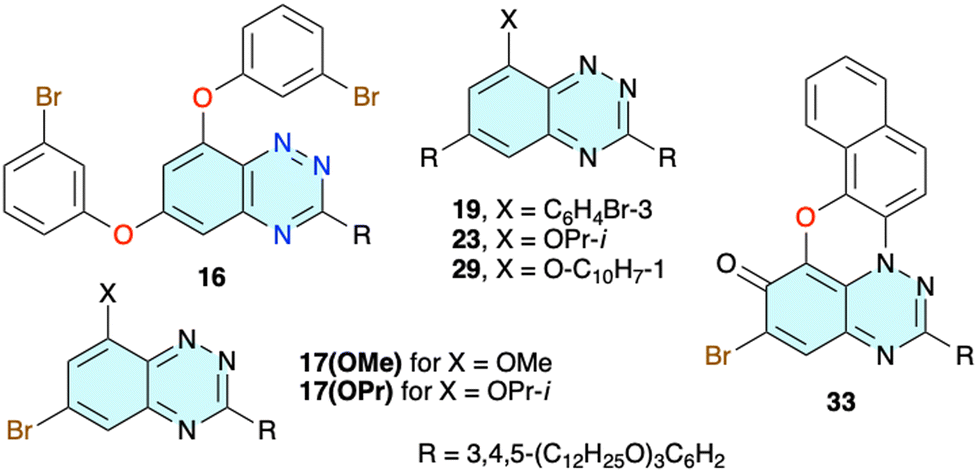 | ||
| Chart 1 | ||
Double Suzuki cross-coupling reactions of 15 with boronic acid 10 and preparation of 18 proved to be problematic. Under several reaction conditions, using a range of catalysts (Pd(Ph3P)4, Pd(OAc)2, Pd(OAc)2 and THPTB, Pd-PEPPSI-iPr, and Pd(t-Bu3P)2) the substitution took place at the C(6) position of the benzo[e][1,2,4]triazine ring, while the bromophenoxy fragment was either unaffected or debrominated, resulting in derivatives 19 and 20, respectively. In order to increase the reactivity of the halogen on the phenoxy substituent, precursor 4 was reacted with 3-iodophenol under i-PrOH/[Me4N]OH × 5H2O conditions, giving iodo derivative 21 in 90% isolated yield (Scheme 2). Double Suzuki coupling with 10 in the presence of the Pd-PEPPSI-iPr catalyst gave the desired 18 in 80% yield (Scheme 3).
Photocyclization of 18 performed in CH2Cl2 solution with a 300 W halogen lamp gave only marginal conversion to the desired 2d (Scheme 3). After several days and tedious chromatographic separation of the reaction mixture, 2d was isolated in 5% yield and unreacted starting 18 was recovered (80%). Multiple recrystallizations gave product 2d, which passed the combustion analysis test.
 | ||
| Scheme 4 Preparation of 2e. Reagents and conditions: (i) for 20: (1) phenol, [Me4N]OH × 5H2O, dry i-PrOH, rt, 45 min; (2) dry i-PrOH, 70 °C, 24 h, 85% yield. For 22: 2-iodophenol (50 equiv.), NaH, 80 °C, 24 h, 60% yield; (ii) CH2Cl2, N2, 300 W halogen lamp, 15 d, 4% yield; and (iii) (1) Bu3SnH, AIBN, PhMe, 80 °C, N2 atm, 18 h; (2) sat. KF, rt, 1 h, 0% yield. | ||
Higher yields of 2e were expected for cyclization of iodo derivative 22 under either photochemical or free radical conditions. Preparation of 22 was ineffective under the conditions used for obtaining 20, and only the product of substitution with i-PrOH 23 (Chart 1) was observed. To avoid formation of the byproduct, the reaction of 5 was conducted neat using excess 2-iodophenol (50 equiv.) as the reaction medium and the desired product 22 was isolated by chromatography in 60% yield on a 200 mg scale of 5 (Scheme 4). Scaling up the reaction gave lower conversions.
The isolated iodo derivative 22 was tested for the formation of radical 2e. Thus, irradiation of 22 in CH2Cl2 solutions with a halogen lamp for six days led to the isolation of 2e in 6% yield (Scheme 4). The rest was the starting material, de-iodinated starting material (compound 20) and intractable decomposition products. Two attempts at converting 22 to 2e using Bu3SnH gave no reaction, and the starting material was fully recovered.
To improve the yield of 2e the strategy was changed and the initial substitution of precursor 4 with a phenoxy group was investigated (strategy B, Fig. 2). Thus, reaction of 4 with sodium 2-aminophenolate in DMSO/THF gave 24 in 58% (Scheme 5). The subsequent aza-Pschorr reaction of 24 with t-BuONO did not give the expected radical 25. Suzuki cross-coupling reactions of 24 with boronic acid 10 were also unsuccessful with Pd(Ph3P)4, Pd(OAc)2 and Pd-PEPPSI-iPr.
 | ||
| Scheme 5 Attempted preparation of 25. Reagents and conditions: (i) (1) 2-aminophenol, NaOH, MeOH, rt, 30 min; (2) DMSO/THF (1:1), rt, 12 h, 58% yield; (ii) t-BuONO, C6H6, rt, 30 min then 70 °C, 2 h, 0% yield. | ||
 | ||
| Scheme 6 Preparation of discotic 3e. Reagents and conditions: (i) (1) 2-naphthol, [Me4N]OH × 5H2O, dry i-PrOH, rt, 45 min; (2) dry i-PrOH, 70 °C, 24 h, X = Br 50–60% yield, X = H 52% yield, X = NO2 0% yield; (ii) CH2Cl2, 300 W lamp. 6–10 d, 4–7% yield. | ||
Irradiation of the isolated derivative 26 or crude 27 dissolved in CH2Cl2 with a halogen lamp led to significant consumption of the starting materials after six days (Scheme 6). The resulting dark-green radical was isolated in both cases in 4–7% yield using neutral Al2O3. The low yield was, in part, due to the instability of the product during chromatographic separation, evident from the presence of highly polar and colored oxidation products.
For comparative purposes 8-(1-naphthyloxy) 29 (Chart 1), an isomer of 26, was considered as a potential precursor to an isomer of 3e. Thus, using reaction conditions developed for 26, the isomeric 29 was obtained in 80% yield from 5 and 1-naphthol. Attempts at photocyclization of 29 under standard conditions failed, and only the starting material was recovered.
The preparation of 3e following strategy B was briefly investigated. Thus, a reaction of intermediate 4 with 2-naphthol and 1-nitro-2-naphthol in the i-PrOH/[Me4N]OH × 5H2O system gave the desired products 30 and 31, respectively, isolated in 66% (X = H) and 36% (X = NO2) yield along with the i-propoxy by product 17(OPr) (Scheme 7). The same substitution run in t-BuOH instead of i-PrOH gave no reaction. Isolation of the product 31 was challenging due to its high photoreactivity, already noted for the parent system.39 Irradiation of the nitro derivative 31 in CH2Cl2 led to full conversion in 3 h, giving a purple-red solution. TLC analysis revealed the presence of only very polar, highly coloured products consistent with oxidation of the expected radical 32, to e.g. a zwitterionic quinonimine, such as 33 (Chart 1).
