
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzochalcogenazolo-based N,O-coordinated boron difluoride complexes as gain media for organic solid-state lasers†
Radosław Pytlarza,
Stepan Kutsiyab,
Andrii Hotynchanc,
Enzo Jean-Woldemard,
Roman Luboradzkie,
Paulina H. Marek-Urbanf,
Georg Merklind,
Sébastien Chénaisd,
Dmytro Volyniukg,
Juozas V. Grazulevicius *g,
Krzysztof Durka*f,
Sébastien Forget*d and
Mykhaylo A. Potopnyk*ac
*g,
Krzysztof Durka*f,
Sébastien Forget*d and
Mykhaylo A. Potopnyk*ac
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: potopnyk@gmail.com; mykhaylo.potopnyk@icho.edu.pl
bDepartment of Electronic Engineering, Lviv Polytechnic National University, pl. Sv. Yura 1, 79013, Lviv, Ukraine
cDepartment of Organic Chemistry, Faculty of Chemistry, Ivan Franko National University of Lviv, Kyryla and Mefodia 6, 79005, Lviv, Ukraine
dLaboratoire de Physique des Lasers, Université Sorbonne Paris Nord, CNRS UMR 7538 99, av. J.B. Clément, F-93430, Villetaneuse, France. E-mail: sebastien.forget@univ-paris13.fr
eInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland
fFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664, Warsaw, Poland. E-mail: krzysztof.durka@pw.edu.pl
gDepartment of Polymer Chemistry and Technology, Kaunas University of Technology, Barsausko 59, LT-51423, Kaunas, Lithuania. E-mail: juozas.grazulevicius@ktu.lt
First published on 1st July 2025
Abstract
Three N,O-coordinated benzochalcogenazolo-based boron difluoride complexes have been designed, synthesized, and spectroscopically characterized using their solutions, solid state, and the films of dye-doped polymers. The structural analysis demonstrated that dyes adopt twisted molecular structures with the torsion angle ranging from 24° to 37°, attributed to the steric effect of the cyano group. The obtained compounds exhibit aggregation-induced emission and blue to cyan emission (455–487 nm) in the solid state with a photoluminescence quantum yield ranging from 21% to 85% and a short excited-state lifetime of 0.80–2.27 ns. These characteristics facilitate the amplified spontaneous emission (ASE) with the progressive reduction in the full width at half maximum observed across the series of benzoxazole → benzothiazole → benzoselenazole derivatives up to 15 nm, 11 nm, and 8 nm, respectively, and also the low ASE threshold values of 18.7–40.3 μJ cm−2. The successful application of the dyes in the fabrication of organic solid-state lasers results in a laser threshold of 46.1 μJ cm−2, 28.0 μJ cm−2, and 58.6 μJ cm−2 for the devices based on benzoxazole, benzothiazole, and benzoselenazole-based boron difluoride complexes, respectively. Our work opens the pathway for a novel class of heavy atom-containing organic laser dyes, which can be used for both organic optically and electrically pumped lasers.
1. Introduction
Organic solid-state lasers (OSSLs) represent a rapidly advancing domain within the field of materials science. Due to their customized molecular tunability, cost-effective production, mechanical flexibility, and superior optical performance, OSSLs have been effectively utilized in information technology, nanotechnology, and biomedicine.1–5 One of the most significant characteristics of laser emission is high coherence, which occurs when the laser operates in a single mode, resulting in emitted photons that are confined to narrow ranges of frequency, phase, and direction. This phenomenon is typically manifested as nearly monochromatic emission and a narrow beam. Consequently, the implementation of organic lasers in the field of optoelectronics holds significant promise and is anticipated to yield a new generation of devices, specifically organic semiconducting laser diodes.6–8Like any other laser, an OSSL requires three main components: a gain medium, an optical feedback structure, and a pump source. Thus, for light-emitting applications, the organic gain material should exhibit intense solid-state emission (SSE). In addition to the chemical structure and conformation factor, the SSE of organic dyes is highly dependent on the molecular packing resulting from intermolecular interactions.9–11 In this context, materials demonstrating aggregation-induced emission (AIE) are particularly demanded.12–16 Furthermore, the lasing properties strongly depend on the type of optical cavity used for amplification. In practice, to minimize the research cost and time, an amplified spontaneous emission (ASE) threshold is usually evaluated instead of a lasing threshold of new gain materials. ASE occurs through the exciton population inversion mechanism under strong optical pumping of the organic gain medium. The measurements of ASE allow for the quantitative characterization of gain materials independently of an optical feedback structure. Lasing can be obtained upon adding a resonator to ASE-active materials, for example, a distributed feedback structure directly onto the film, or by sandwiching the active film between two distributed Bragg reflectors to form a vertical-cavity surface-emitting laser.
Up to now, ASE has been observed for different classes of organic luminophores including polyaromatic hydrocarbons,17–21 oligophenylene vinylene derivatives,22,23 carbazole-containing styryls,24–27 1,2,5-benzothiadiazole derivatives,28 diketopyrrolopyrroles,29 Cibalackrot,30 indigo derivatives,31 phosphorus-containing heterocyclic dyes,32 etc.
In this context, one of the most promising classes of luminescent dyes are boron difluoride complexes. Due to their stability, synthetic variability and tunability of the photophysical properties, they have found numerous applications in bioimaging,33,34 photodynamic therapy,35 organic photovoltaics,36 stimuli-responding materials,37–40 and organic light-emitting diodes.41–45 However, the studies on ASE-active boron complexes remain scarce. They are mostly limited to selected N,N-46–50 and O,O-chelated41,42,51–54 BF2 chromophores. Meanwhile, the thiochromen-4-one-based dye is a sole representative of the ASE-active N,O-chelated BF2 complex.55–57
Our group is involved in the design and synthesis of donor–acceptor type benzothiazole-based N,O-coordinated boron difluoride complexes such as benzo[4,5]thiazolo[3,2-c][1,3,5,2]oxadi-azaborinine and its derivatives.58–60 We have found that the photophysical properties of such compounds can be finely tuned by the attachment of substituents at the benzothiazole unit or by adjusting the electron-donating properties of the π-conjugated donor. These dyes usually exhibit efficient emission in a non-polar environment and solid state. With this in mind, we focused on the discovery of boron difluoride complexes with different chalcogen-containing N,O-chelate ligands. Consequently, in the current study, we present the synthesis of benzochalcogenazole-fused cyano-oxazaborinine (1–3) dyes incorporating benzoxazole, benzothiazole, or benzoselenazole cores (Fig. 1). The photophysical properties of compounds in solutions, as solid samples and as films of their molecular dispersions in a polymer have been comprehensively studied. The experimental results are supplemented by DFT and TD-DFT calculations. In addition, we examine the effect of chalcogen atom exchange (O → S → Se) on the molecular conformation and crystal packing, both governing the photophysical properties of the dyes in the solid state. It is anticipated that the incorporation of a heavier chalcogen atom such as selenium increases the spin–orbit coupling (SOC) between singlet and triplet excited states, facilitating the application of such materials in optoelectronic devices.61–63 Finally, benzochalcogenazole-boron complexes were applied for the construction of organic solid-state lasers.
