
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen-bonded ionic liquid crystals: supramolecular organization and ionic transport†
Mercedes
Marcos
ab,
Alberto
Concellón
 ab,
Almudena
Terrel
ab,
Rosa I.
Merino
ac,
Rosa M.
Tejedor
d,
Joaquín
Barberá
ab,
José L.
Serrano
*ab and
Santiago
Uriel
*ab
ab,
Almudena
Terrel
ab,
Rosa I.
Merino
ac,
Rosa M.
Tejedor
d,
Joaquín
Barberá
ab,
José L.
Serrano
*ab and
Santiago
Uriel
*ab
aInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: joseluis@unizar.es; suriel@unizar.es
bDepartamento de Química Orgánica, Universidad de Zaragoza, 50009 Zaragoza, Spain
cDepartamento de Física de la Materia Condensada, Universidad de Zaragoza, 50009 Zaragoza, Spain
dCentro Universitario de la Defensa Academia General Militar, Zaragoza, Spain
First published on 11th June 2025
Abstract
Ionic liquid crystals (ILCs) are emerging materials that combine the anisotropic self-assembly of liquid crystals with the ionic conductivity of ionic liquids, making them promising candidates for electrochemical applications such as ion-conducting membranes and next-generation electrolytes. In this work, we report the synthesis and characterization of a series of 1-alkyl-3-halopyridinium halides, where both the cationic and anionic components participate in halogen bonding, leading to enhanced mesophase stability and well-defined ionic nanochannels. Compounds with alkyl chains of 12 carbons or longer exhibit Smectic A liquid crystalline phases, with their stability increasing with chain length and halogen bond strength. X-ray diffraction analysis confirms the role of halogen bonding in driving molecular self-assembly and charge segregation, key factors in mesophase formation. Ionic conductivity measurements demonstrate that these ILCs facilitate ion transport through their nanosegregated ionic domains, with conductivities comparable to other liquid crystalline electrolytes. These findings highlight the potential of halogen-bonded ILCs as functional materials for electrochemical devices, providing a tunable platform for the development of advanced ion-conducting materials.
Introduction
Liquid crystals represent a well-established field of research that, in recent years, has been extensively explored for the development of functional materials.1–4 The broad variety of compounds exhibiting mesogenic properties offers remarkable versatility for new applications, spanning from optoelectronics to energy storage and sensing applications. Among the different classes of liquid crystals, ionic liquid crystals (ILCs) have emerged as a fascinating category of hybrid materials that integrate the anisotropic and self-assembling nature of liquid crystals with the ionic character and chemical tunability of ionic liquids.5–9 This unique combination results in materials with macroscopic orientability, miscibility with various species, phase stability over a wide temperature range, nanostructural tunability, and the formation of polar nanochannels.The study of ILCs has gained significant attention in the past two decades due to their potential applications in a broad range of technological fields, including host frameworks for guest recognition, separation membranes, ion-/proton-conducting membranes, reaction media, and optoelectronic materials.10–17 Their intrinsic ability to self-assemble into ordered nanostructures enables the formation of ion-conducting pathways, making them particularly attractive for energy storage and conversion devices such as batteries, fuel cells, and transistors.18 In these applications, the well-ordered nanochannels inherent to ILCs provide efficient and directional conduction of ions and protons, a crucial feature for high-performance electrochemical devices. Furthermore, ILCs have demonstrated promising properties as ion-conducting membranes in lithium-ion batteries, where their nanostructured architecture ensures high ionic conductivity, electrochemical stability, and flexibility suitable for device fabrication.
A recent classification of ILCs has been based on the cationic and anionic moieties that form the ion pair.6,7 Most of the cations belong to nitrogen-containing heterocyclic families such as imidazoliums, pyridiniums, bipyridiniums, pyrimidiniums, quinoliniums, isoquinoliniums, 1,10-phenanthroliniums, and pyrazoliums. The counteranions, on the other hand, include halides, carboxylates, sulfonates, and metallic anions. The general molecular architecture of ILCs involves a charged moiety coupled with an aliphatic chain, which may be introduced in either the cationic or anionic component. Among heterocyclic derivatives, pyridinium-based ILCs are particularly noteworthy. Since their first description in 1938,19 pyridinium salts have been widely investigated due to their ability to form well-ordered liquid crystalline phases.20–26 Their mesomorphic behavior is mainly dictated by the length of the alkyl chain and the nature of the counteranion, with smectic phases being the most commonly observed.27–29
Besides electrostatic interactions, halogen bonding (XB) has emerged as an important supramolecular interaction in ILCs, significantly influencing their mesophase stability and molecular organization.30–34 Halogen bonding is characterized by its high directionality and has been extensively used in crystal engineering.35–38 Notably, halide anions frequently encountered in ionic liquids and ILCs can serve as halogen bond acceptors, further stabilizing the liquid crystalline phase. In the first examples, the electron-donating halogen bond acceptors were neutral Lewis bases such as pyridine groups (Fig. 1a). More recently, a new approach in the field has focused on charge-assisted halogen-bonded liquid crystals (Fig. 1b), involving a 1-alkyl-3-methylimidazolium iodide, where the iodide anion acts as a bidentate halogen-bond acceptor, forming interactions with a neutral halogen bond donor.39–41
 | ||
| Fig. 1 Representative halogen-bonded ionic liquid crystal architectures. | ||
Herein, we present the synthesis and characterization of halogen-bonded ionic liquid crystals, in which the cationic component not only compensates for the charge but also acts as a halogen bond donor, while the anion functions as the halogen bond acceptor (Fig. 1c and Scheme 1). This approach reduces the number of interacting species, thereby strengthening the halogen bonding interactions. Moreover, the strength of the halogen bond can be fine-tuned by selecting the appropriate halogen donor and halide acceptor. To investigate the impact of halogen bonding on the mesomorphic properties and ionic transport characteristics of these materials, we designed a series of 3-halo (Br or I)-1-alkylpyridinium halides (Br− or I−). These compounds were systematically analyzed to determine their mesogenic behavior, with a particular focus on their phase transitions and supramolecular organization. These studies reveal the presence of well-defined ionic channels that facilitate ionic mobility. Given the potential applications of these materials in ion-conducting devices, their ionic conductivity was also measured, and the results were correlated with their supramolecular organization. This work highlights the interplay between halogen bonding, molecular self-assembly, and ionic transport, contributing to the rational design of functional materials for electrochemical applications.
