
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-functional metal–organic framework for efficient removal and fluorescent detection of perfluorooctanoic acid (PFOA) from water†
Rana Dalapati ,
Jiangfan Shi,
Matthew Hunter and
Ling Zang*
,
Jiangfan Shi,
Matthew Hunter and
Ling Zang*
Department of Materials Science and Engineering, University of Utah, Salt Lake City, Utah 84112, USA. E-mail: lzang@eng.utah.edu
First published on 15th July 2025
Abstract
Perfluorooctanoic acid (PFOA), a synthetic compound belonging to the per- and polyfluoroalkyl substances (PFAS) family, is notorious for its environmental persistence, bioaccumulation potential, and adverse health effects, posing a major challenge to environmental safety. Effective removal and detection of PFOA remains a challenge for conventional capture materials due to its unique molecular structure. In this study, we present a dual-functional metal–organic framework (MOF), UiO-66-N(CH3)3+, specifically designed for selective removal and detection of PFOA in water. This MOF is synthesized through the post-synthetic modification of UiO-66-NH2(Zr) with methyl iodide, introducing partially quaternized ammonium groups that enable ion-exchange functionality. The cationic ammonium groups significantly enhance electrostatic interaction with the anionic PFOA, leading to improved affinity and selectivity. As a result, UiO-66-N(CH3)3+ exhibits outstanding adsorption performance, achieving a high adsorption capacity of 1178 mg g−1 as estimated using the Langmuir isotherm model, along with over 99% removal efficiency within 5 minutes from a 50 ppb PFOA solution. Beyond its high sorption capability, the same MOF is also developed into a highly efficient fluorescence “turn-on” sensor for PFOA detection via a straightforward indicator displacement assay (IDA). In this approach, the MOF is initially loaded with the anionic dye sulforhodamine B (SRB), replacing the original iodide counterions. When bound within the MOF, SRB is nonfluorescent; however, upon exposure to PFOA, it is displaced and regains its strong fluorescence in solution. This rapid fluorescence “turn-on” response enables effective detection of PFOA with both high sensitivity and selectivity. The dual-functional MOF system described herein offers a promising strategy for the integrated detection and removal of PFOA from water, providing a simple yet powerful framework for designing multifunctional MOF-based adsorbents and sensors for PFAS pollutants.
Introduction
The per- and polyfluoroalkyl substances (PFAS) constitute a class of synthetic compounds that have attracted widespread scientific attention due to their environmental persistence, global distribution, and potential health and ecological risks.1–3 Perfluorooctanoic acid (PFOA), in particular, is widely used in industrial and consumer products due to its unique chemical stability.4,5 The presence of these compounds in air, water, soil, and biota has raised serious concerns.6Among various remediation strategies, adsorption is considered practical and environmentally sustainable. Commercial sorbents like activated carbon (AC)7 and anion exchange resins (AERs)8 show good PFAS uptake but are limited by slow kinetics,9,10 low reusability,11,12 or poor selectivity.13 To overcome these limitations, synthetic sorbents with functional diversity and tailored architectures have been developed to promote specific PFAS interactions.14,15 However, the mechanisms underlying selectivity remain unclear, and rational design of PFAS-specific sorbents remains an ongoing challenge.
Metal–organic frameworks (MOFs), a class of porous crystalline materials built from metal nodes and organic linkers, have shown strong potential for PFAS removal.16–18 Their high surface area, large internal voids, and tunable chemistry allow for efficient PFAS encapsulation and adsorption via multimodal interactions.19,20 These unique attributes make MOFs highly promising candidates for developing selective and versatile PFAS adsorbents suitable for diverse environmental conditions. Moreover, post-synthetic modification (PSM) offers a powerful strategy for introducing various functional groups into MOFs to enhance their PFAS uptake capacity.21,22 Recent studies on various non-MOF sorbent materials have demonstrated that surface modification with cationic functional groups enhances PFAS capture through strong electrostatic interactions.23–25 Consequently, the design of MOFs incorporating cationic side groups through PSM presents a straightforward and effective approach for enhancing PFAS adsorption performance.
Building on this concept, we developed a dual-functional MOF platform for both PFOA adsorption and detection. Through post-synthetic modification, cationic ammonium groups were introduced into MOF UiO-66-NH2, to create UiO-66-N(CH3)3+ with enhanced ion exchange capacity (Scheme 1). This modified MOF exhibited significantly higher PFOA uptake compared to the unmodified MOF, activated carbon, and other conventional sorbent materials. An impressive adsorption capacity of 1178 mg g−1 was achieved, as estimated by fitting the experimental data to the Langmuir isotherm model, along with fast adsorption kinetics, excellent reusability, and strong salt tolerance.
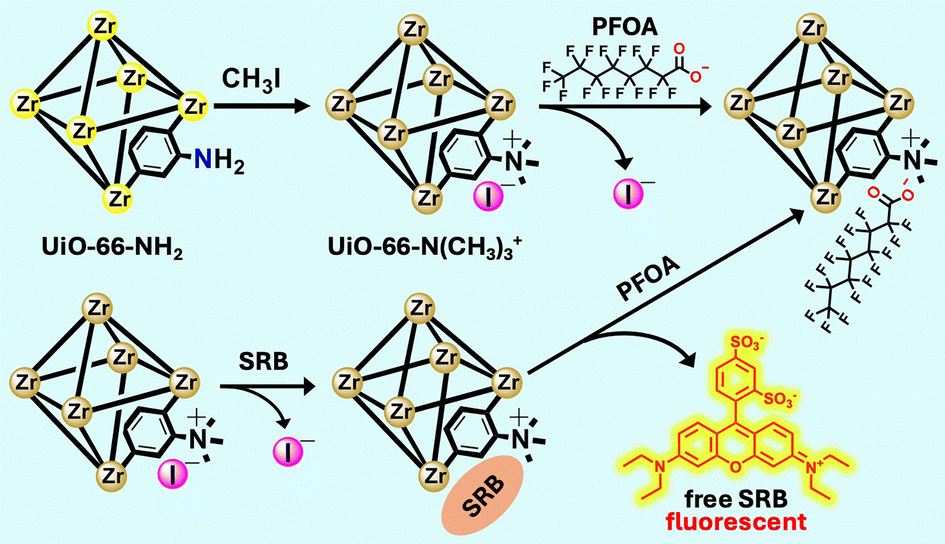 | ||
| Scheme 1 Schematic illustration of the synthesis of UiO-66-N(CH3)3+, adsorption of PFOA via ion exchange, and IDA-based fluorescent sensor for PFOA. | ||
Beyond PFAS removal, the intrinsically high adsorption capacity of cationic MOFs can also be harnessed for developing highly sensitive PFAS sensors. Accurate quantification of PFAS, particularly PFOA, is critical for assessing environmental contamination, understanding exposure pathways, and evaluating health risks. However, conventional detection methods, such as liquid chromatography coupled with mass spectrometry, are limited by complex sample preparation and lengthy analysis times, rendering them impractical for real-time, on-site monitoring.26,27 These challenges have spurred the development of alternative strategies, with fluorescence-based sensors emerging as a promising solution due to their high sensitivity, rapid response, and cost-effectiveness.28–33
MOF-based sensors offer dual functionality by combining PFAS capture with detection. Their high porosity, tunable surface chemistry, and inherent fluorescence responsiveness enable selective binding and real-time monitoring of PFAS via signal modulation. A particularly innovative strategy for designing MOF-based sensors involves the use of indicator displacement assays (IDA), though it remains underexplored for PFAS detection.34–36 In this work, we developed an IDA-based fluorescence sensing strategy using sulforhodamine B (SRB) as an anionic indicator, which is released upon PFOA binding, generating a distinct fluorescence “turn-on” signal (Scheme 1). To the best of our knowledge, this represents the first example of a MOF-based platform that integrates high-capacity PFOA adsorption with IDA-based fluorescence sensing, offering a simple, effective, and dual-functional solution for addressing PFAS contamination.
