
Tailored molecular engineering of mesoporous silica for high concentration Er doping and unconventional 1640 nm luminescence†
Sudipta
Ghosh
 *a,
Shyamal
Mondal
a,
Rajwinder
Kaur
b,
Bishnu
Daripa
cd,
Prakash Chandra
Mondal
b and
Anirban
Dhar
cd
*a,
Shyamal
Mondal
a,
Rajwinder
Kaur
b,
Bishnu
Daripa
cd,
Prakash Chandra
Mondal
b and
Anirban
Dhar
cd
aDepartment of Chemistry, New Alipore College, Kolkata, 700 053, India. E-mail: gsudipta12@gmail.com; Tel: +91 8910463643
bDepartment of Chemistry, Indian Institute of Technology, Kanpur, Uttar Pradesh 208 016, India
cCSIR-Central Glass & Ceramic Research Institute, Kolkata, 700 032, India
dAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201 002, India
First published on 23rd June 2025
Abstract
The incorporation of high concentrations of erbium (Er) into mesoporous silica is crucial for advancing applications in up-conversion lasers, optical amplifiers, biomedical diagnostics, and light detection and ranging (LiDAR) technologies. This study presents a novel fabrication strategy to achieve Er doping levels up to 6.7 wt% within mesoporous KIT-6 silica by functionalizing the matrix with 1,2-phthaloyl diamido-propyltriethoxysilane (1,2-PA-APTS), followed by solid-phase batch extraction and annealing at 1000 °C. Comprehensive characterization using XRD, BET, FE-SEM, HRTEM, XPS, EPMA, UV-vis, and photoluminescence (PL) spectroscopy was carried out to ensure the material formation and structure–property correlation. A well-ordered 3D mesoporous silica (KIT-6) was synthesized with a high surface area of 1410 m2 g−1 and a pore size of 3.5 nm. Er oxide nanoparticles formed in two size regimes (∼3.5 nm and ∼18 nm) and were homogeneously distributed within the matrix, minimizing clustering. PL spectra revealed prominent emission peaks at 1539 nm and an enhanced, unconventional emission at 1640 nm. The formation of ordered Er oxide nanoparticles (Er2O3) within the crystalline silica matrix, coupled with enhanced Er concentration, was the dominant factor contributing to the sharp absorption and broad photoluminescence emission. The unconventional emission at 1640 nm is attributed to Stark splitting of Er3+ energy levels. This approach effectively addresses dopant clustering challenges at high concentrations and provides a scalable method for controlled rare-earth incorporation into mesoporous silica, advancing its potential for diverse photonic applications.
Introduction
Among the rare-earth elements, Er is one of the most widely studied due to its unique optical properties, including high quantum efficiencies, long metastable state lifetimes, and narrow absorption and emission range of wavelengths.1–6 Er-doped silica has gained significant attention in recent years due to its potential applications in optical fiber amplifiers,4 lasers,5 and upconversion (UC) materials for various application fields such as imaging, diagnostics, and therapy.7–10 Over the last few decades, numerous types of Er-doped silica materials have been developed to enhance optical gain by augmenting the doping density leading to adverse outcomes, such as quenching attributable to clustering or Er atom interaction when the density surpasses 1020 cm−3.11,12 So far, the concentration for Er-doping achieved by researchers falls within the range of 1020–1021 cm−3.13–16 The development of high-concentration Er-doped silica has been a subject of limited investigation to date.Several research groups have been using various synthesis methodologies to achieve the goal of high Er concentration with minimum clustering.6,14,15,17–19 Among them, Biggemann et al. utilized reactive radio frequency (RF) sputtering of silicon targets to produce 1 wt% Er-doped silicon nanoparticles, which were annealed at 1250 °C.18 Similarly, Yan et al. employed RF sputtering techniques to develop Er-doped multicomponent phosphate glass waveguides with an Er concentration of 5.3 × 1020 cm−3.2 Hwang et al. fabricated single-mode, high-Er-doped phosphate glass fibers with an Er3+ concentration of 3.7 × 1020 cm−3, measuring a net gain of 2.1 dB cm−1 at 1537 nm with a pump diode power of 110 mW at 980 nm over a 2.15 cm fiber length.14 Other notable efforts by various researchers have shown Er concentrations ranging from 1 to 4.2 × 1020 cm−3 with significant optical gain, but challenges persist today.15,19
To address these challenges, four approaches have been investigated, as explored by several researchers to date: (a) the incorporation of Er2O3 nanoparticles (NPs) into silica composites instead of bulk Er2O3, leveraging their advantages, including improved dispersion, increased surface area, enhanced optical properties, and superior bioavailability.20 (b) The utilization of crystalline host materials, which offer multiple advantages, such as enhancing the effective excitation cross-section of Er by improving the interaction of Er with the host matrix,21,22 and ensuring better biocompatibility and lower cytotoxicity.20,23,24 (c) The encapsulation of Er2O3 nanoparticles within a mesoporous silica host, providing structural protection against aggregation and uncontrolled association of Er ions.25 (d) Molecular engineering through functionalization of 15 mesoporous silica with 1,2-PA-APTS to enhance Er ion uptake within the silica pores.26,27
In the present study, we developed a methodology to integrate the four aforementioned approaches to enhance Er concentration while minimizing clustering. Mesoporous silica (KIT-6) was synthesized with a pore size of 3.5 ± 0.2 nm and subsequently functionalized with 1,2-PA-APTS, creating a suitable binding environment with an optimal bite angle for Er ions, thereby enabling high uptake capacity. Solid-phase batch extraction experiments were conducted to incorporate Er at varying concentrations into the functionalized mesoporous silica, followed by isothermal heat treatment at 1000 °C in a nitrogen atmosphere for 10 hours. Our results demonstrated a homogeneous and well-distributed incorporation of Er2O3 NPs within the mesoporous silica pores, achieving an enhanced Er concentration of 6.7 wt% with minimal clustering. Notably, an unconventional photoluminescence emission at 1640 nm, along with the characteristic emission at 1540 nm, was observed upon excitation with a 980 nm laser. These findings may shed light on long-standing challenges and reveal a potential pathway for the development of Er-doped silica with enhanced Er concentrations. Furthermore, this approach can be extended to other rare-earth nanoparticle-doped silica systems, enabling the design of tailored optical materials for various advanced photonic applications.