 | ||
| Fig. 1 Benzochalcogenazolo-based boron difluoride complexes 1–3. | ||
2. Results and discussion
2.1. Synthesis
Complexes 1–3 were synthesized using 3-oxo-3-phenylpropanenitrile (4) as a starting material (Scheme 1). Firstly, following a literature protocol,64 compound 4 was converted into 2-benzoyl-3,3-bis(methylthio)acrylonitrile (5) (91% yield) via reaction with carbon disulphide in the presence of sodium hydrate with subsequent treatment with methyl iodide. In the next step, disulphide 5 was reacted with 2-aminophenol (6), 2-aminobenzenethiol (7), or 2,2′-diselanediyldianiline (8)65 in refluxing ethanol and in the presence of triethylamine affording benzochalcogenazole derivatives 9–11 in good yields ranging from 63% to 90%. Finally, compounds 9–11 were employed as ligands for the synthesis of boron difluorides 1–3 by reaction with boron trifluoride diethyl etherate (BF3·OEt2) and diisopropylethylamine (DIPEA), in yields of 72–74%. The structure and purity of the synthesized boron dyes and their precursors were confirmed by 1H, 13C, 19F, 77Se NMR spectroscopy and high-resolution mass spectrometry (see the ESI†).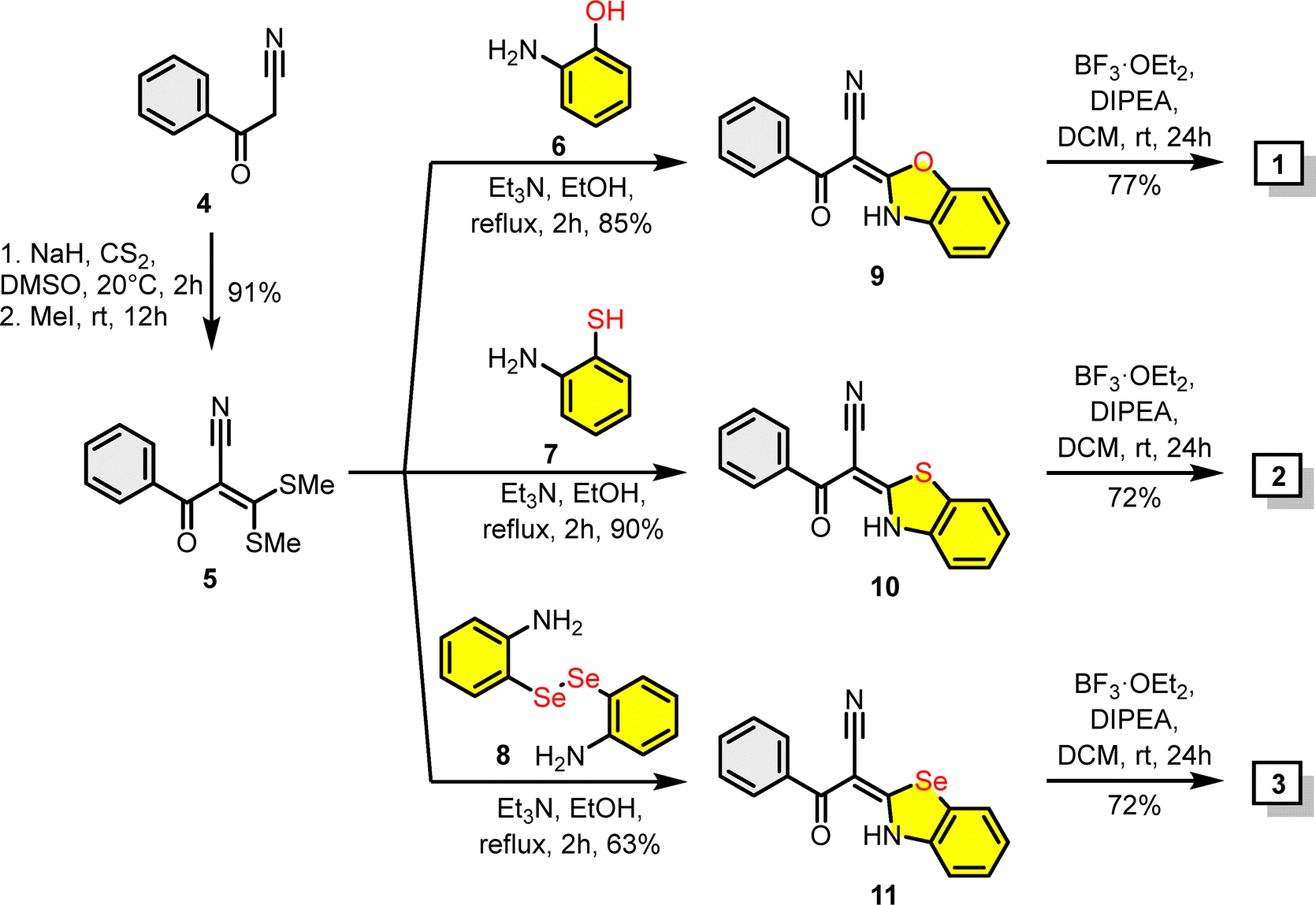 | ||
| Scheme 1 Synthesis of benzochalcogenazolo-based boron difluoride complexes 1–3. | ||
2.2. Crystal structure analysis
The crystal structures of boron difluoride complexes 1–3 were confirmed by single-crystal X-ray diffraction analysis (Fig. S1–S3 in the ESI†). Single crystals of compounds 2 and 3 were obtained by slow evaporation of the corresponding cyclohexane/dichloromethane (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solutions at room temperature. To receive single crystals of boron complex 1, its saturated solution in acetonitrile was slowly evaporated. The phenyl ring C1–C6 in the structure of dye 1 is disordered into two positions each with an occupancy of 50%; thus, two different values of torsion angles can be found in Table S4 (the ESI†). The analysis of molecular geometries revealed that the cyano-group induces a twist between the phenyl ring and heterocyclic unit. This is reflected by the C5–C6–C7–C15 torsion angles (Θ/ΘA in Fig. 2a, e and j) of 23.8°/−32.5° (phenyl ring is disordered over two positions with an equal occupancy ratio), 33.5°/34.0° (two molecules in the asymmetric part of the unit cell, ASU), and 36.5°/37.0° (two molecules in ASU) for structures of dyes 1, 2, and 3, respectively (Table S4, ESI†).
