
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the circularly polarized luminescence of covalent organic frameworks (COFs) through ab initio modelling†
Giovanni
Bella
 *,
Giuseppe
Bruno
,
Francesco
Nicolò
and
Antonio
Santoro
*
*,
Giuseppe
Bruno
,
Francesco
Nicolò
and
Antonio
Santoro
*
Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Viale F. Stagno d’Alcontres 31, 98166 Messina, Italy. E-mail: gbella@unime.it
First published on 1st July 2025
Abstract
Chirality is a radical property largely observed in the universe, arising in objects lacking inversion symmetry, thereby making the objects exist as entities with distinct handedness. After proper interference with asymmetric radiation, such forms can potentially emit chiral light with an excess of right- or left-handed circular polarization, reporting events of circularly polarized luminescence. Recent developments in reticulated nanomaterials, such as covalent organic frameworks, have shown chiroptical responses that far exceed those found in their isolated monomers. However, these recent advances still lack a solid computational foundation for proper interpretation of chiroptical phenomena. The present contribution, through robust yet accessible ab initio theoretical treatment, aimed at precisely decoding the unique CPL spectral features that arise in COFs presenting different types of chirality (stereogenic, axial and intrinsic). A multilevel approach combining DFT benchmark, excited Born–Oppenheimer molecular dynamics and TD-DTF analyses clarified how the effects of dimensionality, reticulation and chirality transfer through bond/space can influence the CPL bands of COFs. Lastly, it was demonstrated that a small set of exchange–correlation functionals (M11L, O3LYP and ωB97xD) is valid for simulating the CPL spectra of all the chiral-emissive pure COFs reported in the literature to date. These results can play a constructive role in the future design of high-performance CPL COFs, stimulating new opportunities for research on chiral luminescent bidimensional architectures.
1. Introduction
Chirality fascinated Alice as she admired the macroscopic world through the looking glass. Her extravagant thoughts on the taste of mirror-world milk proved that the exploration into the significance of chirality is not a merely chemical virtuosity. In the real world, the interference between circularly polarized electromagnetic waves and enantiomeric molecules can activate chiroptical events such as circular dichroism (CD)1–5 and circularly polarized luminescence (CPL).6–12 In both cases, the magnitude of the electric vector of radiation remains constant, whereas its direction changes over time. The phase difference remains unchanged, causing the electric vector to spiral. This spiral can be counterclockwise (left circularly polarized light) or clockwise (right circularly polarized light) as time progresses.13–16 Focusing on chiral-emissive phenomena, two components are necessary to obtain a decent CPL signal: (1) an efficient radiative deactivation process of luminophores17–19 and (2) a valid luminescence dissymmetry factor (glum, which quantifies the asymmetry degree of emission in left- and right-circularly polarized light) promoted by the chiral environment.20–22 Considering these premises, various inorganic/organic materials have been recently explored with the goal of steering the research on chiral functional materials23–27 from small chiral molecules to 2D-chiral materials, exploiting the advent of the reticular chemistry.28–35 Metal organic frameworks (MOFs) and covalent organic frameworks (COFs) undoubtedly represent the maximum expression of such a chemical approach, containing homogeneous pores and discrete cavities in which chirality features can be imparted.36–41 Although there are more studies in the literature on MOFs than on COFs, the complex combination of several MOF key factors (such as the well-defined coordination preferences of metal ions as well as the different topicities and denticities of ligands) is gradually shifting these preferences, favouring the advancement of COFs. Basically, COFs are realized through the covalent integration of organic monomers into two-dimensional networks, reaching highly ordered pore architectures, facile functionalization, superior chemical stability and mechanical processability.42–46 For this class of compounds, the asymmetric induction is the crowning of a judicious selection of organic building blocks incorporating chiral properties (stereogenic, axial or intrinsic chirality) that can be transferred, preserved and amplified into the bidimensional reticular framework.47–51 To guarantee an appropriate CPL profile, a strong balance between a favourable enantiopurity and a controllable luminogenic activity must be accomplished. To achieve this goal, a well-established synthetic strategy uses a C3-symmetric aromatic core with three electrophilic sites (typically aldehyde) that can covalently react with chiral bis-nucleophilic pendants (usually diamines). These imine/hydrazone linkages form an initial three-bladed propeller scaffold, which then serves as a starter for a honeycomb ramification pattern, as shown in Fig. 1. Alternatively, if the two building blocks are achiral, a third chiral component can be a reasonable solution. Until now, 8 examples of CPL-active COFs have been reported in the literature, which can be split in two subgroups: | ||
| Fig. 1 Illustration of the chiral COF construction strategy. Technically, it is also possible to invert the role of the building blocks (triamines and di-aldehydes). | ||
(1) Pure COFs.52–56
(2) Hybrid COFs.
In the latter, the extended chemical buildings were supramolecularly doped with organic dyes to enhance the luminescence process.52,55,57 Notwithstanding the rising interest for the CPL in silico simulations,58–64 in this specific landscape, a theoretical modelling of CPL spectra emerging from 2D chiral COFs is still missing in the literature. With the present contribution, we propose a computational approach to thoroughly investigate the complex relationship between dimensionality and chiroptical properties in reticulated enantiomerically resolved species. Specifically, our modelling workflow involved three preliminary steps:
(1) A mini benchmark for distinguishing the more suitable DFT functional to optimize the COFs hexagonal periodic units in the ground state (S0).
(2) The time-dependent density functional formalism (TD-DFT) integrated in the Born–Oppenheimer molecular dynamic (BOMD), adopted to explore the meshed structures in its excited state (S1), by taking heed of the diverse conformations that COFs of variable size can assume.
(3) An extensive TD-DFT analyses to scrutinize how exchange–correlation functionals perform when different types of COFs are represented by chiral skeletons, which reflect their peculiarities in CPL spectra.