Materials and methods
Chemical and materials
Tetraethoxysilane (TEOS, purity > 98%), (3-aminopropyl)triethoxysilane (APTS, 98%), and N,N-dioctylamine (97%) were purchased from Sigma-Aldrich. Phthaloyl chloride (94%), isophthaloyl dichloride (98%), and terephthaloyl chloride (99%) were purchased from Alfa Aesar and were used without further purification. Analytical-reagent grade hydrochloric acid (37%), nitric acid (65%), and sodium hydroxide were purchased from Merck and were used without further purification. Toluene (Sigma-Aldrich) and triethylamine (Sigma-Aldrich, 99%) were dried and distilled using sodium benzophenone, and stored in molecular sieves under nitrogen. Solutions of Er nitrate (Er(NO3)3.5H2O) were prepared from the reference standard solutions (Plasma, Cal, SCP Science) using double-distilled water.Synthesis of 3D mesoporous silica (KIT-6)
The synthesis of Er NPs-Si materials was carried out with modifications based on previously reported methods.27,28 Pluronic P123 (EO20PO70EO20) (4.50 g) was dissolved in distilled water (162.5 g), and HCl (37%, 8.70 g) was added with stirring. Once the solution was completely dissolved, n-butanol (BuOH) (4.95 g) was added, and the mixture was stirred at 35 °C for 6 hours. Then, tetraethoxysilane (TEOS) (9.70 g) was added to the solution. The molar composition of the mixture was TEOS/P123/HCl/H2O/BuOH = 1.0/0.017/1.83/195/1.31. The mixture was stirred at 35 °C for 24 hours, followed by aging for 24 hours at 100 °C under static conditions. The resulting solid product was filtered and dried at 100 °C for 24 hours to obtain a 3D mesoporous nanomaterial. The template was removed by extraction in an ethanol–HCl mixture, and the material was calcined in air at 550 °C for 5 hours.Synthesis of 1,2-phthaloyl diamido-propyltriethoxysilane (1,2-PA-APTS)
The synthesis of 1,2-phthaloyl diamido-propyltriethoxysilane (1,2-PA-APTS) was performed following the procedure reported by Hu et al.27 Initially, a solution of 1,2-phthaloyl chloride (1.53 mL, 10 mmol) in dry toluene (25 mL) was gradually added to a mixture of APTS (4.9 mL, 21 mmol) and triethylamine (14 mL, 100 mmol) in toluene. The resulting mixture was stirred at room temperature for a full night. Afterward, the reaction mixture was filtered, and the solvent was removed under reduced pressure, giving rise to a solid product with a light-yellow appearance.Functionalization of KIT-6 with 1,2-PA-APTS (KIT-6-APTS)
The synthesis was carried out with modification of previous work,27 where during the grafting process, the KIT-6 support, underwent activation overnight under vacuum at 150 °C. To functionalize KIT-6, 1.0 g of the support material was dispersed in 60 mL of dry toluene in an inert atmosphere. After 20 minutes, a chosen silane-modified ligand (0.46 g of 1,2-PA-APTS) and triethylamine (1.2 mL) were simultaneously added to the suspension. The mixture was stirred under reflux conditions for 24 hours. Following the modification process, the functionalized silica product was filtered and washed thoroughly with toluene. Soxhlet extraction in dichloromethane for at least 12 hours was used to remove any unreacted silane molecules.Solid batch extraction of Er (Er-KIT-6-APTS)
The synthesis was carried out with slight modification based on previously reported methods.28 Before analysis, the sorbents were subjected to outgassing at 80 °C overnight. Standard batch solid-phase extraction (SPE) experiments of rare earth element (REE) solutions were conducted using Er-1,2-PA-APTS in a 10 mL centrifuge tube. The Er nitrate pentahydrate [Er(NO3)3, 5H2O] solutions were prepared at desired concentrations. In each sample, 10 mg of Er-1,2-PA-APTS were placed into a centrifuge tube after undergoing overnight outgassing at 80 °C. Subsequently, 5 mL of rare earth solutions, consisting of 400, 800, and 1600 ppm, were added to the centrifuge tube and agitated thoroughly. The solutions were allowed to undergo adsorption for 2 hours, followed by centrifugation (Model: REMI). The same process was repeated while stirring for 2 hours. The resulting supernatants (clear solutions) were manually discarded, and the residue was dried in an oven at 150 °C for 5 hours. The samples were named Er-400-WHT, Er-800-WHT and Er-1600-WHT.Heat treatment of Er-doped silica
A tube heating furnace (model: YTF-100-17, Yatherm Scientific, 2020) was utilized for heat treatment, and a quartz crucible (M.P.: 1700 °C) was used as a heating base. Er-doped mesoporous silica was subjected to heat treatment in an inert atmosphere of N2 at 1000 °C for 10 hours with a heating rate of 50 °C min−1 and cooled naturally. After heat treatment, a fine white powder was observed. The samples were named Er-400-HT, Er-800-HT and Er-1600-HT, respectively.Results and discussion
Synthesis and characterization
The entire synthesis is presented in the diagram shown in Fig. 1, where mesoporous silica KIT-6 was first synthesized, followed by the synthesis of the chelating ligand 1,2-PA-APTS. Subsequently, KIT-6 was functionalized using the synthesized 1,2-PA-APTS. The solid batch extraction of Er ions by the functionalized KIT-6-1,2-PA-APTS was performed with varying Er concentrations of 400, 800, and 1600 ppm. Finally, isothermal heat treatment at 1000 °C for 10 hours in an inert nitrogen (N2) atmosphere was conducted. A fine white powder sample was observed, and details of the samples are shown in Table S1 (ESI†). In our work, Er encapsulation into the functionalized mesoporous silica nanoparticles is very selective, because the organic ligand 1,2-PA-APTS possesses two non-bridging oxygens, which creates a bite angle appropriate for the Er3+ radius (103 pm).27 | ||
| Fig. 1 Schematic diagram of the synthesis of Er2O3 nanoparticles embedded in mesoporous silica using 1,2-PA-APTS ligands and KIT 6. | ||