Experimental
Materials and physical measurements
All the reagents and solvents were commercially available and used as received unless otherwise specified. Milli-Q water was used for the preparation of the analyte stock solutions. Fourier transform infrared (FT-IR) spectra were collected in the region of 500–4000 cm−1 with a Nicolet iS50 FT-IR spectrometer. Ambient-temperature powder X-ray diffraction (PXRD) patterns were measured on a Bruker D2 Phaser X-ray diffractometer operated at 30 kV and 10 mA by using Cu Kα (λ = 1.5406 Å) radiation. The nitrogen sorption isotherms up to 1 bar were recorded using a Micromeritics 3Flex gas sorption analyzer at −196 °C. Before the sorption measurement, the compound was degassed at 80 °C for 24 h under a dynamic vacuum. Steady-state fluorescence spectral measurements were carried out using an Agilent Cary Eclipse fluorescence spectrophotometer. UV-vis spectra were obtained with an Agilent Cary 100 spectrophotometer. NMR data were recorded on a Varian Mercury 400 MHz spectrometer. Scanning electron microscopy (SEM) images were obtained on an FEI Nova NanoSEM scanning electron microscope. Liquid chromatography-mass spectrometry (LC-MS) analysis was performed using an Agilent 1290 Infinity II LC system coupled with an Agilent 6470B Triple Quadrupole mass spectrometer.Synthesis and post synthetic modification of UiO-66-NH2
UiO-66-NH2 (Zr) was synthesized following the procedure reported by Wang et al.37 The resulting MOF was subsequently modified using a previously described method with slight modificationss.38 Specifically, UiO-66-NH2(Zr) (0.5 g) was dispersed in a mixture of methyl iodide (CH3I, 3 mL) and toluene (15 mL). The suspension was transferred to a Teflon-lined autoclave and heated at 80 °C overnight to facilitate quaternization. The resulting dark yellow product, designated as UiO-66-N(CH3)3+, was collected by filtration, washed thoroughly with methanol, and dried at 60 °C. Notably, this approach significantly reduces the reaction time compared to previously reported methods, which required up to three days.39Adsorption kinetics and isotherm measurements
Adsorption experiments of UiO-66-N(CH3)3+ toward PFOA were conducted in aqueous solutions at both high and low concentration regimes to investigate the adsorption kinetics and isotherm behavior. For high-concentration experiments, a PFOA solution of 1000 mg L−1 (1000 ppm) was treated with UiO-66-N(CH3)3+ at a dose of 500 mg L−1. For low-concentration studies, the PFOA solution was prepared at 50 μg L−1 (50 ppb) and treated with 20 mg L−1 of the MOF. In both cases, the fully dried MOF was added to the PFOA solution in 50 mL polypropylene centrifuge tubes and stirred at room temperature. At predetermined time intervals, the supernatant was collected via centrifugation and filtered using a 0.22 μm syringe filter for quantitative analysis; the concentration thus determined was used for calculating the adsorption uptake. Time zero samples were withdrawn before the addition of the MOF. Control experiments were performed under identical conditions without the MOF. To evaluate the influence of co-existing ions, adsorption experiments were repeated in the presence of 1000 ppm of common salts such as NaCl, KCl, CaCl2, MgSO4, and Zn(NO3)2.Quantitative analysis of PFOA at high concentrations (in the ppm range) was performed using 19F NMR. Briefly, 700 μL of the filtered supernatant was transferred to an NMR tube, followed by the addition of 50 μL of trifluoroacetic acid (TFA) in deuterium oxide (D2O) as an internal standard. The 19F NMR measurements were conducted with a 15-second relaxation delay to ensure accurate signal integration.40 The concentration of PFOA before and after adsorption was determined using eqn (1):41
| [PFOA] = IPFOA/ITFA × NTFA/NPFOA[TFA] | (1) |
For trace-level quantification (50 ppb), PFOA concentrations were determined using LC-MS, for which a calibration curve was established using PFOA standards ranging from 0 to 80 ppb (Fig. S10b, ESI†).
The adsorption kinetic data were fitted using the pseudo-first-order (PFO) model for low PFOA concentrations (<50 ppb) and the pseudo-second-order (PSO) model for higher concentrations in the ppm range, as described by eqn (2) and (3), respectively:
| qt = qe(1 − e(−k1·t)) | (2) |
| qt = (k2·qe2·t)/(1 + k2·qe·t) | (3) |
Adsorption isotherm studies were conducted using a fixed MOF dose of 500 mg L−1 and varying initial PFOA concentrations (0, 100, 200, 400, 600, 800, and 1000 ppm). The mixtures were stirred for 60 minutes to reach the adsorption equilibrium, and the adsorption capacities at equilibrium (qe) were fitted to the Langmuir isotherm model:
| qe = (Qmax·KL·Ce)/(1 + KL·Ce) | (4) |
PFOA removal efficiency of UiO-66-N(CH3)3+ was calculated according to eqn (5):42
 | (5) |
To assess the reusability of UiO-66-N(CH3)3+ for adsorption of PFOA, the MOF was recovered by centrifugation and subsequently treated with 50 mL of a washing solution composed of 0.2 M HCl and methanol (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 v/v). The mixture was sonicated at room temperature for 30 minutes, after which the solid was separated again by centrifugation. This washing process was repeated three times to ensure effective recovery of PFOA through ion exchange with chloride ions. The regenerated UiO-66-N(CH3)3+ was then dried under vacuum at 60 °C for 2 hours before being reused in subsequent adsorption cycles.
:70 v/v). The mixture was sonicated at room temperature for 30 minutes, after which the solid was separated again by centrifugation. This washing process was repeated three times to ensure effective recovery of PFOA through ion exchange with chloride ions. The regenerated UiO-66-N(CH3)3+ was then dried under vacuum at 60 °C for 2 hours before being reused in subsequent adsorption cycles.
