
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the sensing ability of new MoS2 nanoparticles: from fundamental insights into practical applications for nitrites†
Federica
Florio
a,
Angelo
Ferlazzo
 a,
Stefano
Bonforte
a,
Giuseppe
Nicotra
b,
Giovanni
Neri
c,
Iddo
Pinkas
d,
Milko E.
van der Boom
*e and
Antonino
Gulino
*a
a,
Stefano
Bonforte
a,
Giuseppe
Nicotra
b,
Giovanni
Neri
c,
Iddo
Pinkas
d,
Milko E.
van der Boom
*e and
Antonino
Gulino
*a
aDepartment of Chemical Sciences, University of Catania, and I.N.S.T.M. U.d.R. of Catania, Viale Andrea Doria 6, 95125 Catania, Italy. E-mail: antonino.gulino@unict.it
bCNR-IMM, Z.I. VIII Strada 5, Catania, 95121, Italy
cDepartment of Engineering, University of Messina, Contrada Di Dio, 98166 Messina, Italy
dChemical Research Support, Weizmann Institute of Science, Rehovot 7610001, Israel
eDepartment of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 7610001, Israel
First published on 29th April 2025
Abstract
The unique properties of transition metal dichalcogenides (TMDs), particularly molybdenum disulfide (MoS2), have garnered significant attention in various fields including electronics, catalysis, and energy storage. The synthesis of MoS2, along with controlled morphology and properties, remains a crucial aspect because of its practical applications. Here, we present an alternative synthesis approach for MoS2, obtained by a solvothermal method, starting from bis(acetylacetonato)dioxomolybdenum(VI), MoO2(acac)2. Our method results in the formation of a carbon MoS2 (∼75%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :∼25%) composite material. This composite holds promise for advancing our understanding and utilization of MoS2 for sensing. Through detailed characterization and analysis, we elucidate the structure and morphology of the synthesized MoS2, and provide insights into its sensing applications for nitrites. This study not only contributes to the synthesis methodology of MoS2—it also offers valuable insights for the design and development of advanced TMD-based materials.
:∼25%) composite material. This composite holds promise for advancing our understanding and utilization of MoS2 for sensing. Through detailed characterization and analysis, we elucidate the structure and morphology of the synthesized MoS2, and provide insights into its sensing applications for nitrites. This study not only contributes to the synthesis methodology of MoS2—it also offers valuable insights for the design and development of advanced TMD-based materials.
Introduction
Transition metal dichalcogenides (TMDs) are a set of MX2 inorganic compounds, where M belongs to the IV–IX groups, and X is a chalcogen ion (S, Se, Te).1,2 Depending on the number of d-electrons of M and on the X oxidation state, TMDs can be insulators, semiconductors, or metallic.3 Within TMDs, MoS2 exhibits unique properties that mainly develop from its layered structure.4 Therefore, bulk and monolayer MoS2 exhibits an indirect bandgap of ∼1.2 eV or a direct bandgap of ∼1.8 eV, respectively.5,6 MoS2 undergoes the following three main crystalline phases: 1T (one-layered trigonal, metallic), 2H (two-layered hexagonal, semiconductor), and 3R (three-layered rhombohedral, semiconductor), where 1, 2, and 3 indicate the number of MoS2 layers contained in the unit cell, and the letters T, H, and R refer to the lattice systems.7,8MoS2 has recently emerged as a promising material for sensing applications due to its unique electronic, mechanical, and chemical properties.9,10 The layered structure of MoS2, along with its high surface-to-volume ratio and tunable bandgap, makes it an attractive candidate for sensing platforms.11 Considerable research efforts have been directed toward exploring and harnessing the sensing capabilities of MoS2 in environmental monitoring, healthcare diagnostics, and industrial process control.12–14 In this context, the sensing of widely used nitrites as additives in the food industry is important, since, within the human body, nitrites can easily be transformed into nitrosamines. Importantly, these compounds are a probable human carcinogen.15,16 Typical sensing approaches for nitrites involve Raman spectroscopy, chromatography, spectrophotometry, chemiluminescence, supramolecular receptors, and electrochemical measurements.17–19 Electrochemical methods offer a combination of practical advantages regarding sensitivity, selectivity, and possible miniaturization.20 Nanostructured MoS2-based sensors are responsive toward NO2−. Zhang et al. used a flower-like 3D MoS2 microsphere/2D C3N4 nanosheet composite for the electrochemical sensing of nitrite and found a large and linear detection range (0.1–1100 μM), and a detection limit of 0.065 μM.21 Li et al. used high-valence Mo(VI) derived from in situ-oxidized MoS2 nanosheets for nitrite sensing and reported a linear relationship with nitrite concentrations ranging from 1.0 μM to 386.0 μM with an even lower detection limit of 0.028 μM.22 Ghanei-Motlagh used a silver/halloysite nanotube/molybdenum disulfide nanocomposite for nitrite sensing and observed a linear response from 2 to 425 μM with a detection limit of 0.7 μM.23
Nanostructured MoS2 can be synthesized by both bottom-up and top-down approaches.24 We have chosen, among the bottom-up approaches, the solvothermal method that embodies the best compromise between a straightforward synthesis procedure and the desirable properties of the final product.25 In this study, we demonstrated the use of the commercially available MoO2(acac)2 as a precursor for the formation of a composite of MoS2/C and the detection of nitrites. We focused on the electrochemical behavior of this composite to systematically investigate its sensing performance and elucidate its response mechanism. Through comprehensive characterization and analyses, we provide insights into its suitability for practical sensing applications. The detection limit of the composite toward nitrite ions is appreciably below the permissible limit in potable water. We also demonstrate its use for detecting nitrite ions in a meat product.
Experimental
Some chemicals were of reagent grade and were used without further purification. The following compounds were purchased from Sigma Aldrich: bis(acetylacetonato)dioxomolybdenum, MoO2(acac)2 was purified by multiple sublimation procedures at 120 °C under vacuum,26 sulfur, 1-methyl-2-pyrrolidone (NMP), purity ≥99.0%.MoS2 synthesis
1.0770 g (3.302 mmol) of MoO2(acac)2 and 0.318 g (9.906 mmol) of sulfur were dissolved in 60 mL of NMP with two drops of water. Then, the mixture was stirred for 30 minutes, transferred into a 100 mL Teflon-lined stainless steel autoclave and heated at 200 °C for 24 h. Next, the autoclave was left to cool down to room temperature and a black precipitate was collected by centrifugation (14000 rpm) and washed with NMP, ethanol, and water. Finally, the product was dried in a vacuum oven at 60 °C for 24 h; the yield = 0.528 g of overall product (0.396 g C, 0.132 g MoS2), 25% with respect to MoS2.