 | ||
| Scheme 1 Nomenclature of 1-alkyl-3-halopyridinium halides Py-X1-X2-n. | ||
Results and discussion
Synthesis
The preparation of 1-alkyl-3-halopyridinium halides was carried out by refluxing the corresponding 3-halopyridine with an excess of appropriate 1-haloalkane in dry toluene for 20 hours (Scheme 1). After cooling to room temperature, the crude product was isolated by washing with diethyl ether, yielding the 1-alkyl-3-halopyridinium halide with a yield ranging from 60 to 85%.The purity and identity of the final compounds were confirmed using a combination of analytical techniques, including nuclear magnetic resonance (NMR) spectroscopy, infrared (IR) spectroscopy, and high-resolution mass spectrometry (HRMS). The detailed synthetic procedures and full characterization data are provided in the ESI.†
Supramolecular self-assembly organization
The thermal properties of all compounds were examined by polarizing optical microscopy (POM), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). The transition temperatures, enthalpies, phase transition assignments, and structural parameters are listed in Table 1, along with the thermal stability data obtained from TGA. All corresponding TGA and DSC curves are provided in the ESI.† As shown in Table 1, with the exception of compounds bearing the longest chains (C18), all other derivatives exhibit decomposition onsets significantly higher than their clearing points. Additionally, after the first heating cycle, these compounds display reproducible behavior in subsequent DSC scans, both upon heating and cooling. In contrast, the introduction of the octadecyloxy terminal chain leads to a substantial increase in molecular interactions, resulting in decomposition before reaching the clearing point. Consequently, the clearing point for these derivatives was determined via POM observations and is included in Table 1 as the temperature at which decomposition occurs. As expected, following this initial isotropization, the subsequent DSC scans no longer represent the pure compound, and thus the values listed in Table 1 correspond exclusively to the first heating cycle.| Compound | T onset (°C) | Thermal transitionsb | d exp (Å) |
|---|---|---|---|
| a Onset of the decomposition detected in the thermogravimetric curve. b DSC data of the second heating process at a rate of 10 °C min−1 (note: for compounds bearing octadecyloxy alkyl chains, data are taken from the first heating scan). Temperatures (°C) are read at the maximum of the corresponding peaks, and enthalpies (kJ mol−1) are in brackets. g: glass, SmA: smectic A phase, Cr: crystal, I: isotropic liquid. c Isotropization temperature detected by POM which is accompanied by decomposition. d Value calculated according to Bragg's equation. | |||
| Py-Br-Br-10 | 200 | g −4 I | — |
| Py-I-Br-10 | 215 | g −6 I | — |
| Py-Br-Br-12 | 193 | SmA 84 (0.3) I | 29.4 |
| Py-I-Br-12 | 217 | SmA 88 (0.2) I | 31.2 |
| Py-Br-Br-14 | 189 | Cr 84 (22.6) SmA 160 (0.9) I | 31.9 |
| Py-Br-I-14 | 192 | Cr 93 (43.9) SmA 150 (0.9) I | 32.4 |
| Py-I-Br-14 | 217 | Cr 98 (25.9) SmA 178 (0.7) I | 31.2 |
| Py-I-I-14 | 208 | Cr1 70 (3.6) Cr2 89 (33.2) SmA 166 (0.4) I | 32.0 |
| Py-Br-Br-18 | 200 | Cr1 26 (2.7) Cr2 98 (34.8) SmA 218c dec | 37.1 |
| Py-Br-I-18 | 208 | Cr 102 (61.9) SmA 216c dec | 41.2 |
| Py-I-Br-18 | 219 | Cr 109 (42.3) SmA 228c dec | 40.0 |
| Py-I-I-18 | 222 | Cr 103 (65.9) SmA 223c dec | 39.0 |
With the exception of Py-Br-Br-10 and Py-I-Br-10, which possess the shortest alkyl chains, all compounds exhibited liquid crystalline behavior. Under POM, they displayed focal-conic fan textures with homeotropic domains (Fig. 2), characteristic of Smectic A (SmA) mesophases. For homologous liquid crystalline compounds with the same halogen–halide interaction, both the clearing point (TSmA-I) and mesophase range increased with the length of the alkyl chain, indicating that van der Waals interactions between the chains contribute significantly to mesophase stabilization. Moreover, TSmA-I values were primarily influenced by the nature of the 1-halo-pyridine moiety. Compounds containing 1-iodopyridinium units exhibited higher transition temperatures than those bearing 1-bromopyridinium analogues, likely due to stronger electrostatic and packing interactions (e.g., Py-Br-Br-12vs.Py-I-Br-12, Py-Br-Br-14vs.Py-I-Br-14, and Py-Br-I-14vs.Py-I-I-14). Regarding the influence of the counterion, a consistent trend was observed in which compounds bearing iodide (I−) exhibited lower melting points than their bromide (Br−) counterparts. This suggests that the nature of the anion also plays a role in modulating thermal behavior, possibly due to differences in halogen bonding strength and packing efficiency (e.g., Py-Br-Br-14vs.Py-Br-I-14, and Py-I-Br-14vs.Py-I-I-14).
