
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrystalline pseudorotaxane enabling dual-wavelength photothermal mechanical actuation†
Kuan-Hung
Huang
a,
Hong-Kai
Chang
a,
Meng-Che
Tsai
 ac,
Ting-Wei
Liao
ac,
You-Ting
Chen
a,
Chu-Liang
Hsieh
ac,
Pei-Lin
Chen
b,
Kai-Jen
Chen
*ac and
Masaki
Horie
*ac
ac,
Ting-Wei
Liao
ac,
You-Ting
Chen
a,
Chu-Liang
Hsieh
ac,
Pei-Lin
Chen
b,
Kai-Jen
Chen
*ac and
Masaki
Horie
*ac
aDepartment of Chemical Engineering, National Tsing Hua University, 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan
bInstrumentation Center, National Tsing Hua University, 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan
cResearch Institute for Electronic Science (RIES), Hokkaido University, N21W10, Kita-Ward, Sapporo 001-0021, Japan. E-mail: kjchen@es.hokudai.ac.jp; mhorie@es.hokudai.ac.jp
First published on 8th July 2025
Abstract
We report the synthesis and photothermal-mechanical conversion behavior of a photoresponsive pseudorotaxane comprising an ammonium salt bearing ferrocenyl and anthryl groups as the axle and dibenzo[24]crown-8 (DB24C8) as the macrocyclic ring. In the crystalline state, the pseudorotaxane exhibits notable void spaces around the ferrocene and anthryl substituents, with void volume increasing from 12.7% to 17.3% over the temperature range of −173 °C to 77 °C. The crystals display characteristic photoabsorption at 375 nm and 445 nm, attributed to the anthryl and ferrocenyl units, respectively, enabling efficient photothermal conversion. Upon incorporation into a poly(n-butyl methacrylate) (PBMA) matrix, the crystal-dispersed film undergoes significant temperature increases from 27 °C to 72 °C and 69 °C when irradiated at 375 nm and 445 nm, respectively, resulting in light-induced film expansion. This expansion translates into pronounced photoactuated lifting and bending motions, which are significantly greater than those observed in films containing only the axle or lacking the anthryl moiety. These findings demonstrate the utility of ferrocenyl–anthryl-functionalized crystalline pseudorotaxanes in the development of photomechanical materials, soft actuators, and optically responsive mechanical systems.
1. Introduction
Molecular machines and switches have been extensively studied for their capacity to manipulate molecular structures at the nanoscale in response to external stimuli.1–8 Notably, mechanically interlocked rotaxanes and pseudorotaxanes—comprising ring and axle components—undergo relative positional changes upon exposure to stimuli such as light,9–13 pH shifts,14 thermal input,15,16 and chemical signals.5,7 Due to their structural adaptability during such responses, these systems can transmit forces,4,17 motions,5,18 and energy4,19 both within individual molecules and across molecular assemblies, depending on the specific characteristics of the supramolecular architecture. To date, most investigations of molecular machines have been conducted in solution,20–22 where molecular alignment is inherently absent. In contrast, in the crystalline state, molecules are precisely aligned, oriented, and regularly spaced, enabling concerted molecular motions that can produce macroscopic mechanical effects.1,2,23–25Organic photochemical and thermal reactions capable of generating mechanical motion on scales from nanometers to millimeters have attracted significant attention due to the diverse range of molecular systems and available reaction pathways.6,9,26,27 These stimuli-responsive systems offer notable advantages, such as rapid response6,18,28,29 and high elastic modulus,6 positioning molecular crystals as promising candidates for photo- and thermomechanical actuators. Stimulus-induced transformations in these crystals can generate various mechanical motions, including expansion,23,30 bending,27,31 twisting,18,31 and curling.32 In several cases, structural changes associated with these dynamic responses have been elucidated by single-crystal X-ray crystallography.23 Crystalline molecular systems also offer significant potential in applications ranging from optoelectronics,33,34 rotors,29,35 and sensors,36 to drug delivery,25 and catalysis.37,38 This versatility stems from the high density and precise spatial organization of functional components within crystals. These properties are generally difficult to achieve in dilute solutions or thin surface-bound assemblies. While these advantages are notable, most molecular crystals exhibit dense packing, which often limits the free volume necessary for dynamic molecular motion.39 Therefore, careful molecular design and advanced crystal engineering are essential to realizing functional, solid-state dynamic molecular machines.
We have synthesized a range of pseudorotaxanes and complexes consisting of dibenzo-crown ethers (host molecules) and functionalized ammonium salts (guest molecules), and have investigated their structural transformations and dynamic behaviors in response to external stimuli.1,2 For example, crystals composed of dibenzo[24]crown-8 ether (DB24C8) and ferrocene-containing ammonium salts exhibited reversible crystal-to-crystal phase transitions and morphological changes upon heating and photoirradiation.40,41 In another system, crystals formed from dibenzo-crown ethers and azobenzene-functionalized ammonium salts exhibited reversible bending motions upon photoirradiation, driven by cis–trans isomerization of the azobenzene moiety induced by light and heat.42–44 Within these assemblies, each molecular component plays a distinct role. The dibenzo-crown ether serves as the host framework, offering sufficient spatial freedom to accommodate guest mobility.45 In contrast, the functional ammonium salts act as transducers, converting absorbed light energy into mechanical motion. Under certain conditions, intermolecular interactions can immobilize the guest species, resulting in rigid structures that inhibit motion.42–44 To overcome this limitation, structural modifications, such as introducing different functional substituents or extending the axle length, are essential for enhancing molecular mobility within the crystalline matrix.
Anthracene and its derivatives are recognized for their high planarity, extended π-conjugation, and strong fluorescence.46–49 These characteristics have been widely utilized in the development of luminescent materials and organic semiconductors. In addition, certain derivatives undergo [4+4] photodimerization in the crystalline state, which can induce photosalient effects.50–53 For example, bis-o-carborane-substituted anthracene undergoes photodimerization, leading to macroscopic mechanical responses in the crystal such as cracking, debris formation, and jumping, accompanied by decolorization. These phenomena have been observed using fluorescence microscopy, and the molecular structures before and after photodimerization have been determined by single-crystal X-ray crystallography.51 Anthryl groups have also been incorporated into rotaxanes and pseudorotaxanes, enabling photocycloaddition and functioning as photoluminescent units in solution.54–56 Moreover, anthracene-containing compounds have been investigated in thin-film systems.54,55 Despite these advances, no studies have yet reported the functionalization of anthracene units within single-crystal pseudorotaxanes.
Soft actuators composed of flexible polymers, such as poly(n-butyl methacrylate) (PBMA)57 and polyurethane,58 offer several advantages, including high flexibility, adaptability, and reconfigurability, which resemble properties found in biological systems.59,60 These features support a wide range of applications, such as soft electronics, surgical tools, drug delivery systems, artificial organs, and prosthetic devices.60,61 The integration of intelligent materials into soft actuator systems expands their functional capabilities, enabling changes in structure, macroscopic behavior, and shape in response to external stimuli including electric fields, thermal input, and light.60,62 Organic chromophores are commonly used in photothermal conversion materials due to their tunable structures, favorable photophysical properties, strong absorption in the near-infrared region, excellent biocompatibility, and high photostability.63–65 Soft polymers serve as effective host matrices due to their intrinsic elasticity and compatibility with photoresponsive guest molecules, which together facilitate reversible mechanical deformation. In previous work, we developed photoresponsive soft actuators incorporating ferrocene-based and dithienylethene chromophores embedded in PBMA.57,66 These actuators exhibited light-induced bending, vertical lifting motions, and self-healing capabilities following mechanical damage.