 | ||
| Scheme 7 Attempted preparation of 32 as a precursor to 3e. Reagents and conditions: (i) (1) 2-naphthol or 1-nitro-2-naphthol, [Me4N]OH × 5H2O, dry i-PrOH, rt, 30 min; (2) dry i-PrOH, 70 °C, 24 h, X = H 66% yield, X = NO2 36% yield; (ii) CH2Cl2, 300 W lamp, 3 h, 0% yield. | ||
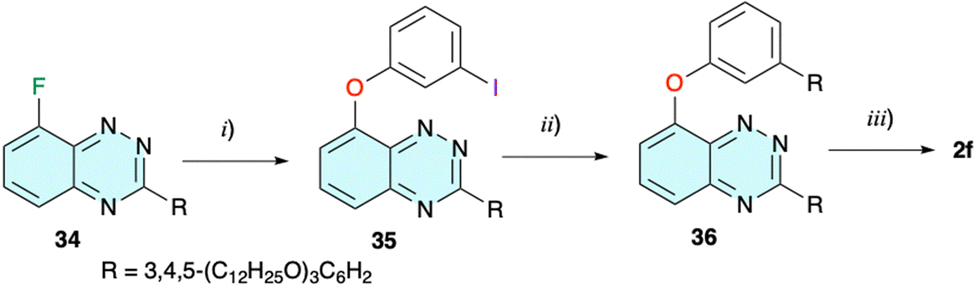 | ||
| Scheme 8 Preparation of 2f. Reagents and conditions: (i) (1) 3-iodophenol, [Me4N]OH × 5H2O, dry i-PrOH, rt, 30 min; (2) dry i-PrOH, 70 °C, 24 h, 92% yield; (ii) 3,4,5-(C12H25O)3C6H2B(OH)2 (10), Na2CO3, Pd-PEPPSI-iPr, THF/H2O (5:2), N2, reflux, 24 h, 73%; and (iii) CH2Cl2, halogen lamp, 300 W, 10 d, 5% yield. | ||
The requisite benzo[e][1,2,4]triazine 34 was obtained in a similar way to the preparation of its 6-bromo analogue 4 by reductive cyclocondensation of 37 (Scheme 9).
 | ||
| Scheme 9 Preparation of 34. Reagents and conditions: (i) 3,4,5-(C12H25O)3C6H2CONHNH2 (7), dry DMSO, N2, 85 °C, 12 h, 91% yield; (ii) (1) Sn, AcOH/THF (5:1), rt, 8 h, 130 °C, 0.5 h. (2) NaIO4, CH2Cl2/MeOH (1:1), 0.5 h, 76% yield. | ||
All radicals 2 and 3 exhibit similar chemical stability during purification and storage. Chromatographic purification of the radicals was most effective using neutral alumina, although passivated silica (eluent with 1–2% of Et3N) also gave good results.
Mechanistic analysis
The photocyclization of 8-(2-naphthyloxy)-3-phenylbenzo[e][1,2,4]triazine (38), the fundamental structural unit in 26, has been determined to undergo a photo-Smiles rearrangement, giving the parent 3 (R1 = Ph; R2 = H) with complete regioselectivity.43,45 This indicates that irradiation of 26 gives radical 3e. So far, such an analysis has not been performed for the phenoxy analogue. For this reason, derivative 39, a model for precursor 18, was obtained from the bromide 40 (Scheme 10), and its photocyclization in AcOEt was investigated experimentally and using DFT methods.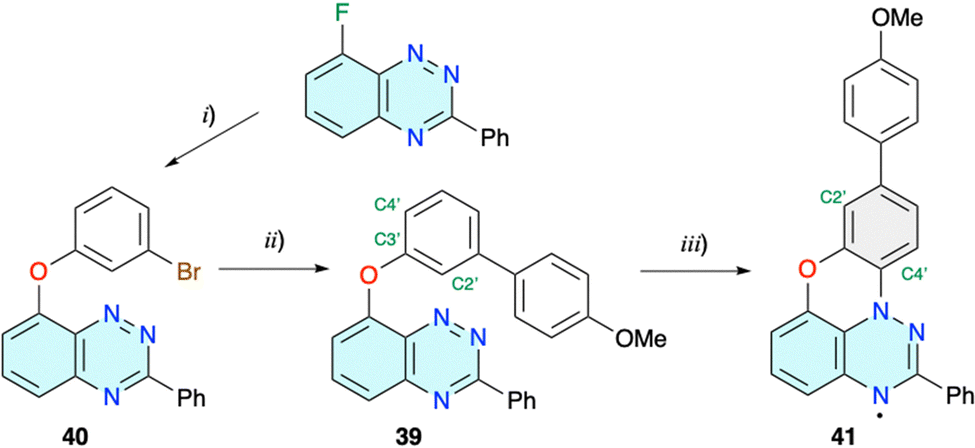 | ||
| Scheme 10 Preparation of model radical 41. Reagents and conditions: (i) 3-bromophenol, NaH, dry DMSO, 100 °C, overnight, 80% yield; (ii) (4-methoxyphenyl)boronic acid, Na2CO3, Pd-PEPPSI-iPr, THF/H2O (5:2), N2, reflux, 24 h, 93%; and (iii) CH2Cl2 or AcOEt, halogen lamp 300 W, 3 d, 3–4% yield. | ||
Single crystal XRD analysis (vide infra) of precursor 39 and product 41 revealed that the photocyclization occurs without rearrangement, which is contrary to findings for naphthyloxy, phenanthrenyloxy, pyrenyloxy and quinolinyloxy analogues investigated to date.44 Correlation NMR analysis of the leuco form of radical 41 demonstrated the structural homogeneity of the bulk sample of the photocyclization product, hence the complete regioselectivity of the cyclization onto the C(4′) position.46 According to the recently proposed mechanism,43 photo-Smiles requires a persistent charge separation, which is necessary for the formation of the transient spirooxazole species. It was postulated that such stable charge separation can be obtained if a π HOMO is solely localized on the aryloxy fragment and a lower energy n MO is associated exclusively with the [1,2,4]triazine ring. Detailed analysis of the electronic structure of the S0 state of 39 at the triplet geometry revealed that unlike in 38, the n MO is delocalized and encompasses not only the lone pairs of the heterocycle, but also the phenoxy π system (Fig. 3). This prevents the formation of persistent charge separation in 39.
