
Balancing the molecular twist and conformational rigidity in imidazo[1,2-a]pyridines to achieve dual-state emissive (DSE) luminogens for applications in OLEDs and cell-imaging†
Chinmay
Thakkar
a,
Seemantini
Kale
a,
Mohammad Amir
Ahemad
b,
Monalisa
Debnath
 c,
Anjali
Tripathi
d,
Saona
Seth
e,
Purav
Badani
d,
Rohit
Srivastava
c,
Sangita
Bose
*b and
Satyajit
Saha
*a
c,
Anjali
Tripathi
d,
Saona
Seth
e,
Purav
Badani
d,
Rohit
Srivastava
c,
Sangita
Bose
*b and
Satyajit
Saha
*a
aDepartment of Speciality Chemicals Technology, Institute of Chemical Technology, Mumbai, Maharashtra-400019, India. E-mail: ss.saha@ictmumbai.edu.in
bSchool of Physical Sciences, UM-DAE Centre for Excellence in Basic Sciences, University of Mumbai, Kalina Campus, Santacruz (E), Mumbai 400098, India. E-mail: sangita@cbs.ac.in
cDepartment of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Mumbai, Maharashtra, India
dDepartment of Chemistry, University of Mumbai, Kalina Campus, Mumbai, Maharashtra, India
eDepartment of Applied Sciences, Tezpur University, Assam, India
First published on 15th April 2025
Abstract
The need for fluorophores that could be emissive in both solution and solid states has led to the development of dual state emissive (DSE) materials, which bridge the gap between the ACQ and AIE and offer omnipresent emission. However, designing DSEgens requires a meticulous balance of the emissive and non-radiative pathways in both states. Despite some advancements, achieving dual-state emission deals with challenges in synthesis, scalability, and application. This study focuses on balancing the molecular twist and electronic rigidity to design DSE molecules using imidazo[1,2-a]pyridine scaffolds, aiming at enhancing emission performance in the solid and solution states. Six novel D–A-structured imidazo[1,2-a]pyridine–arylketone conjugates (SK-1 to SK-6) were synthesized, incorporating bulky aryl rotors like triphenylamine (TPA) and aryl ketones (–COPh) to achieve twisted configurations and impact electronic structures. Experimental results showed promising features, with SK-3 and SK-4 displaying DSE effects and SK-2 and SK-5 exhibiting AIE characteristics with good PLQY values. The luminogens demonstrated high thermal stabilities, stable electrochemical properties, and effective cell imaging capabilities with low cytotoxicity. The calculated ΔEST values and lifetimes of prompt/delayed components feature the potential TADF properties of these luminogens. Furthermore, as a proof of concept, SK-3 and SK-4 were successfully used in OLED device fabrication. SK-4 shows a good EQEmax of ∼10.00%, while SK-3 shows a reasonable EQEmax of ∼6.4% with superior efficiency roll-off as compared to SK-4. This study highlighted the importance of balancing molecular distortion and conjugation in designing multifunctional DSE molecules for optoelectronics and bioimaging applications.
1. Introduction
Organic luminogens have captured tremendous attention over the years due to their impressive fluorescence properties, leading to wide-ranging applications in chemical sensing, bioimaging, optoelectronics, theranostics, and many more.1–9 Concentration-induced emission quenching and emission enhancement have categorized the fluorophores as ACQgens and AIEgens, respectively, which have applications in their respective fields.10–17 Apparently, these two mutually exclusive phenomena lead to the realization of the need for fluorophores that will be emissive in both the solution and the solid state. Ever since dual state emission was first reported by Tang and co-workers in 2015, the development of Dual State Emissive (DSE) materials has attracted significant attention as the same fluorophore can be used for multiple purposes, paving the way for a vast range of applications.18–27However, the technology of embedding fluorophores in polymeric or supramolecular matrices to improve the solid-state photoluminescence quantum yield (PLQY) underlines the urge to develop the DSE concept.28–30 Nevertheless, the difficulties of matrix synthesis and/or fluorophore loading and scalability issues make the process complicated. Additionally, some applications like fluorescence staining of biomolecules in cells are not always accomplishable by the use of fluorescent host–guest materials. Therefore, DSEgens offer a simpler solution to retain fluorescence in both states. However, the design of DSE fluorophores is a challenging task because maintaining a balance between emissive and non-radiative pathways in both the solution and the solid-state requires meticulous designing.31–33
Although several research groups have come up with newer strategies to design DSEgens, we consider that a more general design strategy to achieve dual-state emission is still in its infancy and therefore requires an extensive investigation. Navigating through the molecular structures reveals the critical importance of the delicate balance between the twisted conformation and rigid or interrupted conjugation to achieve the DSE.34–42 It is important to note that although some degree of freedom is needed, an equilibrium should be established because obtaining molecules that experience extremely fast molecular rotations could enable undesired relaxation pathways and render a molecule non-emissive.