:1) solutions at room temperature. To receive single crystals of boron complex 1, its saturated solution in acetonitrile was slowly evaporated. The phenyl ring C1–C6 in the structure of dye 1 is disordered into two positions each with an occupancy of 50%; thus, two different values of torsion angles can be found in Table S4 (the ESI†). The analysis of molecular geometries revealed that the cyano-group induces a twist between the phenyl ring and heterocyclic unit. This is reflected by the C5–C6–C7–C15 torsion angles (Θ/ΘA in Fig. 2a, e and j) of 23.8°/−32.5° (phenyl ring is disordered over two positions with an equal occupancy ratio), 33.5°/34.0° (two molecules in the asymmetric part of the unit cell, ASU), and 36.5°/37.0° (two molecules in ASU) for structures of dyes 1, 2, and 3, respectively (Table S4, ESI†).
 | ||
| Fig. 2 X-ray molecular structures of dyes 1 (a) (for clarity, only one phenyl ring C1–C6 is shown), 2 (e), and 3 (j). Fragments of the crystal structures showing: CH⋯π interactions of structure 1 (b); CH⋯N and CH⋯O hydrogen bonds of structures 1 (c) and 2 (f); CH⋯F hydrogen bonds and F⋯π interactions of structures 1 (d), 2 (g), and 3 (l); n⋯π and S⋯F interactions of structure 2 (h); π⋯π interactions of structures 2 (i) and 3 (n); CH⋯Se, CH⋯N and CH⋯O hydrogen bonds of structure 3 (k); F⋯π and Se⋯F interactions of structure 3 (m); [hydrogen atoms are omitted for a clear view in (h), (j), (m) and (n)]. | ||
Compound 1 crystallizes in the monoclinic crystal system in the P21/c space group [a = 15.0569(3) Å; b = 5.09777(9) Å; c = 17.8384(4) Å; β = 102.0236(19)°] with four molecules in the unit cell (Table S1 and Fig. S4, ESI†). In the molecular packing, a half of the molecules are oriented in a near-to-perpendicular position to the other half, forming a stair-type architecture (Fig. S5, ESI†) by connections via CH⋯π interactions (C1H⋯C2 = 2.891 Å and C11H⋯C11 = 2.847 Å, Fig. 2b) and CH⋯F hydrogen bonding (C12H⋯F1 = 2.631 Å, Fig. 2d). The molecules in the same plate interact with each other by CH⋯N and CH⋯O hydrogen bonds (C13H⋯N2 = 2.587 Å and C14H⋯O2 = 2.591 Å, Fig. 2c), while additional F⋯π interactions (F2⋯C8 = 2.782 Å, Fig. 2d) appear between coplanar molecules from parallel plates. No essential π–π stacking is observed in the structure of dye 1.
Boron difluoride complex 2 crystallizes in the orthorhombic crystal system in the non-centrosymmetric P212121 space group [a = 13.42495(6) Å; b = 14.17067(6) Å; c = 14.59118(6) Å] with eight molecules in the unit cell (due to the presence of two symmetrically independent conformers) (Table S2 and Fig. S6, ESI†).
The molecules are linked via numerous CH⋯N and CH⋯O hydrogen bonds (C14H⋯N2A = 2.691 Å; C14AH⋯N2 = 2.697 Å; C12AH⋯O1 = 2.514 Å; O1A⋯C12H = 2.513 Å; Fig. 2f) forming 2-dimensional molecular layers (Fig. S7, ESI†). The adjacent layers are interconnected by CH⋯F hydrogen bonds (C5AH⋯F2A = 2.450 Å; C2H⋯F1 = 2.569 Å; C5H⋯F2 = 2.338 Å; Fig. 2g) and N⋯π interactions (N2⋯C15A = 3.205 Å; F1A⋯C8 = 2.944 Å; S1⋯C10A = 3.489 Å, Fig. 2h). This is complemented by halogen-type F⋯S interactions (S1⋯F2A = 3.170 Å; S1A⋯F2 = 3.136 Å; Fig. 2h), with a geometry consistent with the σ-hole bonding concept.66 It should be noted that some weak π–π stacking interactions (m = 3.583 Å, Fig. 2i) are observed between the heterocyclic parts of antiparallel oriented neighboring molecules of different conformations.
Boron difluoride complex 3 crystallizes in the triclinic system in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group [a = 9.8999(2) Å; b = 11.7333(3) Å; c = 13.4074(3) Å; α = 73.054(2)°; β = 69.987(2)°; γ = 83.5242°] with four molecules in the unit cell (analogically to structure 2, due to the presence of two symmetrically independent conformers) (Tables S3 and Fig. S8, ESI†). Molecular packing is governed by CH⋯Se, CH⋯N and CH⋯O hydrogen bonds (C4AH⋯Se1 = 3.061 Å; C4H⋯Se1A = 3.056 Å; C14H⋯N2A = 2.694 Å; C14AH⋯N2 = 2.700 Å; C12H⋯O1A = 2.466 Å; C12AH⋯O1 = 2.464 Å; Fig. 2k) resulting in the formation of molecular layers (Fig. S9, ESI†). Neighbouring layers are connected via CH⋯F hydrogen bonds (C5AH⋯F2 = 2.414 Å; C5H⋯F2A = 2.425 Å; Fig. 2l), F⋯π (F1⋯C14A = 3.115 Å; F1A⋯C7A(π) = 2.914 Å, Fig. 2m) and Se⋯F chalcogen-type (Se1⋯F2 = 3.271 Å; Se1A⋯F2A = 3.286 Å; Fig. 2m) interactions. Similar to the structure of compound 2, some π–π stacking (m = 3.631 Å; n = 3.560 Å; Fig. 2n) appears between the heterocyclic parts of the neighbouring molecules of the same conformations.
space group [a = 9.8999(2) Å; b = 11.7333(3) Å; c = 13.4074(3) Å; α = 73.054(2)°; β = 69.987(2)°; γ = 83.5242°] with four molecules in the unit cell (analogically to structure 2, due to the presence of two symmetrically independent conformers) (Tables S3 and Fig. S8, ESI†). Molecular packing is governed by CH⋯Se, CH⋯N and CH⋯O hydrogen bonds (C4AH⋯Se1 = 3.061 Å; C4H⋯Se1A = 3.056 Å; C14H⋯N2A = 2.694 Å; C14AH⋯N2 = 2.700 Å; C12H⋯O1A = 2.466 Å; C12AH⋯O1 = 2.464 Å; Fig. 2k) resulting in the formation of molecular layers (Fig. S9, ESI†). Neighbouring layers are connected via CH⋯F hydrogen bonds (C5AH⋯F2 = 2.414 Å; C5H⋯F2A = 2.425 Å; Fig. 2l), F⋯π (F1⋯C14A = 3.115 Å; F1A⋯C7A(π) = 2.914 Å, Fig. 2m) and Se⋯F chalcogen-type (Se1⋯F2 = 3.271 Å; Se1A⋯F2A = 3.286 Å; Fig. 2m) interactions. Similar to the structure of compound 2, some π–π stacking (m = 3.631 Å; n = 3.560 Å; Fig. 2n) appears between the heterocyclic parts of the neighbouring molecules of the same conformations.