Synthesis of UiO-66-N(CH3)3+@SRB
The cationic MOF preloaded with sulforhodamine B (SRB), denoted as UiO-66-N(CH3)3+@SRB, was prepared via an ion exchange method. Briefly, a thoroughly dried sample of UiO-66-N(CH3)3+ (50 mg) was immersed in 50 mL of a 50 mM aqueous solution of sulforhodamine B (SRB) for 15 minutes. The resulting solid was then washed thoroughly with water until the supernatant exhibited no detectable fluorescence, indicating the complete removal of unbound dye. The final product was isolated by centrifugation.Testing of UiO-66-N(CH3)3+@SRB as an IDA-based fluorescent sensor for PFOA
UiO-66-N(CH3)3+@SRB (10.0 mg) was dispersed in 10 mL of acetonitrile and sonicated for 30 minutes to obtain a homogeneous suspension. A 200 μL aliquot of this suspension was transferred to a quartz cuvette and diluted to a final volume of 3 mL with acetonitrile. Stock solutions of various analytes, including perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS) potassium salt, sodium dodecyl sulfate (SDS), lauric acid, octanoic acid, acetic acid, dodecane, phenol, and nitrobenzene, were prepared at a concentration of 0.5 mM in deionized (DI) water, with a minimal amount of acetonitrile added to aid the dissolution of less soluble analytes. The emission spectra of the MOF suspension were recorded in the presence of varying concentrations (0–5 μM) of each analyte using a 1 × 1 cm quartz cuvette, with the excitation wavelength set to 520 nm. After thorough mixing at room temperature for 10 minutes, the fluorescence emission spectra were measured. The initial emission intensity in the absence of any analyte was recorded as I0, and I denoted the emission intensity of the suspension in the presence of a given analyte.Results and discussion
Synthesis and structural characterization
As previously described, the cationic MOF UiO-66-N(CH3)3+ with iodide as the counterion was synthesized via post-synthetic modification of UiO-66-NH2 (Zr) using iodomethane in toluene. The quaternization process was visually indicated by a color change from pale yellow to dark yellow. Subsequent anion exchange with sulforhodamine B (SRB) in water yielded the IDA-based sensor UiO-66-N(CH3)3+@SRB, which appeared as a pink-colored powder (Fig. S1, ESI†). The resulting material was thoroughly characterized by PXRD, FT-IR, 1H-NMR, SEM, and N2 sorption analysis to confirm its structural and chemical modifications.UiO-66-NH2(Zr) was selected as the starting material due to its well-established chemical stability and the reactivity of its amine functional groups, which enable diverse post-synthetic transformations.43–45 The PXRD pattern of UiO-66-N(CH3)3+ closely matches that of the parent UiO-66-NH2(Zr), with minimal loss of crystallinity, indicating that the MOF retaines its structural integrity throughout the quaternization process (Fig. 1a). Furthermore, the PXRD pattern of UiO-66-N(CH3)3+@SRB remained largely unchanged, confirming that the anion exchange with SRB did not significantly impact the MOF's crystalline framework.
 | ||
| Fig. 1 (a) PXRD patterns, (b) FT-IR spectra, and (c) N2 adsorption isotherms as measured for UiO-66-NH2 and UiO-66-N(CH3)3+ (free or loaded with SRB). | ||
FT-IR analysis was performed to gain insight into the cationic modification of the MOF and the subsequent ion exchange process involving SRB molecules. The FT-IR spectrum of the unmodified UiO-66-NH2(Zr) displays characteristic peaks, including a C–O stretching vibration at 1384 cm−1, an O–Zr–O vibration at 664 cm−1, and prominent bands at 3316 cm−1 and 3480 cm−1 corresponding to the –NH2 functional groups, confirming the successful formation of the MOF framework (Fig. 1b and S2, ESI†).46–48 After quaternization to form UiO-66-N(CH3)3+, a new peak appeared at 1418 cm−1, attributed to –CH3 vibrations. In addition, the C–N stretching vibration, originally observed at 1258 cm−1 in UiO-66-NH2, shifted to 1280 cm−1 in UiO-66-N(CH3)3+, indicating the formation of a new quaternary ammonium group. Importantly, the O–Zr–O vibration at 664 cm−1 remained unchanged, suggesting that the MOF framework remained intact during the quaternization process. The FT-IR spectrum of UiO-66-N(CH3)3+@SRB exhibited new peaks at 1180, 1130, 1075, and 1029 cm−1, which can be attributed to vibrational modes of the SRB molecules. These results confirm the successful incorporation of SRB within the MOF structure (Fig. S3, ESI†).
The Brunauer–Emmett–Teller (BET) surface area was measured via nitrogen sorption experiments to assess changes in porosity of the MOF at different stages: prior to quaternization, after quaternization, and following anion exchange with SRB fluorophores. As shown in Fig. 1c, the MOF retained significant porosity even after the introduction of quaternized amine functional groups. The surface area decreased from 1007 m2 g−1 for UiO-66-NH2(Zr) to 722 m2 g−1 for UiO-66-N(CH3)3+, which is attributed to reduced internal void space resulting from the incorporation of bulky quaternary ammonium groups. A slight further decrease in surface area to 688 m2 g−1 was observed for UiO-66-N(CH3)3+@SRB, compared to the quaternized MOF with iodide as the counterion. This modest reduction is likely attributed to the larger size of SRB relative to iodide.
The extent of the quaternization reaction was confirmed through detailed 1H NMR analysis. For this purpose, the MOF powder was digested using hydrofluoric acid, and the resulting solution was dissolved in DMSO-d6 for NMR measurements. The 1H NMR spectrum of the digested parent UiO-66-NH2(Zr) MOF displayed characteristic peaks at 7.01, 7.36, and 7.75 ppm, corresponding to the three aromatic protons of the aminoterephthalate ligand (Fig. S4, ESI†).49 Following modification, a new peak appeared at 7.19 ppm, consistent with the formation of the quaternized ammonium ligand. Additional peaks at 7.05 and 7.08 ppm were observed, which are indicative of residual primary and secondary amine-containing species, respectively.39 By analyzing the integrated peak areas corresponding to the NH2-BDC ligand and the quaternized products, the conversion efficiency of the quaternization reaction was estimated to be approximately 52%.
The same digestion protocol was employed to verify the successful exchange of SRB within the MOF structure. The 1H NMR spectrum of the digested UiO-66-N(CH3)3+@SRB sample exhibited new peaks at 1.19, 3.62, 6.89, 7.02, 7.14, 7.71, and 8.26 ppm, all of which matched the spectrum of free SRB molecules (Fig. S5, ESI†). These results confirm the successful formation of UiO-66-N(CH3)3+@SRB via ion exchange with the SRB fluorophore.
SEM imaging was used to examine the morphology of both UiO-66-NH2(Zr) and UiO-66-N(CH3)3+. As shown in Fig. S6 (ESI†), both samples exhibited uniform distribution of MOF particles with sizes below 100 nm. The small particle size enables stable and homogeneous dispersion in solution, which is advantageous for sensor testing and facilitates quantitative analysis with high reproducibility. Clearly, as evidenced by the SEM images, the morphology of the MOF is well preserved following post-synthetic modification.