Captivated by the myriad of applications of imidazo[1,2-a]pyridines in diverse areas like pharmaceuticals, imaging, phosphorescent OLEDs, and fluorescence sensing, we delved into the literature to realize that a handful of examples of luminogens with solid-state emissive properties built using imidazo[1,2-a]pyridines are reported, Fig. 1.43–53 Therefore, apprehending the alluring prospect of imidazo[1,2-a]pyridines in the realm of modulating their emissive properties to achieve dual-state emission, we embarked on designing dual-state emissive molecules by intricately balancing the donor–acceptor moieties across the imidazo[1,2-a]pyridine scaffold.54 However, aiming for a more reliable design strategy rather than a circumstantial feat of achieving DSE properties, as well as to gain more insights into DSEgens, in this contribution, we reported the design and synthesis of six D–A-structured imidazo[1,2-a]pyridine–arylketone conjugates as novel luminogens, designated as SK-1 to SK-6, Fig. 2. The design incorporates appending bulky aryl rotors like the triphenylamine (TPA) moiety into the planar imidazo[1,2-a]pyridine skeleton and tethers aryl ketones (–COPh) directly or indirectly through a suitable π bridge to the imidazo[1,2-a]pyridine moiety. The objective behind the design is to integrate the twisted configuration to exert an