The comprehensive analysis of all crystal structures revealed that molecules interact with each other mainly via CH⋯N, CH⋯O and CH⋯F hydrogen-bond interactions, which are accompanied by other weaker interactions such as halogen-type F⋯S and F⋯Se bonds. The presence of the cyano group in 1–3 cause the twist of the molecular structures, which weakens the π–π stacking interactions between neighbouring molecules. It can be expected that such types of intermolecular interactions collectively facilitate efficient solid-state luminescence.
2.3. Electrochemical properties
The electrochemical properties of boron difluoride complexes 1–3 were investigated by cyclic voltammetry (CV) in degassed acetonitrile with tetrabutylammonium hexafluorophosphate ([n-Bu4N]PF6) as the supporting electrolyte and ferrocene as the reference (Fig. 3 and Table S5 in the ESI†). The values of the ionization energies (IEs) of compounds 1, 2, and 3 were determined as 6.49 eV, 6.38 eV, and 6.30 eV, respectively, demonstrating that exchange of the chalcogen atom (O → S → Se) in the benzochalcogenazole unit from oxygen to sulfur and selenium results in the decrease of the IE. This observation indicates that the energy level of the highest occupied molecular orbital (HOMO) of these dyes increase in the line of the derivatives of benzoxazole → benzothiazole → benzoselenazole. In contrast, the values of the electron affinity (EA) [and the corresponding energy levels of the lowest unoccupied molecular orbitals (LUMO)] are not significantly impacted by the chalcogen atom of the azole ring. They are in the short range of 3.35–3.39 eV. Consequently, the energy gap between the HOMO and LUMO is the highest for the benzoxazole-based dye 1 (3.12 eV), and lower for benzothiazole and benzoselenazole analogues 2 (3.03 eV) and 3 (2.91 eV). | ||
| Fig. 3 Cyclic voltammograms of compounds 1–3 in acetonitrile. | ||
2.4. Quantum chemical calculations
To shed light on the electronic properties of the investigated dyes, density functional theory (DFT) computations were performed. Molecular geometries were optimized with the B3LYP/6-31G+(d) method in the Gaussian 16 program. According to quantum mechanical calculations, molecules 1–3 possess twisted conformations with the torsion angles between the phenyl substituent and heterocyclic moiety of 30.1–30.8°. Thus, the strong variations of this angle in crystal structures predominantly arise from the crystal packing effects. This also implies the low rotation barrier of the phenyl group in solution, likely responsible for observed fluorescence quenching.The DFT calculations show that the HOMO and LUMO are spread over the whole molecules with an energy gap of 4.03, 3.87, and 3.82 eV for dyes 1, 2, and 3, respectively (Fig. 4). In order to understand the nature of transitions under UV-Vis irradiation, the time-dependent DFT (TD-DFT) calculations were also performed at the same level of theory. For each molecule, the most intense absorption peak is attributed to the S0 → S1 excitation, which is described with HOMO–LUMO transition with an overall contribution of 99% and the local excited (LE) nature.
 | ||
| Fig. 4 Results of DFT calculations performed at the B3LYP/6-31G+(d) level of theory for compounds 1–3. | ||
In order to reveal the influence of the chalcogen atom on the photophysical properties of compounds 1–3, spin–orbit coupling matrix elements (SOCME) were calculated using Orca 5.0. Calculations were carried out using the PBE0 method and the DEF2-TZVP basis set. The calculation results clearly demonstrate that the SOC increases between excited triplet states and first singlet states in the line of benzoxazole → benzothiazole → benzoselenazole derivatives (Table S7, ESI†) indicating a small participation of triplet states in the photoluminescence nature of selenium-containing compound 3.
2.5. Photophysical properties
The toluene solutions demonstrate broad photoluminescence spectra with negligible photoluminescence quantum yields (PLQY) below 0.1%. The emission maxima of the solutions of compounds 1, 2, and 3 are located at 465 nm, 463 nm, and 471 nm, respectively. Additionally, to test the influence of the presence of oxygen on the emission efficiency, we performed photoluminescence measurements of the solutions in toluene after argon bubbling. The solutions of dyes 1–3 demonstrate a slightly increased emission intensity after deoxygenation (Fig. S17, ESI†); however, due to the negligible PLQY of these dyes, this impact is not essential. Therefore, the excited molecules deactivate predominantly by S1 → S0 internal conversion, with minimal involvement of intersystem crossing.
To examine the ability of aggregation-induced emission of the investigated dyes, we performed photoluminescence measurements of the dispersions of compounds 1–3 (C = 5.0 × 10−6 M) in a tetrahydrofuran (THF)/water mixture with different ratios. Analogous to the solutions in toluene, boron difluoride complexes 1–3 demonstrate negligible emission in THF (Fig. S18 and Table S9, ESI†) and THF/water mixtures up to an 85% of water percentage (fw). However, for all dyes, the emission intensity increases a little for the dispersions in THF/water mixtures with a fraction of water of 90%. In sharp contrast, the mixtures with a water content above 95% demonstrate a very strong increase of the emission intensity (Fig. S19, ESI†). Due to the low solubility of compounds 1–3 in water, they form aggregates in highly aqueous solutions. Therefore, the observed photoluminescence behavior clearly indicates the AIE properties of the investigated dyes.