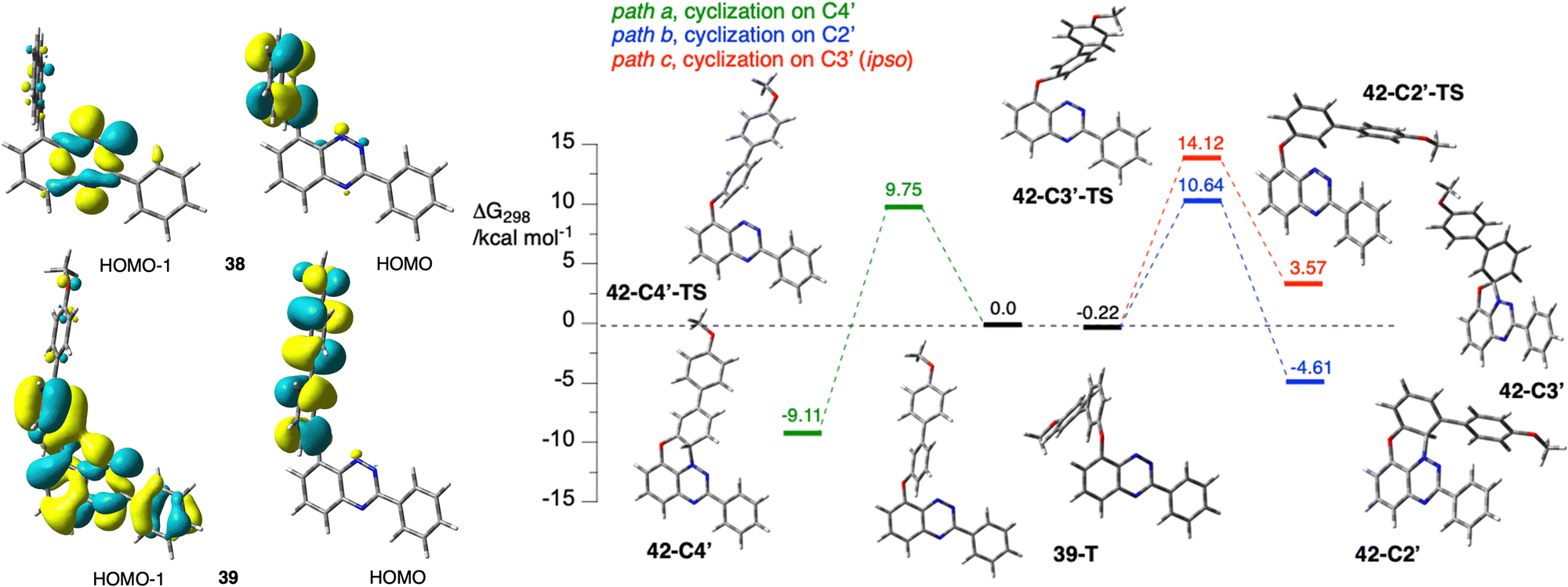 | ||
| Fig. 3 Left: Selected CAM-B3LYP/6-311G(d,p)-derived MO contours and energies relevant to low energy excitations of 38 and 39 at the GS equilibrium geometry (MO isovalue = 0.03). Right: Energies of transition states and cyclization products relative to that of 39 in T1 state (ΔG298 = 0 kcal mol−1, grey line) for the proposed mechanism of photocyclization of model 39 obtained at the UCAM-B3LYP/6-311G(d,p) level of theory in EtOAc dielectric medium in the triplet state. For details, see the text. | ||
In the absence of permanent charge separation, the cyclization takes place in a non-polar triplet state (39-T) onto the ortho position. DFT calculations of three pathways for cyclization of 39-T onto the C(4′) (formation of 42-C4′, path a), the C(2′) (formation of 42-C2′, path b), and C(3′) (ipso, formation of 42-C3′, path c), demonstrate that the observed regioisomer 42-C4′ is the most thermodynamically stable and its formation has the lowest activation energy ΔG‡298 = 9.75 kcal mol−1 (Fig. 3). Considering the relatively low activation energies for forward and reverse processes, the formation of the three products is under thermodynamic control at ambient temperature. Therefore, the product of cyclization at the C(4′) position 42-C4′ (observed experimentally) is formed preferentially to the C(2′) product 42-C2′ (not observed). Based on the calculated ΔΔG298, the ratio of the two products is about 1800:1. The cyclic product 42-C4′ undergoes oxidation with air present in the reaction mixture, giving radical 41.
Analysis of other model derivatives with two or three 3,4,5-trimethoxyphenyl substituents revealed a similar distribution of the relevant MOs (Fig. 3), suggesting that photocyclization of 36 and 20, respectively, also proceeds without Smiles rearrangement.46 The observed significant delocalization of the MOs relevant to the T1 state is a possible reason for the observed inefficient formation of radicals.
Single crystal XRD
Yellow monoclinic crystals of precursors 39 and 40 with the space group Pc and black-brown monoclinic crystals of radical 41 (the P21/c space group) were obtained by slow evaporation of CH2Cl2/EtOH, CH2Cl2/MeCN, and CH2Cl2/benzene solutions, respectively. Their solid-state structures were determined by single crystal XRD analysis, and molecular structures of 39 and 41 are shown in Fig. 4, while all details are provided in the SI.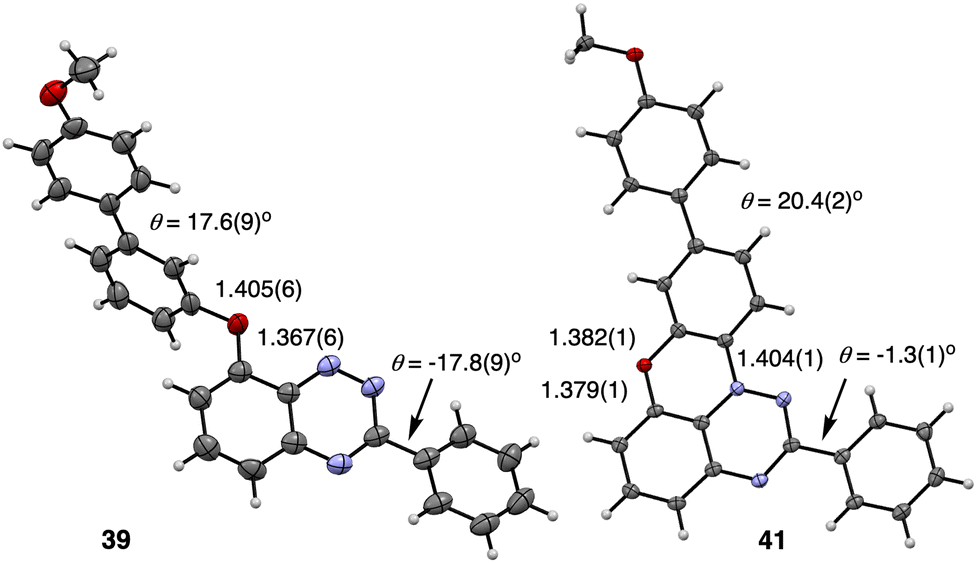 | ||
| Fig. 4 Atomic displacement ellipsoid representation of precursor 39 (left) and radical 41 (right). The ellipsoids are at the 50% probability level. See the text and SI for details. | ||
The benzo[e][1,2,4]triazine in intermediates 39 and 40, and the triazino[5,6,1-kl]phenoxazine fragment in radical 41 are essentially planar with dimensions typical for these heterocycles.35–38,47 The C(3)–Ph group is twisted from the [1,2,4]triazine mean plane in 39 and 40 (18.0° and 11.5°, respectively), while the C(2)–Ph in 41 is nearly co-planar with the heterocycle (3.5°).
In the crystal structure molecules of radical 41 form close centrosymmetric dimers with 3.088 Å separation between mean planes defined by the benzo[e][1,2,4]triazine rings (Fig. 5). The dimers are arranged in slipped stacks with the separation of 3.423 Å and the slippage angle of 51.5° propagating along the [0 1 0] direction. The stacks are separated with rotated isolated dimers.
 | ||
| Fig. 5 The discrete dimer (left) and partial packing diagram (right) for radical 41. | ||
DFT calculations at the UB3LYP/6-311+G(d) level of theory of the fundamental discrete dimer at its crystallographic coordinates using the Yamaguchi formalism48,49 demonstrated significant stabilization of the singlet over triplet state (EST = −3.52 kcal mol−1).