It has been observed from previous work that most of the organo-Er complex has a melting point below 300 °C.20,29 For our case, the organo-Er complex, which was formed inside the mesoporous silica pore, is not isolated; it is an Er-1,2-PA-APTS binding with the pore surface through oxygen linkage. As we have started the annealing process at 1000 °C by raising the temperature under an N2 environment, first the Er-1,2-PA-APTS complex has started to melt in the temperature range 400–500 °C, and further increasing the temperature leads to evaporating the organic ligand, leaving the Er ions in the pores. As we know, up to 600 °C, mesoporous silica nanoparticles’ pore structure remains unchanged, with no shrinking of the pore diameter.30 It is well recognized that pristine mesoporous silica materials such as MCM-41, MCM-48, HMS, FSM-16, KIT-1, PCH, and SBA-15 struggle to retain their nanostructural integrity at high temperatures (e.g., ∼1000 °C). Cassiers et al. extensively investigated the thermal behavior of various mesoporous silicas and reported that materials like MCM-41, MCM-48, KIT-1, HMS, and SBA-15 undergo complete loss of textural properties at around 900 °C.31 In the present work, we employed KIT-6, a three-dimensional mesoporous silica, which has been shown by Kishor et al. to exhibit greater thermal stability compared to other mesoporous analogs (e.g., MCM-41, MCM-48, SBA-1, KIT-1, HMS, and SBA-15).32 This enhanced stability was attributed to the relatively thicker pore walls of KIT-6. Furthermore, previous studies have shown that the incorporation of metal nanoparticles, such as Ce in SBA-15, can significantly improve thermal resilience by preventing thermally induced radial shrinkage of the mesopores. Building on this insight, we propose that the encapsulation of Er2O3 nanoparticles within the KIT-6 matrix contributes to the preservation of its nanostructure at elevated temperatures (>1000 °C), owing to a similar stabilizing mechanism.33,44
XRD and BET analysis
X-ray diffraction (XRD) pattern analysis was conducted on KIT-6, Er-800-WHT (without heat treatment), and Er-800-HT (heat-treated) samples at 1000 °C for 10 hours. Additionally, deconvoluted patterns of Er-800-HT and small-scale XRD of KIT-6 were performed to elucidate the morphology and structure of Er-doped mesoporous silica nanoparticles (Fig. 2). Small-angle XRD analysis revealed that KIT-6 possesses a well-organized mesoporous structure, as evidenced by a prominent and intense peak at the (211) plane and a shoulder at the (220) plane (Fig. 2a). This peak corresponds to the Ia3d bi-continuous space group in KIT-6, consistent with previous reports by Kleitz et al.34 The XRD pattern of KIT-6 exhibited a broad peak at 2θ = 23 ± 2°, confirming the presence of amorphous silica in the pore walls.35 Upon Er doping, a similar broad peak at 2θ = 23 ± 2° was observed, accompanied by a minor hump at 2θ = 41 ± 2° attributed to crystalline SiO2 nanoparticles (Fig. 2b).36 The intensity of the hump at 2θ = 41 ± 2° increased after Er incorporation due to a 5-hour heat treatment at 150 °C aimed at removing water and other moieties completely. Moreover, this intensity further increased in the Er-800-HT sample, annealed at 1000 °C for 10 hours, with a shift in the peak at 2θ = 40 ± 2°. Another minor hump appeared for the Er-800-HT sample at 2θ = 20 ± 1°, attributed to crystalline SiO2 nanoparticles, as indicated by literature XRD peaks at 2θ = 21.8°, 36°, and 40.3°.36 The increased intensity and shift of the peak at 2θ = 41 ± 2°, along with the appearance of the peak at 2θ = 20 ± 1°, were attributed to the enhancement of the crystalline nature of SiO2 in the sample. The XRD pattern of Er-800-HT was deconvoluted (Fig. 2c), revealing three peaks at 2θ = 20.2 ± 1°, 29.7 ± 0.2°, and 38.3 ± 2°, among which the peak at 2θ = 29.7 ± 0.2° was attributed to Er2O3 nanoparticles. This peak exhibited sharpness and higher intensity compared to the other peaks, and a similar observation was reported by other researchers.37,38 | ||
| Fig. 2 Spectroscopic characterization of KIT-6, Er-800-WTH, and Er-800-HT. (a) Small-angle XRD patterns for KIT-6, (b) high-angle XRD patterns for KIT-6, Er-800-WHT, and Er-800-HT, and (c) deconvoluted pattern of Er-800-HT. | ||
The N2 adsorption/desorption experiment was conducted for KIT-6, KIT-6-1,2-PA-APTS and Er-KIT-6-1,2-PA-APTS samples to calculate the surface area, pore volume and pore size and to understand the pore nature (micropore and mesopore) and pore shape (circular, capillary and ink-bottle like) (Fig. 3). The isotherm (Fig. 3a) observed for KIT-6 was described in our previous work,24 while KIT-6-APTS bears a resemblance to H2 (Fig. 3a and b) hysteresis loops as per the IUPAC classification of 1985.39 These loops serve as evidence for the development of intricate pore structures, highlighting the significance of network effects. The observed behaviour may be ascribed to either pore-blocking or percolation within a restricted range of pore necks, or to cavitation-induced evaporation.40 Conversely, the isotherm for Er-KIT-6-APTS exhibits an H1 loop,41 characteristic of materials possessing a narrow and uniform mesopore range. This indicates minimal network effects, and the presence of a steep, narrow loop serves as a distinct indicator of delayed condensation on the adsorption branch. The pore size of KIT-6 was calculated as 3.5 ± 0.2 nm, while the pore size distribution of KIT-6, KIT-6-APTS and Er-KIT-6-APTS (Fig. 3b) provides evidence for micropores (∼1.2 ± 0.1 nm), for all the materials but for Er-KIT-6-APTS an interesting feature of pore size distribution was observed where the pore size increased and spread within a range of 3.8 ± 0.2 nm to 6.6 ± 0.2 nm.
 | ||
| Fig. 3 The N2 adsorption/desorption of KIT-6, KIT-6-APTS and Er-KIT-6-APTS: (a) isotherm, and (b) pore size distribution. | ||
The surface area and pore volume of KIT-6, KIT-6-APTS and Er-KIT-6-APTS were 1451 m2 g−1, 648 m2 g−1, 744 m2 g−1 and 2.23 cc g−1, 1.093 cc g−1, 1.275 cc g−1 as shown in Table 1.