X-ray photoelectron spectroscopy (XPS)
These measurements were made at a 45° take-off angle relative to the surface normal using a PHI 5000 Versa Probe II system (ULVAC-PHI, Inc.). The base pressure of the main chamber was 1 × 10−8 Pa. Samples were excited with monochromatized Al Kα X-ray radiation using a pass energy of 5.85 eV. Spectra were calibrated by fixing the Ag 3d5/2 peak of a clean sample at 368.3 eV. The instrumental energy resolution was ≤0.5 eV. The XPS peak intensities were obtained after Shirley background removal. The atomic concentration was analyzed by considering the relevant atomic sensitivity factors. The fittings of the Mo 3d, S 2s, S 2p, XP spectra were carried out with the XPSPEAK4.1 software using Gaussian envelopes after subtracting the background until there was the highest possible correlation between the experimental spectra and the theoretical profiles. The residual or agreement factor R was defined as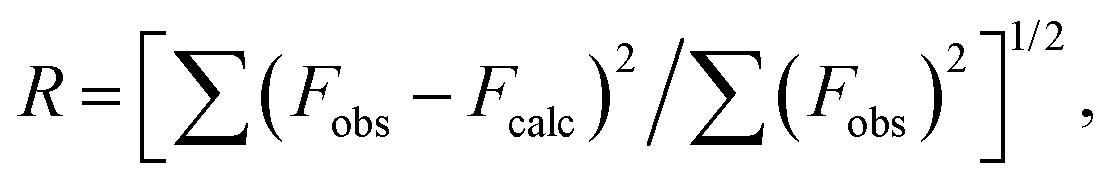 after minimizing the function
after minimizing the function  which converged to a value of 0.03.27–29
which converged to a value of 0.03.27–29
X-Ray diffraction (XRD)
A Rigaku Smartlab diffractometer was used, equipped with a rotating anode of Cu Kα radiation, operating at 45 kV and 200 mA. Bragg–Brentano patterns were acquired with a resolution step of 0.02° 2θ.Raman measurements
The micro-Raman spectra (λ = 532 nm excitation laser) were collected using a Horiba LabRAM HR Evolution (Horiba, France) spectrometer equipped with four laser lines (785, 633, 532, and 325 nm). The system has an 800 mm focal length spectrograph for high-resolution and low stray light, with several interchangeable gratings; it includes an open electrode, front illuminated, cooled CCD detector. The sample was placed under a modular microscope (Olympus BX-FM) with a suitable objective. For this work, a MPlanFL N × 150 NA = 0.9 BD (Olympus Japan) objective with spatial resolution better than 1 μm was used. The Raman scattered light from the sample was dispersed by a 600 g mm−1 grating and the pixel resolution was better than 2 cm−1. Spectra were collected between 100 and 1800 cm−1, with a power of up to 3 mW, and an exposure of 20–100 seconds using 5–20 averages (depending on the signal quality). The system was calibrated using the Si peak at 520.7 cm−1 before every measurement session.Electron microscopy
Scanning electron microscopy (SEM) images were acquired using an Everhart–Thornley detector installed on a Thermo Scientific™ Helios™ 5 UC DualBeam system, equipped with a monochromated SFEG electron gun, operating at 5.00 kV. A JEOL ARM 200F electron microscope was used to characterize the MoS2/C composite by scanning transmission electron microscopy in high annular angle dark field mode (HRSTEM-HAADF) and electron energy-loss spectroscopy (EELS) analyses. Measurements were conducted under gentle STEM conditions (60 keV, a probe size of 1.1 Å, and 5 μA emission current) to prevent the sample from beam damage.30Electrochemistry
Cyclovoltammetry (CV), linear sweep voltammetry (LSV), and differential pulse voltammetry (DPV) measurements were performed using a DropSens μStat 400 potentiostat equipped with Dropview 8400 software. Electrical impedance spectroscopy (EIS) was performed using a Metrohm Autolab galvanostatic potentiostat. A 0.1 M phosphate buffered saline (PBS) solution at pH 7.4 was used to perform the electrochemical measurements. CV tests were performed at a scan rate of 50 mV s−1 in the −0.3 to 0.6 V potential range, using 10 mM potassium ferricyanide (K3[Fe(CN)6]) and 0.1 M KCl standard solutions. LSV tests were conducted using a 0.1 M PBS solution, at a scan rate of 50 mV s−1, in a 0–1 V potential range, to detect 0–1000 μM NaNO2 concentrations, with steps of 10 μM. DPV tests were conducted using an optimized potential step (Estep) of 0.03 V, a potential pulse (Epuls) of 0.09 V, and a time pulse (Tpul) of 200 ms with a scan rate of 40 mV s−1. EIS tests were conducted using 10 mM potassium ferricyanide (K3[Fe(CN)6]) and 0.1 M KCl standard solutions in the 0.1–105 Hz frequency range, an amplitude of 5 mV, and an applied potential of 0.25 V. Measurements were made using commercial screen-printed electrodes, with a carbon working electrode (SPCE) from Methrom DropSens company; the SPCE was modified with MoS2/C composite (hereafter referred to as MoS2/SPCE). This sensor was prepared by depositing 20 μL of a suspension of the MoS2/C composite (1 mg in 1 mL of distilled water) on the working electrode. The resulting sensor was air-dried at room temperature for 24 hours. The sensor's sensitivity was always calculated as the ratio between the slope of the calibration line and the geometric surface area of the used electrode (0.125 cm2).31 The limit of detection (LOD) was calculated by multiplying the ratio between the intercept value and the slope of the calibration line by 3.3. Chronoamperometric curves were obtained by recording the oxidation current, under a constant potential of 0.6 V, while an appropriate volume of 10 mM of the NO2− solution was added to the electrolyte solution (PBS 0.1 M) under magnetic stirring.DPV nitrite sensing measures were also performed at different pH (5–8) and ionic strength (0.1 M, 0.2 M) values.