Thermotropic properties
The mesomorphic properties of planar Blatter discogens 2 and 3e and also their precursors were investigated by differential scanning calorimetry (DSC), polarizing optical microscopy (POM) and powder XRD.Optical microscopy revealed that only two radicals, 2d and 2f, form mesophases with textures characteristic of discotic phases, identified by powder XRD as Colh. DSC analysis demonstrated that planarization of the central Blatter radical in half-disc311a improves packing of the molecules in the crystal phase so effectively that the melting temperature is increased by about 130 K in 2e and mesogenic behavior is eliminated. Powder XRD analysis indicates,46 however, that the crystalline phase of 2e has a structure resembling that of a columnar phase with a rectangular unit cell. Similarly high melting point (170 °C) was observed for the benzo-fused analogue of 2e, compound 3e. An isomer of 2e, compound 2f, does exhibit a columnar hexagonal phase with the same clearing temperature as in the Blatter derivative 1a (Table 1 and Fig. 6). The enthalpy of the Colh–I transition is noticeably lower for 2f relative to 1a. Interestingly, the crystalline polymorph of 2f obtained from the melt has a different structure and exhibits a melting enthalpy that is about half of that of the pristine sample (Fig. 6).
| Compound | –OAr | Phase thermal behavioura | Phase | T/°C | a or a, b, c/Å, γ/°a | Halo/Å | |
|---|---|---|---|---|---|---|---|
| a Transition temperature peak (°C) and transition enthalpy (kJ mol−1) in parentheses. First heating and cooling cycles at 10 K min−1: Cr crystal; Colh columnar hexagonal LC phase; Colx 3D ordered columnar LC phase; X unknown LC phase. b Data from ref. 31. c For XRD of the Cr phase see the SI. d POM observations. | |||||||
| Radical | 2d | Cr 10 Colh 122 (7.5) I | Colh | 130 | 34.4 | 4.56 and 3.53 | |
| 100 | 35.0 | 4.54 and 3.48 | |||||
| Precursor | 18 | OC6H4C6H2(OC12H25)3 | Cr < 25 Colx 100 (13.5) Colh 125 (42.4) I | Colh | 100 | 33.0 | 4.51 and 3.41 |
| Colx | 70 | a: 116.8; b, 67.4; c, 26.5 | 4.45 and 3.24 | ||||
| Blatter | 1a | Cr 46 (49.2) Colh 73 (6.0) I | Colh | 60 | 30.02 | 4.5 and 3.4 | |
| Radical | 2e | Cr 175 (105) I | — | — | — | ||
| Precursor | 20 | OC6H5 | Cr 45 (61.8) Colx 98 (10.0) Colh 140 (38.4) I | Colh | 100 | 29.7 | 4.56 and 3.43 |
| Colx | 70 | a: 104.2; b: 60.2; c: 24.5 | 4.46 and 3.26 | ||||
| Precursor | 22 | OC6H4I-2 | Cr 36 (106.0) Colh 140 (26.2) I | Colh | 100 | 29.6 | 4.45 and 3.44 |
| Colh | 40 | 29.4 | 4.38 and 3.43 | ||||
| Isomer | 19 | OC6H4Br-3 | Cr 49 (99.5) Colx 85 (10.2) Colh 159 (24.8) I | Colh | 140 | 29.9 | 4.54 |
| Colh | 100 | 29.8 | 4.48 | ||||
| Colx | 40 | a: 104.4; b: 60.2; c: 25.1 | 4.37 | ||||
| Radical | 3e | Cr 170 Id | — | — | — | — | |
| Precursor | 26 | OC10H7-2 | Cr < 25 Colx 163 (56.9) I | Colx | 120 | a: 105.2; b: 60.7; c: 24.4 | 4.47 and 3.35 |
| Isomer | 29 | OC10H7-1 | Cr < 25 X 128 (15.0) Colx 156 (21.6) I | Colx | 100 | a: 105.1; b: 61.2; c: 24.2 | 4.54 and 3.33 |
| Radical | 2f | Cr 51 (114.9) Colh 73 (1.2) I | Colh | 46 | 32.7 | 4.47 and 3.46 | |
| Precursor | 36 | OC6H4C6H2(OC12H25)3 | Cr 60 (102.5) I | — | — | — | — |
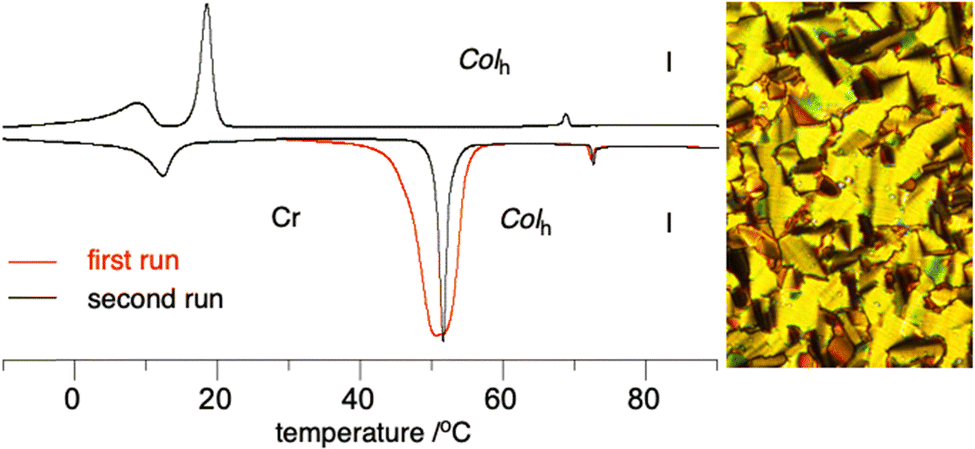 | ||
| Fig. 6 Left: DSC trace for 2f: first cycle (red) and second cycle (black). Heating and cooling rates are 10 K min−1. Right: Optical texture at 30 °C obtained on cooling from the isotropic phase and observed in crossed polarizers (magnification ×20). | ||
The full disc derivative 2d also exhibits a Colh phase. Although there is no direct analogue of 2d in series 1, it can be concluded that planarization and addition of a benzene ring to 1a increase the phase stability by 50 K.
Precursors to the disc-like radicals exhibit two types of columnar mesophases, the common Colh and a more organized 3D phase Colx, according to powder XRD. Only compound 36, a direct precursor to 2f, shows no mesogenic behavior.
The phenoxy derivative 20, a precursor to 2e, exhibits both types of columnar mesophases with the clearing temperature of 140 °C and a significant transition enthalpy. Substitution of the phenoxy group in 20 with Br increases the clearing temperature by 19 K, but destabilizes the 3D columnar phase by 13 K in 19 (Fig. 7). Substitution of the phenoxy in 20 with iodine in 22 has little effect on the stability of the Colh phase, but it completely eliminates the Colx phase.
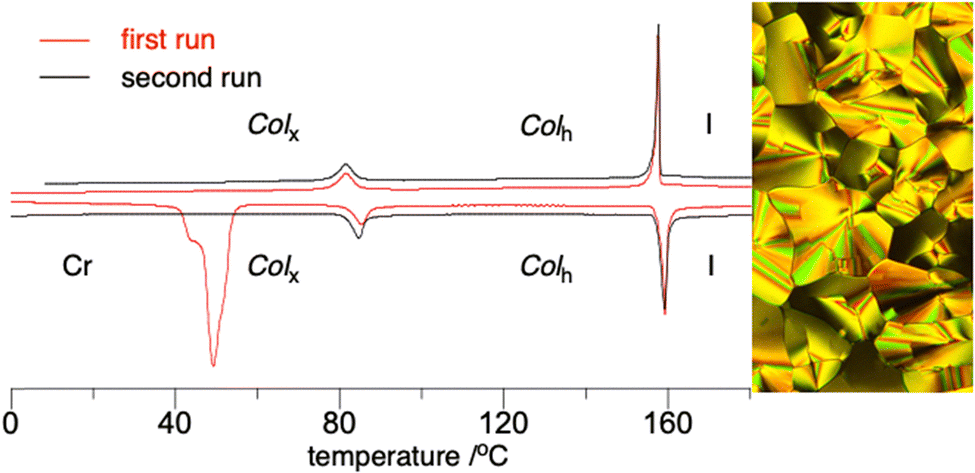 | ||
| Fig. 7 Left: DSC trace for 19: first cycle (red) and second cycle (black). Heating and cooling rates are 10 K min−1. Right: A texture of Colx at 30 °C obtained on cooling from the isotropic phase and observed in crossed polarizers (magnification ×30). | ||
Precursors to 3, naphthyloxy derivative 26 and its isomer 29, exhibit only the 3D Colx phase, with the clearing temperatures of about 160 °C and a significant enthalpy of transition to the isotropic phase. Results indicate that the 1-naphthyloxy derivative 29 has another soft crystalline phase below the Colx phase, but its structure is presently unclear. Data in Table 1 also demonstrate that increasing the size of the aryloxy substituent at the benzo[e][1,2,4]triazine core completely eliminates the Colh phase and increases stability of the 3D Colx phase by about 60 K in 26 and 29, relative to that in 20.