| Materials | S BET (m2 g−1) | V pore (cm3 g−1) | Mesopore diameter (nm) | Micropore diameter (nm) |
|---|---|---|---|---|
| KIT-6 | 1451 | 2.231 | 3.56 | 1.20 |
| KIT-6-APTS | 648 | 1.093 | 3.98 | 1.26 |
| Er-KIT-6-APTS | 744 | 1.275 | 4.36 | 1.24 |
FE-SEM-EDX and EPMA analysis
The elemental analysis of Er-doped silica was performed using field-emission scanning electron microscopy (FE-SEM-EDX; model: INSPECT F50, maker: FEI, Netherlands) and with electron probe microanalyzers (EPMA; model: JXA-8230, manufacturer: JEOL Ltd, Japan) to investigate the presence, distribution, and quantity of elements of Si, O, and Er. The FE-SEM-EDX (Fig. S1, ESI†) confirmed the presence of Er along with Si and O. Furthermore, the elemental mapping of Si, O, and Er from FESEM-EDX (Fig. S2, ESI†) indicates that the Er was evenly distributed throughout the silica matrix. Two important facts came out of this elemental mapping that Er has a close approximation with O rather than Si and a good separation among the Er, leaving few clusters in nature. Furthermore, elemental analysis has been done by EPMA as shown in Fig. 4, to determine the amount of Er doping as well as the fine distribution of the elements Si, O, and Er. From the elemental mapping (Fig. 4a–c), a well homogeneous distribution of Er was observed without a significant clustering phenomenon along with Si and O, and quantitative analysis revealed (Fig. 4d) 6.67 mass% Er intakes for the Er-400HT sample. | ||
| Fig. 4 EPMA analysis (a) elemental mapping of O, (b) Si, (c) Er of the Er-400HT sample, and (d) elemental spectra with their quantitative analysis. | ||
In the solid batch extraction process, the erbium uptake capacity of KIT-6 functionalized with 1,2-PA-APTS exhibits nonlinear behavior and becomes saturated beyond a certain concentration threshold. Hu et al. reported that the adsorption capacity of such functionalized mesoporous silica increases with rare-earth ion concentrations ranging from 5 to 100 mg L−1.27 In our study, erbium nitrate solutions were prepared with concentrations ranging from 400 ppm (400 mg L−1) to 1600 ppm (mg L−1) to enhance Er loading into the mesoporous silica framework. Interestingly, as presented in Table S1 (ESI†), the maximum erbium incorporation (6.7 wt%) was achieved at 400 ppm, whereas higher solution concentrations led to a decline in Er uptake. This counterintuitive trend may be attributed to several limiting factors: saturation of available grafted functional groups, ion clustering or aggregation, possible precipitation at higher concentrations, and reduced diffusivity of Er3+ ions into the mesoporous channels.42 These factors collectively hinder effective incorporation of erbium into the silica matrix. We believe that further in-depth studies are warranted to elucidate the precise mechanisms governing this behavior and to optimize conditions for maximum rare-earth ion loading.
HR-TEM and XPS analysis
HR-TEM analysis was performed using a JEM-2100 microscope (JEOL, Japan) to investigate the surface topology of the silica matrix and to elucidate the nanoscale characteristics of the embedded Er2O3 nanoparticles, including their dimensional attributes, morphology, and spatial distribution. The three-dimensional architecture of the synthesized KIT-6 material was distinctly visualized, with its mesoporous network successfully impregnated with Er2O3 nanoparticles, as indicated by the yellow arrows in Fig. 5a. A notable observation was that the introduced Er species were primarily localized within the mesoporous silica walls, facilitating a structural framework hindrance to Er clustering while leaving certain pore openings exposed. The combined analysis of XRD and FESEM-EDX confirmed that the embedded Er2O3 predominantly comprised Er2O3 nanoparticles. Fig. 5b illustrates the uniform spatial dispersion of Er2O3 nanoparticles, which was an anticipated outcome, attributed to the targeted encapsulation of Er ions within the functionalized mesoporous silica, yielding a well-ordered and homogeneously structured system. Two types of Er2O3 nanoparticles were formed: one larger (marked by a yellow arrow) and the other smaller (marked by a white arrow), both exhibiting an approximately circular shape, as shown in Fig. 5c. A detailed size analysis was performed using ImageJ software (Fig. S3, ESI†), revealing that the average size of the smaller Er2O3 nanoparticles was 3.5 ± 0.2 nm, while the larger ones measured 18 ± 2 nm. The SAED pattern of the Er2O3 nanoparticles, shown in the inset of Fig. 5c, indicates their amorphous nature. The formation of the 3.5 ± 0.2 nm nanoparticles corresponds well with the pore diameter of KIT-6, approximately 3.5 nm as determined by BET analysis, suggesting that Er was encapsulated within the pores, consistent with the measured nanoparticle size. In contrast, the larger Er2O3 nanoparticles likely resulted from the merging of pores during isothermal heat treatment at 1000 °C for 10 hours. Literature reports indicate that upon thermal treatment at temperatures exceeding 600 °C,30,43 mesoporous frameworks such as KIT-6 undergo progressive structural degradation, potentially leading to pore merging. This observation may result in the aggregation of adjacent pores, ultimately forming larger Er2O3 nanoparticles, as evidenced by the observed 18 ± 2 nm nanoparticles (merging typically four to five units), given that our synthesis involved heat treatment at 1000 °C. A study by Mitran et al.44 further substantiates this phenomenon, demonstrating that the crystallite size of CeO2 NPs embedded in SBA-15 mesoporous silica increased to 4.9 ± 0.2 nm, 6.0 ± 0.1 nm, and 12.3 ± 0.3 nm at annealing temperatures of 700 °C, 900 °C, and 1100 °C, respectively. The polycrystalline nature of silica, as previously inferred from XRD analysis, was corroborated by HRTEM imaging (Fig. 5d), where a well-defined crystal lattice plane structure was observed. Additionally, the selected area electron diffraction (SAED) pattern (Fig. 5d, inset) further confirmed the polycrystalline nature of the synthesized material. The X-ray photoelectron spectroscopy (XPS) analysis (Fig. 6) of Er-KIT-6 was undertaken to elucidate the bonding characteristics of the constituent elements in the presence of elevated Er concentrations, aiming to discern the nature of Er bonding within the silica matrix. The XPS spectra of Si 2p exhibited a binding energy (BE) peak at 103.3 eV, which manifested as doublets with BEs at 103.3 eV and 103.7 eV, corresponding to Si 2p3/2 and Si 2p1/2, respectively (Fig. 6a). The presence of the Si 2p BE peak at 103.3 eV is indicative of characteristic features observed in mesoporous silica nanoparticles.45,46 The emergence of doublets in the Si 2p BEs may be attributed to the inclusion of Er in the system, suggesting potential Si–O–Er and Si–O–Si bonding configurations. | ||