In the next step, we studied the photoluminescence properties of crystalline powders of the dyes. In sharp contrast to their solutions, compounds 1–3 demonstrate strong photoluminescence in the solid state. The maxima of the SSE are located at 455 nm, 474 nm, and 478 nm for dyes 1, 2, and 3, respectively (Fig. 5 and Table 1). The solid-state emission intensity is dependent on the chalcogen type: benzoxazole-based compound 1 exhibits a very high PLQY value of 85%, while the solid samples of benzothiazole- and benzselenazole-containing analogues are less emissive with PLQYs of 33% and 21%, respectively. The higher PLQY accompanied by a hypsochromic shift of the crystals of compound 1 can be caused by relatively strong intermolecular CH⋯O hydrogen bonds (Fig. 2c). A similar O-effect was previously observed for the other dyes.67 Additionally, the PLQY of the crystalline samples of dyes 2 and 3 can be reduced by the presence of some π⋯π interactions in these crystals (Fig. 2i and n), which are not observed in the structure of dye 1. The average lifetimes of the excited state of these samples are in the short nanosecond or subnanosecond range (0.80–2.27 ns) (Fig. S20 in the ESI†), confirming the fluorescence nature of the solids. The high PLQYs and the short lifetimes result in high values of the rate constants for radiative (kr) deactivation of 3.47 × 108 s−1, 2.22 × 108 s−1, and 2.65 × 108 s−1 for solids 1, 2, and 3, respectively, indicating their perspective application in solid-state lasers.
 | ||
| Fig. 5 Solid-state photoluminescence spectra of benzochalcogenazole-containing boron difluoride complexes 1–3. | ||
High photoluminescence efficiency of the investigated dyes in the solid state can be attributed to the restricted molecular rotations and vibrations arising from the rigid crystal environment. In addition, the SSE can be enhanced by specific intermolecular interactions. Noticeably, molecules of dyes 1–3 are mostly involved in numerous hydrogen bond interactions, while π–π-stacking interactions are rather avoided. Such a molecular arrangement is particularly advantageous for an efficient photoluminescence process.
:97. The obtained films demonstrate the absorption spectra similar to those of the corresponding toluene solutions. The absorption peak is bathochromically shifted in the line of benzoxazole → benzothiazole → benzselenazole derivatives. The absorption maxima of samples of PPMA doped with dyes 1, 2, and 3 are located at 345 nm, 365 nm, and 372 nm, respectively (Fig. 6 and Table 2). These blend films also demonstrate the broad emission bands maximized at 470–472 nm. The values of the full width at half maximum (FWHM) of the investigated films are larger than 100 nm: 144 nm for the blend with benzoxazole compound 1, 131 nm for the blend with benzothiazole derivative 2, and 124 nm for the blend with benzoselenazole analogue 3. The film of the blend with benzoxazole derivative 1 shows a PLQY of 28%. The decrease of the PLQY value of the film of the dye-doped PMMA relative to that of the corresponding crystalline sample should be caused by the absence of specific intermolecular interactions (CH⋯O hydrogen bonds), which are present in the crystalline state. The corresponding value of the film of PMMA molecularly doped with benzothiazole derivative 2 is even higher (32%). Surprisingly, the PLQY of the film of the PMMA blend with benzoselenazole analogue 3 (28%) is comparable with those of the films of the analogical benzoxazole and benzothiazole compounds 1 and 2.
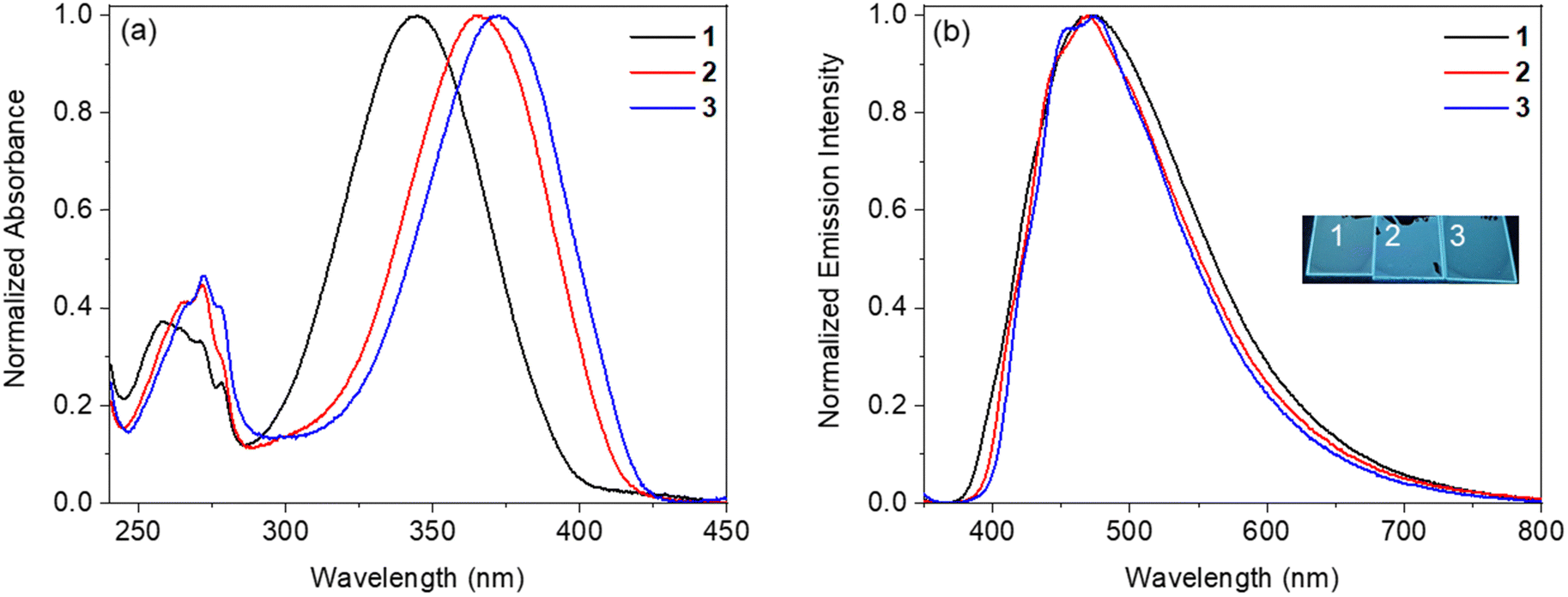 | ||
| Fig. 6 Absorption (a) and emission (λex = 337 nm) (b) spectra of the films of dyes 1–3 doped PMMA. | ||
| Dye | λabsa (nm) | λPLb (nm) | FWHMPLc (nm) | PLQYd (%) | τFle (ns) | τPhf (ms) | λASEg (nm) | FWHMASEh (nm) | Eth-ASEi (μJ cm−2) |
|---|---|---|---|---|---|---|---|---|---|
| a Wavelength of the absorption maximum.b Wavelength of the photoluminescence maximum.c Full width at half maximum of the photoluminescence spectrum.d Photoluminescence quantum yield.e Average excited-state lifetime (nanosecond component).f Average excited-state lifetime (millisecond component).g Wavelength of the maximum of the ASE.h Full width at half maximum of the ASE spectrum.i ASE threshold. | |||||||||
| 1 | 345 | 476 | 144 | 27 | 1.99 | — | 524 | 15 | 20.4 |
| 2 | 365 | 470 | 131 | 32 | 1.89 | — | 506 | 11 | 18.7 |
| 3 | 372 | 475 | 124 | 28 | 1.46 | 0.4 | 484 | 8 | 40.3 |
This observation clearly indicates that the selenium atom in the case of compound 3 does not significantly suppress the radiative deactivation. To verify the possibility of phosphorescence of the samples, we recorded the photoluminescence spectra of the films of the dye-doped PMMA in a vacuum without and with a 50 μs delay. All three films demonstrated a dramatic decrease in the emission intensity after a 50 μs delay (Fig. S22 and S23, ESI†). Only the film of the blend of PMMA with benzoselenazole-containing dye 3 demonstrates some emission recorded with a 50 μs delay at 77K, indicating a very weak phosphorescence, which is consistent with SOCME calculations (Table S7, ESI†).