Finally, compound 18 exhibits similar stability of the Colh phase as the cyclic product 2d. Unlike the cyclic product, 18 also displays the Colx phase below 100 °C.
Powder XRD investigation
Most of the investigated compounds, except for precursors 26 and 29 with the naphthyloxy unit, form a columnar hexagonal phase, Colh, typical for discotic mesogens. XRD patterns for this phase can be indexed assuming a 2D lattice with the p6 symmetry. The resulting unit cell parameter a, corresponding directly to the column diameter, is about 30 Å and consistent with the molecular dimensions. High-angle patterns revealed liquid-like order of molecules along the columns, with two characteristic distances, corresponding to separation between aliphatic chains (about 4.5 Å) and the mesogenic cores (about 3.5 Å), as shown for 20 in Fig. 8. The relatively narrow latter diffraction signal suggests the ordering of the mesogenic cores with a significant correlation length.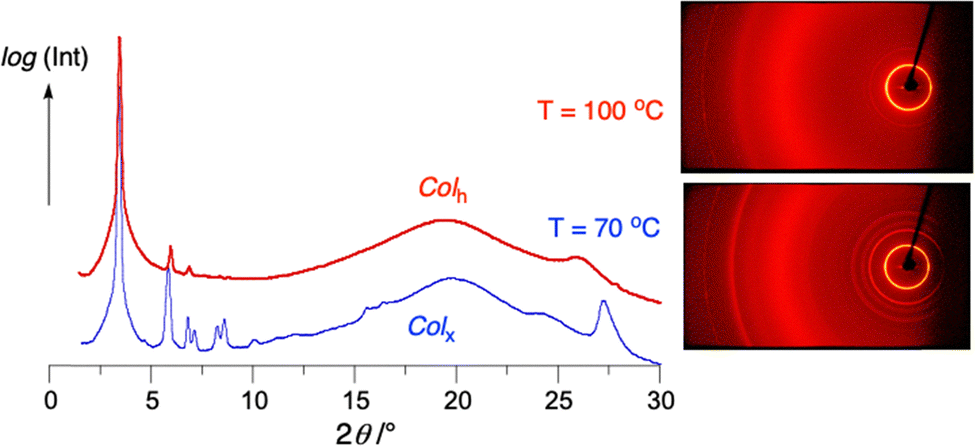 | ||
| Fig. 8 XRD diffractograms for 20 (left) obtained by integration of the 2D patterns (right) taken at 100 °C (red) and 70 °C (blue). | ||
On cooling, the Colh phase is transformed to a more organized columnar LC phase, Colx, in most of these derivatives, except for radicals 2d, 2f and the iodo derivative 22. The XRD patterns observed for this phase are much richer than those of the higher temperature Colh phase and cannot be fitted to any 2D lattice (Fig. 8). It should be stressed, however, that the positions of the main signals corresponding to the hexagonal lattice of columns remain nearly identical. Diffraction patterns taken for aligned samples (Fig. S71) demonstrated that the phase exhibits a 3D crystallographic lattice with an orthorhombic unit cell. The structure resembles columnar phases with the Fddd symmetry, recently reported for linear polycatenar mesogens50,51 and a lath-like discotic quinoxaline derivative.52 However, observation of diffraction signals forbidden for the Fddd space group evidences that either the actual symmetry of the Colx phase is lower or might indicate a considerable level of local disorder and a large number of defects in the structure. Although the size of the columns’ cross-section and their local hexagonal packing is not affected by the Colh–Colx phase transition, the unit cell parameters of the Colx phase are doubled compared to the upper temperature hexagonal phase, i.e. the unit cell (centered) contains 8 columns. The unit cell parameter c, reflecting periodicity of electron density modulations along the columns in the Colx phase, was found to be about 24 Å for all these compounds. Such modulation is ascribed to a helical arrangement of lath-like mesogens (which leads to a circular footprint of the column). The observed complex crystallographic lattice with d-type gliding mirror planes results from a balance between spontaneously formed right- and left-handed helices. It can be assumed that the appearance of the helical periodicity along the column axis drives the observed Colh–Colx phase transition.
XRD data indicate that the two isomeric naphthyloxy derivatives, 26 and 29, exhibit only the 3D Colx phase with similar unit cell parameters (Table 1). Results confirmed that radical 2f forms another crystalline polymorph upon cooling of its Colh phase (Fig. 6).
Magnetic properties
Magnetic properties of neat samples of 2d and 2f were investigated with variable temperature EPR spectroscopy in the temperature range of 230–400 K. Results demonstrate that signal intensity, obtained by double integration (DI, proportional to χp) and peak-to-peak signal width, ΔHpp, response to phase transitions observed in DSC, as shown for 2f in Fig. 9. Both parameters reflect interactions between the spins. Thus, the signal intensity typically increases with decreasing spin–spin interaction, while the ΔHpp follows the opposite trend. Consequently, the increasing ΔHpp with a simultaneous decrease in signal intensity indicates increasing antiferromagnetic interactions. | ||
| Fig. 9 Temperature dependence of the relative EPR intensity DIrel (left) and peak-to-peak signal width ΔHpp for 2f on heating (red) and cooling (blue). The dotted lines indicate the transition temperatures observed by DSC on heating. The minor step observed on heating at 5 °C may be due to the Cr–Cr transition. | ||
Heating of a sample of 2f showed a dramatic 90% decrease of EPR signal intensity at 48 °C, with a simultaneous significant increase of ΔHpp from 1.9 to 6.1 G (Fig. 9). This is consistent with melting of a paramagnetic solid to a fluid mesophase with significant antiferromagnetic interactions. At the Colh to isotropic transition at 73 °C the intensity drops further by an additional 5%, while the signal width increases by an additional 6.2 G. On cooling, the changes in the DI and ΔHpp parameters fully reverse at the I–Colh and only partially at the lower temperature transition, consistent with the DSC results. Thus, at 20 °C, the signal intensity increases by 50%, while the signal width drops by 3.6 G to 2.4 G, short of 1.9 G in the pristine sample. These results are consistent with those obtained with DSC and XRD methods.
Similar investigations of 2d showed less pronounced changes in the DI and ΔHpp parameters. One reason is the lack of crystallization, but rather the glassification of the Colh phase.