| Fig. 5 HR-TEM of Er-doped mesoporous silica (a) Er2O3 nanoparticle encapsulation into mesopores, (b) Er2O3 nanoparticle distribution, (c) shape and size of Er2O3 nanoparticles, and (d) crystalline nature of the host silica. | ||
 | ||
| Fig. 6 Chemical analysis of Er-KIT-6 of the XPS spectra of (a) Si 2p, (b) O 1s, (c) Er 4d, (d) Er 4p, and 4f. XPS signals are deconvoluted, and characteristic peaks are marked. | ||
The O 1s spectrum exhibited a BE centered at 532.5 eV (Fig. 6b), which is generally attributed to oxygen in the Si–O–Si network and matches well with the previous report.47 According to the literature, oxygen vacancies typically manifest at lower binding energies, around 531.0–531.5 eV, due to the reduced coordination environment.48,49 Therefore, the observed BE suggests a low probability of oxygen vacancy formation in the sample. Furthermore, the splitting of the O 1s peak reflects distinct bonding environments of oxygen with silicon and Er, which may correlate with BEs at 532.5 eV for O–Er and 532.7 eV for O–Si, owing to the higher electronegativity of silicon relative to Er.50 To further probe the oxygen environment, we analyzed the O 2s core level, shown in Fig. S3c (ESI†). Although less chemically sensitive than O 1s, the O 2s peak can still reflect variations in local electronic structure. The recorded BE of ∼24.2 eV is consistent with oxygen atoms within the Si–O–Si framework, supporting the conclusion that the silica matrix retains a largely stoichiometric structure with minimal oxygen deficiencies.51 Deconvolution of the XPS spectra of Er 4d revealed six discernible peaks at BEs of 168.9 eV, 169.6 eV, 172.9 eV, 173.9 eV, 174.7 eV, and 177.2 eV, respectively (Fig. 6c). Among these, the peaks at 168.9 eV and 169.6 eV correspond to doublet splitting of Er 4d5/2, while the remaining peaks denote Er 4d3/2.52,53 Specifically, BEs at 168.9 eV and 169.6 eV for Er 4d5/2 are indicative of Er3+ states,54,55 whereas the other peaks signify Er 4d3/2 states.56 Comparison with existing literature suggests that Er 4d BE peaks typically occur at 169.1 eV for nanorods, and 168.4 eV for nanosheets,57 with 168.4 eV corresponding to well-crystallized Er2O3 and 167.5 eV for the Er2O3 film annealed at 900 °C.58 Accordingly, our observations suggest the existence of Er2O3 in the nanoforms, corroborated by the results of our analysis of heat treatment, XRD, and FESEM-EDX elemental mapping. Furthermore, the XPS spectra of Er 4f and Er 5p (Fig. 6d) exhibited BEs of 7.7 eV and 24.2 eV, respectively, slightly higher values than metallic Er, affirming the formation of Er2O3 nanoparticles within the silica matrix.56 The presence of well-defined Er3+ states suggests a stable oxidation state without significant electron deficiency, which correlates with a low tendency for oxygen vacancy formation. However, the additional peaks in the Er 4d3/2 region may arise from minor defect-related states or subtle interactions at the Er2O3–SiO2 interface,56 which could imply localized oxygen non-stoichiometry or vacancy-related sites. The combined analysis of O 1s, O 2s, and Er 4d spectra indicates that oxygen vacancies are unlikely to be dominant in the current high-Er-doped mesoporous silica sample. However, the presence of defect-related Er 4d3/2 features cannot entirely rule out their existence.
Absorption and PL emission analysis
The photophysical properties of Er-doped mesoporous silica were investigated to assess the impact of high Er concentration, Er2O3 nanoparticles, and surface morphology on absorption, emission, and lifetime, as depicted in Fig. 7. The normalized absorption spectra of the Er-400-HT and Er-1600-HT thin films (Fig. 7a) reveal sharp, prominent peaks at 960 nm and 1590 nm, which can be attributed to transitions from the 4I15/2 to 4I11/2 and from 4I13/2 to 4I15/2, respectively.59 The literature suggests that sharp absorption features are typically observed when rare earth ions are embedded in an ordered manner within a crystalline phase.60,61 In this study, both the ordered arrangement of Er ions within the mesoporous silica structure and the formation of a crystalline silica phase contributed to the observed sharp absorption spectra.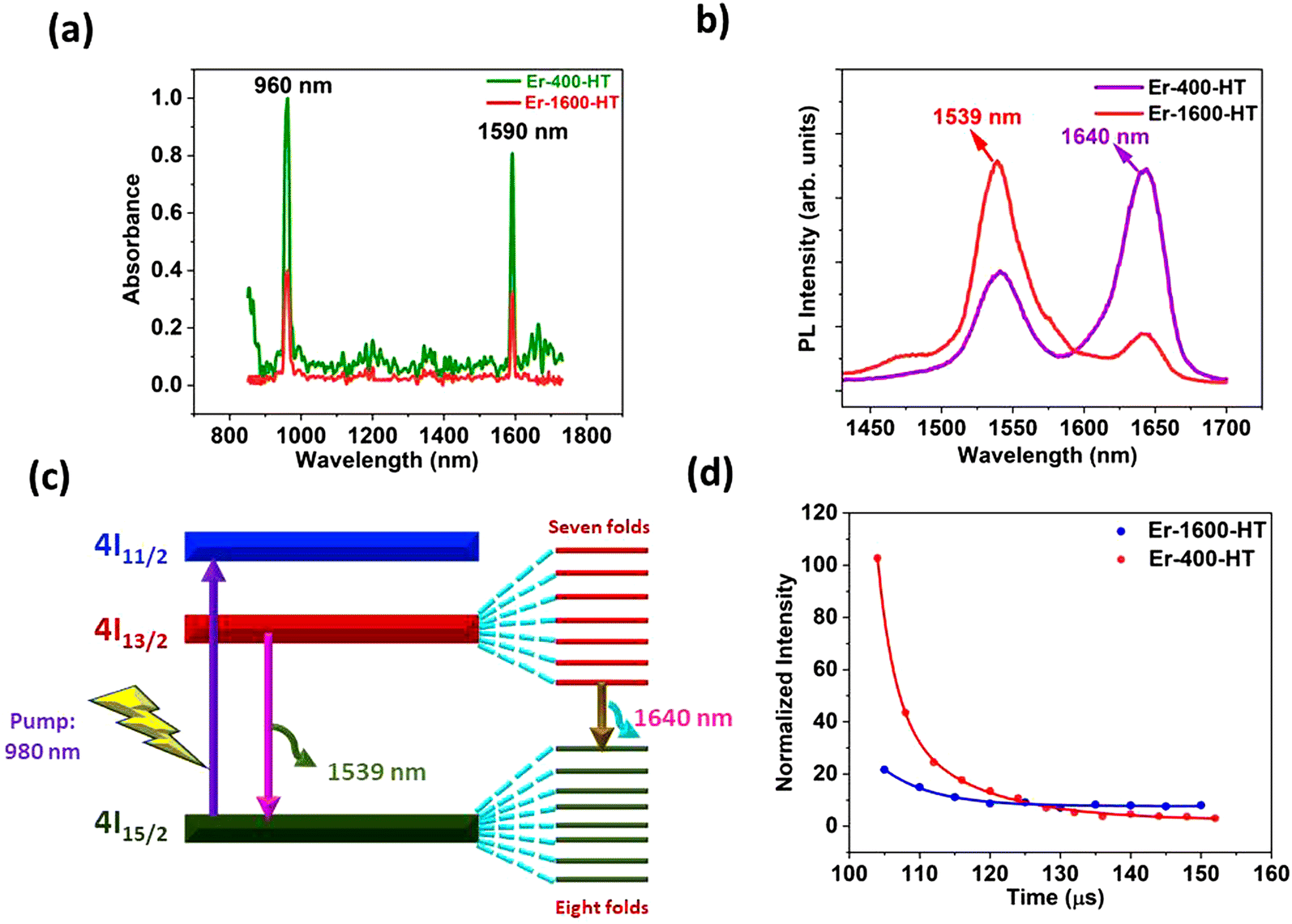 | ||
| Fig. 7 Photophysical properties of Er-doped silica: (a) UV-vis spectra of Er-400-HT and Er-1600-HT, (b) photoluminescence spectra of Er-400-HT and Er-1600-HT excited by 980 nm light, (c) schematic representation of Stark splitting, and (d) lifetime of Er-400-HT and Er-1600-HT at 1540 nm PL emission. | ||