Similar to the samples of the crystalline powders, the films of the dye-doped polymer demonstrate very short lifetimes of the excited states (1.46–1.99 ns, Table 2), which are generally similar when recorded at the different wavelengths of analysis (Fig. S22a, b and S23a, ESI†). The films of dyes 1 and 2 doped PMMA do not exhibit microsecond- and millisecond-scale lifetimes (Fig. S22c–f, ESI†). In turn, the film of the blend of compound 3 and PMMA demonstrates a low-intensity sub-millisecond lifetime of 0.4 ms (Fig. S23b, ESI†), confirming weak phosphorescence. Additionally, we investigated the photophysical properties of the films of the corresponding dye-doped PMMA with lower concentrations of the dyes of 1 wt% and 2 wt%. The decrease of the concentration of the dyes does not significantly change the photoluminescence properties of the samples (Fig. S24, S25 and Table S9, ESI†).
 | ||
| Fig. 7 Evolution of the peak wavelength of the recorded spectra versus pump absorbed energy for compounds 1, 2 and 3. | ||
With the presence of more triplets for compound 3, a higher level of losses is also expected (triplet–triplet absorption): we indeed observe a two time higher ASE threshold for benzoselenazole analogue 3 with 40.3 μJ cm−2 versus 20.4 μJ cm−2 for benzoxazole-containing dye 1 and 18.7 μJ cm−2 for benzothiazole derivative 2 (Fig. 8).
 | ||
| Fig. 8 Normalized spectra of the amplified spontaneous emission of dyes 1 (a), 2 (c), and 3 (e) in PMMA. FWHM values as a function of the absorbed energy density for dyes 1 (b), 2 (d), and 3 (f). | ||
However, excited-state absorption from the first excited singlet state (S1) level cannot be ruled out as the wavelengths of the emission maxima of dye 3 are hipsochromically shifted even at lower excitation energies (<500 μJ cm−2) where the triplet population is expected to be very small.
2.6. Organic solid-state lasers
Finally, we fabricated OSSLs based on dye-doped PMMA films (see the ESI,† for details). The vertical-cavity surface-emitting lasers were realized by sandwiching the active films between two Bragg reflectors so that only one cavity mode persists in the mirror stopband. Here, the laser spectra of the investigated dyes demonstrate single mode emissions at 514 nm, 513 nm, and 476 nm for dyes 1, 2, and 3, respectively (Fig. 9). The corresponding thresholds depend on the material but, unlike ASE measurements, also on the cavities that can have slightly different thicknesses and incorporate various levels of uncontrolled losses due to the fabrication process. The measured lasing threshold values are 46.1 μJ cm−2, 28.0 μJ cm−2, and 58.6 μJ cm−2 for dyes 1, 2, and 3, respectively (Fig. S30, ESI†), which are consistent with the measured ASE thresholds. | ||
| Fig. 9 Spectra of organic solid-state lasers based on dyes 1–3. | ||
3. Conclusions
New benzochalcogenazole-based boron difluoride chromophores 1–3 for solid-state laser applications were discovered. The influence of the chalcogen atom of the benzochalcogenazole core on the photophysical properties of the dyes was investigated. Despite the negligible fluorescence of the solutions, these dyes demonstrate strong aggregation-induced emission behavior and the intense solid-state fluorescence with a photoluminescence quantum yield (PLQY) of up to 85%. The thin films of poly(methylmethacrylate) doped with the synthesized dyes show emission peaking at ca. 470 nm with a PLQY of ca. 30%, indicating that the chalcogen atom does not significantly affect the emission of the compounds molecularly dispersed in the polymeric matrix. The observed PLQY quenching of the solutions presumably results from the rotation of the phenyl group, while the strong enhancement of the solid-state emission for complex 1 can be ascribed to the highly advantageous intermolecular interactions, such as C–H⋯O hydrogen bonding. The investigated boron difluoride complexes molecularly dispersed in the polymer matrix exhibit amplified spontaneous emission with the threshold depending on the type of the chalcogen atom. This work expands the scope of organic laser dyes. The results of this work demonstrate that incorporation of heavy atoms (sulfur and selenium) can be successfully explored in the design of organic laser dyes. Moreover, such incorporation can provide the decrease of ASE and lasing thresholds. This is especially important due to the ability of heavy atoms to increase the spin–orbit coupling between the excited singlet and triplet states, and, as a result, to enhance thermally activated delayed fluorescence/room-temperature phosphorescence of the chromophores.Author contributions
M. A. P. designed the dyes. R. P. and M. A. P. synthesized the materials. R. L. collected the crystallographic data. A. H. performed the theoretical calculations and analysis. S. K. conducted the electrochemical investigation. R. P., S. K., P. H. M.-U. and D. V. performed absorption and photoluminescence studies. E. J.-W., G. M., S. C. and S. F. performed ASE measurements and fabricated OSSLs. A. H., S. F. and M. A. P. wrote the original draft. S. C., J. V. G. and K. D. revised the draft. M. A. P., S. F., D. V. and J. V. G. initiated, coordinated and supervised this research and received funding acquisition. All the authors contributed with discussions, reviewing, and editing and have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of its ESI.† Crystallographic data for dyes 1–3 have been deposited at the CCDC under 2405238, 2405617, and 2405841.† Additionally, all relevant experimental data can be accessed upon request by contacting the corresponding author.Acknowledgements
The authors are grateful for the “Long-term program of support to the Ukrainian research teams at the Polish Academy of Sciences carried out in collaboration with the U.S. National Academy of Sciences with the financial support of external partners” (PAN.BFB.S.BWZ.330.022.2023) for the financial support. S. F. acknowledges the ANR – FRANCE (French National Research Agency) for its financial support through the ANR-23-CE06-0027 project. M. A. P. is also thankful to the University Sorbonne Paris Nord for a visiting academic fellowship. D. V. has received funding from the Research Council of Lithuania (LMTLT) (projects SeSeoS). J. V. G. acknowledges funding from the EU Research and Innovation Funding Programme “Horizon Europe” (HiSOPE project no. 101161573). We gratefully acknowledge the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2023/016879. We are also sincerely grateful to Dr. Katarzyna Rybicka-Jasińska, Dr. Marek Szymański, Dr. Yevgen Poronik, Prof. Agnieszka Szumna, and Prof. Daniel T. Gryko (Institute of Organic Chemistry PAS) for permission to use their instruments.References
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS.