Summary and conclusions
Extensive synthetic studies have opened up access to a new class of paramagnetic discotic liquid crystals with stronger core–core interactions. Four disc-like compounds were obtained in low yields by photocyclization of appropriate precursors and two of them exhibited liquid crystalline behavior. Results obtained for these derivatives demonstrate that planarization of Blatter-based discotic 1a by oxygen peri-annulation markedly improves molecular packing to the point that the melting point is significantly increased and the mesophase is eliminated. There is also indirect evidence that cyclic derivatives exhibit mesophases with increased stability, which is consistent with the results obtained for a pair of bent-core mesogens.40Most of the direct precursors to the disc-shaped radicals exhibit columnar Colh and/or more organized 3D Colx phases. Interestingly, photocyclization of phenoxy 20 and naphthyloxy 22 leads to high melting half-disc products 2e and 3e, respectively, and at least the former has a columnar-like arrangement in the solid state. On the other hand, cyclization of a non-mesogenic 36 induces a Colh phase in 2f. Thus, half-disc planar molecules prefer crystalline phases, while in full-disc molecules, such as 2d, the proper ratio of alkoxy chains to the rigid core allows the formation of a discotic phase.
Among the three cyclization methods tested, only photocyclization gave the radicals, albeit in low yields 2–7%. This low conversion is presumably related to the electronic structure of the benzo[e][1,2,4]triazine core perturbed by the presence of tri(alkoxy)phenyl groups, which changes the distribution of the n MO and HOMO orbitals. Mechanistic and structural studies demonstrated that photocyclization of phenoxy derivatives, such as 18, 20 and model 39, proceeds without the Smiles rearrangement. DFT analysis concluded that in these cases, a persistent zwitterionic T1 state was unattainable. This finding constitutes the first example of photocyclization of an 8-aryloxy benzo[e][1,2,4]triazine without Smiles rearrangement.
Many precursors to radicals 2d, 2e and 3e were obtained from fluoride 5 (strategy A), which appears to be a convenient common intermediate to a larger class of such C(8) substituted derivatives exhibiting mesogenic properties. Application of differently functionalized phenoxy substituents may lead to functional discotic materials with homochiral columns. The substitution of fluorine in 5 is, however, complicated by the presence of six oleophilic dodecyloxy chains. They significantly affect the reactivity of 5, presumably by the formation of micelles and obstructing access of the nucleophiles to the reactive aromatic core in polar solvents, such as DMSO and MeCN. Experiments demonstrated that the most effective solvent in these substitution reactions is i-PrOH.
Further studies are aimed at optimization of the cyclization methods and expanding the pool of paramagnetic mesogens to those exhibiting the Colx phase with homochiral columnar arrangements and their magnetic studies.
Experimental section
General
All chemical operations were performed without contact with metal objects or salts and reactions were carried out under an inert atmosphere (N2 or Ar gas), and subsequent reaction workup was conducted in air. Heat for the reactions requiring elevated temperatures was supplied using oil baths. Reaction mixtures and column eluents were monitored by TLC using commercial aluminium backed thin layer chromatography (TLC) plates (Merck silica gel 60 F254 or Merck Al2O3 60 F254 neutral). Column chromatographic purifications were performed using silica gel 60 (70–230 mm, Merck) or aluminium oxide 60 G neutral type (type E, 70–230 mesh, Merck). Unless stated otherwise, reported yields refer to analytically pure samples.Synthetic details for the preparation and characterization of precursors and closed-shell discotics, and also details of spectroscopic characterization methods (NMR, IR, MS, EPR and UV-vis), are provided in the SI.
Differential scanning calorimetry (DSC) analysis was conducted with a TA DSC 2500 instrument using a sample size of 2–5 mg at a heating rate of 10 K min−1. X-ray diffraction experiments in a broad angle range were performed with a Bruker D8 GADDS (Cu Kα radiation, Göbel mirror, point collimator, Vantec 2000 area detector) equipped with a modified Linkam heating stage. Details of these methods and full results are provided in the SI.
Preparation of radicals by photocyclization: the general method
A solution of the precursor 18, 20, 26 or 36 (0.1 mmol) in CH2Cl2 (200 mL) was placed in a 250 mL borosilicate RB flask fitted with a reflux condenser, N2 was passed through the solution, and the flask was filled with N2. The solution was stirred and irradiated with a 300 W halogen lamp (see the SI), which was set 30 cm from the flask. The irradiation warmed up the reaction mixture to 30–35 °C. The progress of the reaction was monitored by TLC (Al2O3, pet. ether/CH2Cl2, 3:2) and the irradiation was stopped after 7 to 15 days, without the consumption of all starting materials. The solvent was evaporated, and the residue was chromatographed on neutral Al2O3, and the product was separated from the unreacted starting material. The obtained radicals were recrystallized/precipitated from EtOAc/MeCN.
2,5,9-Tris[3,4,5-tri(dodecyloxy)phenyl]-3H-[1,2,4]triazino[5,6,1-kl]phenoxazine-3-yl (2d)
Starting from benzo[e][1,2,4]triazine 18. Irradiation time: 7 d. Chromatographed on Al2O3 (pet. ether/CH2Cl2, 3:2) to give the title compound 2d as a dark brown waxy solid. Yield: 5%; IR (neat) ν 2919, 2851, 1582, 1504, 1467, 1431, 1409, 1260, 1116 cm−1; MALDI-TOF m/z [M]+ calcd for C139H236N3O10: 2109.42, found: 2109.80. Anal. calcd for C139H236N3O10: C, 79.15; H, 11.28; N, 1.99. Found: C, 79.02; H 11.09; N, 2.01.
2,5-Bis[3,4,5-tri(dodecyloxy)phenyl]-3H-[1,2,4]triazino[5,6,1-kl]phenoxazine-3-yl (2e)
Starting from benzo[e][1,2,4]triazine 20. Irradiation time: 15 d. Chromatographed on Al2O3 (pet. ether/CH2Cl2, 3:2) to give the title compound 2e as a dark brown waxy solid. Yield: 4%. IR (neat) ν 2918, 2850, 1587, 1466, 1430, 1407, 1279, 1120, 631, 537 cm−1; TOF (ESI+) m/z 1480 (100, M+); MALDI-TOF m/z [M]+ calcd for C97H160N3O7: 1480.23 found: 1480.24. Anal. calcd for C97H160N3O7: C, 78.70; H, 10.89; N, 2.84. Found: C, 79.02; H 10.93; N, 3.12.
2,9-Bis[3,4,5-tri(dodecyloxy)phenyl]-3H-[1,2,4]triazino[5,6,1-kl]phenoxazine-3-yl (2f)
Starting from benzo[e][1,2,4]-triazine 36. Irradiation time: 7 d. Chromatographed on Al2O3 (pet. ether/CH2Cl2, 3:2) to give the title compound 2f as a brown waxy solid. Yield: 5%. IR (neat) ν 2916, 2849, 1587, 1562, 1466, 1428, 1408, 1336, 1259, 1229, 1148, 1116, 783, 713 cm−1; TOF (ESI+) m/z 1480 (100, M+); MALDI-TOF m/z [M]+ calcd for C97H160N3O7: 1480.23, found: 1480.20. Anal. calcd for C97H160N3O7: C, 78.70; H, 10.89; N, 2.84. Found: C, 78.49; H 11.01; N, 2.82.
2,5-Bis[3,4,5-tri(dodecyloxy)phenyl]-3H-benzo[a][1,2,4]triazino[5,6,1-kl]phenoxazine-3-yl (3e)
Starting from benzo[e][1,2,4]triazine 26. Irradiation time: 15 d. Chromatographed on Al2O3 (pet. ether/CH2Cl2, 3:2) to give the title compound 3e as a dark brown solid. Yield: 4–7%. IR (neat) ν 2920, 2850, 1604, 1569, 1482, 1460, 1449, 1344, 1275, 1261, 1121, 805, 774, 765, 741, 710 cm−1; TOF (ESI+) m/z 1531 (100, [M + H]+); MALDI-TOF m/z [M + H]+, calcd for C101H163N3O7: 1530.25, found: 1530.25. Anal. calcd for C101H162N3O7: C, 79.27; H, 10.67; N, 2.75. Found: C, 79.25; H 10.59; N, 2.71.