The photoluminescence (PL) spectra of the Er-400-HT and Er-1600-HT samples, excited by 980 nm light, are shown in Fig. 7b. Both samples exhibit broad emission bands at 1539 nm and 1640 nm. The Er-400-HT sample, which contains a higher Er concentration (6.71 wt%), shows stronger PL intensity at 1640 nm compared to 1539 nm, whereas the Er-1600-HT sample, with a lower Er concentration (3.78 wt%), exhibits the reverse trend. Previous research has demonstrated that incorporating Si nanostructures with Er2O3 enhances PL emission by mitigating narrow band excitation issues through resonant energy transfer between the nano-crystalline Si structures and Er ions.62–64 Furthermore, dipole–dipole interactions and efficient energy transfer between the crystalline silica and Er are believed to enhance the infrared emission of Er ions, even in the presence of weak narrow-band absorption.65 The formation of Er2O3 nanoparticles in our study further facilitated the interaction between Er2O3 nanoparticles and the crystalline silica matrix due to the increased surface area of the Er2O3 nanoparticles, leading to broader PL emission. The emission at 1539 nm is typically associated with the transition from the 4I13/2 to the 4I15/2 energy levels of Er, while the 1640 nm emission, though not unprecedented, is less commonly observed in Er-doped silica composites.66,67 A few studies have reported PL emissions at higher wavelengths,65,67–69 attributing them to Stark splitting of the 4I13/2 and 4I15/2 energy levels. Notably, B. Schmaul et al.67 suggested that the 1640 nm emission results from a transition between the lower 4I13/2 Stark level and the upper 4I15/2 Stark level, a phenomenon consistent with our findings, as shown schematically in Fig. 7c. According to the literature, this phenomenon is attributed to nanostructural effects, rather than bulk properties. In particular, the formation of Er–oxygen complexes,68 facilitated by thermal annealing,65,66 within the nanocrystalline silica matrix is believed to contribute to the observed 1640 nm emission. This emission indicates Stark splitting, where the 4I13/2 level splits into seven sublevels, and the 4I15/2 level splits into eight sublevels.70 This splitting can only occur in an Er–oxygen complex exhibiting octahedral symmetry, in which an Er ion is surrounded by six oxygen atoms.71 Therefore, the pronounced 1640 nm PL emission in our study is indicative of the formation of such Er–oxygen complexes, corroborating our previous observations of Er–NP formation. It is noteworthy that higher Er concentration enhances the 1640 nm PL emission (Fig. 7b), suggesting that an increased number of Er ions participate in the Stark splitting process. However, further studies are required to fully understand the origin of the PL emission associated with Stark splitting, as this phenomenon is influenced by multiple factors, including the formation of Er2O3 nanoparticles. We believe that the formation of Er2O3 nanoparticles plays a crucial role in the exceptional PL emission observed in this study, a subject that warrants further investigation, which is beyond the scope of the current work.
Recent studies by Johnson et al.72 have introduced a new quenching phenomenon in lanthanide-doped nanocrystals, wherein energy migration to the surface (surface quenching) becomes the dominant quenching mechanism at high dopant concentrations, as opposed to the traditional cross-relaxation between dopant ions. In our case, surface quenching is believed to play a significant role in the observed reduction in PL lifetime, which is on the order of microseconds (Fig. 7d). This hypothesis is supported by high-resolution transmission electron microscopy (HRTEM) images (Fig. 5a), which reveal that Er2O3 nanoparticles are embedded within the mesoporous silica structure, which exhibits a honeycomb-like arrangement (Fig. S1a, ESI†). The second quenching mechanism in our system is likely due to the formation of large Er2O3 nanoparticles, with an average size of 18 ± 2 nm, which leads to concentration quenching as a result of Er ion clustering. Future work is needed to optimize the various parameters of high-Er-concentration-doped mesoporous silica composites, and our next phase of research will focus on gaining a deeper understanding of these phenomena.
Conclusions
In this study, we have developed a novel and integrative methodology for synthesizing high-concentration Er-doped Er2O3 nanoparticle-doped mesoporous silica, successfully addressing the persistent challenge of Er clustering at elevated dopant levels. By leveraging molecular engineering through the functionalization of KIT-6 mesoporous silica with 1,2-PA-APTS, a highly selective and stable ligand system was established. This system utilizes a well-defined bite angle optimized for the Er3+ ionic radius, significantly enhancing Er ion uptake during solid-phase batch extraction. Following isothermal heat treatment, this approach enabled the homogeneous embedding of Er2O3 nanoparticles within a crystalline silica host at concentrations as high as 6.7 wt%, with minimal clustering. Crucially, the study reports the formation of uniformly distributed Er2O3 nanoparticles within the mesoporous matrix and the emergence of a distinctive photoluminescence emission at 1640 nm, alongside the traditional 1540 nm peak, upon 980 nm excitation. This enhanced near-infrared emission is attributed to Stark splitting, facilitated by the crystalline silica matrix and the spatial confinement of Er2O3 nanoparticles with an enhanced Er concentration. The successful stabilization of Er3+ ions in a structurally crystalline and thermally robust silica environment marks a significant advancement in rare-earth doping strategies. This methodology offers a versatile and scalable platform for the controlled doping of rare-earth ions in mesoporous hosts and opens new avenues for high-performance materials in photonics, telecommunications, bioimaging, and sensing applications.Conflicts of interest
The authors declare no conflict of interest.Data availability
Data are available upon request from the corresponding author.Acknowledgements
The authors would like to thank New Alipore College for all kinds of support. S. G. would like to acknowledge Sophisticated Analytical Instrument Facility (SAIF) at North-Eastern Hill University, Shillong, for characterization facilities. R. K. thanks the University Grant Commission (UGC, New Delhi, India) for a junior research fellowship. B. D. thanks DST for Inspire Fellowship (Award No. IF210364).References
- K. Annapoorani, Ch Basavapoornima, N. Suriya Murthy and K. Marimuthu, J. Non-Cryst. Solids, 2016, 447, 273–282 CrossRef CAS.