 | ||
| Fig. 2 Representative POM micrographs of (a) Py-I-I-14 at 150 °C (cool) and (b) Py-I-I-18 at 122 °C (heat). | ||
To confirm the nature of the mesophases and determine their structural parameters, the liquid crystalline compounds were further investigated in their mesomorphic state by powder X-ray diffraction (XRD). All the XRD patterns for the studied compounds are provided in the ESI.† These patterns exhibited one or two sharp maxima at low angles, characteristic of layered SmA mesophases. In cases where two maxima were detected, their reciprocal spacing followed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, indicative of first- and second-order reflections from smectic layers. This pattern is consistent with a Smectic A mesophase, and the measured layer spacing values are provided in Table 1. No additional X-ray scattering was observed, even in the high-angle region where a diffuse halo from alkyl chain disorder typically appears in liquid crystal phases. This absence may be attributed to either high conformational disorder of the alkyl chains or the relatively low X-ray scattering contribution of a single alkyl chain compared to heavier halogen atoms. As expected, the measured layer thickness increased with the length of the alkyl chain. The layer thickness values are consistent with a bilayer packing arrangement, in which alkylpyridinium units are organized in opposite orientations.
:2 ratio, indicative of first- and second-order reflections from smectic layers. This pattern is consistent with a Smectic A mesophase, and the measured layer spacing values are provided in Table 1. No additional X-ray scattering was observed, even in the high-angle region where a diffuse halo from alkyl chain disorder typically appears in liquid crystal phases. This absence may be attributed to either high conformational disorder of the alkyl chains or the relatively low X-ray scattering contribution of a single alkyl chain compared to heavier halogen atoms. As expected, the measured layer thickness increased with the length of the alkyl chain. The layer thickness values are consistent with a bilayer packing arrangement, in which alkylpyridinium units are organized in opposite orientations.
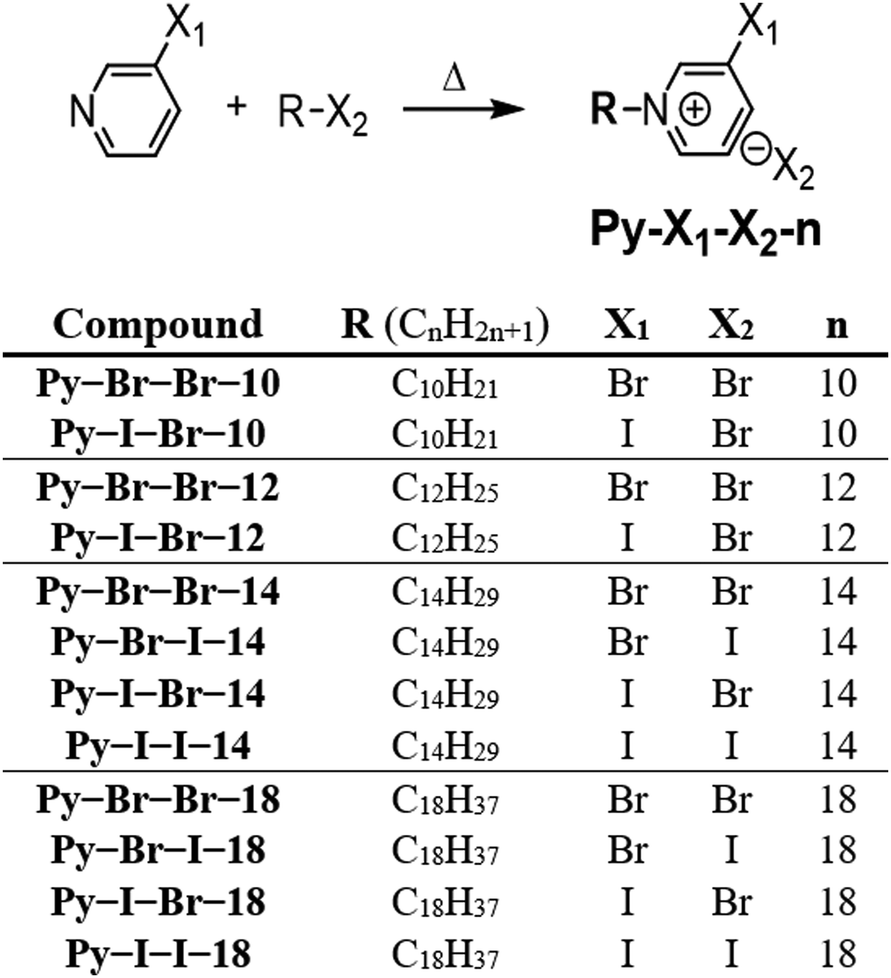
To gain deeper insight into the supramolecular organization and explain the absence of mesogenic behavior in Py-I-Br-10, single-crystal XRD studies were performed on three derivatives: Py-I-Br-10, Py-I-Br-12, and Py-I-Br-18 (Fig. 3). Crystallographic data for these structures are summarized in Table S1 (ESI†). All three structures are characterized by the presence of C–I⋯Br− halogen bonding. The halogen bonding distances and angles are listed in Table S2 (ESI†). The C–I⋯Br− angles are nearly linear, ranging from 169.5° to 179.1° (highlighted in green in the figures), which is consistent with the high directionality expected for halogen bonding.