In this study, we report a novel pseudorotaxane composed of an ammonium salt bearing ferrocenyl and anthryl groups as the axle component and dibenzo[24]crown-8 (DB24C8) as the ring component, which provides flexibility and internal free volume (Fig. 1). We investigate structural changes in the single-crystal state, where the ferrocenyl and anthryl moieties exhibit distinct absorption at 445 nm and 375 nm, respectively, enabling efficient photothermal conversion. Incorporation of the crystals into a PBMA matrix results in pronounced light-induced lifting and bending motions. The photomechanical response exceeds that of films composed solely of the axle molecule or those lacking the anthryl unit. These findings demonstrate the potential of crystalline pseudorotaxanes incorporating functional chromophores for applications in photomechanical materials and soft actuation systems.
 | ||
| Fig. 1 Schematic of ferrocenyl- and anthryl-containing pseudorotaxane for photoinduced motion. | ||
2. Results and discussion
2.1. Synthesis and characterization
Scheme 1 illustrates the synthesis of pseudorotaxane PR1, which consists of a ferrocenyl- and anthryl-functionalized ammonium hexafluorophosphate (PF6−) axle (A1) and DB24C8. The axle molecule A1 was synthesized via condensation of anthracene-2-carbaldehyde with ferrocenemethylamine, followed by hydrogenation and protonation. Single crystals of PR1 were obtained by vapor diffusion of diethyl ether (Et2O) into a dichloromethane–acetone solution containing a mixture of A1 and DB24C8. Obtained compounds were confirmed by 1H NMR and mass spectroscopies (Fig. S1–S11, ESI†). For comparative analysis, the previously reported pseudorotaxane PR2,40,41 bearing a tolyl substituent, and its corresponding axle A2 are also examined. | ||
| Scheme 1 Synthesis of PR1 and PR2. | ||
The structural changes of PR1 at various temperatures were analyzed by single-crystal X-ray crystallography (Fig. 2, 3 and Fig. S12, S13, Tables S3, S4, ESI†). Due to substantial deterioration in data quality above 77 °C (350 K), detailed structural analysis is based on the crystallographic data obtained below 47 °C (320 K). Fig. 2a–3c present the molecular structure of PR1, a detailed view of the ferrocenyl unit, and a summary of the intra- and intermolecular interactions, along with temperature-dependent structural changes. PR1 is primarily stabilized by [N–H⋯O] and [C–H⋯O] hydrogen bonds (2.43–3.06 Å) between the ammonium N–H or N–CH2 protons and the oxygen atoms of DB24C8. An intramolecular [π⋯π] interaction is observed between one catechol unit of DB24C8 and the ferrocenyl moiety of A1, with the centroid–centroid distance increasing from 3.94 Å at −173 °C to 3.98 Å at 24 °C (Fig. 2a). At −173 °C, the two cyclopentadienyl (Cp) rings of the ferrocenyl group adopt a nearly eclipsed conformation with a dihedral angle of 0.1°. Upon heating to 24 °C, this angle increases to 5°, suggesting a slight rotation that reduces steric repulsion between Cp-ring hydrogen atoms. As a result, the Cp–Cp distance decreases from 3.29 Å (Fig. 2b). At 77 °C, further conformational changes are observed, with partial collapse of the ferrocenyl units, indicating increased conformational freedom of the ferrocene group.
 | ||
| Fig. 2 (a) Molecular structure of PR1 determined by single-crystal X-ray crystallography at −173 °C. (b) Top view of the ferrocenyl group at various temperatures. (c) Schematic representation of molecular motion upon heating. (d) Molecular packing at −173 °C. | ||
 | ||
| Fig. 3 Temperature dependence of PR1: (a) [C–H⋯π] interaction distance, (b) anthryl–anthryl distance, (c) relative changes in unit cell parameters, and (d) unit cell volume. | ||
Intermolecular interactions are also temperature dependent. A [C–H⋯π] interaction is observed between the anthryl unit and a neighboring ferrocenyl group, with the interaction distance increasing from 2.69 Å to 2.87 Å between −173 °C and 47 °C (Fig. 2c and 3a). Additionally, the distance between anthryl moieties in adjacent molecules increases by 8.2%, from 5.47 Å to 5.92 Å over the same temperature range (Fig. 2c and 3b). This increase precludes π–π stacking and photocyclization in the crystalline state.
Crystal packing analysis (Fig. 2d) reveals that the top surface corresponds to the (010) plane, with the crystal extending along the a- and c-axes, while the thickness aligns with the b-axis. Thus, the relationship between macroscopic crystal deformation and molecular-level packing changes is considered. Fig. 3c and d illustrate temperature-dependent variations in unit cell parameters and volume. Between −173 °C and 47 °C, the a- and c-axes expand by 1.7%, whereas the b-axis increases only marginally by 0.6%. The overall volume increase is therefore primarily due to elongation along the a- and c-axes, attributed to enhanced ferrocenyl mobility at elevated temperatures. As noted earlier, weakening of [C–H⋯π] interactions facilitates this mobility and contributes to crystal plane expansion. Although the anthryl group remains conformationally rigid, the increasing intermolecular distance along the b-axis slightly contributes to overall crystal expansion.
The volumetric thermal expansion coefficient (αv) is estimated to be 1.88 × 10−4 K−1, exceeding values typically reported for crystalline organic compounds67 and metal–organic materials.68 This temperature-dependent molecular motion and anisotropic expansion play a key role in the photothermal–mechanical conversion discussed in subsequent sections.
Hirshfeld surface and crystal void analyses are performed using CrystalExplorer version 21.569 to visualize intra- and intermolecular interactions in PR1 (Fig. 4). The Hirshfeld surface is generated by partitioning the crystal space into distinct regions based on proximity to atoms. The parameters de and di represent the distances from a point on the surface to the nearest nucleus outside and inside the surface, respectively. The normalized contact distance (dnorm) is used to highlight intermolecular interactions, with its color scale reflecting deviations from van der Waals radii.
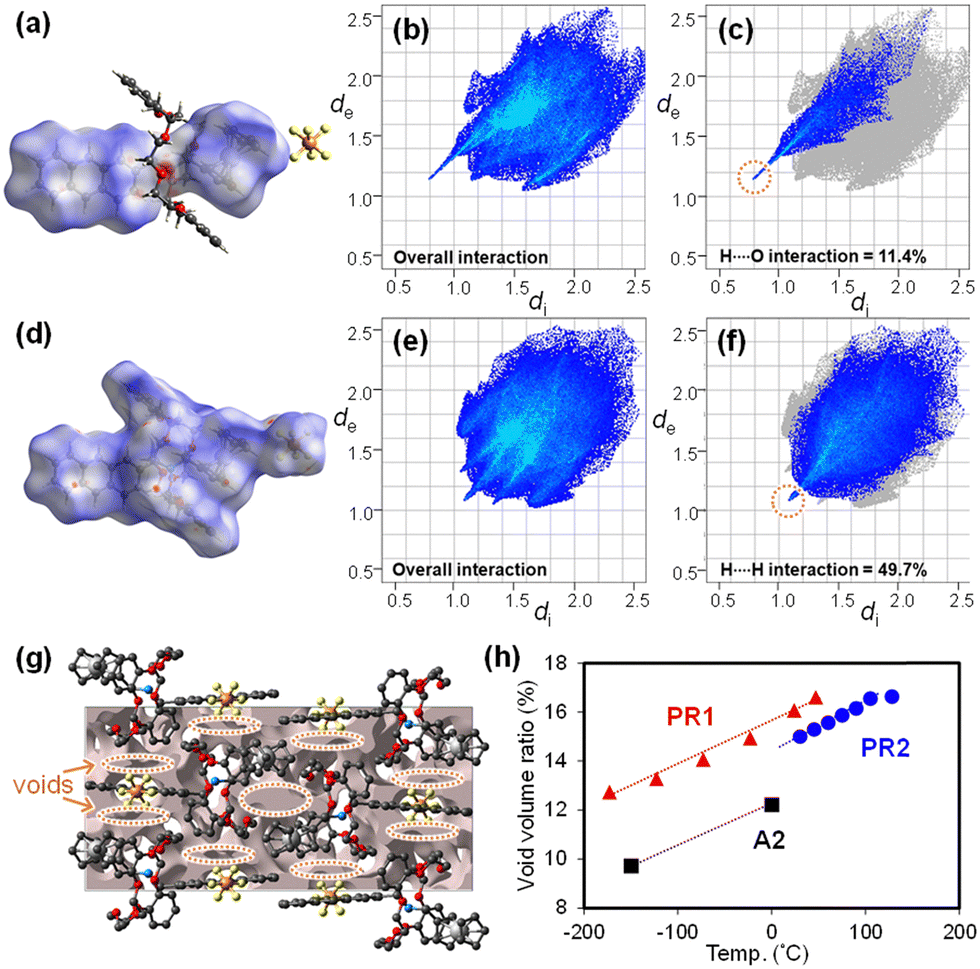 | ||
| Fig. 4 (a) Hirshfeld plot of dnorm for the axle molecule in PR1 at −173 °C. (b) 2D fingerprint plots of overall interactions and (c) dominated interactions for the axle molecule in PR1 at −173 °C. (d) Hirshfeld plot of dnorm for PR1 at −173 °C. (e) 2D fingerprint plots of overall interactions and (f) dominated interaction in PR1 at −173 °C. (g) Graphical representation of voids in the crystal packing of PR1 at 47 °C. (h) Temperature dependence of the void volume ratio for PR1, PR2, and A2 with trend lines. | ||
In the interaction between A1 and DB24C8 (Fig. 4a), intense red regions are observed around the protonated amino group of the cation and the oxygen atoms of DB24C8, indicating strong [N–H⋯O] hydrogen bonding. Fig. 4b presents the 2D fingerprint plot of the axle molecule (also shown in Fig. S15–S18, ESI†), where sharp spikes correspond to significant interactions.70,71 The dominant contribution arises from O⋯H/H⋯O contacts (di + de ∼ 2.0 Å), consistent with [N–H⋯O] interactions (Fig. 4c). White patches between the catechol and ferrocenyl units indicate weak π–π interactions. The largest contribution in the fingerprint plot is from H⋯H contacts (52.7%), concentrated around di = de ≈ 1.2 Å, largely arising from π-stacking tendencies between the ferrocenyl and catechol groups.