To assess the ability of the MoS2/SPCE sensor to detect NO2− anions in a real water sample, we performed the DPV analysis on commercial bottled water (Fontenoce; its physico-chemical analysis indicated the absence of nitrite anions) before and after adding 1, 2, and 20 μM of nitrite anion, respectively. The MoS2/SPCE can be reset by washing with distilled water. In addition, we investigated the nitrite concentration in an Italian sausage (bresaola punta d’anca RIGAMONTI; https://www.rigamontisalumificio.it) by DPV using the already optimized potential step, potential pulse, time pulse, and scan rate. Next, we minced a bresaola slice of 10.1 g using a Beku blender in 100 mL of water and after 2 h, we filtered off the liquid. Finally, we diluted 1 mL of this liquid to 5 mL using 4 mL of distilled water and measured the resulting solution.
Results and discussion
Concerning the solvothermal syntheses of MoS2, it was reported that varying the reaction temperature, from 180 °C to 240 °C, results in different degrees of crystallinity and structures, along with a great variety of morphologies (e.g., nanorods, nanosheets, and nanospheres).32 In our solvothermal process we used MoO2(acac)2 as a metal–organic precursor. A possible reaction mechanism for the MoS2 formation involves a redox process in which Mo(VI) is reduced to Mo(IV) and sulfur is involved in a disproportion reaction:| I. 2S0 + 4e− → 2S2− |
| II. S0 → S6+ + 6e− |
| III. Mo6+ + 2e− → Mo4+ |
| IV. 3S0 + Mo6+ → MoS2 + S6+ |
The graphitic carbon was produced by decomposing Hacac.33
Another key factor in our synthetic method is the choice of NMP as solvent. The surface tension value of 40 mJ m−2 matches the surface energy estimated for few-layered MoS2, namely, 46.5 mJ m−2.34 NMP is often used to exfoliate layered materials and to stabilize the thin sheets, produced by material ultrasonication, after the synthesis.34 However, such a procedure may induce undesired oxidation processes and consequently the formation of undesired sulfur vacancies.35 Taking all this into account, we employed NMP as the solvent for the MoS2/C syntheses to obtain a layered nanostructure without further post-synthetic treatment. The other important parameter in our solvothermal synthesis is the temperature, which was set at a value (200 °C) close to the boiling point of NMP (202 °C).
Fig. 1a presents a comprehensive micro-Raman spectrum of the MoS2/C composite in the 100–1800 cm−1 range, and Fig. 1b provides the 100–500 cm−1 region.
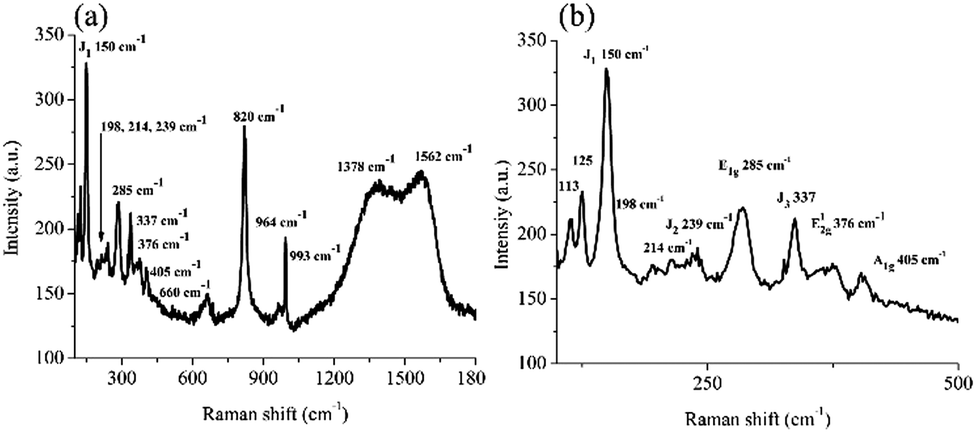 | ||
| Fig. 1 (a) Micro-Raman spectrum of the MoS2/C composite in the 100–1800 cm−1 range. The low frequency shows MoO3 and MoS2; the middle frequencies show other oxides, and the higher frequencies belong to the carbonaceous material; (b) an expanded scale in the 100–500 cm−1 range shows the assignments of the MoS2 vibrations (1T and 2H) and the MoO3 oxide. | ||
The peaks observed at 150, 239, 285, and 337 cm−1 correspond to the characteristic J1, J2, E1g, and J3 MoS2 vibrational modes, respectively, which were indicative of the 1T-phase of MoS2.36 On the other hand, the peaks at 376 and 405 cm−1 correspond to the E12g and A1g vibrational modes of the 2H-phase of MoS2, respectively.3,37 In addition, the peaks at 113 and 125 cm−1 correspond to the B1g and B2g vibrational modes associated with some MoO3 compounds, as well as the peaks at 660, 820, 964, and 993 cm−1.36,38,39 The vibrational modes at 1378 and 1562 cm−1 correspond to the D and G bands of graphitic carbon, respectively.40 Therefore, the Raman data confirm the presence of a composite material consisting of graphitic carbon as well as the MoS2 1T and 2H mixed phases.
XPS was performed on the as-synthesized MoS2/C to obtain important information about its electronic structure. Fig. 2a shows the XP spectrum of the Mo 3d and S 2s states. Three peaks were evident at 226.5, 229.1, and 232.3 eV. Deconvolution of the experimental XP spectrum revealed the presence of a band at 226.5 eV due to the S 2s states. The following six Gaussians correspond to three Mo 3d doublets (Mo 3d5/2,3/2 spin–orbit components). Those Gaussians at 229.0–232.2 eV are consistent with the Mo4+ states of the 1T MoS2 phase (39% of the overall Mo content); those at 230.1–233.3 eV are consistent with the Mo4+ states of the 2H MoS2 phase (56% of the overall Mo content), and those at 232.2–235.2 eV are consistent with a few Mo6+, due to the presence of some sizeable (∼5% with respect to the total Mo content) MoO3.26,41–43 The data are in good agreement with the literature regarding layered structured MoS2.44
 | ||
| Fig. 2 (a) Al-Kα excited XPS of MoS2 in the Mo 3d5/2,3/2, S 2s binding energy region. The olive, cyan, and wine lines denote the 229.0–232.2, 230.1–233.3, and 232.2–235.2 eV components, respectively, of the Mo 3d states, and the purple line denotes the S 2s signal at 226.5 eV. The blue line denotes the background, and the red line superimposed on the experimental black profile denotes the sum of all components. (b) Al-Kα excited XPS of MoS2 in the S 2p3/2,1/2 binding energy region. The purple and the pink lines denote the 162.0–163.2 and 163.5–164.7 eV components, respectively, of the S 2p states. The blue line denotes the background, and the red line superimposed on the experimental black profile denotes the sum of all components. | ||
Fig. 2b shows the XP spectrum of the S 2p states. The spectrum revealed two evident peaks and an additional high binding energy shoulder at 162.1, 162.9, and 164.4 eV. Fitting the spectrum profile disclosed the superimposition of two S 2p3/2,1/2 doublets at 162.0–163.2 and 163.5–164.7 eV (1.2 eV spin–orbit coupling), whose single doublet components overlapped. According to the reported results, the lower binding energy doublet refers to both the terminal S22− and S2− basal plane ions, whereas the higher binding energy doublet is due to apical S2− and bridging S22− ions.45
The SEM analysis of MoS2/C reveals granular structures that are highly dispersed in size, irregularly faceted, and with rounded surfaces. These features indicate the absence of well-defined crystallinity and the presence of large grains (Fig. 3).