- J. Gierschner, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2016, 4, 348–364 CrossRef CAS.
- F. Matino, L. Persano, A. Camposeo and D. Pisignano, Adv. Opt. Mater., 2019, 7, 1900192 CrossRef.
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC.
- X. Tang, C. A. M. Senevirathne, T. Matsushima, A. S. D. Sandanayaka and C. Adachi, Adv. Mater., 2024, 36, 2211873 CrossRef CAS PubMed.
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, S. Terakawa, W. J. Potscavage, Jr., C. Qin, T. Fujihara, K. Goushi, J.-C. Ribierre and C. Adachi, Appl. Phys. Express, 2019, 12, 061010 CrossRef CAS.
- C. Adachi and A. S. D. Sandanayaka, CCS Chem., 2020, 2, 1203–1216 CrossRef CAS.
- K. Yoshida, J. Gong, A. L. Kanibolotsky, P. J. Skabara, G. A. Turnbull and I. D. W. Samuel, Nature, 2023, 621, 746–752 CrossRef CAS PubMed.
- S. K. Behera, S. Y. Park and J. Gierschner, Angew. Chem., Int. Ed., 2021, 60, 22624–22638 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- A. Huber, J. Dubbert, T. D. Scherz and J. Voskuhl, Chem. – Eur. J., 2023, 29, e202202481 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2019, 58, 8632–8639 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- F. Wurthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef.
- V. Bonal, R. Muñoz-Mármol, F. G. Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 3327 CrossRef PubMed.
- Y. Zou, V. Bonal, S. M. Quintero, P. G. Boj, J. M. Villalvilla, J. A. Quintana, G. Li, S. Wu, Q. Jiang, Y. Ni, J. Casado, M. A. Díaz-García and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 14927–14934 CrossRef CAS PubMed.
- Y. Oyama, M. Mamada, A. Kondo and C. Adachi, J. Mater. Chem. C, 2021, 9, 4112–4118 RSC.
- Y. Gu, V. Vega-Mayoral, S. Garcia-Orrit, D. Schollmeyer, A. Narita, J. Cabanillas-González, Z. Qiu and K. Müllen, Angew. Chem., Int. Ed., 2022, 61, e202201088 CrossRef CAS PubMed.
- P. J. Welscher, D. Straub, F. Stümpges, A. L. Respondek, B. Esser and A. J. C. Kuehne, Adv. Funct. Mater., 2025, 35, 2417129 CrossRef CAS.
- M. Mamada, H. Nakanotani and C. Adachi, Mater. Adv., 2021, 2, 3906–3914 RSC.
- P. A. Praveen, T. Kanagasekaran, C. Ma, M. Terada, T. Jin, Y. Wakabayashi and H. Shimotani, J. Mater. Chem. C, 2024, 12, 15995–16003 RSC.
- A. S. D. Sandanayaka, K. Yoshida, M. Inoue, C. Qin, K. Goushi, J.-C. Ribierre, T. Matsushima and C. Adachi, Adv. Opt. Mater., 2016, 4, 834–839 CrossRef CAS.
- M. Mamada, T. Fukunaga, F. Bencheikh, A. S. D. Sandanayaka and C. Adachi, Adv. Funct. Mater., 2018, 28, 1802130 CrossRef.
- B. S. B. Karunathilaka, U. Balijapalli, C. A. M. Senevirathne, Y. Esaki, K. Goushi, T. Matsushima, A. S. D. Sandanayaka and C. Adachi, Adv. Funct. Mater., 2020, 30, 2001078 CrossRef CAS.
- B. Donoso, V. Bonal, I. Torres-Moya, P. G. Boj, J. A. Quintana, J. M. Villalvilla, J. Herrera, P. Prieto and M. A. Díaz-García, J. Mater. Chem. C, 2022, 10, 16004–16015 RSC.
- R. Martín, P. Prieto, J. R. Carrillo, A. M. Rodríguez, A. de Cozar, P. G. Boj, M. A. Díaz-García and M. G. Ramírez, J. Mater. Chem. C, 2019, 7, 9996–10007 RSC.
- A. Shukla, S. K. M. McGregor, R. Wawrzinek, S. Saggar, E. G. Moore, S.-C. Lo and E. B. Namdas, Adv. Funct. Mater., 2021, 31, 2009817 CrossRef CAS.
- N. R. Wallwork, A. Shukla, R. B. Roseli, I. Allison, S. K. M. McGregor, M. Coles, I. Gale, V. P. Rahane, V. Entoma, E. G. Moore, E. H. Krenske, E. B. Namdas and S.-C. Lo, Small, 2024, 20, 2406817 CrossRef CAS.
- A. Shukla, N. R. Wallwork, X. Li, J. Sobus, V. T. N. Mai, S. K. M. McGregor, K. Chen, R. J. Lepage, E. H. Krenske, E. G. Moore, E. B. Namdas and S.-C. Lo, Adv. Opt. Mater., 2020, 8, 1901350 CrossRef CAS.
- X. Tang, U. Balijapalli, D. Okada, B. S. B. Karunathilaka, C. A. M. Senevirathne, Y.-T. Lee, Z. Feng, A. S. D. Sandanayaka, T. Matsushima and C. Adachi, Adv. Funct. Mater., 2021, 31, 2104529 CrossRef CAS.
- A. H. Ashoka, P. Ashokkumar, Y. P. Kovtun and A. S. Klymchenko, J. Phys. Chem. Lett., 2019, 10, 2414–2421 CrossRef CAS PubMed.
- S. Bou, A. S. Klymchenko and M. Collot, Mater. Adv., 2021, 2, 3213–3233 RSC.