impact on the electronic structures and hence on the charge transfer properties within the π system, which further influences the emission performances of the luminogens. The propeller-like TPA group with its twisted rotors and electron-donating attributes is expected to assist in restricting the intramolecular rotations (RIR) in the solid state and enhance the electronic transition within the distorted π-frameworks to improve the emission efficiency of the luminogens in solutions.
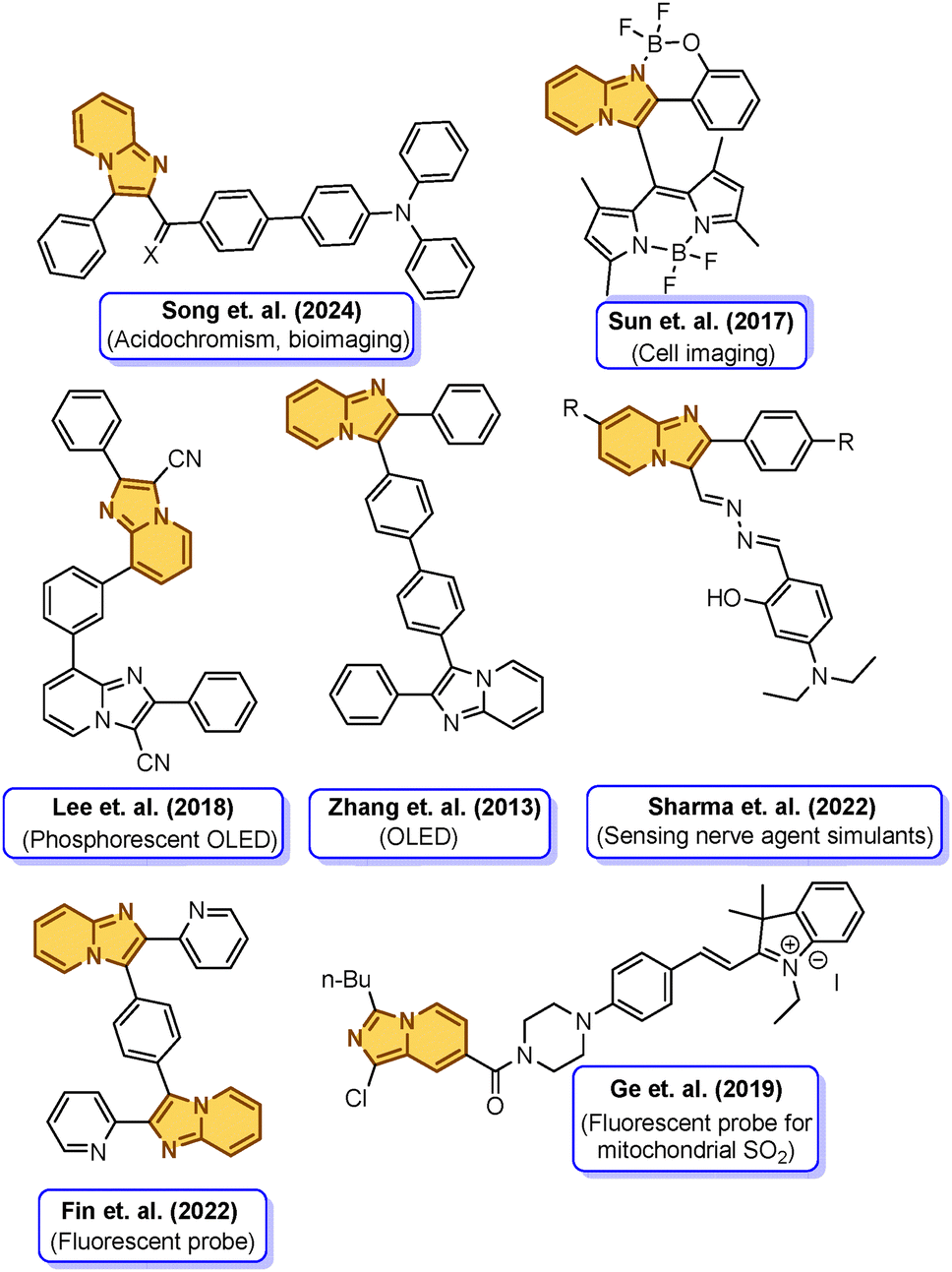 | ||
| Fig. 1 A few reported imidazo[1,2-a] pyridine-derived luminescent molecules and their applications. | ||
 | ||
| Fig. 2 Designed luminogens SK-1 to SK-6. | ||
The distorted –COPh moiety is anticipated to prohibit the disadvantageous π–π stacking between imidazo[1,2-a]pyridine molecules and prevent close packing in the crystal. Meanwhile, a phenyl linker between imidazo[1,2-a]pyridine and –COPh units was incorporated to further tune the electronic structures and spatial conformations of the molecules. In addition to that, molecular tethering to achieve planarization by replacing diaryl ketones with fluorenone was also sought to understand the influence of the –COPh group in achieving solid-state emission. This strategy is expected to achieve color tunability with intriguing DSE features of the luminogens.