 | ||
| Fig. 3 SEM images of the MoS2/C composite. | ||
HRSTEM-HAADF imaging of the MoS2/C composite (Fig. 4a–c) revealed a nanostructured specimen made by a randomly oriented nanoribbon-like matrix very visible in the thinner regions.46 Layered structures are visible with an average length of 2.79 nm, and typical interplanar distances of 0.60 and 0.90 nm, consistently with the 2H and 1T-MoS2 phases, respectively, which coincide with the Raman analysis.36
 | ||
| Fig. 4 HRSTEM-HAADF images of the MoS2/C composite showing (a) some nanoribbons of 3.50 nm at 0.90 nm; (b) some nanoribbons of 2.00 and 1.32 nm at 0.60 and 0.90 nm, respectively; (c) nanoribbons of 3.00 and 3.34 nm (d = 0.6 nm); (d) a selected area electron diffraction (SAED) pattern and its intensity profile (the cyan line) of the MoS2/C composite. | ||
Both the XRD spectra and the SAED patterns obtained from HRSTEM show broadened diffraction peaks and continuous rings, respectively, which are characteristics of partially ordered crystals with a short-range order of <10 nm for which one observes relevant peak broadening due to a small crystal size.
From the SEM and TEM analyses, it emerges that these nanostructures have a matrix with an intermediate structure between amorphous and crystalline. There are locally ordered regions that exhibit a layered structure with nanoribbon-like morphologies.
Fig. S1 (ESI†) shows the XRD pattern of the MoS2/C composite. As shown, apart from a broad band covering the 30–60 2θ range, no well-defined peaks were observed throughout the 2θ range. This observation reinforces the absence of a long-range order or a defined interlayer spacing. Such a pattern characterizes nanostructures with dimensions well below 10 nm, which lack a long-range order yet retain short-range periodicity.47 This observation is in good agreement with literature XRD data for a similar nanosized, destacked MoS2 material.35,48,49 Raman spectroscopy is highly sensitive to local atomic arrangements, allowing the detection of localized phonon modes even under conditions where XRD shows no long-range order.50 Indeed, the SAED pattern (Fig. 4d) and the corresponding intensity profile (the cyan line in Fig. 4d), which is in close agreement with the XRD profile (Fig. S1, ESI†), exhibited a “poor crystalline” diffraction signature with blurry halo rings.4,51
The EELS spectrum (Fig. S2, ESI†) confirmed that MoS2 is homogeneously dispersed within the carbon matrix.
The electrochemical properties of the MoS2/C composite indicated a band gap of 1.68 eV, estimated from the difference between the LUMO (−4.077 eV) and HOMO (−5.757 eV) energy levels.52 These HOMO–LUMO levels were calculated from the oxidation and reduction onset from the cyclic voltammogram shown in Fig. 5 using eqn (1) and (2):
| HOMO = −[Eonset,ox + 4.637 eV] | (1) |
| LUMO = −[Eonset,red + 4.637 eV] | (2) |
 | ||
| Fig. 5 Cyclic voltammogram versus the Ag/AgCl reference electrode of the synthesized MoS2/C composite. The ΔV of the standard hydrogen electrode (SHE) vs. vacuum = 4.440 V; the ΔV of the Ag/AgCl vs. SHE = 0.197 V; the ΔV of the Ag/AgCl vs. vacuum = 4.637 V. | ||
Fig. S3–S5 (ESI†) show the characterization of the MoS2/C composite/SPCE electrode. We performed DPV measurements with the MoS2/SPCE sensor using increasing NO2− concentrations (Fig. 6a). Fig. 6b shows the calibration curve of the MoS2/SPCE sensor; it exhibits excellent responsiveness to nitrite concentrations, showing a sensitivity of 4.993 μA μM−1 cm−2.53
 | ||
| Fig. 6 (a) DPV at different nitrite concentrations (0–1100 μM, initial step 1 μM) in 0.1 M PBS (pH 7.4); (b) calibration curve for anodic peak current (Ipa) versus the nitrite concentration (SD ≤ 1.17 for 5 repeated whole cycles). Inset: Expanded scale in the 0–50 μM range of NaNO2. | ||
The reported oxidative peak potential of MoS2 is 1.3 V.5,6 In our carbon MoS2 composite we observed this oxidative peak at 1.6 V.
The Food and Drug Administration (FDA) states that the nitrite anion concentration in water must be lower than 1.0 mg L−1 (20 μM); however, we succeeded in detecting much lower concentrations (0.085 μM). Concerning the sensing mechanism for nitrite anions by MoS2, it has been reported that some Mo6+, obtained upon applying a potential of about 1 V, may be responsible for the following reaction:22
| Mo6+ + NO2− + H2O → Mo4+ + NO3− + 2H+ |
In this context, we detected ∼5% of MoO3 (Mo6+) in our composite (see above the Raman and XPS data); therefore, the sensing activity of our material was intrinsically possessed. Note that our present study is not really concerned with identifying the nature of compensating defects such as cation vacancies, interstitial sulfur, more complex defect clusters, or defects at the grain boundaries.54,55
Concerning the specific role of the 1T and 2H MoS2 phases for the electrochemical sensing of nitrites, the metallic 1T-MoS2 (highly active at both edges and basal planes) represents the most active phase for electrochemical sensing, since possesses excellent electrical conductivity, while the semiconducting 2H-MoS2 phase (that has only active edges) is more stable. Therefore, the synergic contribution of both 1T (metallic) and 2H (higher stable) phases are crucial factors in the development of sensible and stable electrochemical sensors.56,57
The repeatability of the MoS2/SPCE sensing ability for NO2− was demonstrated by replicating five times the analyses at a concentration of 20 μM (Fig. S6a, ESI†); the calculated standard deviation was 0.78. Chronoamperometry was also applied to better assess the NO2− sensing limit of MoS2/SPCE using a constant applied potential (0.6 V vs. Ag/AgCl). Fig. 7 shows the current response upon changing the nitrite concentration. The increase in oxidation current reaches a plateau in 2 s upon increasing the [NO2−], up to the tested 1500 μM concentration. The corresponding calibration curve, shown in Fig. 8, shows the linear fit of the current vs. the nitrite μM concentration, which yields a sensitivity of 259 μA μM−1 cm−2 and a LOD value of 0.085 μM.