PFOA adsorption capacity and selectivity
UiO-type MOFs are well known for their exceptional mechanical, chemical, and thermal stability.50,51 The unique combination of the highly stable UiO-66-type framework, high crystallinity, inherent porosity, and the incorporation of cationic ammonium functional groups, along with recent progress in MOF-based PFOA adsorbents,51–53 positions the cationic MOF UiO-66-N(CH3)3+ as a highly promising candidate for PFOA adsorption. To evaluate its adsorption performance, UiO-66-N(CH3)3+ was tested against an aqueous PFOA solution with an initial concentration of 1000 ppm at room temperature. The adsorption experiments were performed using a UiO-66-N(CH3)3+ dosage of 500 mg L−1. After 30 minutes of stirring, the PFOA-loaded MOF (PFOA@UiO-66-N(CH3)3+) was separated, and the residual PFOA concentration in the supernatant was quantified via 19F NMR spectroscopy using trifluoroacetic acid (TFA) as an internal standard (Fig. 2). The resulting data revealed a PFOA uptake of 665 mg g−1 within 30 minutes. Importantly, the PFOA uptake from the 1000 ppm solution was found to decrease with reduced MOF dosage, as shown in Fig. S7 (ESI†), indicating a dosage-dependent removal efficiency. For comparative analysis, other benchmark adsorbents, including UiO-66, UiO-66-NH2, ZIF-8, and activated carbon, were also evaluated under identical conditions. Among them, UiO-66-N(CH3)3+ exhibited the highest PFOA uptake (Fig. 3a). Remarkably, despite having a lower surface area, the quaternized UiO-66-N(CH3)3+ showed a 3.4-fold enhancement in adsorption capacity compared to its parent framework, UiO-66-NH2. This substantial improvement underscores the pivotal role of surface functionalization in enhancing MOF-based adsorbents for PFOA removal. This promising adsorption performance prompted us to explore the kinetics of the adsorption process in greater detail. As depicted in Fig. 3b, the adsorption follows a pseudo-second-order kinetic model, as fitted to eqn (3) with an excellent correlation coefficient (R2 = 0.99). The fitting yielded a high rate constant (k2) of 5.6 ± 0.2 × 10−3 g mg−1 h−1, indicating a rapid kinetics. Notably, the rate constant is nearly four times greater than that of a recently reported zirconium-based MOF (PCN-999) employed for PFOA adsorption.52 It is important to note that maintaining molecular dispersion of PFOA is essential for accurate concentration measurements, and consequently, for reliable assessment of MOF adsorption performance. At elevated concentrations, PFOA can form micelles in aqueous solution, potentially leading to aggregation near the adsorbent surface and affecting measurement accuracy.13,25 Given that the critical micelle concentration (CMC) of PFOA in water exceeds 10000 ppm,54 all PFOA concentrations used in this study were maintained at least tenfold below the CMC to ensure stable molecular dispersion of PFOA.
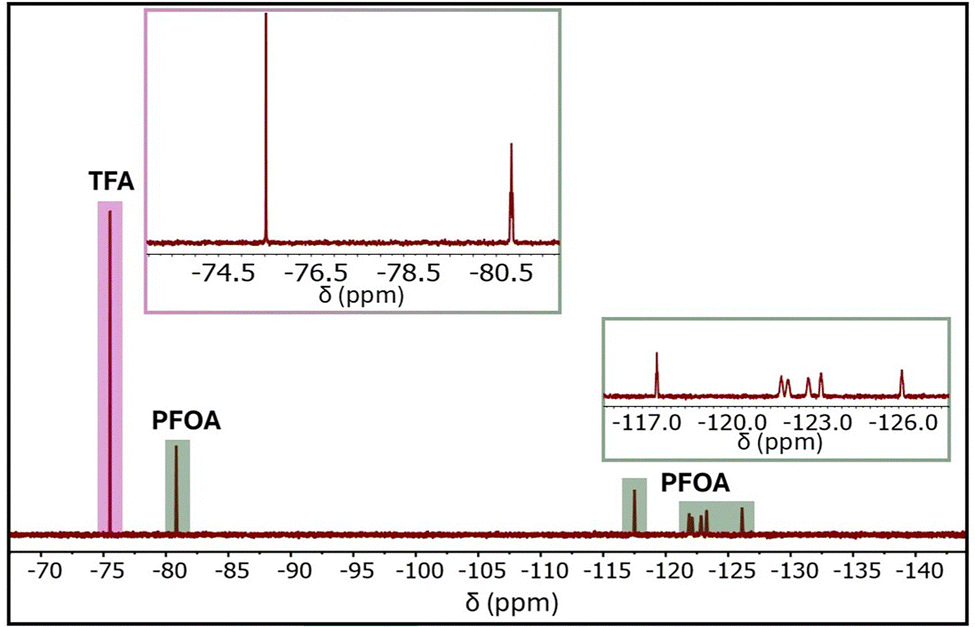 | ||
| Fig. 2 Representative 19F NMR spectrum of PFOA with TFA as the internal standard, where the peak at −75.53 ppm (–CF3 of TFA) and the peak at −80.82 ppm (–CF3 of PFOA) were used for quantitative analysis. Additional multiplets between −117 and −126 ppm correspond to the –CF2 groups along the PFOA chain. Insets show magnified views of the –F3 region (top left) and the –CF2 region (bottom right), highlighting the chemical shifts used for signal assignment. | ||
 | ||
| Fig. 3 (a) The PFOA uptake obtained for different adsorbents. (b) Adsorption kinetics of PFOA by UiO-66-N(CH3)3+ (500 mg L−1) with an initial concentration of 1000 ppm, fitted with a pseudo-second-order model. (c) Equilibrium PFOA uptake as a function of PFOA concentration (Ce), fitted using the Langmuir isotherm model. | ||
The adsorption isotherm of PFOA on UiO-66-N(CH3)3+ was established using experimental data collected over an initial concentration range of 0 to 1000 ppm. The data were fitted to the Langmuir isotherm model (Fig. 3c), yielding a high correlation coefficient (R2 = 0.99), indicating excellent model agreement. The maximum adsorption capacity for PFOA was calculated to be 1178 mg g−1 from the Langmuir model, demonstrating the outstanding performance of the material. To the best of our knowledge, this remarkably high uptake surpasses that of most previously reported adsorbents, including other MOF-based materials.53,55–58
To assess the performance of UiO-66-N(CH3)3+ in the presence of salts commonly found in real water samples, the same PFOA adsorption experiments were conducted in the presence of various potentially interfering salts (1000 ppm), including KCl, CaCl2, MgCl2, Na2SO4, and Zn(NO3)2. As shown in Fig. S8 (ESI†), UiO-66-N(CH3)3+ maintained a high PFOA adsorption capacity despite the presence of these salts at high concentrations. This result highlights the MOF's exceptional selectivity for PFOA, even in complex aqueous environments with high ionic strength.