- Y. C. Yan, A. J. Faber, H. De Waal, P. G. Kik and A. Polman, Appl. Phys. Lett., 1997, 71, 2922–2924 CrossRef CAS.
- B. J. Ainslie, J. Lightwave Technol., 1991, 9, 220–227 CrossRef CAS.
- A. Bellemare, Prog. Quantum Electron., 2003, 27(4), 211–266 CrossRef CAS.
- J. D. B. Bradley and M. Pollnau, Laser Photonics Rev., 2011, 5, 368–403 CrossRef CAS.
- P. Araichimani, K. M. Prabu, G. S. Kumar, G. Karunakaran, N. V. Minh, S. Karthi, E. K. Girija and E. Kolesnikov, Ceram. Int., 2020, 46, 18366–18372 CrossRef CAS.
- M. Fujii, M. Yoshida, Y. Kanzawa, S. Hayashi and K. Yamamoto, Appl. Phys. Lett., 1997, 71, 1198–1200 CrossRef CAS.
- J. H. Zeng, J. Su, Z. H. Li, R. X. Yan and Y.-D. Li, Adv. Mater., 2005, 17(17), 2119–2123 CrossRef CAS.
- G. S. Yi and G. M. Chow, Adv. Funct. Mater., 2006, 16, 2324–2329 CrossRef CAS.
- M. Y. Hossan, A. Hor, Q. Luu, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. C, 2017, 121(30), 16592–16606 CrossRef CAS.
- E. Talbot, R. Lardé, P. Pareige, L. Khomenkova and F. Gourbilleau, Nanoscale Res. Lett., 2013, 8, 39 CrossRef.
- K. Hijazi, R. Rizk, J. Cardin, L. Khomenkova and F. Gourbilleau, J. Appl. Phys., 2009, 106, 024311 CrossRef.
- W. J. Miniscalco, J. Lightwave Technol., 1991, 9, 234–250 CrossRef CAS.
- B. C. Hwang, S. Jiang, T. Luo, J. Watson, S. Honkanen, Y. Hu, F. Smektala, J. Lucas and N. Peyghambarian, Electron. Lett., 1999, 35, 1007–1009 CrossRef CAS.
- J. D. B. Bradley, L. Agazzi, D. Geskus, F. Ay, K. Wörhoff and M. Pollnau, J. Opt. Soc. Am. B, 2010, 27, 187–196 CrossRef CAS.
- S. S. Nafee, T. A. Hamdalla and A. A. A. Darwish, Opt. Laser Technol., 2020, 129, 106282 CrossRef CAS.
- J. H. Shin, M. J. Kim, S. Y. Seo and C. Lee, Appl. Phys. Lett., 1998, 72, 1092 CrossRef CAS.
- D. Biggemann, D. Mustafa and L. R. Tessler, Opt. Mater., 2006, 28, 842–845 CrossRef CAS.
- H. Sun, L. Yin, Z. Liu, Y. Zheng, F. Fan, S. Zhao, X. Feng, Y. Li and C. Z. Ning, Nat. Photon., 2017, 11, 589–593 CrossRef CAS.
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS.
- D. Pacifici, A. Irrera, G. Franzo, M. Miritello, F. Iacona and F. Priolo, Physica E, 2003, 16, 331–340 CrossRef CAS.
- J. John, J. L. Coffer, Y. Chen and R. F. Pinizzotto, J. Am. Chem. Soc., 1999, 121, 1888–1892 CrossRef.
- F. Vetrone and J. A. Capobianco, Int. J. Nanotechnol., 2008, 5, 1306–1339 CrossRef CAS.
- D. Dosev, M. Nichkova and I. M. Kennedy, J. Nanosci. Nanotechnol., 2008, 8, 1052–1067 CrossRef CAS.
- P. Escribano, B. Julian-Lopez, J. Planelles-Arago, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23–40 RSC.
- Y.-S. Lin, Y. Hung, J.-K. Su, R. Lee, C. Chang, M.-L. Lin and C.-Y. Mou, J. Phys. Chem. B, 2004, 108, 15608–15611 CrossRef CAS.
- Y. Hu, E. Drouin, D. Larivière, F. Kleitz and F. G. Fontaine, ACS Appl. Mater. Interfaces, 2017, 9, 38584–38593 CrossRef CAS.
- S. Ghosh, S. Mondal, R. Kaur, D. Mondal, B. Daripa, P. K. Sinha, P. C. Mondal, S. Das and A. Dhar, Chem. – Asian J., 2024, 19, 202400921 CrossRef.
- G. A. Kumar, R. E. Riman, L. A. Diaz Torres, O. B. Garcia, S. Banerjee, A. Kornienko and J. G. Brennan, Chem. Mater., 2005, 17(20), 5130–5135 CrossRef CAS.
- F. Silencieux, M. Bouchoucha, O. Mercier, S. Turgeon, P. Chevallier, F. Kleitz and M.-A. Fortin, Langmuir, 2015, 31(47), 13011–13021 CrossRef CAS.
- K. Cassiers, T. Linssen, M. Mathieu, M. Benjelloun, K. Schrijnemakers, P. Van Der Voort, P. Cool and E. F. Vansant, Chem. Mater., 2002, 14(5), 2317–2324 CrossRef CAS.
- R. Kishor and A. K. Ghoshal, Microporous Mesoporous Mater., 2017, 242, 127–135 CrossRef CAS.