 | ||
| Fig. 7 Current–time response of the MoS2/SPCE electrode upon successive additions of nitrite to the 0.1 M PBS electrolyte at 0.6 V; the inset shows the response with 0–10 μM of nitrite. | ||
 | ||
| Fig. 8 Calibration line for detecting and quantifying nitrite. The inset shows the calibration line for 0–3 μM of nitrite. | ||
To assess the ability of the MoS2/SPCE sensor to detect nitrite anions in real samples (without the use of PBS), we performed DPV measurements on commercial nitrite-free bottled water, before and after adding 1, 2, and 20 μM of NO2− (Fig. S6b, ESI†). The calculated recoveries ranged between 99.3% and 104.4% (Table 1).58 Such results indicate that the MoS2/SPCE sensor selectively detects nitrite anions even in the presence of other potential interfering species (those stated by the Fontenoce factory: Cl−, Ca2+, Mg2+, K+, SO42−, F−, SiO2, HCO3−, Na+, and NO3−).59 Furthermore, we investigated the nitrite concentration by DPV in an Italian sausage (bresaola) (Fig. 9). The measured current variation at the exact expected nitrite oxidation potential (0.6 V), using the already optimized potential step, potential pulse, time pulse, and scan rate (see Experimental), was 5.8 μA. Using the calibration line shown in Fig. 6b, and considering the performed dilution (see Experimental), we obtained 72.8 mg nitrite per kg sausage. This value is very close to the limit indicated by European legislation (80 mg kg−1) for this kind of food product.
| Sensor | [NO2−] (μM) | ΔCurrent (μA) | Recovery (%) |
|---|---|---|---|
| MoS2/SPCE | 1 in 0.1 M PBS | 1.47 | 100 |
| “ | 2 in 0.1 M PBS | 2.56 | 100 |
| “ | 20 in 0.1 M PBS | 5.54 | 100 |
| ” | 1 in bottled water | 1.46 | 99.3 |
| “ | 2 in bottled water | 2.67 | 104.4 |
| “ | 20 in bottled water | 5.57 | 100.5 |
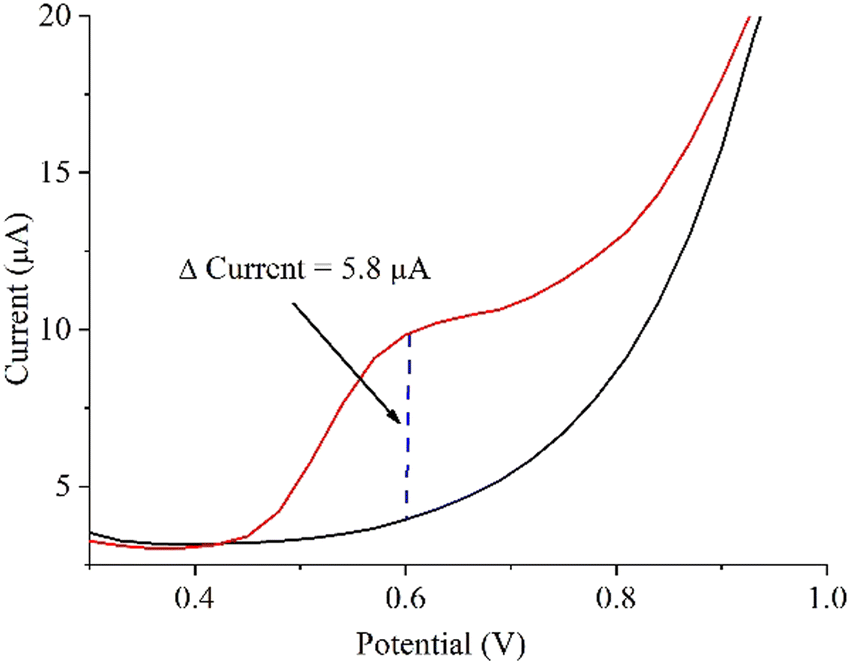 | ||
| Fig. 9 DPV analyses to determine the nitrite in an Italian sausage (bresaola). | ||
In addition, to further investigate the ability of our MoS2/SPCE sensor to detect nitrite anions in real samples, we measured the DPV in water containing single interferents (100 μM each), as shown in Fig. 10. The results further show the selectivity of the material.
 | ||
| Fig. 10 The histogram shows the response of the MoS2/SPCE toward possible interferents (100 μM each) and nitrite in water. | ||
Furthermore, the MoS2/SPCE sensor exhibits good stability that extends at least six months after its initial use (Fig. S6c, ESI†).
The response of the MoS2/SPCE sensor to 200 μM nitrite at different pH values (5.0–8.0) is shown in Fig. S7 (ESI†) and indicates a small shift toward lower oxidation potentials (0.67–0.53 V) as the pH increases from 5.0 to 8.0. Fig. S8 (ESI†) shows virtually no change in current or potential as the ionic strength increases from 0.1 to 0.2 M.
Electrochemical and optical methods are often used for nitrite sensing in water and food.21,60–69 Catalytic and electrocatalytic nitrite determination in aqueous solution have been reported as well.70,71 Therefore, the already reported LODs for nitrite sensing (Table 2) seem to span a large range, with values as low as ∼0.065 μM; however, often the sensing platforms required multi-step syntheses or rather complex apparatuses other than the one we reported.72–79
| Electrode modifier | Method | Linear range (μM) | LOD (μM) | Ref. |
|---|---|---|---|---|
| Ag/HNTs/MoS2/CPE | CA | 2–425 | 0.7 | 23 |
| Ag/H-C3N4/CC-1 | CA | 5–1000 | 0.9 | 80 |
| Au/NiO/rGO/SPCE | DPV | 1–500 | 0.2 | 81 |
| Pd–Cu–Mo2C/GCE | CA | 5–165 | 0.35 | 63 |
| Ni/MoS2/GCE | DPV | 20–1000 | 2.74 | 82 |
| AuNPs/GCE | DPV | 10–3800 | 2.4 | 83 |
| NBC180/SPCE | DPV | 1–1400 | 2.08 | 16 |
| 3DMoS2/2DC3N4/GCE | DPV | 0.1–1100 | 0.065 | 21 |
| CoBIM/CTAB | ECL | 1–1500 | 0.67 | 84 |
| MoS2/SPCE | CA | 1–1000 | 0.085 | This work |
It turns out that the LOD value obtained with our rather simple sensor (0.085 μM, see the present chronoamperometry results) is within the state of-the art.