2. Results and discussion
We decided to limit our research to the 5 cases of pure chiral COFs present in the literature (considering that the three existing examples of hybrid chiral COFs can be seen as an evolution of pure COFs in which a secondary photochemical path, such as the Förster resonance energy transfer, associates a dye donor with a COF acceptor to achieve a higher photoluminescence). Before immediately acceding to the chiroptical trends of pure chiral COFs, the strategy behind our workflow implicated a computational screening approach to capture the structure–property relationships based on a well-defined geometrical parameter. In light of the complexity of conformational effects regarding meshed structures like chiral COFs, it clearly appears that the chiroptical signatures are intimately dependent on the location of the extended 2D architecture within the hypersurface of potential energy. Commonly, in DFT theory, the quality of energy-minimized conformations is significantly affected by the selection of exchange–correlation functionals. These are typically evaluated by focusing on the benchmarks aimed at accurately reproducing the atomic positions expressed by single-crystal X-ray diffraction (SC-XRD) data collection.65,66 Although COFs are structurally crystalline while the relative polymers are largely amorphous in nature, no carbon skeleton of the 5 chiral-emissive COFs have been characterized through SC-XRD. Keeping in mind the wealth of possible COFs structural parameters, we preferred to choose the pore diameter as the geometrical descriptor to identify an adequate level of theory. This kind of choice provides a more global description of the hexagonal pattern than local measurements like bond lengths and angles. Additionally, from an experimental point of view, the pore sizes are often divulged as they serve as an important dimensional indicator. To appropriately imitate this pore size, we decided to build our DFT set with the consideration that the invoked functionals: (1) were already profitably utilized to optimize COFs;67–71 (2) are advantageous for the energy-minimization of carbon-based systems.72–75 Accordingly, B3LYP, M062X, CAM-B3LYP, PBEPBE and ωB97xD exchange–correlation functionals were interrogated in the gas phase to compare the cavity diameter between the DFT-minimized systems and the reported pore sizes. Hence, the Pople basis set was coupled with the aforementioned functionals, as it is proven to be quite advisable in modelling the second-period elements.76–78 In this part, we intentionally omit the chiral characteristics (which will be thoroughly discussed in the next sections), and we summarized the five COFs in Fig. 2, specifying their relative building blocks. Contemplating the very high number of atoms in COFs, we opted to use the single hexagonal frame as a “computational probe” to analyse the pore dimensions between the theoretical and experimental species. In detail, the systems were built via a two-fold condensation between the related partners. The chemical buildings were thus truncated by leaving an unreacted electrophile site for each component (right part, Fig. 2). It is worth noting that COF2 was not treated in this benchmark because there is no theoretical or experimental information about the cavity diameter available in the literature.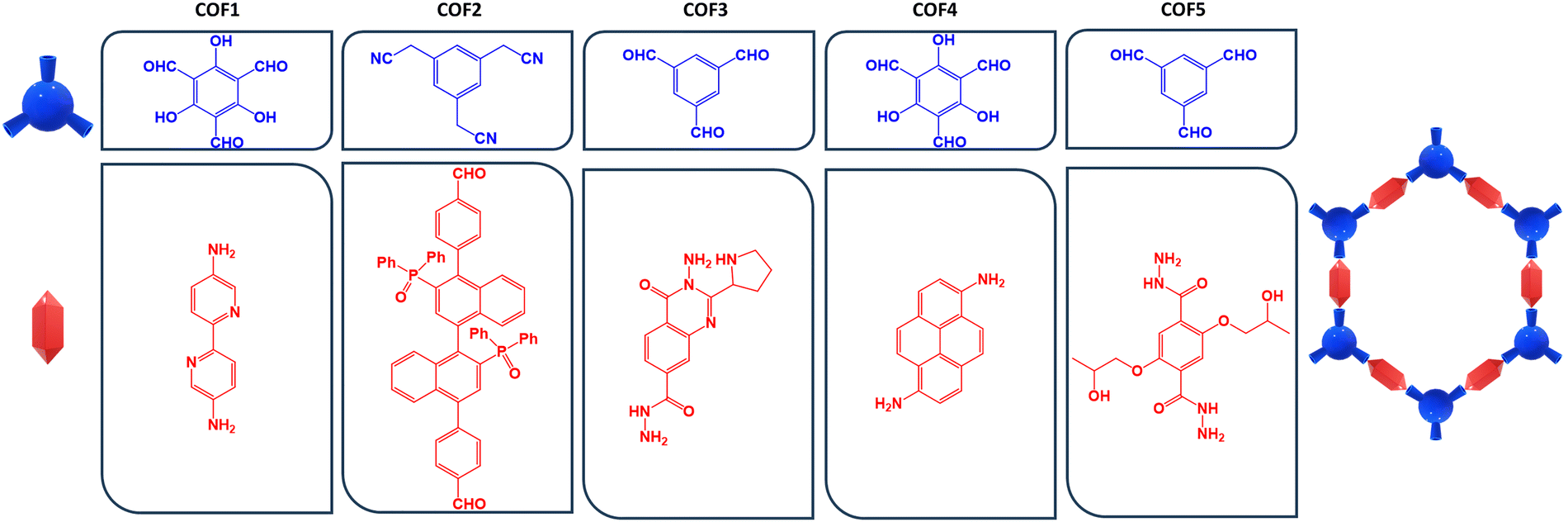 | ||
| Fig. 2 (Left) COFs schematization with the corresponding monomers. (Right) Hexagonal periodic unit. | ||
The collected geometrical data presented in Fig. 3 (left part) shows the similar functionals performances for each COF, and no exaggerated fluctuation was recorded. Condensing the outcomes of Fig. 3 and Table 1, it appears that the dispersion embedded ωB97xD functional displayed more accurate pore diameter values for all of the COFs. The superiority of this functional can be attributed to its tendency of competently cloning highly-conjugated molecular systems and non-covalent interactions, owing to the long-range corrections.79–81 Optimized COFs in Fig. 3 (right part) evidence that they share almost the same electrophilic building block (blue miniatures, Fig. 2), and the pore spacing is directly influenced by the flexibility and length of the nucleophilic partner (red miniatures, Fig. 2).
 | ||
| Fig. 3 (Left) Cumulative pore diameter delta histograms (nm) at the functional/6-311G(d,p) level in the gas phase for the studied COFs. (Right) Optimized structures at the ωB97XD/6-311G(d,p) level. | ||
2.1. Case 1: COF1
Once the leader functional for the COFs optimization in the ground state was established as ωB97xD, we focussed our attention on COF1 which was tested as a computational reference to inspect the dimensionality–chiropticity relationship. As excellently described by Chen et al.,52 a bottom-up approach combining 1,3,5-triformyl phloroglucinol (Tp) with 2,2′-bipyridine-5,5′-diamine (Bpy) (Fig. 2) was shown to guarantee an anisotropic growth of the chemical nanosheet. In this situation, a third chiral monodentate component ((R)- or (S)-1-(1-naphthyl)ethylamine, R/S-NEA) was mixed to achieve a double benefit: (1) introducing a chiral inducer to extend the chirality effect along with the planar directions; (2) preventing the fluorescence quenching caused by π–π stacking.84 Although this category of COFs (also including COF4) is intended as “2D imine-linked”, tautomerism drives the reaction toward the β-ketoenamine form instead of the enol–imine one85 (Fig. S1, ESI†). Despite experimental IR absorbance bands and 13C NMR resonances certifying the presence of the chiral NEA molecules,52 their exact distribution along the COF chemical building still remains unknown. Following this background, and conscious that the three building blocks were (approximately) equimolarly blended, we hypothesized a starting hexagonal prototype model in which the peripheral aldehyde sites were saturated with the chiral NEA (Fig. 4 and Fig. S2, ESI†). Interestingly, although a conjugated frame could be used to predict a planar skeleton, the related DFT-optimization showed a distorted saddle-shape structure (Fig. S3, ESI†). We retain that this peculiar arrangement can be due to the limited dimensionality of the hexagonal aperture, which can fluctuate its six edges to accomplish a global conformational preference (mainly governed by intramolecular hydrogen bonding, Fig. S4, ESI†). With the intention of understanding how the scale-effect can impact the geometrical package, we proposed three supplementary models (two 1D and a 2D). Surprisingly, the COF extension along an arbitrary direction (COF1[2] and COF1[3]) led to a helical topology, which still reflects a relevant conformational freedom (Fig. 4 and Fig. S5, S6, ESI†). When a transition from a mono-directional (1D) axis to a bi-directional (2D) plane occurs, the drastic change impacts the atomic COF disposition. A de facto COF1[7] exhibited a planar and rigid orientation, consequently resulting in a restriction of torsional-flexible movements (Fig. 4 and Fig. S7, ESI†).