- M. Won, S. Koo, H. Li, J. L. Sessler, J. Y. Lee, A. Sharma and J. S. Kim, Angew. Chem., Int. Ed., 2021, 60, 3196–3204 CrossRef CAS.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Métivier, C. Allain and I. Ciofini, Adv. Mater., 2018, 30, 1800817 CrossRef.
- M. A. Potopnyk, J. Mech-Piskorz, G. Angulo, M. Ceborska, R. Luboradzki, E. Andresen, A. Gajek, A. Wisniewska and U. Resch-Genger, Chem. – Eur. J., 2024, 30, e202400004 CrossRef CAS PubMed.
- D.-H. Kim, A. D’Aleo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Bredas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- H. Ye, D. H. Kim, X. Chen, A. S. D. Sandanayaka, J. U. Kim, E. Zaborova, G. Canard, Y. Tsuchiya, E. Y. Choi, J. W. Wu, F. Fages, J.-L. Bredas, A. D’Aleo, J.-C. Ribierre and C. Adachi, Chem. Mater., 2018, 30, 6702–6710 CrossRef CAS.
- P. Xue, X. Wang, W. Wang, J. Zhang, Z. Wang, J. Jin, C. Zheng, P. Li, G. Xie and R. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 47826–47834 CrossRef CAS.
- L. Zhou, F. Ni, N. Li, K. Wang, G. Xie and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202203844 CrossRef CAS.
- S. Kutsiy, D. Volyniuk, S. R. Sahoo, M. Ceborska, A. Wisniewska, P. Stakhira, J. V. Grazulevicius, G. V. Baryshnikov and M. A. Potopnyk, ACS Appl. Mater. Interfaces, 2024, 16, 60633–60647 CrossRef CAS.
- B. Sui, M. V. Bondar, D. Anderson, H. J. Rivera-Jacquez, A. E. Masunov and K. D. Belfield, J. Phys. Chem. C, 2016, 120, 14317–14329 CrossRef CAS.
- Y. Kage, S. Kang, S. Mori, M. Mamada, C. Adachi, D. Kim, H. Furuta and S. Shimizu, Chem. – Eur. J., 2021, 27, 5259–5267 CrossRef CAS.
- Y. Wang, L. Xie, Q. Suna and L. Liu, New J. Chem., 2023, 47, 12287–12295 RSC.
- Y. Wang, Q. Sun, L. Xie, L. Chen, F. Zhu and L. Liu, ACS Omega, 2023, 8, 28376–28386 CrossRef CAS.
- L. Cui, A. Horioka, R. Ishimatsu, M. Mamada, C. Adachi, K. Tahara, Y. Hoshino and T. Ono, Adv. Opt. Mater., 2024, 12, 2302803 CrossRef CAS.
- A. D’Aléo, M. H. Sazzad, D. H. Kim, E. Y. Choi, J. W. Wu, G. Canard, F. Fages, J.-C. Ribierre and C. Adachi, Chem. Commun., 2017, 53, 7003–7006 RSC.
- R. Aoki, R. Komatsu, K. Goushi, M. Mamada, Y. Ko, J. W. Wu, V. Placide, A. D’Aléo and C. Adachi, Adv. Opt. Mater., 2021, 9, 2001947 CrossRef CAS.
- H. Huang, Z. Yu, D. Zhou, S. Li, L. Fu, Y. Wu, C. Gu, Q. Liao and H. Fu, ACS Photonics, 2019, 6, 3208–3214 CrossRef CAS.
- Z. Lv, Z. Man, Z. Xu and H. Fu, Adv. Opt. Mater., 2024, 12, 2301246 CrossRef CAS.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS.
- S. Li, Z. Yu, X. Xiao, H. Geng, K. Wang, X. Jin, Q. Liao, Y. Liao, Y. Wu, J. Yao and H. Fu, Laser Photonics Rev., 2019, 13, 1900036 CrossRef.
- S. Li, X. Jin, Z. Yu, X. Xiao, H. Geng, Q. Liao, Y. Liao, Y. Wu, W. Hub and H. Fu, J. Mater. Chem. C, 2021, 9, 7400–7406 RSC.
- M. A. Potopnyk, D. Volyniuk, M. Ceborska, P. Cmoch, I. Hladka, Y. Danyliv and J. V. Gražulevičius, J. Org. Chem., 2018, 83, 12129–12142 CrossRef CAS.
- M. A. Potopnyk, D. Volyniuk, R. Luboradzki, M. Ceborska, I. Hladka, Y. Danyliv and J. V. Gražulevičius, J. Org. Chem., 2019, 84, 5614–5626 CrossRef CAS PubMed.
- M. A. Potopnyk, D. Volyniuk, R. Luboradzki, A. Lazauskas and J. V. Grazulevicius, Eur. J. Org. Chem., 2021, 2772–2781 CrossRef CAS.
- D. R. Lee, K. H. Lee, W. Shao, C. L. Kim, J. Kim and J. Y. Lee, Chem. Mater., 2020, 32, 2583–2592 CrossRef CAS.
- S. Goto, Y. Nitta, N. O. Decarli, L. E. de Sousa, P. Stachelek, N. Tohnai, S. Minakata, P. de Silva, P. Data and Y. Takeda, J. Mater. Chem. C, 2021, 9, 13942–13953 RSC.
- J. W. Campbell, M. T. Tung, K. N. Robertson, A. A. Beharry and A. Thompson, J. Org. Chem., 2023, 88, 10655–10661 CrossRef CAS PubMed.
- T. Amelia, J. P. D. van Veldhoven, M. Falsini, R. Liu, L. H. Heitman, G. J. P. van Westen, E. Segala, G. Verdon, R. K. Y. Cheng, R. M. Cooke, D. van der Es and A. P. Ijzerman, J. Med. Chem., 2021, 64, 3827–3842 CrossRef CAS PubMed.
- D. Göbel, P. Rusch, D. Duvinage, N. C. Bigall and B. J. Nachtsheim, Chem. Commun., 2020, 56, 15430–15433 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- K. Durka, P. H. Marek-Urban, K. Nowicki, J. Drapała, K. N. Jarzembska, P. Łaski, A. Grzelak, M. Dąbrowski, K. Woźniak and S. Luliński, Chem. – Eur. J., 2022, 28, e202104492 CrossRef CAS.
- S. J. Stricker and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef.
- S. Forget and S. Chénais, Organic Solid-State Lasers, Springer-Verlag, Berlin, Germany, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2405238, 2405617 and 2405841. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc02236c |
| This journal is © The Royal Society of Chemistry 2025 |