Conclusions
We synthesized a composite material having a graphitic MoS2/C composition and a nanoribbon morphology well-tailored for the current sensing applications.67 The MoS2/SPCE sensor was found to be highly sensitive to nitrite concentrations, showing a sensitivity of 4.993 μA μM−1 cm−2 and a LOD value of 0.085 μM. The sensor is selective and can be reset by simply washing in distilled water. The overall performance is comparable or even better than that of the previously reported nitrite sensors.Author contributions
A. Gulino: conceptualization, device design, writing, editing, and funding; F. Florio: calibration analysis, device design, experimental and statistical analyses, and writing; A. Ferlazzo: calibration analysis, device design, experimental and statistical analyses, and writing; S. Bonforte: experimental analyses; G. Nicotra: experimental analyses, writing; G. Neri: experimental and statistical analyses; M. E. V. D. Boom: conceptualization, writing, and editing; I. Pinkas: experimental analyses.Data availability
The data supporting this article have been included in the main text and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union (NextGeneration EU) through the MUR-PNRR project SAMOTHRACE (ECS00000022). The authors also thank the University of Catania for the financial support of the PIA.CE.RI. and the B.R.I.T. laboratory at the University of Catania for the availability of the XPS facility. M. V. D. B. is the incumbent of the Bruce A. Pearlman Professorial Chair. IP is the incumbent of the Sharon Zuckerman research fellow chair.Notes and references
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef.
- J. L. Musfeldt, Y. Iwasa and R. Tenne, Phys. Today, 2020, 73, 42–48 CrossRef CAS.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- D. Kozawa, P. Liu, Y. Zeng, V. B. Koman, M. Kuehne and M. S. Strano, Nano Lett., 2020, 20, 3067–3078 CrossRef CAS.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- D. S. Schulman, D. May-Rawding, F. Zhang, D. Buzzell, N. Alem and S. Das, ACS Appl. Mater. Interfaces, 2018, 10, 4285–4294 CrossRef CAS PubMed.
- Y. Dong, M.-M. Yang, M. Yoshii, S. Matsuoka, S. Kitamura, T. Hasegawa, N. Ogawa, T. Morimoto, T. Ideue and Y. Iwasa, Nat. Nanotechnol., 2023, 18, 36–41 CrossRef CAS PubMed.
- P. S. Kiran, K. V. Kumar, N. Pandit, S. Indupuri, R. Kumar, V. V. Wagh, A. Islam and A. K. Keshri, Adv. Funct. Mater., 2024, 34, 2316266 CrossRef CAS.
- H. Wang, C. Li, P. Fang, Z. Zhang and J. Z. Zhang, Chem. Soc. Rev., 2018, 47, 6101–6127 RSC.
- L. Li, Q. Wang, F. Wu, Q. Xu, J. Tian, Z. Huang, Q. Wang, X. Zhao, Q. Zhang, Q. Fan, X. Li, Y. Peng, Y. Zhang, K. Ji, A. Zhi, H. Sun, M. Zhu, J. Zhu, N. Lu, Y. Lu, S. Wang, X. Bai, Y. Xu, W. Yang and G. Zhang, Nat. Commun., 2024, 15, 1825 CrossRef CAS.
- A. Dodda, D. Jayachandran, A. Pannone, N. Trainor, S. P. Stepanoff, M. A. Steves, S. S. Radhakrishnan, S. Bachu, C. W. Ordonez, J. R. Shallenberger, J. M. Redwing, K. L. Knappenberger, D. E. Wolfe and S. Das, Nat. Mater., 2022, 21, 1379–1387 CrossRef CAS.
- V. D. Dang, R. Putikam, M. C. Lin and K. H. Wei, Small, 2024, 20, 2305220 CAS.
- T. X. Huang, X. Cong, S. S. Wu, J. B. Wu, Y. F. Bao, M. F. Cao, L. Wu, M. L. Lin, X. Wang, P. H. Tan and B. Ren, Nat. Catal., 2024, 7, 646–654 CrossRef CAS.
- J. Sengupta and C. M. Hussain, TrAC, Trends Anal. Chem., 2024, 117742 CrossRef CAS.
- D. Zhang, M. Li, Y. Yang, H. Yu, F. Xiao, C. Mao, J. Huang, Y. Yu, Y. Wang, B. Wu, C. Wang, L. Shu, Z. He and Q. Yan, Water Res., 2022, 220, 118637 CrossRef CAS.
- A. Ferlazzo, V. Bressi, C. Espro, D. Iannazzo, E. Piperopoulos and G. Neri, J. Electroanal. Chem., 2023, 928, 117071 CrossRef CAS.
- N. Ibrahim, M. A. Hefnawy, S. A. Fadlallah and S. S. Medany, Food Chem., 2024, 140962 Search PubMed.
- C. N. Carroll, J. J. Naleway, M. M. Haley and D. W. Johnson, Chem. Soc. Rev., 2010, 39, 3875–3888 RSC.
- I. H. Abidi, S. P. Giridhar, J. O. Tollerud, J. Limb, M. Waqar, A. Mazumder, E. L. H. Mayes, B. J. Murdoch, C. Xu, A. Bhoriya, A. Ranjan, T. Ahmed, Y. Li, J. A. Davis, C. L. Bentley, S. P. Russo, E. Della Gaspera and S. Walia, Adv. Funct. Mater., 2024, 34, 2402402 CrossRef CAS.
- J. F. Tan, A. Anastasi and S. Chandra, Curr. Opin. Electrochem., 2022, 32, 100926 CrossRef CAS.
- L. Wang, Z. Fan, F. Yue, S. Zhang, S. Qin, C. Luo, L. Pang, J. Zhao, J. Du, B. Jin and H. Zhang, Food Chem., 2024, 430, 137027 CrossRef CAS PubMed.