For intermolecular interactions between PR1 molecules (Fig. 4d), the fingerprint plots indicate that H⋯H contacts dominate (di + de ∼ 2.2 Å), corresponding to [C–H⋯π] interactions between ferrocenyl and anthryl groups (Fig. 4e, f, and Fig. S19–S22, ESI†). With increasing temperature, the [N–H⋯O] intramolecular interactions between the axle and ring remain essentially constant (di + de ∼ 2.0 Å), while the [C–H⋯π] interactions between PR1 molecules weaken, increasing the sum of distances to approximately 2.4 Å.
Fig. 4g (also shown in Fig. S23, ESI†) shows the crystal void distribution in the unit cell of PR1. Fig. 4h plots the temperature-dependent void volume for PR1 in comparison with PR2 and A2. Voids are primarily localized around the ferrocenyl and anthryl groups. The void volume of PR1 increases linearly with temperature, rising from 12.7% at −173 °C to 17.3% at 77 °C, closely following the unit cell volume expansion trend (Fig. 3d). Notably, the void volume of PR1 is approximately 1.5% greater than that of PR2 and 3.5% greater than that of its axle molecule A2. These values also exceed the typical void fraction for organic crystals (7.3–8.6%).72,73 These results suggest that incorporation of both the pseudorotaxane structure and ferrocenyl/anthryl functionalities effectively enhances free volume within the crystal, which is considered advantageous for promoting photomechanical conversion.
2.2. Film preparation and characterization
PR1 was incorporated into PBMA to prepare free-standing films, as illustrated in Scheme 2. Single crystals of PR1 were ground and dispersed in 300 μL of a PBMA solution in diethyl ether. The concentration of PR1 in PBMA was adjusted to 7 mol% (35 wt%), as summarized in Table S6 (ESI†). The dispersion was cast onto a PTFE film placed on a glass slide and allowed to dry. The dried film was then sandwiched between another PTFE film and a 70 μm spacer, followed by hot pressing at 50 °C. After molding, the film was peeled off, cut into 1 × 1 mm2 pieces, and designated as Cr.PR1-PBMA. For comparison, a PBMA film containing crystalline PR2 (Cr.PR2-PBMA) was prepared using the same procedure. In addition, a film referred to as PR1-PBMA was fabricated by casting a cosolvent mixture of chloroform and acetone containing A1, DB24C8, and PBMA in a molar ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:86 (19:16:65 wt%). According to the 1H NMR spectra (Fig. S11, ESI†), the complexation ratio of PR1 in CDCl3:acetone-d6 (70:30, vol/vol) is estimated to be approximately 60 mol% (PR1-complex:PR1-added), suggesting that PR1 is the predominant species in the film. For further comparison, a film doped with A1 alone is prepared and referred to as A1-PBMA.
:7:86 (19:16:65 wt%). According to the 1H NMR spectra (Fig. S11, ESI†), the complexation ratio of PR1 in CDCl3:acetone-d6 (70:30, vol/vol) is estimated to be approximately 60 mol% (PR1-complex:PR1-added), suggesting that PR1 is the predominant species in the film. For further comparison, a film doped with A1 alone is prepared and referred to as A1-PBMA.
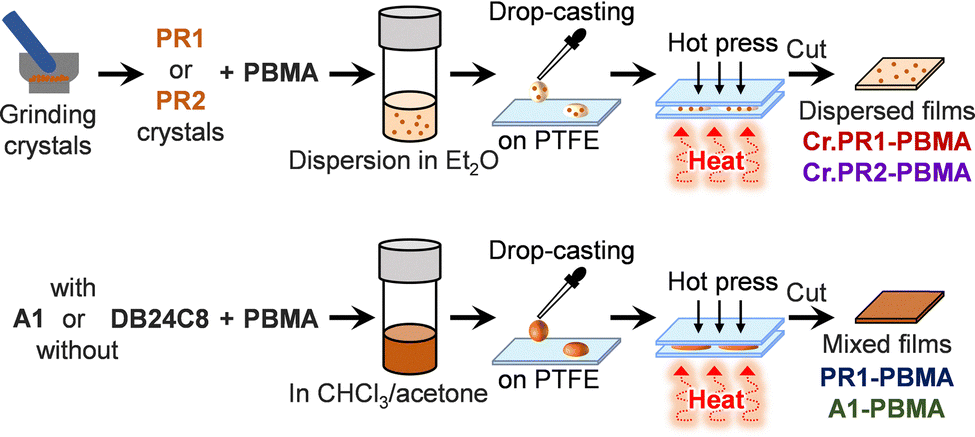 | ||
| Scheme 2 Preparation of PBMA films. | ||
The surface morphology of the films was analyzed using polarized optical microscopy, scanning electron microscopy (SEM), and two-dimensional X-ray diffraction (2D XRD). Fig. 5a and b present polarized optical micrographs of crystal-dispersed Cr.PR1-PBMA and pseudorotaxane-containing PR1-PBMA. Under polarized light, Cr.PR1-PBMA exhibits interference colors, indicating the presence of microcrystalline domains, whereas PR1-PBMA appears dark, suggesting an amorphous and uniform distribution within the PBMA matrix. A similar trend is observed for other films: Cr.PR2-PBMA contains microcrystals, while A1-PBMA appears uniformly dark (Fig. S24–S27, ESI†).
 | ||
| Fig. 5 Polarized optical micrographs of (a) Cr.PR1-PBMA and (b) PR1-PBMA. SEM images of (c) Cr.PR1-PBMA and (d) PR1-PBMA. (e) XRD patterns of the films. | ||
SEM images of Cr.PR1-PBMA and PR1-PBMA (Fig. 5c and d; see also Fig. S28–S31, ESI†) reveal clear morphological differences. Cr.PR1-PBMA displays evenly distributed microcrystals approximately 1–2 μm in diameter, whereas PR1-PBMA shows minimal evidence of crystallization. Elemental distribution is further confirmed by energy-dispersive X-ray spectroscopy (EDX). Elemental mappings of Fe, P, and F confirm the presence of PR1 (C50H56F6FeNO8P) in both Cr.PR1-PBMA and PR1-PBMA.
Fig. 5e shows the 2D XRD patterns of Cr.PR1-PBMA and PR1-PBMA. A broad diffraction peak appears at 2θ = 20–30° for Cr.PR1-PBMA, indicative of crystalline domains, while PR1-PBMA exhibits no such diffraction, confirming its amorphous nature. Similar diffraction behavior is observed for Cr.PR2-PBMA and A1-PBMA (Fig. S32, ESI†).
2.3. Photothermal conversion
Fig. 6a presents the UV-vis absorption spectrum of PR1 in acetone, showing three absorption peaks at λabs = 340, 358, and 378 nm, corresponding to the anthryl moiety, and a weak absorption at λabs = 440 nm, attributed to the ferrocenyl group. The photoluminescence (PL) spectrum of PR1 in acetone (Fig. S33, ESI†) exhibits an emission band at λPL = 432 nm, with a PL quantum yield (PLQY) below 1% (Table S7, ESI†). This value is considerably lower than those reported for anthracene (36%), polycarbosilane grafted with 9-ethylanthracene (66–80%), and terphenyl–anthryl–pyrenoimidazole derivatives (89%).74–76 The PL quenching is possibly attributed to photoinduced electron transfer from the anthryl to the ferrocenyl unit, which facilitates non-radiative decay and is favorable for photothermal conversion.77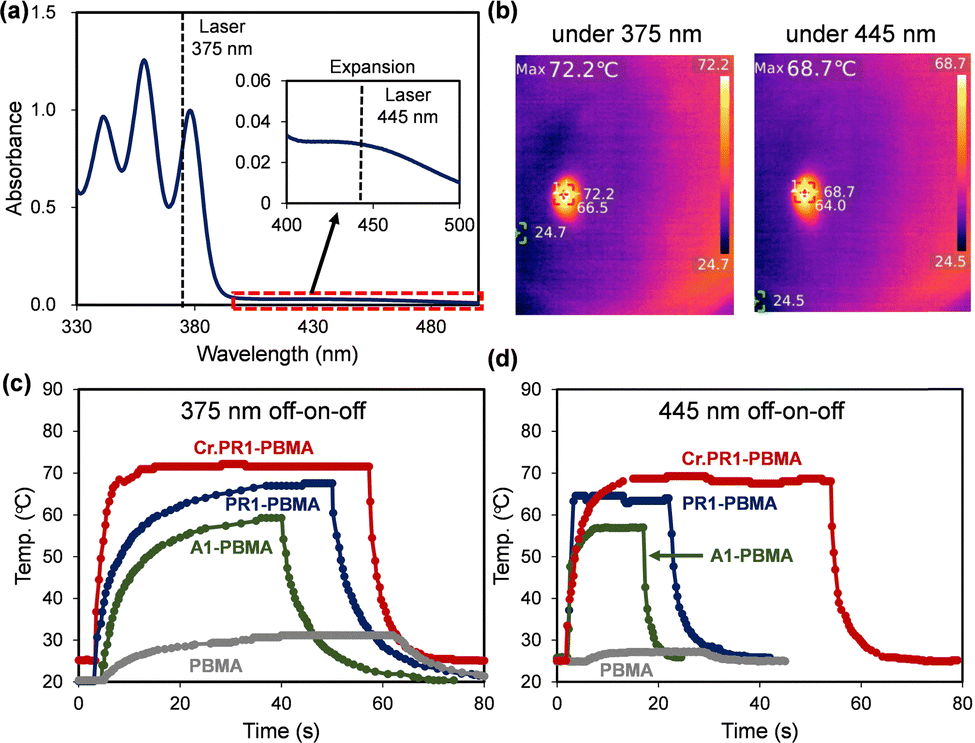 | ||
| Fig. 6 (a) UV-vis absorption spectrum of PR1 in acetone solution, with laser wavelengths at 375 nm and 445 nm indicated. (b) Thermographic images of Cr.PR1-PBMA with irradiation at 375 nm (15 mW cm−2) or 445 nm (61 mW cm−2). Time-dependent temperature profiles of PBMA films under (c) 375 nm irradiation (15 mW cm−2) and (d) 445 nm irradiation (61 mW cm−2). | ||
Based on the absorption characteristics of PR1, the 375-nm and 445-nm lasers were selected for further investigation. Fig. 6b shows thermographic images of the Cr.PR1-PBMA film under continuous laser irradiation at 375 nm (15 mW cm−2) or 445 nm (61 mW cm−2). The surface temperature increases from 27 °C to 72 °C and 69 °C, respectively. The time-dependent temperature profile under 375-nm irradiation with an off–on–off cycle is shown in Fig. 6c for Cr.PR1-PBMA and comparison films. As summarized in Table 1, Cr.PR1-PBMA reaches the highest temperature (72 °C), followed by PR1-PBMA (65 °C), A1-PBMA (57 °C), and PBMA alone (31 °C). A similar trend is observed under 445-nm irradiation (Fig. 6d). After the light source is switched off, the temperature returns to ambient (20–25 °C) within 15–20 s.