The exceptional PFOA adsorption capacity of UiO-66-N(CH3)3+ prompted further evaluation of its removal efficiency. As shown in Fig. 4, the MOF demonstrated an outstanding removal efficiency of over 97% for a PFOA solution with an initial concentration of 100 ppm. To assess its performance under more environmentally relevant and scalable conditions, a lower PFOA concentration of 50 ppb was also tested using a reduced adsorbent dosage of 20 mg L−1. Remarkably, LC-MS analysis confirmed that over 99% of PFOA was removed from water within just 5 minutes (Fig. S9, ESI†), underscoring the material's rapid and highly effective adsorption capability at trace levels. The time-dependent adsorption behavior at low PFOA concentrations followed a pseudo-first-order kinetic model (Fig. S10, ESI†), in contrast to the pseudo-second-order kinetics observed at higher concentrations. This shift likely arises from the change in the adsorption equilibrium. At low PFOA concentrations, the number of available MOF adsorption sites greatly exceeds the number of PFOA molecules, allowing the concentration of adsorption sites to be treated as effectively constant. Under these conditions, the adsorption rate is primarily governed by the PFOA concentration, resulting in pseudo-first-order kinetics. In contrast, at higher PFOA concentrations, the assumption of constant adsorption site concentration no longer holds, and the adsorption behavior is better described by the pseudo-second-order model.
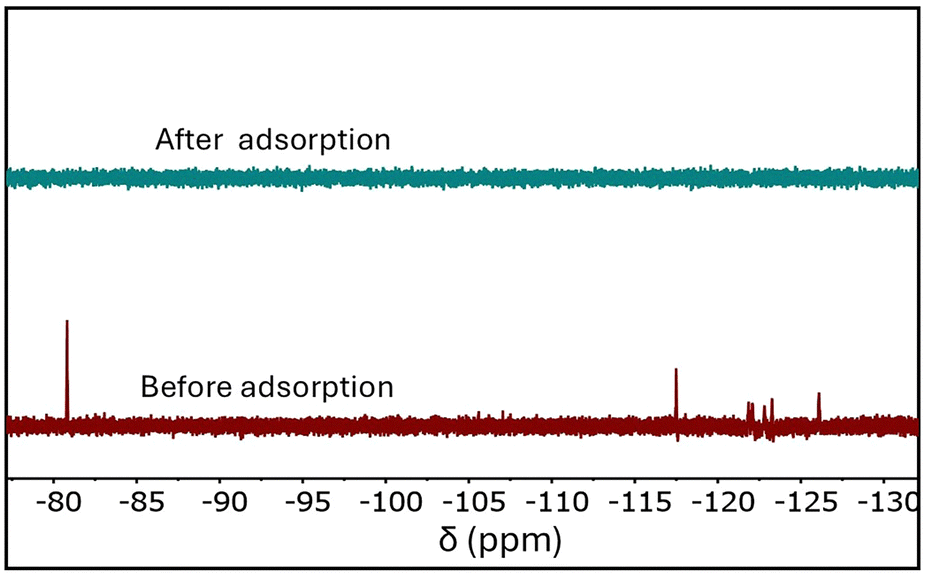 | ||
| Fig. 4 19F NMR spectrum of a 100 ppm PFOA solution before (bottom) and after (top) adsorption by UiO-66-N(CH3)3+ (500 mg L−1). | ||
To assess its practical applicability, the reusability of UiO-66-N(CH3)3+ was also investigated through repeated adsorption–desorption cycles. After each adsorption step, the recovered MOF was washed with a 30:70 (v/v) mixture of 0.2 M HCl and methanol, then dried for reuse. As illustrated in Fig. S11a (ESI†), the material retained over 93% of its original PFOA adsorption capacity after five consecutive cycles. The slight decrease in efficiency was primarily attributed to minor material losses during washing and centrifugation. Notably, the removal efficiency remained consistently above 97% throughout all cycles, as confirmed by 19F NMR analysis (Fig. S11b, ESI†).
These results underscore the excellent reusability, chemical stability, and structural integrity of UiO-66-N(CH3)3+ for PFOA removal. The material's consistent performance across multiple cycles highlights its robustness and strong potential for real-world water purification and continuous environmental remediation applications.
Mechanism of PFOA adsorption
To gain deeper insight into the mechanism of PFOA uptake by UiO-66-N(CH3)3+, particularly via anion exchange, a series of adsorption experiments were performed using varying concentrations of PFOA (0–1000 ppm). The structural integrity of UiO-66-N(CH3)3+ remained intact even after treatment with 1000 ppm PFOA, as confirmed by PXRD analysis (Fig. S12, ESI†), indicating the robustness of the MOF framework during the adsorption process. FT-IR spectra of UiO-66-N(CH3)3+ after PFOA adsorption from a 100 ppm solution revealed the emergence of new peaks at approximately 1240, 1204, and 1149 cm−1, corresponding to the asymmetric stretching of –CF2, asymmetric stretching of –CF3, and symmetric stretching of –CF2 groups, respectively (Fig. S13, ESI†).59,60 These characteristic signals confirm the successful incorporation of PFOA within the MOF. Notably, the intensities of these peaks increased progressively with higher PFOA concentrations, further supporting the enhanced uptake capacity at elevated concentrations. Importantly, the Zr–O vibrational band at 664 cm−1 remained unchanged after PFOA adsorption, indicating that the coordination environment around the zirconium centers was not affected. Additionally, the symmetric C–O stretching band (1415–1380 cm−1) associated with the ligand coordination at the metal center did not exhibit any noticeable shift, suggesting that the binding of PFOA primarily occurs via electrostatic interactions with the cationic ammonium groups rather than coordination with the Zr nodes. These observations support an anion exchange mechanism as the primary mode of PFOA adsorption.Further elemental mapping and EDS analysis were conducted to investigate the anion exchange between PFOA and iodide. Fig. S14 and S15 (ESI†) present the elemental mapping and EDS spectra of UiO-66-N(CH3)3+ prior to PFOA uptake, confirming the presence of iodide as the initial counterion. Following treatment with 1000 ppm PFOA, the elemental mapping and EDS spectra (Fig. S16 and S17, ESI†) revealed a strong fluoride signal, indicating substantial adsorption of PFOA via replacement of the original iodide anions. As summarized in Table S1 and Fig. S18 (ESI†), the iodine content within the MOF decreased progressively with increasing PFOA concentration, while the fluorine content simultaneously increased. Consequently, the fluoride-to-iodide (F/I) ratio showed a consistent upward trend with higher PFOA loading, providing further evidence for an anion exchange process. Collectively, these results strongly support the conclusion that PFOA displaces iodide in UiO-66-N(CH3)3+ due to its stronger and more competitive binding affinity for the quaternized ammonium groups. This anion exchange mechanism underpins the material's high selectivity and efficiency for PFOA adsorption.