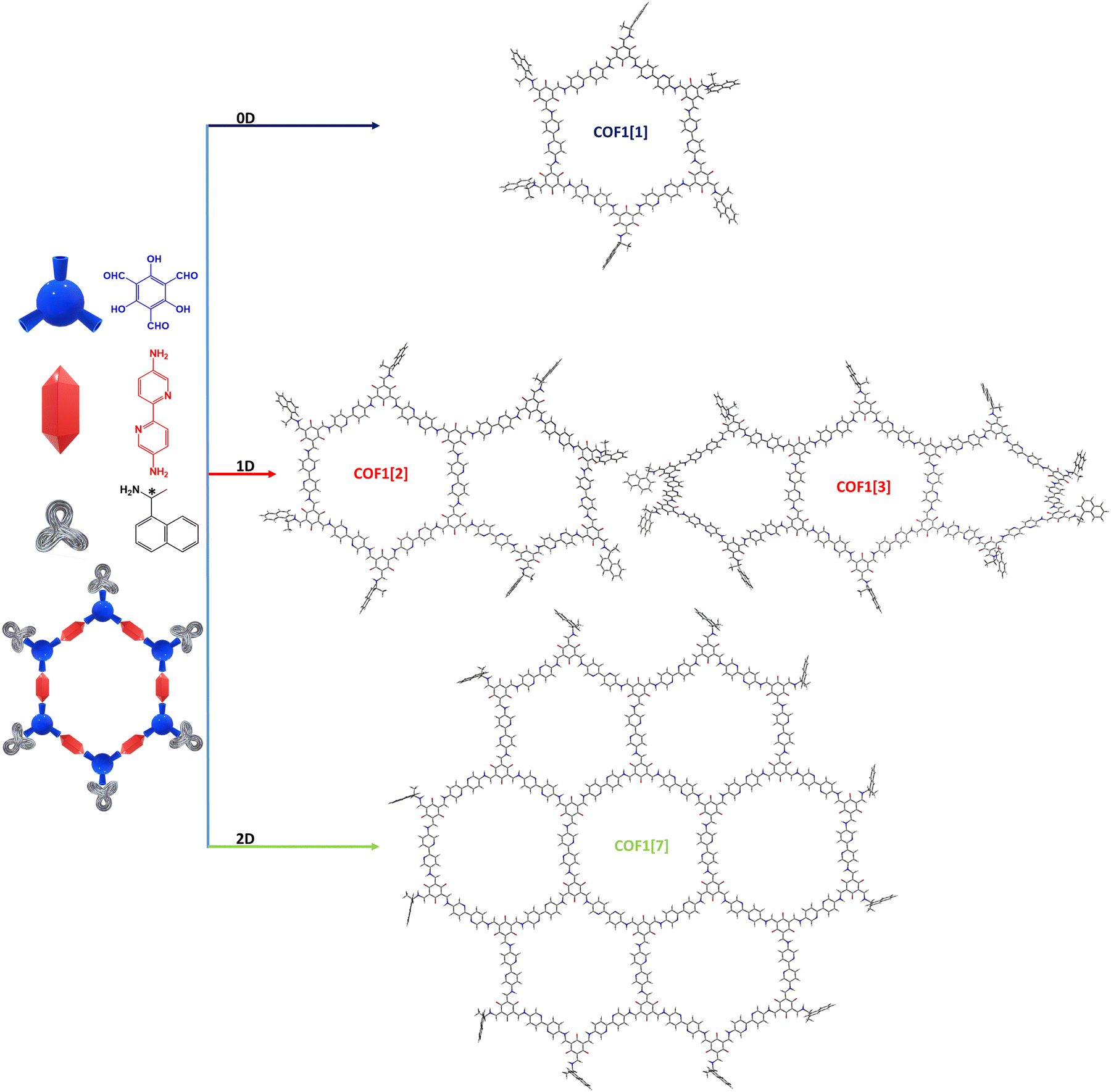 | ||
| Fig. 4 Tube display format of the 0D, 1D and 2D chiral models optimized at the ωB97xD/6-311G(d,p) level in the gas phase, digits in parentheses refer to the number of hexagonal units in the COFs. For convenience, only the (R) enantiomers are depicted here. | ||
Furthermore, from an experimental perspective, the presence of mono-amino chiral groups (R/S-NEA) can randomly generate mono-condensed bipyridines. To attain such a realistic situation, multiple reticular defects were included inside COF1[7] (COF1[X], Fig. S8, ESI†). As revealed by the found DFT energy minimum, such chemical cracks unequivocally alter the π-extended system because of the incorporated irregularities, which modify the initial planar architecture through a bent configuration.
At this point, the question of how the chiral dimensionality influences the circularly polarized luminescence effect arises. By assembling the previous conformational results, it emerges that thermal fluctuations along the entire COF chemical scaffold become significant and surely affect the final spectral signatures of the CPL profile. In this sense, the enantiomeric COF models were examined in their lowest-lying excited state by associating thermal-dependent movements with light irradiation along the radiative deactivation pathway. Adhering to this, the ab initio excited molecular dynamics (Born–Oppenheimer molecular dynamic, BOMD) for COF1[1], COF1[2], COF1[3], COF1[7] and COF1[X] enabled us to execute a valid phase space conformational sampling of the first excited state (S1), allowing the assortment of an ensemble of statistically pertinent conformers. The final CPL spectra were thus defined as a weighted sum of the spectra calculated at the TD-DFT level on equi-temporally interspersed molecular configuration snapshots extracted from BOMD trajectories. The implemented time-dependent formalism in the theoretical workflow is indispensable for catching how the emission energies (band position) and the rotatory strengths (magnitude and sign of the CPL signal) vary when size-dependent effects occur. Motivated by our DFT survey, we employed the ωB97XD/6-311G(d,p) level for an exhaustive characterization of the potential energy surfaces in S1 over a picosecond scale. BOMD productions elucidated valuable information on how the pore apertures behave when the first excited state is scanned. In fact, the S1 trajectories of COF1[1], COF1[2], and COF1[3] showed a perceptible shrinkage of hexagonal distances compared to the S0 condition (Fig. S9 and S10, ESI†). A narrower deviation was detected for COF1[X], while COF1[7] supplied an almost immutable gap. This picture indicates that for S1, the COFs flexibility directly controls the structural setting, illustrating how the more conformationally-free COF1[1], COF[2] and COF[3] are readily subjected to the excited state geometrical distortion. Meanwhile, the rigid COF[7] maintains the ground state geometrical identity. COF1[X] constituted an intermediate case in which the effect of the chemical defects (which tends to augment the skeleton mobility) is partially compensated by the extended frame. Moreover, no proton transfer was observed during the S1 molecular dynamics, confirming that the ketoenamine bond is also in the first exited state. Once the structural responses of the COFs populated excited state were analysed, a noticeable list of exchange–correlation functionals at the TD-DFT level was debated to understand how the size-effect quantitatively/qualitatively determines the circularly polarized luminescence features. After a careful computational evaluation, B3LYP, PBE1PBE, N12SX, HSEH1PBE, LC-ωHPBE, ωB97xD, O3LYP, MN12L, M11L and M06L were selected. Conscious of the functional performances transferability between the COFs models and taking into account the required CPU time, we selected COF1[1] for our TD-DFT study. In view of this, Fig. 5(A) shows the variable abilities of the functionals in duplicating the CPL spectral profile in terms of the shape, sign, intensity and band position. Proceeding from the premise that the experimental CPL maximum is situated at 630 nm (Fig. 5(B)), it is possible to note that all of the functionals correctly distinguished the sign of the chiral emissions (negative for the R-COF enantiomer and positive for the S-COF one) with an evident heterogeneity in the peak position. In particular, B3LYP, PBE1PBE, N12SX, HSEH1PBE, LC-ωHPBE, ωB97xD and O3LYP manifested a notable blue-shift (∼370–500 nm) and an excessively stretched band shape. In contrast, the three Minnesota functionals (MN12L, M11L and M06L) offered much more precise peak locations, maintaining a good symmetry in the CPL spectral line. Specifically, M06L and M11L with 620 nm and 626 nm, respectively, were able to closely emulate the experimental line shape. Ultimately, in virtue of a better CPL signal intensity and peak position (Fig. 5(C)), M11L was elected as the best candidate for the chiral emission simulation of COF1[1]. Confident with the transferability of our computational plan, we applied this modus operandi (TD-DFT calculations at M11L/6-311G(d,p) level) to COF1[2], COF1[3], COF1[7] and COF1[X] with