- H. L. Zou, L. Y. Qin, H. Q. Luo, B. L. Li and N. B. Li, Sens. Actuators, B, 2021, 337, 129812 CrossRef CAS.
- M. Ghanei-Motlagh and M. A. Taher, Biosens. Bioelectron., 2018, 109, 279–285 CrossRef CAS PubMed.
- N. Abid, A. M. Khan, S. Shujait, K. Chaudhary, M. Ikram, M. Imran, J. Haider, M. Khan, Q. Khan and M. Maqbool, Adv. Colloid Interface Sci., 2022, 300, 102597 CAS.
- S. H. Choi, S. J. Yun, Y. S. Won, C. S. Oh, S. M. Kim, K. K. Kim and Y. H. Lee, Nat. Commun., 2022, 13, 1484 CrossRef CAS PubMed.
- A. Gulino, G. G. Condorelli and I. Fragalà, J. Mater. Chem., 1996, 6, 1335–1338 CAS.
- D. Briggs and J. T. Grant, Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, IMP, Chichester, UK, 2003 Search PubMed.
- A. Gulino, Anal. Bioanal. Chem., 2013, 405, 1479–1495 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 59, 5002–5006 CrossRef CAS PubMed.
- O. L. Krivanek, N. Dellby, M. F. Murfitt, M. F. Chisholm, T. J. Pennycook, K. Suenaga and V. Nicolosi, Ultramicroscopy, 2010, 110, 935–945 CAS.
- A. Ferlazzo, A. Gulino and G. Neri, Environ. Sci.: Adv., 2024, 3, 1392–1399 CAS.
- P. Phalswal, P. K. Khanna, H.-G. Rubahn and Y. K. Mishra, Mater. Adv., 2022, 3, 5672–5697 RSC.
- R. C. Mehrotra, R. Bohra and D. P. Gaur, Metal β-Diketonates and Allied Derivatives, Academic Press, London, 1978 Search PubMed.
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2016, 28, 337–348 CAS.
- H. Ma, S. Ben, Z. Shen, X. Zhang, C. Wu, S. Liao and F. An, Appl. Surf. Sci., 2020, 512, 145588 CrossRef CAS.
- J. Strachan, A. F. Masters and T. Maschmeyer, J. Mater. Chem. A, 2021, 9, 9451–9461 RSC.
- Y. Zhang, J. Li, X. Li, L. Shan, W. Zhao, J. Wang, Q. Gao, Z. Cai, C. Zhou, B. Han, K. Amine and R. Sun, Nano Lett., 2024, 24, 3331–3338 CrossRef CAS PubMed.
- J. Bai, B. Zhao, J. Zhou, J. Si, Z. Fang, K. Li, H. Ma, J. Dai, X. Zhu and Y. Sun, Small, 2019, 15, 1805420 CrossRef PubMed.
- G. Mestl, P. Ruiz, B. Delmon and H. Knozinger, J. Phys. Chem., 1994, 98, 11269–11275 CrossRef CAS.
- M. Sarkar, R. Hossain and V. Sahajwalla, Carbon, 2023, 213, 118274 CrossRef CAS.
- H. Cao, Z. Bai, Y. Li, Z. Xiao, X. Zhang and G. Li, ACS Sustainable Chem. Eng., 2020, 8, 7343–7352 CrossRef CAS.
- D. Scirè, R. Macaluso, M. Mosca, S. Mirabella, A. Gulino, O. Isabella, M. Zeman and I. Crupi, Solid-State Electron., 2021, 185, 108135 CrossRef.
- A. Gulino, S. Parker, F. H. Jones and R. G. Egdell, J. Chem. Soc., Faraday Trans., 1996, 92, 2137–2141 RSC.
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1801345 CrossRef.
- D. Tsikritzis, N. Tsud, T. Skála and L. Sygellou, Appl. Surf. Sci., 2022, 599, 153896 CrossRef CAS.
- Y. Wang, W. Zhai, Y. Ren, Q. Zhang, Y. Yao, S. Li, Q. Yang, X. Zhou, Z. Li, B. Chi, J. Liang, Z. He, L. Gu and H. Zhang, Adv. Mater., 2024, 36, 2307269 CrossRef CAS PubMed.
- X. Guan, L. Zhao, P. Zhang, J. Liu, X. Song and L. Gao, Mater. Today Energy, 2020, 16, 100379 CrossRef.
- J. Ekspong, R. Sandström, L. P. Rajukumar, M. Terrones, T. Wågberg and E. Gracia-Espino, Adv. Funct. Mater., 2018, 28, 1802744 CrossRef.
- W. Han, J. Wang, X. Li, L. Zhou, Y. Yang, M. Tang and H. Ge, Catal. Commun., 2019, 124, 86–91 CrossRef CAS.
- Y.-H. Choi, J. Cho, A. M. Lunsford, M. Al-Hashimi, L. Fang and S. Banerjee, J. Mater. Chem. A, 2017, 5, 5129–5141 RSC.
- L. Fei, S. Lei, W.-B. Zhang, W. Lu, Z. Lin, C. H. Lam, Y. Chai and Y. Wang, Nat. Commun., 2016, 7, 12206 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS.
- K. Abid, A. Ferlazzo and G. Neri, FlatChem, 2024, 46, 100673 CrossRef CAS.
- S. Millesi, M. R. Catalano, G. Impellizzeri, I. Crupi, G. Malandrino, F. Priolo and A. Gulino, Mater. Sci. Semicond. Process., 2017, 69, 32–35 CrossRef CAS.
- A. Gulino, A. E. Taverner, S. Warren, P. Harris and R. G. Egdell, Surf. Sci., 1994, 315, 351–361 CrossRef CAS.
- S. Ren, W. Cui, Y. Liu, S. Cheng, Q. Wang, R. Feng and Z. Zheng, Sens. Actuators, A, 2022, 345, 113772 CrossRef CAS.
- S. Ren, R. Feng, S. Cheng, L. Huang, Q. Wang and Z. Zheng, Microchem. J., 2022, 175, 107129 CrossRef CAS.
- J. H. Zhang, Q. Shen and Y. G. Zhou, ACS Sens., 2021, 6, 2320–2329 CrossRef CAS PubMed.
- Chemical and physico-chemical analyses stated for the Fontenoce bottled water: Specific electrical conductivity measured at 20 °C = 147 μS cm−1, source temperature 9.3 °C, pH at the source 7.69, fixed residue at 180 °C = 103.4 mg L−1, NO2− absent, Ca2+ 16.2 mg L−1, Mg2+ 5.9 mg L−1, Na+ 7.2 mg L−1, K+ 1.6 mg L−1, HCO3− 75.3 mg L−1, SO42− 8.9 mg L−1, Cl− 6.9 mg L−1, NO3− 1.7 mg L−1, F− 0.13 mg L−1, SiO2 16.9 mg L−1.