- N. Jayakodiarachchi, P. G. Evans, C. L. Ward and C. H. Winter, Organometallics, 2021, 40(9), 1270–1283 CrossRef CAS.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- S. Sankar, S. K. Sharma, N. Kaur, B. Lee, D. Y. Kim, S. Lee and H. Jung, Ceram. Int., 2016, 42(4), 4875–4885 CrossRef CAS.
- A. Ali, S. Saeed, R. Hussain, G. Afzal, A. B. Siddique, G. Parveen, M. Hasan and G. Caprioli, ACS Omega, 2023, 8(23), 20900–20911 CrossRef CAS.
- A. Bakhsh and A. Maqsood, Electron. Mater. Lett., 2012, 8, 605–608 CrossRef CAS.
- G. Safwat, E. S. M. Soliman and H. R. H. Mohamed, Sci. Rep., 2022, 12, 16333 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Rodriguez-Reinoso, F. Rouquerol, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CrossRef CAS.
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18(25), 9830–9837 CrossRef CAS.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size, and Density, Springer, Dordrecht, 2004, vol. 16 Search PubMed.
- R. J. Umpleby, S. C. Baxter, Y. Chen, R. N. Shah and K. D. Shimizu, Anal. Chem., 2001, 73(19), 4584–4591 CrossRef CAS.
- J. B. Lowe and R. T. Baker, J. Nanomater., 2014, 754076 CrossRef.
- R. A. Mitran, D. C. Culita and I. Atkinson, Microporous Mesoporous Mater., 2020, 306, 110484 CrossRef CAS.
- S. Shen, J. Chen, R. T. Koodali, Y. Hu, Q. Xiao, J. Zhou, X. Wang and L. Guo, Appl. Catal., B, 2014, 150, 138–146 CrossRef.
- V. Sales, R. Moura, A. B. F. Câmara, E. Rodríguez-Castellón, J. A. B. Silva, S. B. C. Pergher, L. M. A. Campos, M. M. Urbina, T. C. Bicudo and L. S. de Carvalho, Catalysts, 2019, 9, 651 CrossRef.
- A. S. Lenshin, V. M. Kashkarov, E. P. Domashevskaya, A. N. Bel’tyukov and F. Z. Gil’mutdinov, Appl. Surf. Sci., 2015, 359, 550–559 CrossRef CAS.
- J. L. Clabel, H. Iram, T. Awan, G. Lozano, M. A. Pereira-da-Silva, R. A. Romano, V. A. G. Rivera, S. O. Ferreira and E. Marega Jr., Phys. Chem. Chem. Phys., 2020, 22, 15022–15034 RSC.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 CrossRef.
- Y. Zhang, L. Zhu, L. Chen, L. Liu and G. Ye, Rev. Adv. Mater. Sci., 2019, 58(1), 32–37 Search PubMed.
- W. Ranke and H. J. Kuhr, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 1595 CrossRef CAS.
- K. Inyawilert, A. Sukee, M. Siriwalai, A. Wisitsoraat, J. Sukunta, A. Tuantranont, S. Phanichphant and C. Liewhiran, Sens. Actuators, B, 2021, 328, 129022 CrossRef CAS.
- T.-D. Nguyen, C.-T. Dinh and T.-O. Do, ACS Nano, 2010, 4(4), 2263–2273 CrossRef CAS.
- F.-H. Chen, J.-L. Her, Y.-H. Shao, Y. H. Matsuda and T.-M. Pan, Nanoscale Res. Lett., 2013, 8, 18 CrossRef.
- S. Bhaumik, S. K. Ray and A. K. Das, Phys. Status Solidi, 2013, 210, 2146–2152 CrossRef CAS.
- H. J. Yoon, J. Lee, Y. Kim, D. W. Cho and Y. Sohn, Ceram. Int., 2017, 43(2), 2069–2075 CrossRef CAS.
- C. H. Kao, H. Chen, Y. T. Pana, J. S. Chiu and T. C. Lu, Solid State Commun., 2012, 152, 504–508 CrossRef CAS.
- B. D. Padalia, W. C. Lang, P. R. Norris, L. M. Watson and D. J. Fabian, Proc. R. Soc. London, Ser. A, 1977, 354, 269–290 CAS.
- Y. Yamasaki, R. Azuma, Y. Kagebayashi, K. Fujioka and Y. Fujimoto, J. Non-Cryst. Solids, 2020, 543, 120149 CrossRef CAS.
- A. Vedda, et al. , IEEE Trans. Nucl. Sci., 2010, 57(3), 1361–1369 CAS.
- S. Brovelli, A. Baraldi, R. Capelletti, N. Chiodini, A. Lauria, M. Mazzera, A. Monguzzi and A. Paleari, Nanotechnology, 2006, 17, 4031–4036 CrossRef CAS.
- P. G. Kik, M. L. Brongersma and A. Polman, Appl. Phys. Lett., 2000, 76, 2325 CrossRef CAS.
- M. Fujii, M. Yoshida, S. Hayashi and K. Yamamoto, J. Appl. Phys., 1998, 84, 4525 CrossRef CAS.
- T. Kimura, A. Yokoi, H. Horiguchi, R. Saito, T. Ikoma and A. Sato, Appl. Phys. Lett., 1994, 65, 983 CrossRef CAS.
- T. Hoang, N. Elhalawany, B. Enders, E. Bahceci, L. Abuhassan and M. H. Nayfeh, Appl. Phys. Lett., 2016, 109(26), 261103 CrossRef.
- R. Plugaru, J. Piqueras, E. Nogales, B. Méndez, J. A. García and T. J. Tate, J. Optoelectron. Adv. Mater., 2002, 4(4), 883–892 CAS.
- B. Schmaul, G. Huber, R. Clausen, B. Chai, P. LiKamWa and M. Bass, Appl. Phys. Lett., 1993, 62(6), 541–543 CrossRef CAS.
- S. B. Aldabergenova, H. P. Strunk, P. C. Taylor and A. A. Andreev, J. Appl. Phys., 2001, 90(6), 2773–2780 CrossRef CAS.
- J. O. White, M. Dubinskii, L. D. Merkle, I. Kudryashov and D. Garbuzov, J. Opt. Soc. Am. B, 2007, 24, 2454–2460 CrossRef CAS.
- S. D. Setzler, M. P. Francis, Y. E. Young, J. R. Konves and E. P. Chicklis, IEEE J. Sel. Top. Quantum Electron., 2005, 11(3), 645–657 CAS.
- G. Glimann, Encyclopedia of Physical Science and Technology, 1987, vol. 7, p. 210 Search PubMed.
- N. J. J. Johnson, S. He, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139(8), 3275–3282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01617g |
| This journal is © The Royal Society of Chemistry 2025 |