 | ||
| Fig. 3 (a) Packing diagram of Py-I-Br-10 crystal structure (the two crystallographically independent cations are shown in blue and red). (b) Packing diagram of Py-I-Br-12 crystal structure. (c) (i) Ribbons formed by the combination of halogen and hydrogen bonds in Py-I-Br-18 structure, (ii) packing diagram of Py-I-Br-18. | ||
In addition to halogen bonding, the halide coordination sphere is further stabilized by hydrogen bonding. The hydrogen bond distances (ranging from 2.73 to 3.04 Å) and angles are provided in Table S3 (ESI†). Bromide anions display a variety of coordination environments, interacting with between three to six cations and forming up to two halogen bonds, though the most common number is one.
The crystal packing analysis reveals that all studied structures exhibit microphase segregation, except for Py-I-Br-10 (Fig. 3). This lack of microphase segregation prevents Py-I-Br-10 from displaying mesogenic properties. Fig. 3a illustrates the packing diagram of Py-I-Br-10, showing a distinct supramolecular organization compared to the other structures. In the Py-I-Br-12 crystal structure (Fig. 3b), the molecules arrange into ac-layers, with bromide anions and pyridinium cations forming the central polar region. These polar layers, measuring 8.4 Å in width, are surrounded by alkyl chains oriented along the b-axis. The alkyl chains interdigitate, creating a 17.3 Å non-polar region.
The Py-I-Br-18 crystal structure consists of two crystallographically independent anions and cations (Fig. 3c). The four ions form ribbons in which one bromide establishes two halogen bonds (highlighted in green in Fig. 3c), whereas the other bromide forms only hydrogen bonds. The alkyl chains form a 21.2° angle with the mean plane of the pyridinium rings. These ribbons stack through hydrogen bonding to form layers with a width of 31.3 Å. The interdigitated layers result in a 10.7 Å polar region separated by 20.6 Å wide non-polar zones. Notably, in both Py-I-Br-12 and Py-I-Br-18, the ratio between the charged and non-polar regions remains approximately 0.50, indicating a well-ordered supramolecular organization that facilitates mesophase formation. These findings highlight the crucial role of microphase segregation in mesogenic behavior, explaining why Py-I-Br-10, with its non-lamellar packing arrangement, fails to exhibit liquid crystalline properties.
Theoretical calculations
The computed electrostatic potential (ESP) surfaces of the optimized 1-methyl-3-bromopyridinium and 1-methyl-3-iodopyridinium cations (Fig. 4) reveal an anisotropic charge distribution around the halogen atoms, characterized by a reduced electron density region along the covalent bond extension (σ-hole).42 The maximum electrostatic potential associated with the halogens follows the expected trend, increasing with atomic size: iodine (459.0 kJ mol−1) exhibits a higher value than bromine (427.6 kJ mol−1). These values align with those reported for similar imidazolium-based cations, confirming the enhanced electrophilicity of iodine, which favors stronger halogen bonding interactions.43 | ||
| Fig. 4 Computed electrostatic potential mapped on the respective isosurfaces (0.002 electron Å−1) of electron density of the DFT optimized 1-methyl-3-bromopyridinium cation (left) and 1-methyl-3-iodopyridinium cation (right), colored according to the electrostatic potential in the range [−0.100 to 0.220] a.u. in a RGB scale where red corresponds to more negative values and blue to more positive ones. Maximum electrostatic potential associated to halogen atoms, Br and I, are also displayed. | ||
DFT optimization of the ion pairs consistently converges to structures featuring halogen bonding between the cationic halogen and the halide anion. The calculated intermolecular distances (Table S5, ESI†) are shorter than the sum of the van der Waals and ionic radii, and also slightly shorter than those observed experimentally (Table S1, ESI†), likely due to additional stabilizing interactions in the solid state. As in the crystal structures, the reduction ratio is lower for bromide-containing ion pairs than for those with iodide, indicating stronger halogen bonding in the latter. Additionally, the calculated C–X⋯X− angles are close to 180°, highlighting the directional nature of the halogen bond. The interaction energies range from −352.7 to −294.2 kJ mol−1, with a general trend suggesting that bromide-based pairs form stronger halogen bonds than iodide-based ones (Table S5, ESI†).