Based on the initial temperature decay profile, the time constant (τ) for each film was calculated and summarized in Table 1, as well as in Fig. S35 (ESI†). The τ values range from 1.5 to 4.7 s, which are approximately two orders of magnitude shorter than previously reported values (∼230 s) for conjugated oligoelectrolytes.78 This rapid photothermal response is considered advantageous for efficient photomechanical conversion.
Dynamic mechanical analysis (DMA) was performed to evaluate the thermal behavior of the films. The DMA profiles of A1-PBMA and PR1-PBMA reveal glass transition temperatures (Tg) of 25 °C and 47 °C, respectively, with a common tanδ peak at 57 °C (Fig. S37, ESI†). The Tg corresponds to the onset of local segmental motion of the polymer chains, while the tanδ peak reflects larger-scale molecular mobility within the network. These results indicate that the films maintain a rubbery state within the operational temperature range, and that laser-induced heating increases polymer chain mobility, facilitating deformation. Additionally, PR1-PBMA remains stable at room temperature, resisting deformation, but undergoes a smooth transition to a flexible state upon reaching temperatures beyond Tg. The coefficient of thermal expansion (CTE) was determined to be 5.7 × 10−3 for A1-PBMA and 5.8 × 10−3 for PR1-PBMA in the temperature range of 50–75 °C, as measured by laser scanning confocal microscopy. Previous studies report that the CTE of PBMA above its Tg is approximately 14 times greater than below Tg.79 These findings support the conclusion that film deformation arises from photoabsorption by the anthryl or ferrocenyl groups, followed by photothermal conversion and thermal activation of polymer chain mobility.
2.4. Photoinduced deformation and mechanical output
Photoinduced deformation of chromophore-doped PBMA films (Cr.PR1-PBMA, PR1-PBMA, and A1-PBMA) and single crystals of PR1 and PR2 was investigated using optical and confocal laser microscopy under 375 nm or 445 nm irradiation (Fig. 7a–c and Fig. S38–S40, ESI†). Upon 375-nm laser irradiation at 28 mW cm−2, Cr.PR1-PBMA expands in width, length, and thickness by 0.8%, 1.0%, and 7.8%, respectively (Fig. 7a). Similarly, under 445-nm irradiation at 40 mW cm−2, the corresponding expansion values are 0.6%, 0.5%, and 5.7%. After cessation of irradiation, the film rapidly contracts to its original dimensions. Comparable photoinduced expansion is observed for the other PBMA-based films and for the PR1 and PR2 single crystals. The relative area changes of the top surfaces of the films and crystals under both wavelengths are summarized in Fig. 7b and c. In all cases, the extent of deformation follows the trend: PR1 crystal ≈ Cr.PR1-PBMA > PR1-PBMA > A1-PBMA > PR2 crystal. The larger deformation observed under 375-nm irradiation compared to 445-nm is attributed to the superior photothermal conversion efficiency of the anthryl group relative to the ferrocenyl group. Moreover, the PBMA film containing the pseudorotaxane (PR1) shows greater deformation than the film containing only the axle molecule (A1), highlighting the cooperative effect of the pseudorotaxane structure in enhancing mechanical output. The significantly enhanced mechanical output of Cr.PR1-PBMA is likely due to the increased free volume induced by the crown ether component, which facilitates greater molecular mobility and contributes to the cooperative effect. | ||
| Fig. 7 (a) Optical and confocal micrographs of Cr.PR1-PBMA without and under 375-nm (28 mW cm−2) or 445-nm (40 mW cm−2) laser irradiation. Photoinduced relative area change under (b) 375-nm laser and (c) 445-nm laser irradiation at different laser powers. (d) Photograph and optical micrographs of force output measurement using a μ-force analyzer. μ-Force analyzer signal response under (e) 375-nm and (f) 445-nm laser irradiation. | ||
The mechanical forces generated by the PR1 crystal, Cr.PR1-PBMA film, and PR1-PBMA film on a glass substrate are measured using a microforce analyzer under focused 445-nm laser irradiation (Fig. 7d–f and Fig. S42, S43, ESI†). Upon exposure to 445-nm light at 0.18 W cm−2, both the PR1 crystal and Cr.PR1-PBMA film generate lifting forces of approximately 100 mg. In contrast, the PR1-PBMA film exhibits a lower lifting force of approximately 40 mg under slightly higher irradiation intensity (0.23 W cm−2). The sample thicknesses are 51 μm for the crystal, 20 μm for Cr.PR1-PBMA, and 21 μm for PR1-PBMA.
The mechanical output of the films and crystals was evaluated using a microforce analyzer equipped with a strain gauge connected to a cantilever, which was placed in contact with the surface of the PBMA films or a single PR1 crystal oriented on the (010) plane (Fig. 7d). For PBMA-based films, a double-layer structure comprising the PBMA film and a transparent cellophane tape as a passive layer was employed to ensure stable force measurement. The generated upward force was recorded under 375-nm or 445-nm laser irradiation at varying power, with the laser alternately switched on and off (Fig. 7e, f and Fig. S42, S43, ESI†). The measured force is found to be proportional to the laser power. Among all samples, the PR1 single crystal produces the highest mechanical output under both irradiation conditions, which is attributed to its high density and ordered molecular packing.
Under 375-nm irradiation at 23 mW cm−2, the PR1 crystal generates a force of 125 milligram-force (mgf, 1 mgf = 9.81 × 10−6 N), corresponding to approximately 67000 times its own weight. Under 445-nm irradiation at 236 mW cm−2, the force reaches 53 mgf, or about 23000 times its own weight (Fig. S42c and d, ESI†). These values represent the highest mechanical output reported in our studies to date.80,81 Furthermore, the PR1 crystal demonstrates excellent reversibility, undergoing repeated expansion and contraction over more than 50 on/off cycles without significant performance degradation (Fig. S42e, ESI†).
Next, the force outputs of various double-layer PBMA films and single crystals are compared under 375-nm or 445-nm laser irradiation (Fig. 7e, f and Fig. S43a, b, ESI†). Under 375-nm irradiation, the force output follows the order: PR1 crystal > Cr.PR1-PBMA > PR1-PBMA > A1-PBMA > Cr.PR2-PBMA. Under 445-nm irradiation, the order is: PR1 crystal > Cr.PR1-PBMA > Cr.PR2-PBMA ≈ PR1-PBMA > A1-PBMA. Among the PBMA-based films, Cr.PR1-PBMA exhibits the highest mechanical output under both wavelengths. This performance is attributed to the photothermal expansion of the embedded microcrystals and the combined light absorption of the anthryl and ferrocenyl moieties at 375 nm and 445 nm, respectively. Notably, Cr.PR1-PBMA, which incorporates DB24C8, generates a significantly greater force than A1-PBMA, which lacks the crown ether. This enhancement suggests that the pseudorotaxane structure contributes to efficient deformation by spatially isolating A1 molecules within the PBMA matrix and promoting cooperative motion. Repeatability testing on PR1-PBMA under repeated laser on/off cycling confirms stable and reversible mechanical response over more than 30 cycles (Fig. S43e, ESI†), supporting its durability for repeated actuation applications.