the aim of verifying if and (eventually) how the chiral dimensionality can modify the CPL spectral fingerprint (we arbitrarily chose the R enantiomer). Moving along a single axis (1D models), we found the following incremental trend for the CPL signal magnitude (Fig. 5(D)), COF1[1] < COF1[2] < COF1[3], which relies on an enlargement of the rotatory strengths (Table S1, ESI†). This sort of proportionality can be justified by looking at the monodirectional growth of the chiral groups (6, 8 and 10 R-NEA fragments for COF1[1], COF1[2] and COF1[3], respectively), which amplifies the chiral transmission on a more extended periodic material. When the bidimensional model was treated (COF1[7]), a further accretion in the signal intensity appeared. This extra contribution in the R-length values can be mutually accredited to both R-NEA (12 units) and the reticulation effect. The latter expands φE–M (accordingly with the Rosenfeld equation,86R10 = |μe10| × |μm10|cosφEM, the angle between the electric dipole transition moment and the magnetic dipole transition moment) during the S1 → S0 transition (Table S2, ESI†). Obviously, the cosine term is more negative with a greater φE–M obtuse angle, which impacts the CPL intensity. Despite COF1[X] bearing 15 R-NEA arms, its increase in relative rotatory strengths were modest compared to those of COF1[7], highlighting how the chemical interruptions in the COF can reduce φE–M, resulting in an almost superimposable CPL spectrum (Fig. 5(D)). It is worth mentioning that, in progressing from COF1[1] to other bigger models, their electronic properties (orbital order, orbital energies, HOMO–LUMO band gap, Fig. S11, ESI†), which characterize the fluorescent radiative process, are coherently maintained between the variable-size COFs skeletons. Specifically, only micro-fluctuations were recognized in the wavelength emission among the different R-COF1 systems (Fig. 5(D)). Moreover, the HOMO and LUMO orbitals are completely confined within the COF backbone with no contributions from asymmetric groups (Fig. S12, ESI†), emphasizing how NEA chiral entities primarily contribute to chiral activity rather than to photoluminescence (Tp-Bpy-based COFs are themselves luminescent52).
 | ||
| Fig. 5 (A) Assemblage of the TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the BOMD trajectories for R/S-COF1[1]. (B) Experimental CPL spectra of R/S-COF1 in solid. Adapted with permission from ref. 52 © 2022 American Chemical Society. (C) Zoomed vision of the CPL spectrum performed at the M11L/6-311G(d,p) level along the BOMD trajectories for R/S-COF1[1]. (D) Overview of the TD-DFT calculated CPL spectra at the M11L/6-311G(d,p) level along the BOMD trajectories for R-COF1[1], R-COF1[2], R-COF1[3], R-COF1[7] and R-COF1[X]. | ||
2.2. Case 2: COF2
Motivated by the previous outcomes (Section 2.1), a different study was contemplated to try to model diverse types of chirality transfer mechanism. Structurally speaking, COF2 diverges from COF1, COF4 and COF5 (Fig. 1) because it does not support the imine/enamine bond. In fact, Du et al.53 ingeniously applied the Knoevenagel condensation to fabricate a cyano-substituted olefinic COF through the reaction between 1,3,5-benzenetriacetonitrile and a formyl-functionalized R/S-[1,1′-binaphthalene]-2,2′-diylbis-diphenylphosphine oxide (BINAPO) linker (Fig. S13, ESI†). In this context, the axial chirality of the BINAPO module can be transmitted to the adjacent covalently-connected luminophore, causing a chiral through-bond propagation along the reticulated mesh. Also, for COF2, the geometrical optimization recorded a semi-distorted hexagonal pattern, confirming that independent from the chemical nature of the monomers, the minima of low-dimensional (0D/1D) models furnish non-planar arrangements when they are energy-minimized (Fig. S14, ESI†). Inspecting the optimized COF2, it is also clear that the chirality transfer is favoured by cyclic intramolecular hydrogen bonds, which limit the fluxionality in the six bis-naphthyl linkers, promoting a distinct axial asymmetry (Fig. S15, ESI†). Remembering that the experimental CPL peak appears at 475 nm, Fig. 6 provides a balanced picture of how the performances of functionals change when a different CPL-active COF is investigated. In fact, for COF2, Minnesota functionals (M11L, M06L and MN12L) failed as a consequence of silent signals (very low R-length values, Table S3, ESI†). The collection of CPL simulated spectra can be ideally divided into three regions, a blue-shifted section (350–350 nm) populated by LC-ωHPBE and ωB97xD functionals, which display an energetic chiral luminescence that is too intense. Meanwhile, B3LYP, PBE1PBE, N12SX and HSEH1PBE expressed decent emission energies with variable CPL intensities within the crowded central interval of 400–450 nm. Finally, with an almost symmetric Gaussian band shape and an excellent band position (488 nm), O3LYP ably captured the CPL opposite signals of R/S-COF2 (Fig. 6). Conforming to the experimental data (Fig. 6(C), blue line), we tried to authenticate the reticulation effect in the chiral transmission mechanism by testing the bis-reacted compound (R-BIS2, Fig. S16, ESI†). Table S4 (ESI†) shows that, notwithstanding the coherent emission wavelength values, its rotatory strengths dramatically collapse and result in a silent CPL signal. Thus, although the luminescence wavelength is (approximately) preserved between the bis-condensed unit and reticulated materials, the COF formation greatly improves the chiroptical activity. | ||
| Fig. 6 (A) Assemblage of the TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the BOMD trajectories for R/S-COF2. (B) Zoomed vision of the CPL spectra performed at the O3LYP/6-311G(d,p) level along the BOMD trajectories for R/S-COF2 and R-BIS2. (C) Experimental CPL spectra of R/S-COF2 (solid dispersion in ethylene glycol). Adapted with permission from ref. 53 © 2022 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
2.3. Case 3: COF3