To further explore the role of halogen bonding in determining the thermal properties of these compounds, the NCI method44 was applied to the DFT-optimized ion pairs. The 2D and 3D NCI plots (Fig. 5) confirm the presence of strong halogen–halide interactions, evidenced by a pronounced spike in the reduced density gradient around −0.040 a.u. The most intense interactions were observed in ion pairs where bromide acts as the halogen bond acceptor (−0.042 and −0.045 a.u. for Py-Br-Br-1 and Py-I-Br-1, respectively), which supports the experimental observation that mesophase stability decreases when the halogen bond acceptor changes from bromide to iodide. This trend is consistent with the higher σ-hole potential of iodine compared to bromine, as shown in the ESP calculations (Fig. 4). The correlation between halogen bond strength and phase transition temperatures suggests that, in homologous compounds with the same alkyl chains, those forming stronger halogen bonds (e.g., Py-I-Br-1) exhibit higher melting and clearing points. This behavior is reflected in the comparisons between Py-Br-Br-12vs.Py-I-Br-12, Py-Br-Br-14vs.Py-I-Br-14, and Py-Br-Br-18vs.Py-I-Br-18.
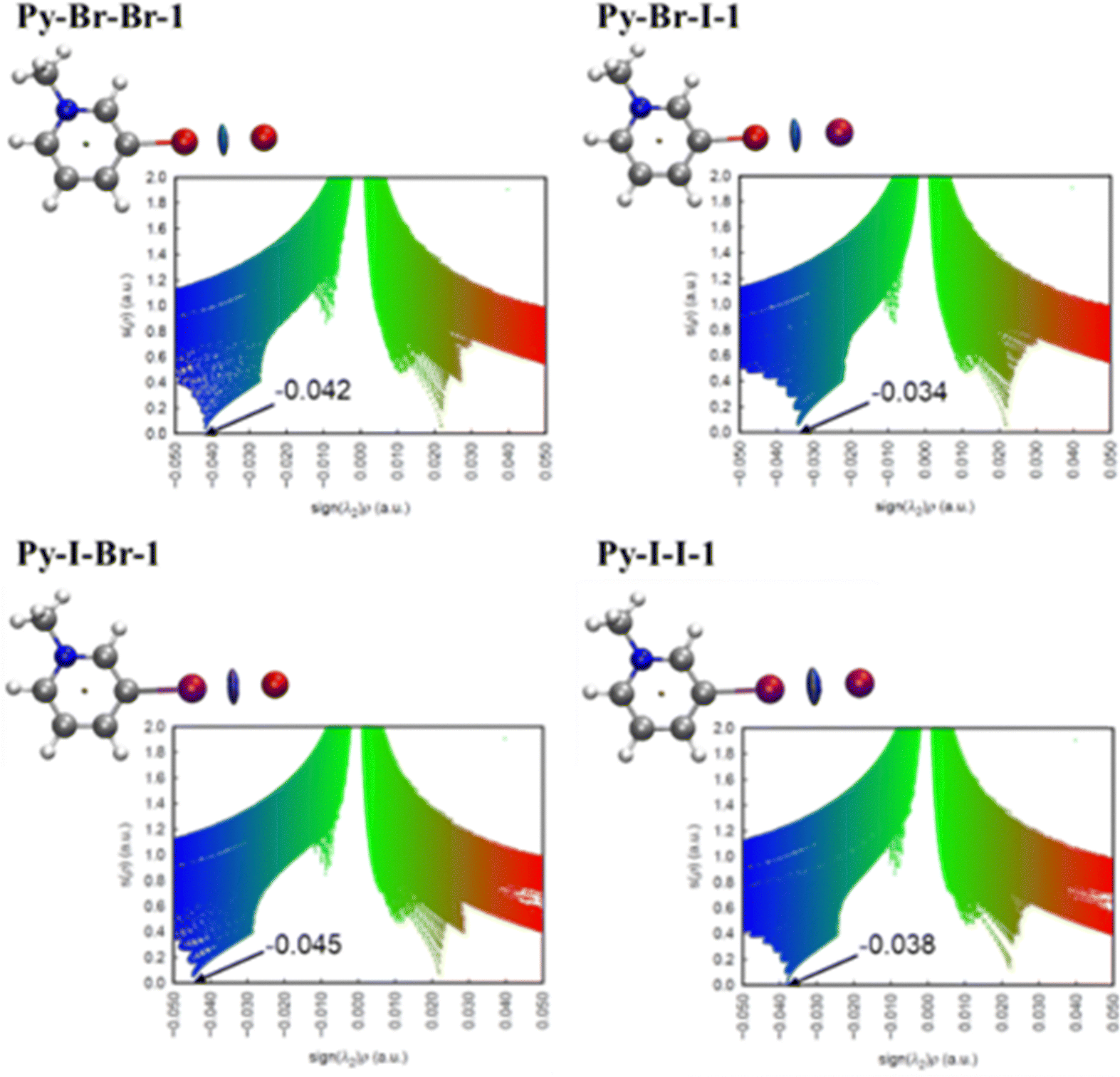 | ||
| Fig. 5 Three-dimensional NCI plot (isosurfaces for s = 0.40 a.u.) of ion pairs colored in the [−0.050, 0.050] a.u. and 2D NCI plots with the electron density multiplied by the sign of the second Hessian eigenvalue corresponding to the halogen bonding. | ||
Interestingly, among the compounds with 14 carbon atoms, Py-Br-I-14, which has the lowest stabilization energy, shows the narrowest mesophase range (56 °C), whereas Py-I-Br-14, with the strongest halogen bond, exhibits higher thermal stability and the widest mesophase range (80 °C). This observation further supports the significant role of halogen bond strength in tuning the mesomorphic behavior of these systems.