Photoinduced bending of the double-layer films was examined under laser irradiation (Fig. 8a). In this configuration, the crystal-dispersed active layer expands upon light exposure, while the attached passive layer restricts three-dimensional expansion, resulting in two-dimensional deformation and macroscopic bending of the film. Fig. 8b shows side-view optical microscopy images of Cr.PR1-PBMA with the passive layer. The film displays a larger bending displacement of 0.17 mm under 375-nm irradiation compared to 0.11 mm under 445-nm irradiation.
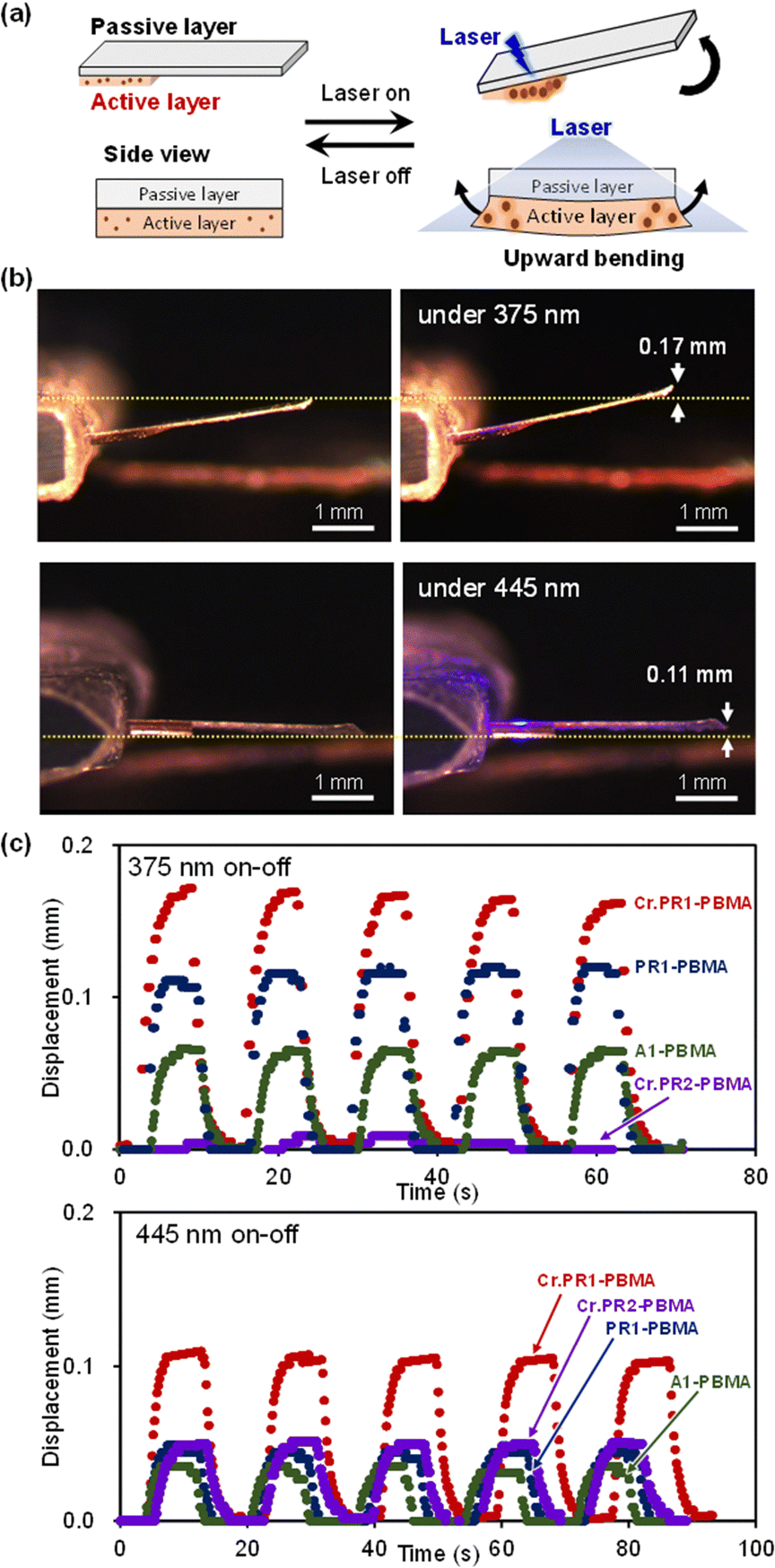 | ||
| Fig. 8 (a) Illustration of the bending motion of a double-layer film. (b) Photographs of Cr.PR1-PBMA without and with 375-nm (4.9 mW cm−2) or 445-nm (13 mW cm−2) laser irradiation. (c) Time-dependent displacement of PBMA films under 375-nm (4.9 mW cm−2) and 445-nm (13 mW cm−2) laser irradiation. | ||
Time-dependent displacement profiles under repeated on/off laser cycles are presented in Fig. 8c for various PBMA-based films. Under 375-nm irradiation, Cr.PR1-PBMA achieves the largest displacement, reaching a maximum within 5.3 s and returning to its original position within 4.8 s after irradiation ceases. The displacement magnitudes follow the order: Cr.PR1-PBMA > PR1-PBMA > A1-PBMA. In contrast, Cr.PR2-PBMA shows minimal response due to its poor absorption at this wavelength. Under 445-nm irradiation, Cr.PR1-PBMA again exhibits the greatest displacement, with expansion occurring within 2.4 s and recovery within 3.9 s. Other PBMA-based films also show light-induced bending, though the magnitude and response speed are lower compared to Cr.PR1-PBMA.
3. Conclusions
In this study, we have developed a photoresponsive pseudorotaxane composed of anthryl and ferrocenyl moieties as axle components and a crown ether as the ring. The incorporation of anthryl and ferrocenyl groups increases the internal free volume of the crystal, facilitating structural deformation, and enhances UV-vis absorption, enabling activation at both 375 nm and 445 nm. The single crystal of PR1 exhibits the highest photoinduced mechanical output among the materials studied and maintains high performance even when embedded in a soft PBMA matrix. This crystal-based molecular system shows rapid responsiveness and excellent stability, making it a promising photoactuator. These findings provide insights into the design of photoresponsive materials and offer guiding principles for the development of next-generation functional systems. The moldability of the films into various forms further broadens their application potential. This approach enables the realization of light-driven, electricity-free devices and energy-efficient mechanical systems, with potential contributions to emerging fields such as soft robotics and adaptive materials.4. Experimental section
4.1. Materials and general methods
PR2 was synthesized according to a previously reported procedure.40 Poly(n-butyl methacrylate) (PBMA; number-average molecular weight = 337000 g mol−1, intrinsic viscosity = 0.47–0.56 dL g−1), sodium acetate (CH3COONa) and dibenzo[24]crown-8 (DB24C8) were purchased from Sigma-Aldrich. n-Butyllithium (n-BuLi), hydroxylamine hydrochloride, 3 Å molecular sieves, ferrocenecarboxaldehyde, and N,N,N′,N′-tetramethylethylenediamine (TMEDA) were obtained from Thermo Scientific. Hydrochloric acid aqueous solution (37 wt%) was purchased from Honeywell. 2-Bromoanthracene and ammonium hexafluorophosphate (NH4PF6) were purchased from Combi. Sodium borohydride (NaBH4) and lithium aluminum hydride (LiAlH4) were purchased from Alfa Aesar. Anhydrous methanol, ethanol, and tetrahydrofuran (THF) were purchased from Sigma-Aldrich in Sure/Seal containers and stored under nitrogen atmosphere. Samples were purified by medium-pressure liquid chromatography (NextGen 300+, EZ Prep), flash column chromatography, solvent flushing, and bi-solvent recrystallization.
1H NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer. Chemical shifts were referenced to acetone (δ = 2.05 ppm), acetonitrile (δ = 1.93 ppm), or CDCl3 (δ = 7.26 ppm). High-resolution (HR) field-desorption (FD) mass spectrometry (MS) was carried out using a JEOL JMS-T200GC AccuTOF GCx instrument. Anion detection was performed by HPLC/MS-MS, using a VARIAN system (ProStar 210 Solvent Delivery Module, ProStar 410 AutoSampler, ProStar 335 Photodiode Array Detector) coupled with a VARIAN 901-MS (FT-ICR mass spectrometer). UV-vis absorption spectra were recorded using a JASCO V-630 spectrophotometer. Fluorescence spectra were measured using a JASCO FP-8500 spectrofluorometer.