IDA-based fluorescence turn-on detection of PFOA
Building on the exceptional PFOA uptake demonstrated by UiO-66-N(CH3)3+ via ion exchange, we harnessed this property to develop a novel fluorescence-based sensing platform for PFOA detection. The cationic framework of UiO-66-N(CH3)3+ was strategically employed to construct a fluorescent indicator displacement assay (IDA) with high selectivity toward PFOA.61 Specifically, the iodide counterions in UiO-66-N(CH3)3+ were first replaced with sulforhodamine B (SRB), an anionic fluorescent dye widely used in cell biology.62 Efficient dye loading was facilitated by electrostatic interactions between the positively charged quaternary ammonium groups of the MOF and the negatively charged sulfonate groups of SRB.Following ion exchange with SRB, the resulting composite, UiO-66-N(CH3)3+@SRB, exhibited a fluorescence “turn-off” state, marked by substantial quenching of SRB emission. This quenching effect prompted us to explore its potential as a “turn-on” fluorescence sensor for PFOA. Fluorescence emission spectra of the UiO-66-N(CH3)3+@SRB suspension in acetonitrile were recorded upon the incremental addition of minimal volume of aqueous stock solution of PFOA (0.5 mM). As shown in Fig. 5a, a clear fluorescence “turn-on” response was observed with increasing PFOA concentrations (0–5 μM). The obtained emission spectra closely resemble those of free SRB molecules in acetonitrile solution, confirming that SRB molecules are released into the solution upon adsorption of PFOA. In contrast, PFOS, one of the top two concerning PFAS compounds identified by the EPA, did not induce any fluorescence recovery (Fig. S19, ESI†). Notably, the system exhibited a 14-fold higher selectivity for PFOA over PFOS, despite their structural similarities.63 The fluorescence enhancement ratio (I/I0) at 574 nm reached up to 21, indicating a strong signal-to-background ratio. The fluorescence increase was also readily visible under UV illumination. The detection limit (LOD) for PFOA was determined to be 0.22 μM within the 0-3 μM dynamic concentration range, based on the standard IUPAC method (3σ) (Fig. 5b).
 | ||
| Fig. 5 (a) Emission spectra of UiO-66-N(CH3)3+@SRB in the presence of increasing concentrations of PFOA (0–5 μM). Plot of the emission intensity at 574 nm as a function of PFOA concentration. (c) Fluorescence turn-on response of UiO-66-N(CH3)3+@SRB toward various analytes tested at the same concentration, highlighting its selectivity for PFOA. | ||
To further assess the sensor's selectivity, we evaluated its response to a range of potentially interfering analytes commonly present in aquatic environments, including lauric acid, octanoic acid, acetic acid, trifluoroacetic acid, sodium dodecyl sulfate (SDS), dodecane, phenol, and nitrobenzene. Interestingly, structurally similar non-fluorinated alkyl carboxylic acids such as lauric and octanoic acid did not induce a “turn-on” fluorescence response in the UiO-66-N(CH3)3+@SRB system (Fig. 5c and Fig. S20, ESI†). Similarly, smaller carboxylic acids (e.g., acetic and trifluoroacetic acid) and other small organic molecules (e.g., phenol and nitrobenzene) failed to generate any detectable fluorescence enhancement under the same conditions. Collectively, these results demonstrate that UiO-66-N(CH3)3+@SRB is highly selective for PFOA, supporting its potential as a sensitive and robust platform for detecting this high-priority PFAS compound identified by the EPA.
In addition to the fluorescence measurements presented in Fig. 5a, the IDA-based fluorescent sensor was further characterized using UV-vis absorption and NMR spectroscopy. Initially, UV-vis absorption spectra were measured for the centrifuged supernatant of UiO-66-N(CH3)3+@SRB suspensions before and after PFOA addition. Prior to adding PFOA, the supernatant exhibited no visible color or characteristic absorption peaks corresponding to free SRB molecules. However, following PFOA addition, the supernatant turned distinctly pink and showed a strong absorption peak at 553 nm, accompanied by a shoulder near 514 nm (Fig. 6). This absorption spectrum closely matched that of a standard SRB molecular solution, confirming that PFOA addition induces the release of SRB molecules into the solution.
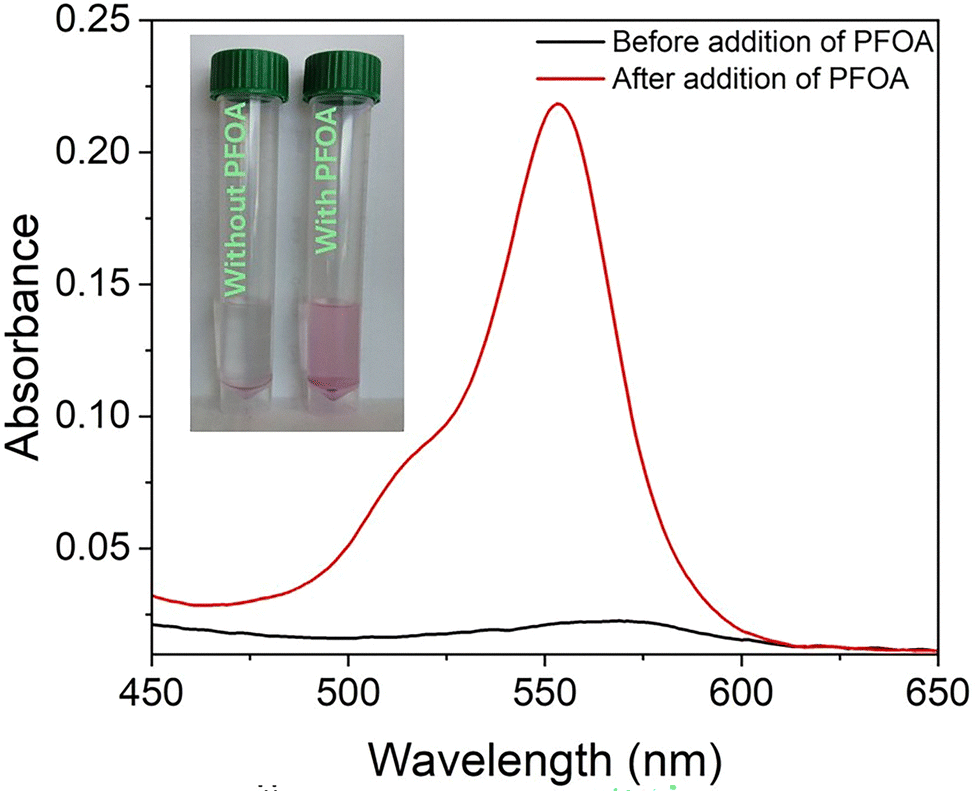 | ||
| Fig. 6 UV-Vis absorption spectra of UiO-66-N(CH3)3+@SRB with and without PFOA. Inset shows the release of SRB dye into solution upon exposure to PFOA. | ||
The same supernatant samples obtained from the UiO-66-N(CH3)3+@SRB suspensions before and after PFOA addition were further analyzed using 1H-NMR spectroscopy. Prior to analysis, the supernatants were dried through solvent evaporation to isolate any residual compounds. The resulting residues were then dissolved in DMSO-d6 for 1H-NMR spectral analysis. As illustrated in Fig. S21 (ESI†), the presence of PFOA yielded prominent SRB-characteristic peaks at 8.2, 7.7, 7.1, and 6.9 ppm, whereas no significant signals appeared in the absence of PFOA. These findings, combined with the UV-vis and fluorescence spectral data, robustly confirm the IDA-based sensing mechanism. As outlined in Scheme 1, this mechanism involves the displacement-driven release of SRB molecules upon adsorption of PFOA, underscoring the sensor's effective and selective detection capability for PFOA.