Ion conduction properties
The ionic conductivity of the synthesized compounds was evaluated using electrochemical impedance spectroscopy (EIS), with measurements performed on thin films sandwiched between FTO-coated electrodes. The electrodes covered both the sample and the thermoplastic separator used to maintain contact (see ESI,† for details). As a result, the dielectric response of the thermoplastic is included in the high-frequency region of the measured impedance. The typical complex impedance plots exhibited a slightly depressed semicircle in the high-frequency region, with capacitance values around 100 pF, attributed to the sample's bulk conduction. This was followed by a second, more depressed arc at lower frequencies, characterized by significantly higher resistance and a capacitance in the range of 1 μF. In many cases, only the onset of this highly resistive contribution was visible, appearing as a nearly straight, sloped line at low frequencies. This low-frequency behavior is associated with electrode polarization effects. The strong polarizing nature of the FTO electrodes indicates that the primary charge carriers are ionic. In these systems, the mobile species are expected to be halide ions (I− and Br−).45,46Due to the anisotropic nature of liquid crystalline phases, the measured conductivity strongly depends on the macroscopic molecular alignment and the orientation of the phase relative to the electrodes. In freshly prepared cells, the samples initially exhibited a random polydomain organization, which can hinder efficient ion transport. To promote long-range molecular order and enable anisotropic ion conduction, various alignment techniques, including mechanical shearing and thermal annealing, were applied to achieve a uniform planar alignment of the SmA phase. This alignment ensures that the nanosegregated ionic channels are oriented perpendicular to the electrodes, optimizing charge transport.
The conductivities of selected compounds (Py-Br-Br-10, Py-I-Br-10, Py-Br-Br-14, Py-Br-I-14, Py-I-Br-14, and Py-I-I-14) were measured as a function of temperature (Fig. 6). In all cases, ionic conductivity increased with temperature from 30 °C to 190 °C (303 K to 463 K), consistent with enhanced molecular dynamics and increased ion mobility. Conductivities were below 3 × 10−5 S cm−1 for the amorphous compounds with the shortest alkyl chains (C10), and below 1 × 10−6 S cm−1 for the C14 series in their crystalline state, with a rapid decline in conductivity observed as temperature decreased. This behavior suggests that ion transport is significantly hindered in the rigid, densely packed crystalline state. Upon reaching the melting transition, a sharp increase in conductivity was observed, attributed to the enhanced mobility of the charge-carrying species.
 | ||
| Fig. 6 Ion conductivities as a function of the temperature. | ||
For the non-mesogenic compound Py-Br-Br-10, the activation energy was approximately 1 eV, with a similar value observed for Py-I-Br-10. Notably, significant differences in ionic conductivity were observed between the two: at 75 °C (348 K), both in the isotropic state, Py-Br-Br-10 (2.2 × 10−5 S cm−1) exhibited over two orders of magnitude higher conductivity than Py-I-Br-10 (1.9 × 10−7 S cm−1). This difference suggests that Br-based salts possess a more favorable ion-transporting network.
For the mesogenic derivatives with C14 alkyl chains, ionic conductivities were also remarkably similar across the series. These compounds did not exhibit measurable conductivity at room temperature, as the values were below the detection limit of the instrument. Nevertheless, all compounds exhibited a rapid increase in conductivity as they approached the SmA transition from the crystalline phase, followed by a more gradual temperature dependence in the SmA region. In this phase, activation energies were approximately 0.65 eV. At 125 °C (398 K), where all compounds exist in the SmA phase, Py-Br-I-14 showed the highest ionic conductivity (3.6 × 10−5 S cm−1), slightly exceeding that of Py-I-Br-14 and Py-Br-Br-14 (1.2 × 10−5 S cm−1), while Py-I-I-14 displayed the lowest value (1.0 × 10−5 S cm−1). These differences remain relatively small, indicating that ionic conduction in the SmA phase is governed more by the structural organization of the mesophase than by specific halogen bonding interactions. The high degree of molecular order in the SmA phase likely results in tightly packed structures with nanosegregated ionic domains that act as ion highways. This is supported by the observed decrease in conductivity near the isotropic transition in Py-I-I-14.
These results indicate that, while halogen bonding may influence ionic transport, the dominant factor in these systems appears to be the supramolecular organization within the liquid crystalline phase. The nanosegregated ionic channels in the SmA phase form a compact and efficient network for ion conduction, resulting in only minor conductivity variations among the compounds. Although the measured conductivities are much lower than those of benchmark electrolytes used in batteries or fuel cells (∼10−1 S cm−1),47,48 they are they are consistent with values reported for other liquid crystalline electrolytes,49–53 placing them among the highest reported values for this class of materials.