9-(4-Methoxyphenyl)-2-phenyl[1,2,4]triazino[5,6,1-kl]phenoxazine (41)
Starting from benzo[e][1,2,4]triazine 39. Irradiation time: 3 d. The obtained brown radical was recrystallized from MeCN. Yield: 3%. M.p. 208–212 °C (MeCN); IR (neat) ν 2958, 2830, 1604, 1576, 1562, 1478m, 1436m, 1402, 1264, 1117 cm−1; TOF (ESI+) m/z 405 (100, [M + H]+); HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H19N3O2: 405.1477, found 405.1472. Anal. calcd for C26H18N3O2: C, 77.21; H, 4.49; N, 10.39. Found: C, 77.23; H 4.51; N, 10.28.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Additional experimental procedures, analytical data and computational details are available within the manuscript and its SI. Supplementary information available: Synthesis and characterization details of compounds, 1D and 2D NMR spectra, IR and EPR spectra, XRD analysis, DSC, POM, powder XRD data, VT EPR analysis, and DFT modelling details. See DOI: https://doi.org/10.1039/d5tc02401cCCDC 2422011–2422013 contain the supplementary crystallographic data for this paper.53–55
Acknowledgements
Financial support was provided by the Polish National Science Center (NCN, 2020/38/A/ST4/00597). Dr Sławomir Kaźmierski is thanked for recording the 2D NMR spectra for 41-H.References
- Functional Organic Materials: Syntheses, Strategies and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, 2007 Search PubMed.
- Stable radicals: fundamentals and applied aspects of odd-electron compounds, ed. R. Hicks, Wiley & Sons, 2010 Search PubMed.
- Z. X. Chen, Y. Li and F. Huang, Persistent and stable organic radicals: Design, synthesis, and applications, Chem., 2021, 7, 288–332 CAS.
- Y. Xiong, Y. Li, X. Cui, S. Li, X. Peng, Y. Ju, T. Zhou, R. Feng, Y. Zhang, Z. Wang, Q. Wang and L. Dong, Stable radical polymers as new electroactive materials: Synthesis, properties, and emerging applications, Adv. Funct. Mater., 2024, 24, 2419661 Search PubMed.
- A. Zhou, Z. Sun and L. Sun, Stable organic radical qubits and their applications in quantum information science, Innovation, 2024, 5, 100662 CrossRef CAS.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Air-stable organic radicals: New-generation materials for flexible electronics?, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- A. Mizuno, R. Matsuoka, T. Mibu and T. Kusamoto, Luminescent radicals, Chem. Rev., 2024, 124, 1034–1121 CrossRef CAS PubMed.
- P. Kaszyński, S. Kapuściński and S. Ciastek-Iskrzycka, Liquid crystalline derivatives of heterocyclic radicals, Adv. Heterocycl. Chem., 2019, 128, 263–331 CrossRef.
- R. Tamura, Y. Uchida and K. Suzuki, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. F. Gleeson and P. Raynes, Wiley-VCH, Weinheim, 2014, vol. 8, pp. 837–864 Search PubMed.
- C. Xue and Q. Li in Liquid Crystals Beyond Displays: Chemistry, Physics and Applications, ed. Q. Li, Wiley&Sons, Hoboken, N.J., 2012, pp. 29–82 Search PubMed.
- T. Seki, N. Kawatsuki and M. Kondo, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. F. Gleeson and P. Raynes, Wiley-VCH, Weinheim, 2014, vol. 8, pp. 539–580 Search PubMed.
- M. Funahashi, T. Yasuda and T. Kato, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. F. Gleeson and P. Raynes, Wiley-VCH, Weinheim, 2014, vol. 8, pp. 675–708 Search PubMed.
- H. M. Blatter and H. Lukaszewski, A new stable free radical, Tetrahedron Lett., 1968, 9, 2701–2705 CrossRef.
- F. J. M. Rogers, P. L. Norcott and M. L. Coote, Recent advances in the chemistry of benzo[e][1,2,4]triazinyl radicals, Org. Biomol. Chem., 2020, 18, 8255–8277 RSC.
- F. A. Neugebauer and I. Umminger, 1,4-Dihydro-1,2,4-benzotriazin-Radikalkationen, Chem. Ber., 1981, 114, 2423–2430 CrossRef CAS.
- F. A. Neugebauer and G. Rimmler, ENDOR and triple resonance studies of 1,4-dihydro-1,2,4-benzotriazinyl radicals and 1,4-dihydro-1,2,4-benzotriazine radical cations, Magn. Reson. Chem., 1988, 26, 595–600 CrossRef CAS.
- A. A. Berezin, C. P. Constantinides, S. I. Mirallai, M. Manoli, L. L. Cao, J. M. Rawson and P. A. Koutentis, Synthesis and properties of imidazolo-fused benzotriazinyl radicals, Org. Biomol. Chem., 2013, 11, 6780–6795 RSC.
- A. A. Berezin, G. Zissimou, C. P. Constantinides, Y. Beldjoudi, J. M. Rawson and P. A. Koutentis, Route to benzo- and pyrido-fused 1,2,4-triazinyl radicals via N′-(het)aryl-N′-[2-nitro(het)aryl]hydrazides, J. Org. Chem., 2014, 79, 314–327 CrossRef CAS PubMed.
- A. Bodzioch, M. Zheng, P. Kaszyński and G. Utecht, Functional group transformations in derivatives of 1,4-dihydrobenzo[1,2,4]triazinyl radical, J. Org. Chem., 2014, 79, 7294–7310 CrossRef CAS.
- G. Karecla, P. Papagiorgis, N. Panagi, G. A. Zissimou, C. P. Constantinides, P. A. Koutentis, G. Itskos and S. C. Hayes, Emission from the stable Blatter radical, New J. Chem., 2017, 41, 8604–8613 RSC.
- J. S. Steen, J. L. Nuismer, V. Eiva, A. E. T. Wiglema, N. Daub, J. Hjelm and E. Otten, Blatter radicals as bipolar materials for symmetrical redox-flow batteries, J. Am. Chem. Soc., 2022, 144, 5051–5058 CrossRef CAS PubMed.
- A. Saal, C. Friebe and U. S. Schubert, Blatter radical as a polymeric active material in organic batteries, J. Power Sources, 2022, 524, 231061 CrossRef CAS.
- C. Friebe and U. S. Schubert, High-power-density organic radical batteries, Top. Curr. Chem., 2017, 375, 19 CrossRef PubMed.
- J. Z. Low, G. Kladnik, L. L. Patera, S. Sokolov, G. Lovat, E. Kumarasamy, J. Repp, L. M. Campos, D. Cvetko, A. Morgante and L. Venkataraman, The environment-dependent behavior of the blatter radical at the metal–molecule interface, Nano Lett., 2019, 19, 2543–2548 CrossRef CAS.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. van der Zant, C. Rovira, E. Burzurí and N. Crivillers, Robust organic radical molecular junctions using acetylene terminated groups for C–Au bond formation, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- F. Ciccullo, A. Calzolari, K. Bader, P. Neugebauer, N. M. Gallagher, A. Rajca, J. van Slageren and M. B. Casu, Interfacing a potential purely organic molecular quantum bit with a real-life surface, ACS Appl. Mater. Interfaces, 2019, 11, 1571–1578 CrossRef CAS PubMed.
- Y. Zheng, M.-S. Miao, G. Dantelle, N. D. Eisenmenger, G. Wu, I. Yavuz, M. L. Chabinyc, K. N. Houk and F. Wudl, A solid-state effect responsible for an organic quintet state at room temperature and ambient pressure, Adv. Mater., 2015, 27, 1718–1723 CrossRef CAS PubMed.