Dissimilar from the other COFs (Fig. 1), COF3 carries two different types (hence, two diverse reactivity modes) of nucleophilic sites in its monomer. The use of (S) and (R)-3amino-4-oxo-2-(pyrrolidin-2-yl)-3,4-dihydroquinazoline-7carbohydrazide and 1,3,5-benzenetricarboxaldehyde was performed by Thang et al.55 to brilliantly engineer a chiral framework through a peculiar aminal linkage induced by an unexpected ring-closure process (Fig. S17, ESI†). Practically, the presence of a β-NH at the ortho-position of the amine was capitalized for a diastereoselective cyclocondensation, leading to L/D-COF3 where each COF segment has a dissymmetric acyl–hydrazone chain bearing a quinazoline unit decorated with a chiral pyrrolidinyl side ring. Both enantiomers were optimized, and the related minima exposed a sigmoid disposition due to the tetrahedral carbons in the cyclic aminals, which impose a non-planar frame (Fig. S18, ESI†). The combination between the luminophoric and chiral characteristics culminated with a cyan (510 nm) and weak mirror-imaged CPL signal in the solid state (Fig. 7(B)). Our TD-DFT protocol (Fig. 7(A)) suggests some similarities between COF2 (Section 2.2) and COF3 functionals performances. In fact, M11L, M06L and MN12L were also unsuccessful, exhibiting a silent spectrum (Table S5, ESI†). B3LYP, PBE1PBE, N12SX, HSEH1PBE, LC-ωHPBE and ωB97xD suffered an exaggerated blue-shift in the peak position and anomalous band shapes (too weak for LC-ωHPBE and ωB97xD, and too sharp for B3LYP, PBE1PBE, N12SX and HSEH1PBE). Again, O3LYP emerged with an appreciable spectral profile, evidencing an emission wavelength at 529 nm. | ||
| Fig. 7 (A) Assemblage of the TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the BOMD trajectories for L/D-COF3. (B) Experimental CPL spectra of L/D-COF3 in the solid state. Adapted with permission from ref. 55 © 2024 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
2.4. Case 4: COF4
Equivalent to COF1, COF4 required the insertion of a third component to gain a compromise between a chiral and emissive monolayer. Indeed, Hu et al.54 ably integrated a luminescent 2D polymeric axle and β-cyclodextrins acting as chiral wheels to strategically obtain a bidimensional chiral polyrotaxane via a mechanically interlocked approach. The 2D polymerization was realized by means of a formation of β-ketoenamine bonds between 1,3,5-triformylphloroglucinol and β-cyclodextrin hosted 1,6-diaminopyrene (Fig. S19, ESI†). In this host–guest COF, β-cyclodextrins played a dual role: (1) avoiding the π–π stacking to prevent the aggregation-caused quenching effect; (2) triggering an asymmetric environment (through its intrinsic chirality) by mechanical bonds. Additionally, the CPL intensity of the whole COF can be tuned by regulating the feeding amount of β-cyclodextrin macrocycle (Cy) interlaced in the pyrene axle. Computationally, the geometrical optimization of naked COF4 resulted in a semi-planar conformation in which the pyrene carbocycles are misaligned to favour the intramolecular hydrogen bonds in the β-ketoenamine segments (Fig. S20, ESI†). To cover all of the possible experimental feeding ratios (β-cyclodextrin/pyrene, mol/mol) until an equilibrium condition was achieved, we defined four different models in which a variable number of β-cyclodextrins are entrapped in the hexagonal pattern: COF4Cy[1], COF4Cy[2], COF4Cy[3] and COF4Cy[4] (Fig. S21, ESI†). Taking COF4Cy[1] as a standard reference, it can be seen that, in its S0 minimum, the main body of the cyclodextrin wheel is centered around the hydrophobic region of pyrene. Meanwhile, the hydroxyl groups of the smaller rim directly interact with the oxygen and with the positively polarized C–H of the β-ketoenamine fragment (Fig. S22, ESI†). Beyond the myriad of less intense electrostatic interactions (which usually accompany all of the hydrogen bonded aggregates87–89) between the cyclodextrin cavity and pyrene cloud, we retain that the previously mentioned hydrogen bonds are predominantly responsible for the supramolecular chirality transfer. Such a through-space propagation mechanism was photochemically examined by preliminarily executing our theoretical protocol for COF4Cy[1]. Fig. 8 evidently depicts how most of the functionals struggled in reproducing the experimental negative CPL band at 450 nm. M11L, M06L and MN12L conferred very weak rotatory strengths, which caused silent spectra (Table S6, ESI†). In contrast, O3LYP, N12SX and HSE1HPBE processed a positive and weak CPL band in the red region (600–700 nm), while B3LYP determined a bisignate CPL profile with two peaks at 562 nm (negative sign) and 611 nm (positive sign). PBE1PBE occupies an intermediate position at 533 nm with a negative line. Only LC-ωHPBE and ωB97xD successfully delineated the experimental CPL sign and band shape, with a perfect match for the band position from ωB97xD (450 nm). Once the reference system (COF4Cy[1]) was chiroptically validated, we tried to emulate how the CPL performances improve by increasing the Cy units until the feeding ratio equilibrium condition is reached. Experimentally, about 8% of Cy was loaded in the COF (∼0.5 Cy unit for each hexagonal frame) when feed = 1, about 43% of Cy was loaded in the COF (∼2.6 Cy unit for each hexagonal frame) when feed = 2, and about 73% of Cy was loaded in the COF (∼4.3 Cy unit each hexagonal frame, equilibrium condition) when feed = 4. Feed = 8, feed = 10 and feed = 16 were not considered because feed = 8 is very similar to feed = 4 (about 78% of Cy was loaded), while the last two activate the hyperbranched polymer formation due to the exceeding wheels in a limited space, preventing the formation of an intact hexagonal skeleton (decreasing the CPL intensities, Fig. 8(D)). Fig. 8(C) modelled how the CPL intensities progressively increased in progressing from COF4Cy[1] to COF4[4], perfectly adhering with the experimental trend (Fig. 8(D)). This evidence explains that in reinforcing the number of interlocked dyads (cyclodextrin/pyrene) until the equilibrium point (COF4Cy[1] → COF4Cy[4]), the related rotatory strengths are boosted (Table S7, ESI†) as a result of an amplified confined chirality transfer mediated by a codified hydrogen bonding pattern.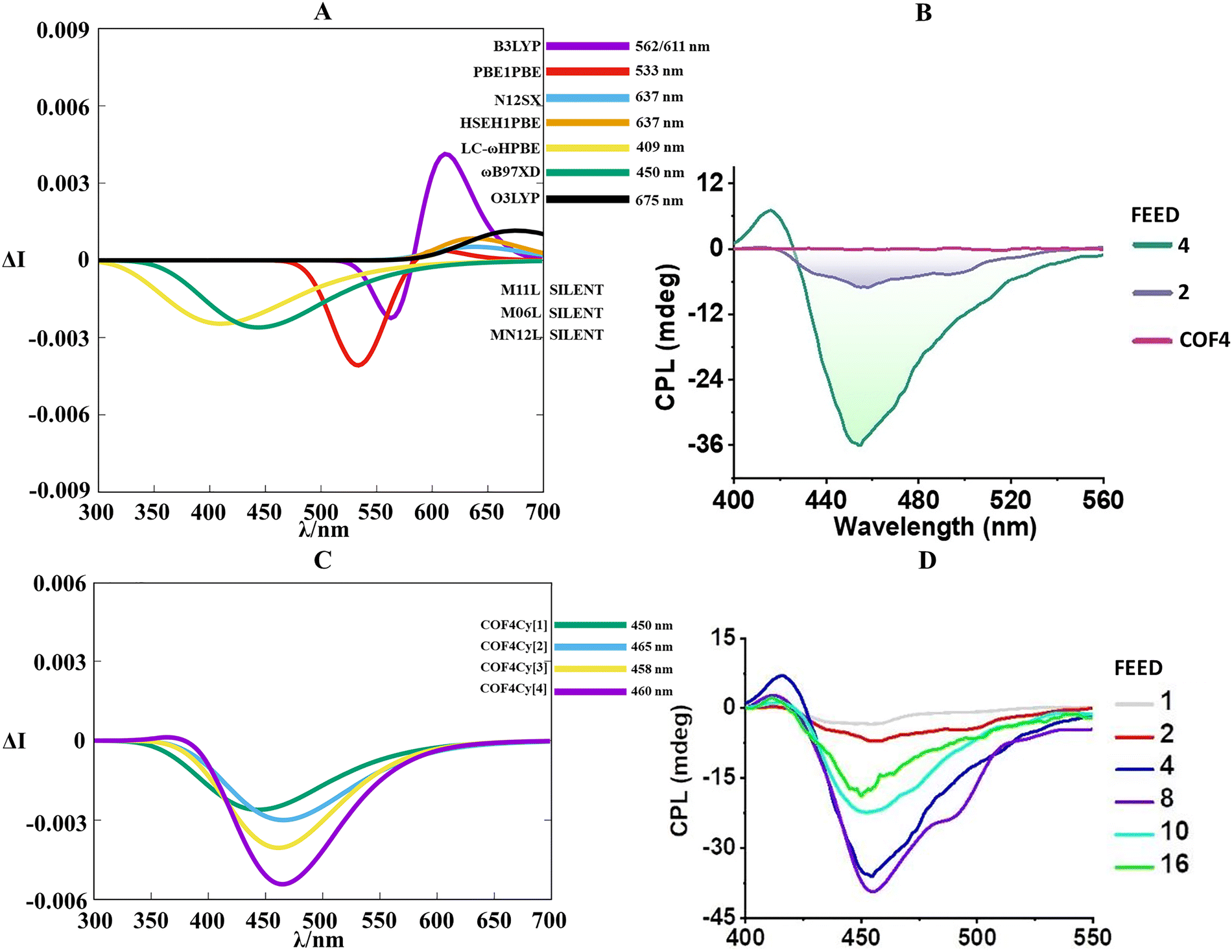 | ||