- D. Thatikayala and B. Min, Biosens. Bioelectron., 2023, 241, 115659 CrossRef CAS PubMed.
- L. Britschgi, K. Villez, P. Schrems and K. M. Udert, Water Res. X, 2020, 9, 100055 CrossRef CAS PubMed.
- S. Velmurugan, S. Palanisamy and T. C.-K. Yang, Sens. Actuators, B, 2020, 316, 128106 CrossRef CAS.
- A. T. E. Vilian, R. Umapathi, S.-K. Hwang, Y. S. Huh and Y.-K. Han, J. Hazard. Mater., 2021, 408, 124914 CrossRef CAS PubMed.
- W. Zhu, Y. Zhang, J. Gong, Y. Ma, J. Sun, T. Li and J. Wang, ACS Sens., 2019, 4, 2980–2987 CrossRef CAS PubMed.
- T. Zhang, Y. Liu, J. Li, W. Ren and X. Dou, Sens. Actuators, B, 2023, 379, 133261 CrossRef CAS.
- L. Singh and N. Ranjan, J. Am. Chem. Soc., 2023, 145, 2745–2749 CrossRef CAS PubMed.
- J. Zhang, T. Hu, B. Xiong, X. Zheng, R. Wang, P. Zhu, J. Chen, T. Cong, Y. Li and X. Wang, Chem. Eng. J., 2023, 475, 146311 CrossRef CAS.
- Z. Ma, J. Li, X. Hu, Z. Cai and X. Dou, Adv. Sci., 2020, 7, 2002991 CrossRef CAS PubMed.
- L. Tan, C. Xie, Q. Yang, K. Luo and L. Zhou, Food Chem., 2023, 405, 134949 CrossRef CAS PubMed.
- V. C. A. Ficca, C. Santoro, E. Marsili, W. Da Silva Freitas and A. Serov, Electrochim. Acta, 2022, 402, 139514 CrossRef CAS.
- A. Shaikh, B. K. Singh and S. Parida, Mater. Chem. Phys., 2019, 235, 121744 CrossRef CAS.
- Y. Dong, F. Liu, S. Liu, S. Feng and S. Peng, ACS Appl. Nano Mater., 2024, 7, 16620–16629 CrossRef CAS.
- T. Zhe, F. Li, M. Liu, K. Ma, Q. Luo and W. Li, Chem. Eng. J., 2024, 488, 150942 CrossRef CAS.
- M. J. Askari, J. D. Kallick and C. C. L. McCrory, J. Am. Chem. Soc., 2024, 146, 7439–7455 CrossRef CAS PubMed.
- L. Bai, F. Franco, J. Timoshenko, C. Rettenmaier, F. Scholten, H. S. Jeon, A. Yoon, M. Rüscher, A. Herzog, F. T. Haase, S. Kühl, S. W. Chee, A. Bergmann and R. C. Beatriz, J. Am. Chem. Soc., 2024, 146, 9665–9678 CrossRef CAS PubMed.
- M. Ghosh, S. E. Braley, R. Ezhov, H. Worster, J. A. Valdez-Moreira, Y. Losovyj, E. Jakubikova, Y. N. Pushkar and J. M. Smith, J. Am. Chem. Soc., 2022, 144, 17824–17831 CrossRef CAS.
- P. Huang, Y. Yan, P. Martinho Ricardo, L. Lefferts, B. Wang and J. F. Albanese, J. Am. Chem. Soc., 2024, 4, 2656–2665 CAS.
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed.
- S. K. Kailasa, M. R. Patel, J. R. Koduru and T. J. Park, Chem. Rev., 2024, 501, 215595 CAS.
- Y. Shen, C. Ma, S. Zhang, P. Li, W. Zhu, X. Zhang, J. Gao, H. Song, D. Chen, D. Pang and A. Li, Sci. Total Environ., 2020, 742, 140622 CrossRef CAS PubMed.
- K. Xu, Q. Chen, Y. Zhao, C. Ge, S. Lin and J. Liao, Sens. Actuators, B, 2020, 319, 128221 CrossRef CAS.
- Y. Yang, J. Zhang, Y. W. Li, Q. Shan and W. Wu, Colloids Surf., A, 2021, 625, 126865 CrossRef CAS.
- H. Shi, L. Fu, F. Chen, S. Zhao and G. Lai, Environ. Res., 2022, 209, 112747 CrossRef CAS PubMed.
- Y. X. Dai, Y. X. Li, X. J. Zhang, R. S. Marks, S. Cosnier and D. Shan, ACS Sens., 2024, 9, 337–343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 optimized reaction method and conditions; Fig. S1 XRD pattern of the synthesized MoS2/C composite; Fig. S2 EELS spectrum of the MoS2/C composite; Fig. S3(a) electrochemical behavior of SPCE, and MoS2/SPCE; (b) cyclic voltammogram of SPCE and MoS2/SPCE in the presence of 10 mM K3[Fe(CN)6]; (c) EIS of SPCE and MoS2/SPCE; (d) cyclic voltammogram of MoS2/SPCE in the presence of 10 mM K3[Fe(CN)6] at different scan rates; Fig. S4 (a) Electrochemical behavior (0–1 V potential range, in PBS) of: SPCE (black dotted line) and MoS2/SPCE (red dotted line), SPCE (black solid line) and MoS2/SPCE (red solid line) in the presence of both PBS and 200 μM of NaNO2; (b) comparison between the responses of SPCE and MoS2/SPCE in the presence of 200 μM of NaNO2; Fig. S5(a) LSV of MoS2/SPCE at different nitrite concentrations; (b) graph of the anodic peak current (Ipa) versus the NO2− concentration; Fig. S6(a) repeatability of the performance of MoS2/SPCE; (b) DPV analyses to determine NaNO2 in a real sample; (c) DPV analyses at t = 0, and at [t = 6 months]; Fig. S7 DPV analyses a different pH (5.0, 6.0, 7.4, 8.0); Fig. S8 DPV analyses in PBS 0.1 M, and PBS 0.1 M and NaCl 0.1 M. See DOI: https://doi.org/10.1039/d5tc01165e |
| This journal is © The Royal Society of Chemistry 2025 |