Conclusions
In conclusion, we have synthesized a series of 1-alkyl-3-halopyridinium halides, through a simple, one-step procedure using commercially available reagents. This straightforward and scalable synthesis provides an efficient route to obtaining halogen-bonded ionic liquid crystals with tunable mesophase behavior and ion transport properties, making them attractive candidates for soft-matter functional materials.The thermotropic behavior of these compounds reveals that those with a dodecyl alkyl chain or longer exhibit Smectic A (SmA) liquid crystalline phases, confirming their ability to self-assemble through electrostatic interactions, halogen bonding, and alkyl chain nanosegregation. Notably, the stability and extent of the SmA phase increase with alkyl chain length, emphasizing the role of van der Waals interactions in stabilizing the mesophase. The range of the mesophase is primarily dictated by the strength of the halogen bond, demonstrating that halogen bonding provides a versatile tool for fine-tuning liquid crystalline properties.
Single-crystal X-ray diffraction analysis further validates the structural impact of halogen bonding in these systems. The cationic pyridinium units, acting as halogen bond donors, engage with the halide anions, which function as halogen bond acceptors, forming a robust network of noncovalent interactions. Notably, only salts with alkyl chains of 12 carbon atoms or longer exhibit a layered packing with charge segregation, a feature that is essential for liquid crystalline behavior.
Ionic conductivity measurements confirm that these halogen-bonded ionic liquid crystals support ion transport within their SmA phases. While their conductivity values are lower than those of benchmark electrolytes for batteries or fuel cells (∼10−1 S cm−1), they fall within the high range for liquid crystalline electrolytes. The nanosegregated ionic domains in the SmA phase create well-defined ion transport pathways, enabling conduction while maintaining structural order. However, the highly compact molecular arrangement of the SmA phase results in relatively small conductivity differences among derivatives.
These findings underscore the critical interplay between halogen bonding, mesophase organization, and ionic transport, providing valuable insights for designing functional soft materials for applications in electrochemical devices, ion-conducting membranes, and next-generation electrolytes.
Author contributions
M. Marcos: investigation, conceptualization; A. Concellón: investigation, validation, writing – review & editing; A. Terrel: investigation; R. I. Merino: investigation, validation; R. M. Tejedor: investigation, writing – original draft; J. Barberá: investigation; J. L. Serrano: supervision, writing – review & editing; S. Uriel: supervision, writing – original draft.Data availability
The data that support the findings of this study are available in the ESI,† of this article. The crystallographic information has been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession codes 2443040–2443042.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the projects PID2023-146811NA-I00, PID2021-122882NB-I00, PID2021-126132-NB-I00, and CEX2023-001286-S, funded by MCIN/AEI/10.13039/501100011033 and by “ERDF: A way of making Europe,” and by the Gobierno de Aragón-FSE (research groups E47_23R and T02_23R). A. C. acknowledges grant RYC2021-031154-I, funded by MICIU/AEI/10.13039/501100011033 and by the European Union NextGenerationEU/PRTR. The authors would like to acknowledge the use of the SAI (UNIZAR), CEQMA (UNIZAR-CSIC), and the Crystallography Service of the UPV/EHU. R. M. T. acknowledges the resources provided by the supercomputer Memento and the technical assistance of BIFI-ZCAM.References
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 CrossRef CAS PubMed.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef CAS PubMed.
- A. Concellón, Angew. Chem., Int. Ed., 2023, 62, e202308857 CrossRef PubMed.
- M. Mansueto and S. Laschat, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2014, ch. 8, vol. 6, pp. 231–280 Search PubMed.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- K. Salikolimi, A. A. Sudhakar and Y. Ishida, Langmuir, 2020, 36, 11702–11731 CrossRef CAS PubMed.
- A. Concellón and V. Iguarbe, in Supramolecular Assemblies Based on Electrostatic Interactions, ed. M. A. Aboudzadeh and A. Frontera, Springer International Publishing, Cham, 2022, pp. 85–118.
- N. Kapernaum, A. Lange, M. Ebert, M. A. Grunwald, C. Haege, S. Marino, A. Zens, A. Taubert, F. Giesselmann and S. Laschat, ChemPlusChem, 2022, 87, e202100397 CrossRef CAS PubMed.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
- T. Kobayashi, T. Ichikawa, T. Kato and H. Ohno, Adv. Mater., 2017, 29, 1604429 CrossRef PubMed.
- A. Concellón, T. Liang, A. P. H. J. Schenning, J. L. Serrano, P. Romero and M. Marcos, J. Mater. Chem. C, 2018, 6, 1000–1007 RSC.
- T. Sakamoto, T. Ogawa, H. Nada, K. Nakatsuji, M. Mitani, B. Soberats, K. Kawata, M. Yoshio, H. Tomioka, T. Sasaki, M. Kimura, M. Henmi and T. Kato, Adv. Sci., 2018, 5, 1700405 CrossRef PubMed.
- N. Uemura, T. Kobayashi, S. Yoshida, Y.-X. Li, K. Goossens, X. Zeng, G. Watanabe and T. Ichikawa, Angew. Chem., Int. Ed., 2020, 59, 8445–8450 CrossRef CAS PubMed.
- I. Marín, R. I. Merino, J. Barberá, A. Concellón and J. L. Serrano, Mater. Adv., 2023, 4, 5564–5572 RSC.
- M. A. Stolberg, B. A. Paren, P. A. Leon, C. M. Brown, G. Winter, K. Gordiz, A. Concellón, R. Gómez-Bombarelli, Y. Shao-Horn and J. A. Johnson, J. Am. Chem. Soc., 2023, 145, 16200–16209 CrossRef CAS PubMed.