| Fig. 8 (A) Assemblage of the TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the BOMD trajectories for COF4Cy[1]. (B) Experimental CPL spectra of COF4 with different feed ratios (2 and 4). The standalone COF4 label refers to free cyclodextrin COF4. Adapted with permission from ref. 54 © 2022 Wiley-VCH Verlag GmbH & Co. KGaA. (C) Zoomed-in vision of the CPL spectra performed at the ωB97xD/6-311G(d,p) level along the BOMD trajectories for COF4Cy[1], COF4Cy[2], COF4Cy[3] and COF4Cy[4]. (D) Experimental CPL spectra of COF4 with different feed ratios. Adapted with permission from ref. 54 © 2022 Wiley-VCH Verlag GmbH & Co. KGaA. It must be noted that about 8% of Cy was loaded in COF4 when feed = 1, about 43% of Cy was loaded in COF4 when feed = 2, about 73% of Cy was loaded in COF4 (equilibrium condition) when feed = 4, about 75% of Cy was loaded in COF4 when feed = 8, about 78% of Cy was loaded in COF4 when feed = 10, and about 81% of Cy was loaded in COF4 when feed = 16. | ||
2.5. Case 5: COF5
According to the literature, COF5 was the first (chronologically) published CPL-active covalent organic framework. In this respect, Yan et al.56 used the Schiff-base reaction between (R) and (S) 2,5-bis (2-hydroxypropoxy)terephthalohydrazide and 1,3,5,-benzenetricarboxyaldehyde to synthesize an enantiomeric pair of 2D hydrazone-linked chiral COFs (Fig. S23, ESI†). Conceptually, COF5 is analogous to COF1 and COF3, in which chirality was imparted through stereogenic sites. However, for COF5, the asymmetric monomer bears two pre-installed stereogenic carbons instead of one (see COF1 and COF3 in Fig. S2 and S17, ESI† respectively). The optimized structure of COF5 reported a slightly positively-curved architecture (like a buckybowl), in which all the 2-hydroxypropoxy lateral chains equipped in the terephthalate derivative formed a cyclic six-member H-bond with the hydrazonic proton (see Fig. S24; ESI† this situation is also reinforced by secondary electrostatic interactions). Bearing in mind that the experimental CPL measurement in the solid phase spotlighted a mirror-image spectrum centered at 480 nm (Fig. 9(B)), a heterogeneous scenario prevailed when COF5 was studied through our TD-DFT protocol. The acquired functional performances were discordant. In fact, M11L, M06L and MN12l calculated rotatory strengths that were too low (Table S8, ESI†), while PBE1PBE and LC-ωHPBE were affected by a net bisignate line shape (356/418 nm and 327/370 nm, respectively, Fig. 9(A)). At about 385 nm, HSEH1PBE and N12SX produced a superimposable spectrum with a moderate change of sign between 420 nm and 500 nm. Although B3LYP decently approached the correct experimental band shape, its peak position was exceedingly blue-shifted (386 nm). Thus, only O3YLP managed to efficiently duplicate both the emission wavelength (478 nm) and the CPL spectral profile with a good symmetry between the R and S bands (S band was a bit more intense than the R band). | ||
| Fig. 9 (A) Assemblage of the TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the BOMD trajectories for COF5. (B) Experimental CPL spectra of COF5 in the solid phase. Adapted with permission from ref. 56 © 2021 Elsevier publisher. | ||
3. Computational details
For the preliminary DFT benchmark, the COF computational models proposed in Fig. 2 were optimized in the gas phase, adopting the following exchange–correlation functionals: B3LYP, M062X, CAM-B3LYP, PBEPBE and ωB97xD, coupled with 6-311G(d,p) basis set, using an ultrafine grid and a tight criterion for energy and geometry optimization convergence. Once the best DFT-performer was established, the energy minima of COF1[1], COF1[2], COF1[3], COF1[7], COF1[X], COF2, COF3, COF4Cy[1], COF4Cy[2], COF4Cy[3], COF4Cy[4] and COF5 (with their relative enantiomers) were searched at the ωB97xD/6-311G(d,p) level in the gas phase. The Born–Oppenheimer molecular dynamics (ωB97xD/6-311G(d,p) level) in the first excited state (S1) of all the aforementioned COF systems were performed by integrating the equation of motion with a time step of 2 fs in a total simulation time of 1 ps. The isothermal conditions were fixed at 298 K. After the production runs, 25 snapshot configurations for each COF were retrieved (temporally-interspersed every 40 fs) and TD-DFT submitted to recover excitation energies, rotatory strengths and chiroptical signs at B3LYP, PBE1PBE, N12SX, HSEH1PBE, LC-ωHPBE, ωB97xD, O3LYP, MN12L, M11L and M06L (associated with 6-311G(d,p) Pople basis set) levels in the gas phase. The CPL intensities ΔI were determined as follows:90 | (1) |
4. Conclusions
The theoretical investigation of chiral covalent organic frameworks featuring intriguing circularly polarized luminescence properties is of paramount significance, but remains a challenging task. The present research represents the first attempt to computationally decodify the chiral emissions of asymmetric COF systems. In this context, we investigated all of the pure CPL-active COFs reported so far in the literature, in order to rationalize how the size-scale effect and the various chirality transfer mechanisms can bias the CPL spectral properties. Encouraged by a mini-benchmark on COFs prototype models, we found a reliable level of theory (ωB97xD combined with Pople basis set) to elucidate the more complex extended frame of COF materials. The COF1 family, assumed as a computational reference, revealed how the dimensionality of the chemical building dominates the final atomic dispositions. In fact, the 0D and 1D models were arranged in distorted configurations, while the 2D model preserved a planar layout. The transferability of this effect was also validated by optimizing the hexagonal units of the other COFs (COF2, COF3, COF4 and COF5), which exhibited curved or semi-curved frames. The chiroptical inspection of COF1 set revealed how the accumulation of chiral groups and the transition from a 0/1D wire to a 2D platform achieved better performances in the CPL signal intensity as a consequence of a capillary propagation of chirality along the whole structure and stronger rotational strengths (bigger φE–M), respectively. COF2 offered us the possibility to scrutinize how the axial chirality is particularly dependent on the reticulation effect, which in turn alters the CPL spectral band. COF3 and COF5 verified that COFs with multiple stereogenic sites (in the starting monomer or in the condensed arms) could be reproducible in terms of the CPL profile by our DFT/TD-DFT workflow. COF4Cy[1] and its COF parents (COF4Cy[2], COF4Cy[3] and COF4Cy[4]) allowed us to study the through-space chirality transfer in mechanically interlocked 2D polyrotaxane. Our computational outputs underlined that specific hydrogen bonds between β-cyclodextrins and the β-ketoenamine segments governed the chirality transmission between the macrocyclic wheel and the COF axle. The resulting calculated CPL magnitudes were proportional to the amount of cyclodextrins (until the equilibrium point) inside the hexagonal unit. This multi-variegated picture ranging from COF1 to COF5 clarified that, notwithstanding that a single level of computation cannot be versatile enough to cover CPL bands from the red region to the blue one, M11L (although M06L is also a robust alternative) worked very well for less-energetic CPL (COF1, 630 nm). O3LYP succeeded in the simulation of CPL signatures standing in an intermediate region (COF2, COF3 and COF5, 475–510 nm), while ωB97xD was the best functional to clone the asymmetric luminescence when the mechanical bond regulates the CPL spectral lines (COF4, 450 nm). In such complex scenarios, our study intends to be an initial but valid guide for the ab initio computational identification of CPL spectra for future chiral-emissive covalent organic frameworks. In parallel, this first theoretical attempt can help experimentalists anticipate the chiroptical features (CPL intensity, dissymmetry ratio, chiral sign and emission wavelength) for ad hoc programmed synthetic COFs through computational screenings.Author contributions
Giovanni Bella: conceptualization, methodology, software, validation, investigation, project administration, writing – original draft. Giuseppe Bruno: resources, supervision, writing – review & editing. Francesco Nicolò: resources, supervision, writing – review & editing. Antonio Santoro: supervision, writing – review & editing visualization, resources, data curation.Conflicts of interest
There are no conflicts to declare.Data availability
The experimental CPL spectra were adapted with permission from the following: (1) https://doi.org/10.1021/jacs.2c00285 (Fig. 2(B) in the main article). (2) https://doi.org/10.1002/anie.202113979 (Fig. 2(E) in the main article). (3) https://doi.org/10.1002/anie.202413171 (Fig. S24A in the ESI†). (4) https://doi.org/10.1002/anie.202114759 (Fig. 3(E) in the main article and Fig. S25D in the ESI†). (5) https://doi.org/10.1016/j.cclet.2020.11.063 (Fig. 3(C) in the main article). The datasets supporting this article and the relative code/software are detailed in the “Computational details” section and the ESI.†Acknowledgements
We acknowledge the CINECA award under the ISCRA initiative for the availability of high-performance computing resources and support.Notes and references
- G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC.
- A. J. Miles, R. W. Janes and B. A. Wallace, Chem. Soc. Rev., 2021, 50, 8400–8413 RSC.
- B. Ranjbar and P. Gill, Chem. Biol. Drug Des., 2009, 74, 101–120 CrossRef CAS PubMed.
- S. Daly, F. Rosu and V. Gabelica, Science, 2020, 368, 1465–1468 CrossRef CAS.
- T. A. Keiderling and A. Lakhani, Chirality, 2018, 30, 238–253 CrossRef CAS PubMed.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875 CrossRef CAS.
- J.-F. Chen, Q.-X. Gao, H. Yao, B. Shi, Y.-M. Zhang, T.-B. Wei and Q. Lin, Chem. Commun., 2024, 60, 6728–6740 RSC.
- X. Yang, X. Gao, Y.-X. Zheng, H. Kuang, C.-F. Chen, M. Liu, P. Duan and Z. Tang, CCS Chem., 2023, 5, 2760–2789 CrossRef CAS.
- S.-Y. Li, L. Xu, R.-T. Gao, Z. Chen, N. Liu and Z.-Q. Wu, J. Mater. Chem. C, 2023, 11, 1242–1250 RSC.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef.
- A. Eisfeld, R. Kniprath and J. S. Briggs, J. Chem. Phys., 2007, 126, 104904 CrossRef CAS PubMed.
- A. O. Govorov, Z. Fan, P. Hernandez, J. M. Slocik and R. R. Naik, Nano Lett., 2010, 10, 1374–1382 CrossRef CAS PubMed.
- W. R. Kitzmann, J. Freudenthal, A.-P. M. Reponen, Z. A. VanOrman and S. Feldmann, Adv. Mater., 2023, 35, 2302279 CrossRef CAS.
- S. C. J. Meskers, ChemPhotoChem, 2022, 6, e202100154 CrossRef CAS.
- Y. Zhang, Y. Wei, C. Li, Y. Wang, Y. Liu, M. He, Z. Luo, X. Chang, X. Kuang and Z. Quan, Chem. Commun., 2025, 61, 85–88 RSC.
- L. Du, Q. Zhou, Q. He, Y. Liu, Y. Shen, H. Lv, L. Sheng, T. Cheng, H. Yang, L. Wan, Y. Fang and W. Ning, Adv. Funct. Mater., 2024, 34, 2315676 CrossRef CAS.
- C. Li, Y. Wei, Y. Zhang, Z. Luo, Y. Liu, M. He and Z. Quan, Angew. Chem., Int. Ed., 2024, 63, e202403727 CrossRef CAS PubMed.
- L. Chen, J. Yuan, X. He, F. Zheng, X. Lu, S. Xiang and Q. Lu, Small Methods, 2024, 8, 2301517 CrossRef CAS PubMed.
- T. He, W. Zhao, M. Lin, B. Sun, Y. Chen, H.-L. Zhang and G. Long, J. Phys. Chem. Lett., 2024, 15, 9844–9851 CrossRef CAS.
- C. Li, X. Jin, J. Han, T. Zhao and P. Duan, J. Phys. Chem. Lett., 2021, 12, 8566–8574 CrossRef CAS PubMed.
- J. Zhao, K. Zeng, T. Jin, W.-T. Dou, H.-B. Yang and L. Xu, Coord. Chem. Rev., 2024, 502, 215598 CrossRef CAS.
- Y. Liu, X. Gao, B. Zhao and J. Deng, Nanoscale, 2024, 16, 6853–6875 RSC.
- Y. Deng, M. Wang, Y. Zhuang, S. Liu, W. Huang and Q. Zhao, Light: Sci. Appl., 2021, 10, 76 CrossRef CAS.
- Z. Wang, A. Li, Q. Cao, W. Liang, J. Feng, K. T. Chang, Z. Xu, A. K. Srivastava and H. Lu, Adv. Opt. Mater., 2025, 13, 2402310 CrossRef CAS.
- A. Kunnummal, R. A. Parambil, S. Maniappan and J. Kumar, ACS Appl. Opt. Mater., 2024, 2, 2451–2458 CrossRef CAS.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS.
- H. Jiang, D. Alezi and M. Eddaoudi, Nat. Rev. Mater., 2021, 6, 466–487 CrossRef CAS.
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS PubMed.
- J. Liang and K. Liang, Coord. Chem. Rev., 2024, 501, 215572 CrossRef CAS.
- O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1295–1300 CrossRef CAS.