4.2. Synthesis of ferrocene carbaldehyde oxime
Ferrocenecarboxaldehyde (1.00 g, 4.67 mmol, 1.0 eq.) was dissolved in ethanol (70 mL) in a 250 mL Schlenk tube. Sodium acetate (CH3COONa, 1.95 g, 16.3 mmol, 3.5 eq.) was dissolved in water and slowly added to the Schlenk tube. After the mixture was thoroughly combined, hydroxylamine hydrochloride (650 mg, 9.34 mmol, 2.0 eq.) was added gradually. The resulting mixture was stirred at 50 °C for 4 h. Upon cooling to room temperature, the reaction mixture was quenched with water, and completion of quenching was confirmed using pH test paper. The mixture was extracted three times with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure using a rotary evaporator. The crude product, ferrocene carbaldehyde oxime, was obtained as a dark red solid (1.20 g, 4.40 mmol, 94% yield). Due to minimal byproduct formation, further purification was not required. Thin-layer chromatography using a 9:1 mixture of CH2Cl2 and ethyl acetate as the eluent showed two distinct spots (Rf = 0.33 and 0.50), attributed to the presence of stereoisomers. 1H NMR (500 MHz, CDCl3): δ 7.94 (s, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 4.49–4.50 (m, 2H, Cp), 4.32–4.35 (m, 2H, Cp), 4.18–4.19 (m, 5H, Cp).
N), 4.49–4.50 (m, 2H, Cp), 4.32–4.35 (m, 2H, Cp), 4.18–4.19 (m, 5H, Cp).
4.3. Synthesis of ferrocene methylamine
LiAlH4 (1.00 g, 26.4 mmol, 6.0 eq.) was placed in a double-neck round-bottom flask under a nitrogen atmosphere. Ferrocene carbaldehyde oxime (1.20 g, 4.40 mmol, 1.0 eq.) was dissolved in dry THF (60 mL) and slowly added to the flask. The reaction mixture was stirred and refluxed at 70 °C for 5 h. After cooling to room temperature, the reaction was quenched with water under an ice bath to control exothermicity. The resulting lithium salts were removed by suction filtration. The aqueous phase was extracted three times with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure using a rotary evaporator. Ferrocenemethylamine was obtained as a dark orange oil (832 mg, 3.87 mmol, 88% yield). 1H NMR (500 MHz, CDCl3): δ 3.53 (s, 2H, CH2), 4.09–4.10 (m, 2H, Cp), 4.12 (m, 5H, Cp), 4.14–4.15 (m, 2H, Cp).4.4. Synthesis of anthracene-2-carbaldehyde
In a Schlenk tube, 2-bromoanthracene (1.00 g, 3.89 mmol, 1.0 eq.) was dissolved in diethyl ether (40 mL) and sonicated for 3 min. TMEDA (2.71 g, 23.3 mmol, 6.0 eq.) was then added, and the mixture was cooled to −70 °C for 3 min. n-BuLi (1.99 g, 31.1 mmol, 8.0 eq.) was added dropwise under stirring. After the addition of n-BuLi, the reaction mixture was allowed to warm to room temperature and stirred for 2 h. The flask was then cooled in an ice bath, and DMF (5.69 g, 77.8 mmol, 20.0 eq.) was added dropwise. Stirring continued for an additional 5 h at room temperature. Upon completion, the reaction was quenched with hydrochloric acid solution, and complete quenching was confirmed using pH test paper. The mixture was extracted three times with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (hexane:CH2Cl2 = 1:1) to afford anthracene-2-carbaldehyde as a yellow powder (352 mg, 1.70 mmol, 44% yield). 1H NMR (500 MHz, CDCl3): δ 7.52–7.57 (m, 2H), 7.88–7.90 (d, 1H, J = 9 Hz), 8.02–8.04 (d, 1H, J = 9 Hz), 8.05–8.06 (d, 2H, J = 9 Hz), 8.45 (s, 1H), 8.49 (s, 1H), 8.61 (s, 1H), 10.17 (s, 1H, CHO).
4.5. Synthesis of A1
Ferrocenemethylamine (832 mg, 3.87 mmol, 2.5 eq.) and anthracene-2-carbaldehyde (319 mg, 1.55 mmol, 1.0 eq.) were dissolved in a 1:1 mixture of THF and ethanol (100 mL), and 3 Å molecular sieves were added. The reaction mixture was stirred at 50 °C for 48 h. After the reaction, the molecular sieves were removed by suction filtration. The filtrate was extracted three times with CH2Cl2. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield a mixture of ferrocenemethylamine and the imine intermediate. This mixture was dissolved in methanol (62 mL), and NaBH4 (935 mg, 24.8 mmol, 16 eq.) was slowly added. The reaction was stirred at room temperature for 7 h. Then, 6 N hydrochloric acid (3.8 mL, 6.0 eq.) was added dropwise and stirring continued for 1 h at room temperature. The reaction was quenched with water and extracted three times with CH2Cl2. The resulting aqueous layer contained protonated ferrocenemethylamine, while the organic layer contained ferrocenyl–anthryl ammonium chloride. The organic layer was dried over MgSO4 and the solvent was removed under reduced pressure to afford [ferrocenyl–anthryl ammonium]+Cl− as a crude product (358 mg, 0.81 mmol, 52.4%). The crude [ferrocenyl–anthryl ammonium]+Cl− was dissolved in acetone (40 mL), and NH4PF6 (579 mg, 4.05 mmol, 5 eq.) was added. The mixture was stirred at room temperature for 2 h and extracted three times with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The resulting residue was washed with diethyl ether (15 mL) and toluene (3 mL) to yield A1 as a brown powder (209 mg, 0.38 mmol, 47%). 1H NMR (500 MHz, CD3CN): δ 3.99 (s, 2H, CH2), 4.19 (s, 5H, Cp), 4.23–4.24 (s, 2H, Cp), 4.25 (s, 2H, CH2), 4.36–4.37 (m, 2H, Cp), 7.48–7.50 (d, 1H, J = 9 Hz), 7.52–7.55 (m, 2H), 8.06–8.11 (m, 4H), 8.54 (s, 2H). HRMS (FD): calcd for C26H24FeN [M]+: 406.1252, found: m/z 406.1249 (error 0.74 ppm). HRMS (ESI): calcd for PF6− [M]−: 144.9642, found: m/z 144.9642 (error 0.07 ppm).
4.6. Synthesis of PR1
A1 (50.0 mg, 0.0907 mmol, 1.0 eq.) and DB24C8 (48.8 mg, 0.1088 mmol, 1.2 eq.) were dissolved in acetone:CH2Cl2 (2 mL, 51:49 v/v) and stirred at room temperature for 4 h. After basic filtration to remove insoluble impurities, the resulting solution was transferred to a small vial and placed inside a larger container filled with diethyl ether for vapor diffusion recrystallization over 2 days. Yellow single crystals of PR1 were obtained. 1H NMR (500 MHz, CD3CN): δ = 3.63–3.65 (m, 8H, OCH2), 3.77–3.79 (m, 8H, OCH2), 3.97 (s, 2H, CH2), 4.12–4.15 (m, 8H, OCH2), 4.20–4.21 (s, 5H, Cp), 4.29 (s, 2H, CH2), 4.31 (s, 2H, CH2), 4.40 (s, 2H, Cp), 6.93–6.99 (m, 8H, aromatic H), 7.46–7.48 (d, 1H, J = 9 Hz, aromatic H), 7.53–7.56 (m, 2H, aromatic H), 8.07–8.14 (m, 4H, aromatic H), 8.56 (s, 2H, aromatic H). HRMS (FD): calcd for C26H24FeN [M]+: 406.1252, found: m/z 406.1249 (error 0.74 ppm).
4.7. Single-crystal X-ray crystallography
Single-crystal X-ray crystallography of the PR1 was carried out using Rigaku XtaLAB Synergy-DW system. The crystallographic data of PR1, PR2 and A1 have been deposited with the CCDC under deposition number CCDC 2452984, 1500708 and 2142250.4.8. Film preparation and characterization
Single crystals of PR1 were ground and dispersed in 300 μL of a PBMA solution in diethyl ether. The concentration of PR1 in PBMA was adjusted to 7 mol% (34.8 wt%), as summarized in Table S7 (ESI†). The dispersion was cast onto a PTFE film placed on a glass slide and allowed to dry. The resulting film was sandwiched between another PTFE film and a 70 μm spacer, followed by hot pressing at 50 °C. After molding, the film was peeled off, cut into 1 × 1 mm2 pieces, and designated as Cr.PR1-PBMA. A PBMA film containing crystalline PR2 (Cr.PR2-PBMA) was also prepared using the same procedure. Additionally, a film referred to as PR1-PBMA was fabricated by casting a cosolvent mixture of chloroform and acetone containing A1, DB24C8, and PBMA in a molar ratio of 7:7:86 (19:16:65 wt%).