Conclusions
In summary, we report the synthesis and comprehensive evaluation of quaternized UiO-66-N(CH3)3+ as a dual-functional MOF material for both adsorption and fluorescence-based detection of PFOA in water, offering a promising strategy for PFOA remediation. The MOF exhibits a remarkable maximum adsorption capacity of 1178 mg g−1, along with high selectivity and sensitivity for PFOA detection through an IDA-based fluorescence “turn-on” mechanism driven by electrostatic interactions and ion exchange. The material demonstrates excellent stability and reusability, retaining over 93% of its adsorption efficiency after five regeneration cycles, even in the presence of common salts found in aqueous environments. This highlights its practical potential for real-world water treatment applications. Moreover, the integration of adsorptive and sensing functionalities underscores the versatility of this MOF platform for environmental monitoring.This work further emphasizes the utility of post-synthetic modification in MOF design, enabling the development of advanced PFAS adsorbents and selective fluorescence probes. The findings presented here can be readily extended to other MOF systems, paving the way for future enhancements in adsorption capacity, kinetics, selectivity, and sensitivity. Efforts such as tailoring functional groups and crystal structures will contribute to the advancement of scalable, efficient technologies for water purification and environmental sensing.
Author contributions
R. D. performed the synthesis of materials, characterization, PFAS adsorption, and fluorescence studies. J. S. and M. H. carried out additional characterizations. R. D. and L. Z. contributed to manuscript writing. R. D. and L. Z. were responsible for project design, conceptualization, supervision, data interpretation, and editing. All authors approved the final version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
Ling Zang has a significant financial interest in Gentex Corporation, which funded this research.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Acknowledgements
The authors are very grateful to Gentex Corporation for research sponsorship under award #10072348, and University of Utah for instrumental facilities and other support.References
- M. T. Islam, J. Cheng, A. A. Sadmani, D. Reinhart and N.-B. Chang, J. Hazard. Mater., 2024, 474, 134646 CrossRef CAS PubMed.
- M.-E. Dimitrakopoulou, M. Karvounis, G. Marinos, Z. Theodorakopoulou, E. Aloizou, G. Petsangourakis, M. Papakonstantinou and G. Stoitsis, npj Sci. Food, 2024, 8, 80 CrossRef PubMed.
- R. Lohmann, I. T. Cousins, J. C. DeWitt, J. Gluge, G. Goldenman, D. Herzke, A. B. Lindstrom, M. F. Miller, C. A. Ng and S. Patton, Environ. Sci. Technol., 2020, 54, 12820–12828 CrossRef CAS PubMed.
- C. M. Teaf, M. M. Garber, D. J. Covert and B. J. Tuovila, Soil Sediment Contam., 2019, 28, 258–273 CrossRef CAS.
- K. Sznajder-Katarzyńska, M. Surma and I. Cieślik, J. Chem., 2019, 2019, 2717528 Search PubMed.
- S. Garg, P. Kumar, V. Mishra, R. Guijt, P. Singh, L. F. Dumée and R. S. Sharma, J. Water Process Eng., 2020, 38, 101683 CrossRef.
- B. Cantoni, A. Turolla, J. Wellmitz, A. S. Ruhl and M. Antonelli, Sci. Total Environ., 2021, 795, 148821 CrossRef CAS PubMed.
- Y. Fang, A. Ellis, Y. J. Choi, T. H. Boyer, C. P. Higgins, C. E. Schaefer and T. J. Strathmann, Environ. Sci. Technol., 2021, 55, 5001–5011 CrossRef CAS PubMed.
- A. K. Ilango, T. Jiang, W. Zhang, M. N. Pervez, J. I. Feldblyum, H. Efstathiadis and Y. Liang, ACS ES&T Water, 2023, 3, 3708–3715 Search PubMed.
- A. Maimaiti, S. Deng, P. Meng, W. Wang, B. Wang, J. Huang, Y. Wang and G. Yu, Chem. Eng. J., 2018, 348, 494–502 CrossRef CAS.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- C. C. Murray, R. E. Marshall, C. J. Liu, H. Vatankhah and C. L. Bellona, J. Water Process Eng., 2021, 44, 102342 CrossRef.
- Y. Wang, S. B. Darling and J. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 60789–60814 CrossRef CAS PubMed.
- Y. Zhi and J. Liu, Chemosphere, 2016, 144, 1224–1232 CrossRef CAS PubMed.
- P. S. Pauletto and T. J. Bandosz, J. Hazard. Mater., 2022, 425, 127810 CrossRef CAS PubMed.
- N. Ilić, K. Tan, F. Mayr, S. Hou, B. M. Aumeier, E. M. C. Morales, U. Hübner, J. Cookman, A. Schneemann and A. Gagliardi, Adv. Mater., 2025, 37, 2413120 CrossRef PubMed.
- J. Pala, T. Le, M. Kasula and M. R. Esfahani, Sep. Purif. Technol., 2023, 309, 123025 CrossRef CAS.
- Y. Qi, F. Li, C. Zhang, X. Tang, C. Wang, Y. Liang, H. Sun and B. Xing, ACS ES&T Eng., 2025, 5, 1405 Search PubMed.
- X. Xu, M. Ma, T. Sun, X. Zhao and L. Zhang, Biosensors, 2023, 13, 435 CrossRef CAS PubMed.
- Z. Chang, Coord. Chem. Rev., 2023, 476, 214921 CrossRef.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Adv. Funct. Mater., 2021, 31, 2006291 CrossRef CAS.
- W. He, D. Lv, Y. Guan and S. Yu, J. Mater. Chem. A, 2023, 11, 24519–24550 RSC.
- X. Liu, C. Zhu, J. Yin, J. Li, Z. Zhang, J. Li, F. Shui, Z. You, Z. Shi and B. Li, Nat. Commun., 2022, 13, 2132 CrossRef CAS PubMed.
- S. Tafazoli, D. B. Shuster, A. Shahrokhinia, S. Rijal, D. M. Ruhamya, K. A. Dubray, D. J. Morefield and J. F. Reuther, ACS Appl. Mater. Interfaces, 2025, 17, 10732–10744 CrossRef CAS PubMed.
- A. Jrad, G. Das, N. Alkhatib, T. Prakasam, F. Benyettou, S. Varghese, F. Gándara, M. Olson, S. Kirmizialtin and A. Trabolsi, Nat. Commun., 2024, 15, 1–17 Search PubMed.
- M. C. Corviseri, A. Polidoro, M. De Poli, C. Stevanin, T. Chenet, C. D’Anna, A. Cavazzini, L. Pasti and F. A. Franchina, Talanta, 2025, 292, 127944 CrossRef CAS PubMed.
- S. R. Newton, J. A. Bowden, N. Charest, S. R. Jackson, J. P. Koelmel, H. K. Liberatore, A. M. Lin, C. N. Lowe, S. Nieto and K. J. Godri Pollitt, Environ. Sci. Technol., 2025, 12, 104–112 CAS.