- Z. Zheng, N. Rampal, T. J. Inizan, C. Borgs, J. T. Chayes and O. M. Yaghi, Nat. Rev. Mater., 2025, 10, 369–381 CrossRef.
- D.-W. Sun, L. Huang, H. Pu and J. Ma, Chem. Soc. Rev., 2021, 50, 1070–1110 RSC.
- K. E. Cordova and O. M. Yaghi, Mater. Chem. Front., 2017, 1, 1304–1309 RSC.
- W. Gong, Z. Chen, J. Dong, Y. Liu and Y. Cui, Chem. Rev., 2022, 122, 9078–9144 CrossRef CAS PubMed.
- A. Hubber and C. Hua, Chem. – Eur. J., 2024, 30, e202400071 CrossRef CAS PubMed.
- M. Chafiq, A. Chaouiki, J. Ryu and Y. G. Ko, Nano Today, 2024, 56, 102227 CrossRef CAS.
- X. Kang, E. R. Stephens, B. M. Spector-Watts, Z. Li, Y. Liu, L. Liu and Y. Cui, Chem. Sci., 2022, 13, 9811–9832 RSC.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 RSC.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- X. Tang, J. Zha, X. Wu, J. Tong, Q. Gu, K. Zhang, Y. Zhang, S. Zheng, J. Fan, W. Zhang, Q. Zhang, C. Tan and S. Cai, Angew. Chem., Int. Ed., 2025, 64, e202413675 CrossRef CAS.
- Y. Zhao, H. Liu and B. Sun, Sens. Actuators, B, 2022, 354, 131253 CrossRef CAS.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- Q. Gu, J. Zha, C. Chen, X. Wang, W. Yao, J. Liu, F. Kang, J. Yang, Y. Y. Li, D. Lei, Z. Tang, Y. Han, C. Tan and Q. Zhang, Adv. Mater., 2024, 36, 2306414 CrossRef CAS.
- M. Xue, L. Zhang, X. Meng, J. Yang, Y. He, C.-S. Lee, J. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202412289 CrossRef CAS.
- H. Chen, Z.-G. Gu and J. Zhang, J. Am. Chem. Soc., 2022, 144, 7245–7252 CrossRef CAS.
- C. Du, X. Zhu, C. Yang and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202113979 CrossRef CAS PubMed.
- L. Hu, X. Zhu, C. Yang and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114759 CrossRef CAS PubMed.
- X. Tang, K. Zhang, R. Xue, Y. Zheng, S. Chen, S. Zheng, J. Fan, Y. Zhang, W. Ye, W. Zhang, S. Cai and Y. Liu, Angew. Chem., Int. Ed., 2024, 63, e202413171 CrossRef CAS.
- Y. Yan, X. Li, G. Chen, K. Zhang, X. Tang, S. Zhang, S. Zheng, J. Fan, W. Zhang and S. Cai, Chin. Chem. Lett., 2021, 32, 107–112 CrossRef CAS.
- X. Tang, X. Liao, X. Cai, J. Wu, X. Wu, Q. Zhang, Y. Yan, S. Zheng, H. Jiang, J. Fan, S. Cai, W. Zhang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202216310 CrossRef CAS.
- Q. Yang, M. Fusè and J. Bloino, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.00801.
- G. Xia, L. Wang, H. Xia, Y. Wu, Y. Wang, H. Hu and S. Lin, Chin. Chem. Lett., 2022, 33, 4253–4256 CrossRef CAS.
- P. Wu, A. Pietropaolo, M. Fortino, M. Bando, K. Maeda, T. Nishimura, S. Shimoda, H. Sato, N. Naga and T. Nakano, Angew. Chem., Int. Ed., 2023, 62, e202305747 CrossRef CAS.
- G. Bella, G. Bruno and A. Santoro, J. Mol. Liq., 2023, 391, 123268 CrossRef CAS.
- G. Bella, G. Bruno and A. Santoro, FlatChem, 2023, 42, 100567 CrossRef CAS.
- G. Bella, M. Milone, G. Bruno and A. Santoro, J. Mater. Chem. C, 2024, 12, 3005–3012 RSC.
- G. Bella, G. Bruno and A. Santoro, J. Mater. Chem. C, 2024, 12, 17913–17924 RSC.
- É. Brémond, M. Savarese, N. Q. Su, Á. J. Pérez-Jiménez, X. Xu, J. C. Sancho-García and C. Adamo, J. Chem. Theory Comput., 2016, 12, 459–465 CrossRef PubMed.
- G. Bella, M. Milone, G. Bruno and A. Santoro, Phys. Chem. Chem. Phys., 2022, 24, 26642–26658 RSC.
- M. A. Alkhalifah, M. Yar, I. Bayach, N. S. Sheikh and K. Ayub, Materials, 2022, 15, 7425 CrossRef CAS PubMed.
- H. Hashemzadeh and H. Raissi, J. Phys. D: Appl., 2018, 51, 345401 CrossRef.
- H. Li and C. Zhang, Comput. Theor. Chem., 2024, 1233, 114503 CrossRef CAS.
- B. Dash, J. Mol. Model., 2018, 24, 120 CrossRef.
- A. Kuc, M. A. Springer, K. Batra, R. Juarez-Mosqueda, C. Wöll and T. Heine, Adv. Funct. Mater., 2020, 30, 1908004 CrossRef CAS.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- G. Bella and A. Rotondo, Chem. Phys. Lipids, 2020, 232, 104973 CrossRef CAS PubMed.
- G. Bella, G. Bruno and A. Santoro, FlatChem, 2023, 40, 100509 CrossRef CAS.
- B. C. Ferrari and C. J. Bennett, J. Phys.: Conf. Ser., 2019, 1290, 012013 CrossRef CAS.
- G. Bella, A. Santoro, M. Cordaro, F. Nicolò and G. Bruno, Chin. J. Chem., 2020, 38, 163–168 CrossRef CAS.
- A. Santoro, G. Bruno, F. Nicolò, G. Bella and G. Messina, J. Mol. Struct., 2020, 1202, 127231 CrossRef CAS.
- M. K. Ravva, T. Wang and J.-L. Brédas, Chem. Mater., 2016, 28, 8181–8189 CrossRef CAS.
- A. Swain, K. Radacki, H. Braunschweig and P. Ravat, Chem. Sci., 2024, 15, 11737–11747 RSC.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- P. Albacete, J. I. Martínez, X. Li, A. López-Moreno, S. A. Mena-Hernando, A. E. Platero-Prats, C. Montoro, K. P. Loh, E. M. Pérez and F. Zamora, J. Am. Chem. Soc., 2018, 140, 12922–12929 CrossRef CAS.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS.
- D. Sidler, P. Bleiziffer and S. Riniker, J. Chem. Theory Comput., 2019, 15, 2492–2503 CrossRef CAS.
- R. Kullmann, M. Delbianco, C. Roth and T. R. Weikl, J. Phys. Chem. B, 2024, 128, 12114–12121 CrossRef CAS.
- S. C. C. van der Lubbe and C. Fonseca Guerra, Chem. – Asian J., 2019, 14, 2760–2769 CrossRef CAS.
- A. Giannetto, F. Nastasi, F. Puntoriero, G. Bella, S. Campagna and S. Lanza, Dalton Trans., 2021, 50, 1422–1433 RSC.
- G. Longhi, E. Castiglioni, S. Abbate, F. Lebon and D. A. Lightner, Chirality, 2013, 25, 589–599 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc02158h |
| This journal is © The Royal Society of Chemistry 2025 |