- T. Kato, K. Imamura, T. Sakamoto and Y. Hoshino, Chem. Commun., 2025, 61, 3998–4001 RSC.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- G. A. Knight and B. D. Shaw, J. Chem. Soc., 1938, 682–683 RSC.
- E. J. R. Sudholter, J. B. F. N. Engberts and W. H. De Jeu, J. Phys. Chem., 1982, 86, 1908–1913 CrossRef CAS.
- C. J. Bowlas, D. W. Bruce and K. R. Seddon, Chem. Commun., 1996, 1625–1626 RSC.
- S. Kumar and S. K. Pal, Tetrahdron Lett., 2005, 46, 4127–4130 CrossRef CAS.
- G. Kohnen, M. Tosoni, S. Tussetschläger, A. Baro and S. Laschat, Eur. J. Org. Chem., 2009, 5601–5609 CrossRef CAS.
- V. Cîrcu, in Ionic Liquids – Progress and Developments, ed. S. Handy, IntechOpen, Rijeka, 2017 Search PubMed.
- D. Devadiga and T. N. Ahipa, RSC Adv., 2019, 9, 23161–23228 RSC.
- S. Kong, X. Wang, L. Bai, Y. Song and F. Meng, J. Mol. Liq., 2019, 288, 111012 CrossRef CAS.
- A. F. M. Santos, C. Cruz, M. H. Godino, M. Dionisio, J. L. Figueirinhas and L. C. Branco, Liq. Cryst., 2022, 49, 1809–1821 CrossRef CAS.
- A. F. M. Santos, Liq. Cryst. Today, 2023, 32, 4–11 CrossRef CAS.
- A. F. M. Santos, J. L. Figueirinhas, C. J. Dias, M. H. Godinho, L. C. Branco and M. Dionísio, J. Mol. Liq., 2023, 377, 121456 CrossRef CAS.
- L. González, N. Gimeno, R. M. Tejedor, V. Polo, M. B. Ros, S. Uriel and J. L. Serrano, Chem. Mater., 2013, 25, 4503–4510 CrossRef.
- L. J. McAllister, C. Präsang, J. P. W. Wong, R. J. Thatcher, A. C. Whitwood, B. Donnio, P. O'Brien, P. B. Karadakov and D. W. Bruce, Chem. Commun., 2013, 49, 3946–3948 RSC.
- H. Wang, H. K. Bisoyi, A. M. Urbas, T. J. Bunning and Q. Li, Chem. – Eur. J., 2019, 25, 1369–1378 CrossRef CAS PubMed.
- D. Devadiga and T. N. Ahipa, J. Mol. Liq., 2021, 333, 115961 CrossRef CAS.
- C. Präsang and D. W. Bruce, Helv. Chim. Acta, 2023, 106, e202300008 CrossRef.
- R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- G. Cavallo, G. Terraneo, A. Monfredini, M. Saccone, A. Priimagi, T. Pilati, G. Resnati, P. Metrangolo and D. W. Bruce, Angew. Chem., Int. Ed., 2016, 55, 6300–6304 CrossRef CAS PubMed.
- M. Saccone, F. F. Palacio, G. Cavallo, V. Dichiarante, M. Virkki, G. Terraneo, A. Priimagi and P. Metrangolo, Faraday Discuss., 2017, 203, 407–422 RSC.
- G. Cavallo, A. Abate, M. Rosati, G. Paolo Venuti, T. Pilati, G. Terraneo, G. Resnati and P. Metrangolo, ChemPlusChem, 2021, 86, 469–474 CrossRef CAS PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- A. Riccobono, A. C. Whitwood, R. R. Parker, S. Hart, A. Pace, J. M. Slattery, I. Pibiri and D. W. Bruce, CrystEngComm, 2022, 24, 7852–7860 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- R. A. Carpio, L. A. King, R. E. Lindstrom, J. C. Nardi and C. L. Hussey, J. Electrochem. Soc., 1979, 126, 1644–1950 CrossRef CAS.
- D. S. Newman, W. Rohr, D. Kirklin and H. D. Frame, J. Electrochem. Soc., 1972, 119, 797–800 CrossRef CAS.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- A. Concellón, S. Hernández-Ainsa, J. Barberá, P. Romero, J. L. Serrano and M. Marcos, RSC Adv., 2018, 8, 37700–37706 RSC.
- D.-J. Mulder, T. Liang, Y. Xu, J. ter Schiphorst, L. M. W. Scheres, B. M. Oosterlaken, Z. Borneman, K. Nijmeijer and A. P. H. J. Schenning, J. Mater. Chem. C, 2018, 6, 5018–5024 RSC.
- T. Liang, H. P. C. van Kuringen, D. J. Mulder, S. Tan, Y. Wu, Z. Borneman, K. Nijmeijer and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2017, 9, 35218–35225 CrossRef CAS PubMed.
- A. Concellón, I. Marín, J. Barberá, M. Marcos and J. L. Serrano, Helv. Chim. Acta, 2023, 106, e202300010 CrossRef.
- I. Dominguez-Candela, I. Zulkhairi, I. Pintre, N. F. K. Aripin, J. Lora-Garcia, V. Fombuena, M. B. Ros and A. Martinez-Felipe, J. Mater. Chem. C, 2022, 10, 18200–18212 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2443040–2443042. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc01507c |
| This journal is © The Royal Society of Chemistry 2025 |