- A. Concellón, J. Castro-Esteban and T. M. Swager, J. Am. Chem. Soc., 2023, 145, 11420–11430 CrossRef PubMed.
- R. Dalapati, S. Manickam, J. Shi, M. Hunter and L. Zang, Anal. Chim. Acta, 2025, 1341, 343670 CrossRef CAS PubMed.
- E. E. Harrison and M. L. Waters, Chem. Sci., 2023, 14, 928–936 RSC.
- Y.-D. Yang, Q. Zhang, X. Jin, C. V. Chau, J. Yang, G. Henkelman, X. Chi, H.-Y. Gong, R. J. Hooley and J. L. Sessler, Angew. Chem., Int. Ed., 2025, e202501245 CAS.
- B. Chen, Z. Yang, X. Qu, S. Zheng, D. Yin and H. Fu, ACS Appl. Mater. Interfaces, 2021, 13, 47706–47716 CrossRef CAS PubMed.
- R. Dalapati, M. Hunter, M. Sk, X. Yang and L. Zang, ACS Appl. Mater. Interfaces, 2024, 16, 32344–32356 CrossRef CAS PubMed.
- Q. Chen, M. Han, J. Chen, H. Zhou, X. Song, Y. Wu and W. Liu, Sens. Actuators, B, 2024, 136660 Search PubMed.
- X. Cong, J. Han, J. Cuan, Z. Wu, S. Zhu, Q. Li and Y. Zhou, ACS Appl. Nano Mater., 2023, 6, 18394–18402 CrossRef CAS.
- N.-N. Yang, L.-J. Zhou, P. Li, Q. Sui and E.-Q. Gao, Chem. Sci., 2019, 10, 3307–3314 RSC.
- X.-Y. Xu, C. Chu, H. Fu, X.-D. Du, P. Wang, W. Zheng and C.-C. Wang, Chem. Eng. J., 2018, 350, 436–444 CrossRef CAS.
- G. Wu, J. Ma, S. Li, S. Wang, B. Jiang, S. Luo, J. Li, X. Wang, Y. Guan and L. Chen, Environ. Res., 2020, 186, 109542 CrossRef CAS PubMed.
- X. Tang, X. Wu, H. Wu, X. Zhang, M. Feng, T. Ouyang, H. Wang and R. Ou, Chem. Commun., 2023, 59, 4507–4510 RSC.
- J. R. Gauthier and S. A. Mabury, ACS ES&T Water, 2024, 4, 4615 Search PubMed.
- D. Camdzic, R. A. Dickman, A. S. Joyce, J. S. Wallace, P. L. Ferguson and D. S. Aga, Anal. Chem., 2023, 95, 5484–5488 CrossRef CAS PubMed.
- P. McCleaf, S. Englund, A. Östlund, K. Lindegren, K. Wiberg and L. Ahrens, Water Res., 2017, 120, 77–87 CrossRef CAS PubMed.
- R. Dalapati and S. Biswas, Sens. Actuators, B, 2017, 239, 759–767 CrossRef CAS.
- L. Mohammadi, R. Taghavi, M. Hosseinifard, M. R. Vaezi and S. Rostamnia, Sci. Rep., 2023, 13, 9051 CrossRef CAS PubMed.
- Z. Wang, Q. Liu, X. Sun, C. Xia, Q. Yin, M. Wang, X. Chen, H. Zhang, S. Wei and X. Lu, ACS Appl. Polym. Mater., 2024, 7, 386–395 CrossRef.
- M.-A. Rezaie, A. Khojastehnezhad and A. Shiri, Sci. Rep., 2024, 14, 24644 CrossRef CAS PubMed.
- F. Moghadaskhou, A. Tadjarodi and A. Maleki, ACS Appl. Mater. Interfaces, 2024, 16, 41048–41059 CrossRef CAS PubMed.
- L. Jin, H. Liu, A. Xu, Y. Wu, J. Lu, J. Liu, S. Xie, Y. Yao, L. Dong and M. Zhang, Microporous Mesoporous Mater., 2021, 317, 110997 CrossRef CAS.
- H. Guo, J. Liu, Y. Li, J. Caro and A. Huang, Microporous Mesoporous Mater., 2021, 313, 110823 CrossRef CAS.
- J. Winarta, B. Shan, S. M. Mcintyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2019, 20, 1347–1362 CrossRef.
- R. Li, S. Alomari, T. Islamoglu, O. K. Farha, S. Fernando, S. M. Thagard, T. M. Holsen and M. Wriedt, Environ. Sci. Technol., 2021, 55, 15162–15171 CrossRef CAS PubMed.
- R.-R. Liang, S. Xu, Z. Han, Y. Yang, K.-Y. Wang, Z. Huang, J. Rushlow, P. Cai, P. Samorì and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 9811–9818 CrossRef CAS PubMed.
- R. R. Liang, Y. Yang, Z. Han, V. I. Bakhmutov, J. Rushlow, Y. Fu, K. Y. Wang and H. C. Zhou, Adv. Mater., 2024, 36, 2407194 CrossRef CAS PubMed.
- T. Sahara, D. Wongsawaeng, K. Ngaosuwan, W. Kiatkittipong, P. Hosemann and S. Assabumrungrat, Sci. Rep., 2023, 13, 13210 CrossRef CAS PubMed.
- K. Liu, S. Zhang, X. Hu, K. Zhang, A. Roy and G. Yu, Environ. Sci. Technol., 2015, 49, 8657–8665 CrossRef CAS PubMed.
- K. Sini, D. Bourgeois, M. Idouhar, M. Carboni and D. Meyer, Mater. Lett., 2019, 250, 92–95 CrossRef CAS.
- M.-J. Chen, A.-C. Yang, N.-H. Wang, H.-C. Chiu, Y.-L. Li, D.-Y. Kang and S.-L. Lo, Microporous Mesoporous Mater., 2016, 236, 202–210 CrossRef CAS.
- L. Xiao, Y. Ling, A. Alsbaiee, C. Li, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 7689–7692 CrossRef CAS PubMed.
- W. Chen, X. Zhang, M. Mamadiev and Z. Wang, RSC Adv., 2017, 7, 927–938 RSC.
- H.-Q. Yin, K. Tan, S. Jensen, S. J. Teat, S. Ullah, X. Hei, E. Velasco, K. Oyekan, N. Meyer and X.-Y. Wang, Chem. Sci., 2021, 12, 14189–14197 RSC.
- A. C. Sedgwick, J. T. Brewster, T. Wu, X. Feng, S. D. Bull, X. Qian, J. L. Sessler, T. D. James, E. V. Anslyn and X. Sun, Chem. Soc. Rev., 2021, 50, 9–38 RSC.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- Z. Zheng, H. Yu, W.-C. Geng, X.-Y. Hu, Y.-Y. Wang, Z. Li, Y. Wang and D.-S. Guo, Nat. Commun., 2019, 10, 5762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01765c |
| This journal is © The Royal Society of Chemistry 2025 |