- Y. Ji, L. Long and Y. Zheng, Recent advances of stable Blatter radicals: synthesis, properties and applications, Mater. Chem. Front., 2020, 4, 3433–3443 RSC.
- S. Kapuściński, A. Gardias, D. Pociecha, M. Jasiński, J. Szczytko and P. Kaszyński, Magnetic behaviour of bent-core mesogens derived from the 1,4-dihydrobenzo[e][1,2,4]triazin-4-yl, J. Mater. Chem. C, 2018, 6, 3079–3088 RSC.
- S. Ciastek-Iskrzycka, J. Szczytko, H. Monobe, D. Pociecha, M. Jasiński and P. Kaszyński, Paramagnetic ionic liquid crystals: Ion conductive bent-core derivatives of stable radicals, J. Mol. Liq., 2021, 337, 116028 CrossRef CAS.
- M. Jasiński, J. Szczytko, D. Pociecha, H. Monobe and P. Kaszyński, Substituent-dependent magnetic behavior of discotic benzo[e][1,2,4]triazinyls, J. Am. Chem Soc., 2016, 138, 9421–9424 CrossRef PubMed.
- M. Jasiński, K. Szymańska, A. Gardias, D. Pociecha, H. Monobe, J. Szczytko and P. Kaszyński, Tuning the magnetic properties of columnar benzo[e][1,2,4]triazin-4-yls with the molecular shape, ChemPhysChem, 2019, 20, 636–644 CrossRef PubMed.
- S. Kapuściński, J. Szczytko, D. Pociecha, M. Jasiński and P. Kaszyński, Discs, dumbbells and superdiscs: Molecular and supermolecular architecture dependent magnetic behavior of mesogenic Blatter radical derivatives, Chem. Mater. Front., 2021, 5, 6512–6521 RSC.
- M. Jasiński, S. Kapuściński and P. Kaszyński, Stability of a columnar liquid crystalline phase in isomeric derivatives of the 1,4-dihydrobenzo[e][1,2,4]triazin-4-yl: Conformational effects in the core, J. Mol. Liq., 2019, 277, 1054–1059 CrossRef.
- P. Kaszyński, C. P. Constantinides and V. G. Young, Jr., The planar blatter radical: Structural chemistry of 1,4-dihydrobenzo[e][1,2,4]triazin-4-yls, Angew. Chem., Int. Ed., 2016, 55, 11149–11152 CrossRef PubMed.
- P. Bartos, A. A. Hande, A. Pietrzak, A. Chrostowska and P. Kaszyński, Substituent effects on the electronic structure of the flat Blatter radical: Correlation analysis of experimental and computational data, New J. Chem., 2021, 45, 22876–22887 RSC.
- P. Bartos, B. Anand, A. Pietrzak and P. Kaszyński, Functional planar Blatter radical through Pschorr-type cyclization, Org. Lett., 2020, 22, 180–184 CrossRef CAS PubMed.
- P. Bartos, M. Celeda, A. Pietrzak and P. Kaszyński, Planar Blatter radicals through Bu3SnH and TMS3SiH–assisted cyclization of aryl iodides: Azaphilic radical addition, Org. Chem. Front., 2022, 9, 929–938 RSC.
- P. Bartos, V. G. Young, Jr. and P. Kaszyński, Ring-fused 1,4-dihydro[1,2,4]triazin-4-yls through photocyclization, Org. Lett., 2020, 22, 3835–3840 CrossRef CAS PubMed.
- K. I. Shivakumar, D. Pociecha, J. Szczytko, S. Kapuściński, H. Monobe and P. Kaszyński, Photoconductive bent-core liquid crystalline radicals with a paramagnetic polar switchable phase, J. Mater. Chem. C, 2020, 8, 1083–1088 RSC.
- M. Cigl, D. Pociecha, R. Jakubowski, S. Kapuściński and P. Kaszyński, Paramagnetic liquid crystals with close π–π packing: The effect of Blatter radical planarization on behavior of bent-core mesogens, Chem. – Eur. J., 2023, e202203288 CrossRef CAS PubMed.
- G. A. Zissimou, P. Bartos, A. Pietrzak and P. Kaszyński, “Upper” ring expansion of the planar Blatter radical via photocyclization: limitations and impact on the electronic structure, J. Org. Chem., 2022, 87, 4829–4837 CrossRef CAS PubMed.
- H. K. Singh, S. Kaźmierski and P. Kaszyński, A photo-Smiles rearrangement: Mechanistic Investigation of the formation of Blatter radical helicenes, J. Org. Chem., 2025, 90, 2386–2392 CrossRef CAS PubMed.
- H. K. Singh, S. Kaźmierski and P. Kaszyński, Photocyclization of 8-aryloxybenzo[e][1,2,4]triazines revisited: Unambiguous structural assignment of planar Blatter radicals by correlation NMR spectroscopy, J. Org. Chem., 2025, 90, 9425–9430 CrossRef CAS PubMed.
- H. K. Singh, A. Bodzioch, A. Pietrzak and P. Kaszyński, π-Curved Blatter radicals: Blatter helicenes, Chem. Commun., 2025, 61, 496–499 RSC.
- For details see the SI.
- A. Bodzioch, D. Pomikło, M. Celeda, A. Pietrzak and P. Kaszyński, 3-Substituted benzo[e][1,2,4]triazines: Synthesis and electronic effects of the C(3) substituent, J. Org. Chem., 2019, 84, 6377–6394 CrossRef CAS PubMed.
- K. Yamaguchi, F. Jensen, A. Dorigo and K. N. Houk, A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals, Chem. Phys. Lett., 1988, 149, 537–542 CrossRef CAS.
- K. Yamaguchi, Y. Takahara, T. Fueno and K. Nasu, Ab initio MO calculations of effective exchange integrals between transition-metal ions via oxygen dianions: Nature of the copper-oxygen bonds and superconductivity, Jpn. J. Appl. Phys., 1987, 26, L1362 CrossRef CAS.
- Y. Li, H. Gao, R. Zhang, K. Gabana, Q. Chang, G. A. Gehring, X. Cheng, X. Zeng and G. Ungar, A case of antiferrochirality in a liquid crystal phase of counter-rotating staircases, Nat. Commun., 2022, 13, 384 CrossRef CAS PubMed.
- Y. Wang, Y.-X. Li, L. Cseh, Y.-X. Chen, S.-G. Yang, X. Zeng, F. Liu, W. Hu and G. Ungar, Enantiomers self-sort into separate counter-twisted ribbons of the Fddd liquid crystal–antiferrochirality and parachirality, J. Am. Chem. Soc., 2023, 145, 17443–17460 CrossRef CAS PubMed.
- R. Rybak, A. Krowczynski, J. Szydlowska, D. Pociecha and E. Gorecka, Chiral columns forming a lattice with a giant unit cell, Soft Matter, 2022, 18, 2006–2011 RSC.
- H. K. Singh, G. A. Zissimou, D. Pociecha, M. Cigl, A. Pietrzak and P. Kaszyński, CCDC 2422011: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2m99dn.
- H. K. Singh, G. A. Zissimou, D. Pociecha, M. Cigl, A. Pietrzak and P. Kaszyński, CCDC 2422012: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2m99fp.
- H. K. Singh, G. A. Zissimou, D. Pociecha, M. Cigl, A. Pietrzak and P. Kaszyński, CCDC 2422013: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2m99gq.
| This journal is © The Royal Society of Chemistry 2025 |