XRD measurements of the PBMA films were carried out using a powder X-ray diffractometer (D2 Phaser; Bruker, Karlsruhe, Germany) with Cu-Kα radiation (λ = 1.5406 Å), operated at 30 kV and 10 mA. SEM was conducted using a Hitachi SU8010 to investigate the surface morphology of the films. Elemental composition and mapping were obtained using an energy-dispersive X-ray spectroscopy (EDS) system (Oxford Instruments X-MaxN 6857) coupled to the SEM, operated at an accelerating voltage of 15 kV. Dynamic mechanical analysis (DMA) was performed using a DMA Q800 instrument (TA Instruments) to evaluate the viscoelastic properties of the films.
4.9. Photoirradiation experiments
Photoinduced mechanical motions of crystals and free-standing PBMA films were observed using an optical microscope (Olympus BX51) equipped with a temperature-controlled stage (HCS302-mk100U and HCS302 heating/cooling unit) and a CCD camera (DFK 51AU02). Continuous-wave diode-pumped solid-state lasers emitting at 445 nm (TAN-YU, LSR445FP-1W) and 375 nm (MDL-D-375-800 mW) were used as the irradiation sources. The laser light was delivered via a multi-optical fiber cable and collimated using an SMA 905 collimator (for the 445 nm laser) or an FC/PC collimator (for the 375 nm laser). Laser power was adjusted by controlling the current from the respective controllers and measured using a power meter (Ophir, Nova II, P/N 7Z01550). Side-view images and videos of deformation behavior were captured using a Lumenera INFINITY 1-3C digital camera and analyzed with INFINITY ANALYZE software.Deformation analysis was performed using ImageJ software. The height change was analyzed using a Python program built with OpenCV (cv2) and NumPy. Each video frame was processed individually. Every frame was first converted to grayscale (using cv2.cvtColor), then binarized into black and white using a fixed threshold (cv2.threshold). To reduce noise and make the shapes clearer, morphological operations (cv2.morphologyEx and cv2.dilate) were applied. A specific region in the image (i.e., the actuator position) was selected, and the highest white pixel in that area was found using np.argmax. The movement was calculated by comparing this highest point to the one in the first frame (first_highest_position–highest_position), and the value was converted to millimeters using a known scale factor.
The dimensions of crystals and films were measured with a confocal laser microscope (Keyence VK9500) using a 10× objective lens. Photoinduced mechanical force was measured using the same microscope system equipped with a microforce detector module (CHIEF SI, μ-force) operated via Bridge DAQ software. The force detection module consisted of a cantilever with strain gauges mounted on both sides to detect small forces. All measurements were conducted on a vibration-isolated optical table to minimize external disturbances. Thermal imaging under photoirradiation was carried out using an infrared thermal imaging camera (UTI-260B).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available as follows: Crystallographic data for compounds PR1, PR2 and A1 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the accession numbers CCDC 2452984, 1500708 and 2142250, respectively. All other data, including raw experimental data, spectral data, and analysis scripts used for photomechanical actuation characterization, are available in the ESI.†Acknowledgements
This work was financially supported by Ministry of Science and Technology Taiwan (110-2221-E-007-006-MY3 and 110-2113-M-007-013-MY3) and Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Research Start-up for Returnees (24K23944).References
- M. Horie and C.-H. Wang, Mater. Chem. Front., 2019, 3, 2258–2269 RSC.
- C.-H. Wang and M. Horie, Mater. Today Chem., 2022, 24, 100852 CrossRef CAS.
- S. Krause and B. L. Feringa, Nat. Rev. Chem., 2020, 4, 550–562 CrossRef CAS.
- A. Mondal, R. Toyoda, R. Costil and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202206631 CrossRef CAS PubMed.
- L. Zhang, Y. Qiu, W.-G. Liu, H. Chen, D. Shen, B. Song, K. Cai, H. Wu, Y. Jiao and Y. Feng, Nature, 2023, 613, 280–286 CrossRef CAS PubMed.
- W. M. Awad, D. W. Davies, D. Kitagawa, J. M. Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. F. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Y. Zhang, C. C. Sun, C. Bardeen, R. O. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 RSC.
- L. Zhang, H. Wu, X. Li, H. Chen, R. D. Astumian and J. F. Stoddart, Nat. Rev. Methods Primers, 2024, 4, 13 CrossRef.
- F. Nishino, P. Krüger, C. H. Wang, R. Nemoto, Y. H. Chang, T. Hosokai, Y. Hasegawa, K. Fukutani, S. Kera, M. Horie and T. K. Yamada, Small, 2025, 21, 2408217 CrossRef CAS PubMed.
- S. Corra, M. Curcio, M. Baroncini, S. Silvi and A. Credi, Adv. Mater., 2020, 32, 1906064 CrossRef CAS PubMed.
- J.-S. Geng, L. Mei, Y.-Y. Liang, L.-Y. Yuan, J.-P. Yu, K.-Q. Hu, L.-H. Yuan, W. Feng, Z.-F. Chai and W.-Q. Shi, Nat. Commun., 2022, 13, 2030 CrossRef CAS PubMed.
- G. Liu, J. Zhu, Y. Zhou, Z. Dong, X. Xu and P. Mao, Org. Lett., 2018, 20, 5626–5630 CrossRef CAS PubMed.
- H.-G. Fu, Y. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 16117–16122 CrossRef CAS PubMed.
- P. Q. Nhien, J.-H. Tien, T. T. K. Cuc, T. M. Khang, N. T. Trung, C.-H. Wu, B. Thi Buu Hue, J. I. Wu and H.-C. Lin, J. Mater. Chem. C, 2022, 10, 18241–18257 RSC.
- L. M. Zhao, L. S. Zheng, X. Wang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202214296 CrossRef CAS PubMed.
- X. Ge, Y. He, X. Liang, L. Wu, Y. Zhu, Z. Yang, M. Hu and T. Xu, Nat. Commun., 2018, 9, 2297 CrossRef PubMed.
- Q. y Jin, L. Meng, Z. h Zhang, Y. y Liang, W. Jin, K. q Hu, L. y Yuan, Z. F. Chai, L. Mei and W. q Shi, Adv. Funct. Mater., 2023, 33, 2303530 CrossRef CAS.
- V. García-López, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567–572 CrossRef PubMed.
- F. Lancia, A. Ryabchun and N. Katsonis, Nat. Rev. Chem., 2019, 3, 536–551 CrossRef CAS.
- C. Biagini and S. Di Stefano, Angew. Chem., 2020, 132, 8420–8430 CrossRef.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- Y. Feng, M. Ovalle, J. S. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5569–5591 CrossRef CAS PubMed.
- C. Zazza, S. Borocci and N. Sanna, Phys. Chem. Chem. Phys., 2024, 26, 5399–5407 RSC.
- A. Colin-Molina, D. P. Karothu, M. J. Jellen, R. A. Toscano, M. A. Garcia-Garibay, P. Naumov and B. Rodríguez-Molina, Matter, 2019, 1, 1033–1046 CrossRef.
- I. Liepuoniute, M. J. Jellen and M. A. Garcia-Garibay, Chem. Sci., 2020, 11, 12994–13007 RSC.
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, Adv. Mater., 2020, 32, 1906036 CrossRef CAS PubMed.
- T. Uchikura, M. Oshima, M. Kawasaki, K. Takahashi and N. Iwasawa, Angew. Chem., Int. Ed., 2020, 59, 7403–7408 CrossRef CAS PubMed.
- W. Cho, J. Jeon, W. Eom, J. G. Lee, D. G. Kim, Y. S. Kim, T. H. Han and J. J. Wie, Adv. Funct. Mater., 2021, 31, 2102106 CrossRef CAS.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS PubMed.
- M. E. Howe and M. A. Garcia-Garibay, J. Org. Chem., 2019, 84, 9835–9849 CrossRef CAS PubMed.
- M. Jin, S. Yamamoto, T. Seki, H. Ito and M. A. Garcia-Garibay, Angew. Chem., Int. Ed., 2019, 58, 18003–18010 CrossRef CAS PubMed.
- D. Kitagawa, H. Tsujioka, F. Tong, X. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS PubMed.
- H. Liu, Z. Lu, B. Tang, C. Qu, Z. Zhang and H. Zhang, Angew. Chem., 2020, 132, 13044–13050 CrossRef.
- Y. Yao, L. Zhang, E. Orgiu and P. Samorì, Adv. Mater., 2019, 31, 1900599 CrossRef PubMed.
- N. Grabicki, O. Dumele, H. Sai, N. E. Powers-Riggs, B. T. Phelan, M. H. Sangji, C. T. Chapman, J. V. Passarelli, A. J. Dannenhoffer and M. R. Wasielewski, Chem. Mater., 2021, 33, 706–718 CrossRef CAS.
- S. D. Karlen, H. Reyes, R. Taylor, S. I. Khan, M. F. Hawthorne and M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14973–14977 CrossRef CAS PubMed.
- A. Hasija, A. J. Thompson, L. Singh, S. N. Megha, K. S. Mangalampalli, J. C. McMurtrie, M. Bhattacharjee, J. K. Clegg and D. Chopra, Small, 2023, 19, 2206169 CrossRef CAS PubMed.
- T. Pan and J. Liu, ChemPhysChem, 2016, 17, 1752–1758 CrossRef CAS PubMed.
- L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. Bunce and S. P. Fletcher, Nat. Rev. Chem., 2018, 2, 0117 CrossRef CAS.
- L. Catalano and P. Naumov, CrystEngComm, 2018, 20, 5872–5883 RSC.
- M. Horie, Y. Suzaki, D. Hashizume, T. Abe, T. D. Wu, T. Sassa, T. Hosokai and K. Osakada, J. Am. Chem. Soc., 2012, 134, 17932–17944 CrossRef CAS PubMed.
- K.-J. Chen, Y.-C. Tsai, Y. Suzaki, K. Osakada, A. Miura and M. Horie, Nat. Commun., 2016, 7, 13321 CrossRef CAS PubMed.
- S.-C. Cheng, K.-J. Chen, Y. Suzaki, Y. Tsuchido, T.-S. Kuo, K. Osakada and M. Horie, J. Am. Chem. Soc., 2018, 140, 90–93 CrossRef CAS PubMed.
- S.-C. Cheng, C.-H. Wang, Y.-C. Lin, Y. Tsuchido, Y. Suzaki, Y. Sei, T.-S. Kuo and M. Horie, ACS Appl. Mater. Interfaces, 2020, 12, 50002–50010 CrossRef CAS PubMed.
- C.-H. Wang, Y.-C. Lin, S. Bhunia, Y. Feng, P. Kundu, C. L. Stern, P.-L. Chen, J. F. Stoddart and M. Horie, J. Am. Chem. Soc., 2023, 145, 21378–21386 CrossRef CAS PubMed.
- C.-H. Wang, K.-J. Chen, T.-H. Wu, H.-K. Chang, Y. Tsuchido, Y. Sei, P.-L. Chen and M. Horie, Chem. Sci., 2021, 12, 3871–3875 RSC.
- H. Lim, S. J. Woo, Y. H. Ha, Y. H. Kim and J. J. Kim, Adv. Mater., 2022, 34, 2100161 CrossRef CAS PubMed.
- L. Xing, Z.-L. Zhu, J. He, Z. Qiu, Z. Yang, D. Lin, W.-C. Chen, Q. Yang, S. Ji and Y. Huo, J. Chem. Eng., 2021, 421, 127748 CrossRef CAS.
- X. Zeng, D. Zhang, Y. Zhu, M. Chen, H. Chen, S. Kasai, H. Meng and O. Goto, J. Mater. Chem. C, 2019, 7, 14275–14283 RSC.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- T. Salzillo and A. Brillante, CrystEngComm, 2019, 21, 3127–3136 RSC.
- K. Yuhara and K. Tanaka, Angew. Chem., 2024, 63, e202319712 CrossRef CAS PubMed.
- J. Zhong, J. Sun, K. Ye and R. Lu, J. Mater. Chem. C, 2023, 11, 16452–16472 RSC.
- C. J. Perry and G. J. Beran, Cryst. Growth Des., 2023, 23, 8352–8360 CrossRef CAS.
- Y. Sagara, M. Karman, A. Seki, M. Pannipara, N. Tamaoki and C. Weder, ACS Cent. Sci., 2019, 5, 874–881 CrossRef CAS PubMed.
- S. Teka, A. Jebnouni, M. Chemli, G. Attia, Z. Baatout, N. S. Jaballah, M. Ghorbel and M. Majdoub, J. Mol. Struct., 2023, 1289, 135802 CrossRef CAS.
- A. Garci, Y. Beldjoudi, M. S. Kodaimati, J. E. Hornick, M. T. Nguyen, M. M. Cetin, C. L. Stern, I. Roy, E. A. Weiss and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 7956–7967 CrossRef CAS PubMed.
- C.-H. Wang, H.-K. Chang, K.-J. Chen, D.-H. Huang, C.-J. Chang, K.-H. Huang, Y.-D. Chiu and M. Horie, ACS Appl. Mater. Interfaces, 2023, 15, 38846–38856 CrossRef CAS PubMed.
- C. Song, Y. Zhang, J. Bao, Z. Wang, L. Zhang, J. Sun, R. Lan, Z. Yu, S. Zhu and H. Yang, Adv. Funct. Mater., 2023, 33, 2213771 CrossRef CAS.
- M. Li, A. Pal, A. Aghakhani, A. Pena-Francesch and M. Sitti, Nat. Rev. Mater., 2022, 7, 235–249 CrossRef PubMed.
- I. Apsite, S. Salehi and L. Ionov, Chem. Rev., 2021, 122, 1349–1415 CrossRef PubMed.
- Z. Han, P. Wang, G. Mao, T. Yin, D. Zhong, B. Yiming, X. Hu, Z. Jia, G. Nian and S. Qu, ACS Appl. Mater. Interfaces, 2020, 12, 12010–12017 CrossRef CAS PubMed.
- Y. Yang, E. M. Terentjev, Y. Zhang, Q. Chen, Y. Zhao, Y. Wei and Y. Ji, Angew. Chem., Int. Ed., 2019, 58, 17474–17479 CrossRef CAS PubMed.
- B. Zhou, Y. Li, G. Niu, M. Lan, Q. Jia and Q. Liang, ACS Appl. Mater. Interfaces, 2016, 8, 29899–29905 CrossRef CAS PubMed.
- B. Guo, Z. Sheng, D. Hu, C. Liu, H. Zheng and B. Liu, Adv. Mater., 2018, 30, 1802591 CrossRef PubMed.
- Y. Jiang, J. Li, X. Zhen, C. Xie and K. Pu, Adv. Mater., 2018, 30, 1705980 CrossRef PubMed.
- H.-K. Chang, C.-C. Huang, P.-R. Wu, K.-H. Lin and M. Horie, Mater. Today Chem., 2025, 45, 102663 CrossRef CAS.
- A. van Der Lee and D. G. Dumitrescu, Chem. Sci., 2021, 12, 8537–8547 RSC.
- T. J. Jacobsson, L. J. Schwan, M. Ottosson, A. Hagfeldt and T. Edvinsson, Inorg. Chem., 2015, 54, 10678–10685 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. Ashfaq, K. S. Munawar, M. N. Tahir, N. Dege, M. Yaman, S. Muhammad, S. S. Alarfaji, H. Kargar and M. U. Arshad, ACS Omega, 2021, 6, 22357–22366 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, J. Joubert, O. Blacque, N. H. Al-Shaalan and A. A. El-Emam, Sci. Rep., 2019, 9, 19745 CrossRef PubMed.
- H. Aziz, A. Saeed, C. J. McAdam, J. Simpson, T. Hökelek, E. Jabeen, A. Khurshid, M. Saleem and H. R. El-Seedi, J. Mol. Struct., 2023, 1281, 135108 CrossRef CAS.
- N. Tyagi, N. Sinha, H. Yadav and B. Kumar, RSC Adv., 2016, 6, 24565–24576 RSC.
- F. Liu, Z. Cheng, L. Wan, L. Gao, Z. Yan, D. Hu, L. Ying, P. Lu and Y. Ma, J. Chem. Eng., 2021, 426, 131351 CrossRef CAS.
- R. Yao, L. Zhong, Y. Han, C. Jin, J. Lu, F. Chen, L. Liao, Z. Chen, Y. Zhang and G. Chen, J. Phys. Chem. C, 2020, 124, 8885–8893 CrossRef CAS.
- M. C. Drummer, V. Singh, N. Gupta, J. L. Gesiorski, R. B. Weerasooriya and K. D. Glusac, Photosynth. Res., 2022, 151, 163–184 CrossRef CAS PubMed.
- H. Niu, J. Liu, H. M. O’Connor, T. Gunnlaugsson, T. D. James and H. Zhang, Chem. Soc. Rev., 2023, 52, 2322–2357 RSC.
- B. Wang, G. Feng, M. Seifrid, M. Wang, B. Liu and G. C. Bazan, Angew. Chem., Int. Ed., 2017, 56, 16063–16066 CrossRef CAS PubMed.
- H. Li, L. Li, Y. Sha, Y. Lu, C. Teng, D. Zhou, W. Chen and G. Xue, Polymers, 2022, 14, 1652 CrossRef CAS PubMed.
- K.-J. Chen, P.-L. Chen and M. Horie, Sci. Rep., 2017, 7, 14195 CrossRef PubMed.
- K.-J. Chen, A. C. Tan, C.-H. Wang, T.-S. Kuo, P.-L. Chen and M. Horie, Cryst. Growth Des., 2018, 19, 17–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization, crystallographic data, fundamental characteristics, and movies of thermography and film bending. CCDC 2452984. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc02111a |
| This journal is © The Royal Society of Chemistry 2025 |