The extension of the conjugated system and variation of the nature and number of substituents allowed fine-tuning of the emission wavelengths of these compounds ranging from blue to red in tetrahydrofuran (THF) solution (λem = 474–566 nm) and from cyan to the deep red region in the solid state (λem = 450–591 nm). The experimental findings revealed promising features of the luminogens, with SK-3 and SK-4 displaying DSE effects and SK-2 and SK-5 displaying AIE characteristics. Interestingly, SK-1 and SK-6 displayed ACQ features with minimal solid-state emission in SK-6, while no emission in SK-1. The emitters exhibited high thermal stabilities and stable electrochemical properties, suitable for their applications in optoelectronic materials. Moreover, the molecular distortion created by tuning the dihedral angles in the donor–acceptor design can lead to a desirable HOMO–LUMO spatial separation. This separation is beneficial for reducing the energy gap between a molecule's lowest excited singlet and triplet states (ΔEST). A small ΔEST is crucial for molecules to show TADF features, an attribute that harnesses 100% internal quantum efficiency through the reverse intersystem crossing (RISC) mechanism by harvesting energy from the triplet to the singlet excited states. The calculated ΔEST values are 0.4604 eV, 0.3908 eV, 0.1014 eV, 0.0246 eV, and 0.068 eV for SK-2, SK-3, SK-4, SK-5, and SK-6 respectively. A small ΔEST (DEST) of the above luminogens suggests easy photon upconversion from T1 to S1 via RISC, confirming their TADF properties. All these emitters showed double exponential decay curves consisting of prompt and delayed components. The lifetimes of prompt/delayed components were 0.115/4879 ns for SK-2, 4.74/13.89 ns for SK-3, 4.23/8.321 ns for SK-4, 9.17/60.3 ns for SK-5, and 1.76/24 ns for SK-6. As a proof of concept, SK-3 and SK-4 were used for OLED device fabrication considering their TADF features and strong emission in the solid-state. The SK-4 device looked yellowish green (CIE: 0.33, 0.44) and the SK-3 device looked yellow (CIE: 0.50, 0.45) in color with low turn voltage, VON. SK-4 shows a good EQEmax of ∼10.00% and higher efficiency roll-off as compared to SK-3 with an EQEmax value of ∼6.4%.
The cytotoxicity study revealed that all of these investigated luminogens possess concentration-dependent anti-metastatic potential towards SiHa cells at a concentration ranging from 0.1 to 1 μg mL−1. The calculated IC50 values of SK-3, SK-4, and SK-6 are 0.58 μg mL−1, 0.52 μg mL−1, and 0.68 μg mL−1 respectively. Furthermore, luminogens SK-3, SK-4, and SK-6, having more than 90% cell viability below 1 μg mL−1 concentration, were successfully applied for live cell imaging. SK-6 was found to evenly stain both the cytosol and the nucleus, while luminogen SK-4 was evenly distributed in the cytosol with higher fluorescence intensity and SK-3 entered the cytoplasm with low fluorescence intensity. This study demonstrated the critical role of balance between molecular distortion and restricted conjugation across donor–acceptor units in achieving dual-state emissive properties. Furthermore, this work also features new avenues for designing multifunctional solid-state emissive molecules based on imidazo[1,2-a]pyridines for optoelectronics and cell imaging.
2. Results and discussion
2.1. Synthetic procedures
As demonstrated in Scheme 1, all the designed molecules SK-1–SK-6 were synthesized starting from 2-amino pyridine 1 and the synthetic procedures are detailed in the section on experimental methods. The synthesis of SK-1 was achieved in one step by heating (E)-chalcone with 2-aminopyridine 1 in the presence of CuCl2·2H2O in toluene. Afterward, luminogens SK-2–SK-6 were readily accessed through the multistep synthesis in good yields starting from 2-amino pyridine 1. The key steps involved in the process are the cyclo-condensation reaction followed by the Heck coupling and Knoevenagel condensation reactions. Mechanochemical grinding of 2-aminopyridine 1 with phenacyl bromide 2 and 2-bromo-1-(4-bromophenyl)ethenone 3 separately afforded 2-phenylimidazo[1,2-a]pyridine 4 and 2-(4-bromophenyl)imidazo[1,2-a]pyridine 5, respectively, in excellent yields within a very short time. The intermediate 2-phenylimidazo[1,2-a]pyridine 4 was subjected to Heck coupling with (4-bromophenyl)(phenyl)methanone 9 to obtain SK-2. The subsequent Knoevenagel condensation reaction of SK-2 with malononitrile offered the designed luminogen SK-3 in good yield. Buchwald Hartwig amination of 2-(4-bromophenyl)imidazo[1,2-a]pyridine 5 with diphenylamine afforded 4-(imidazo[1,2-a]pyridin-2-yl)-N,N-diphenylamine 7 in good yield, which was then subjected to Heck coupling reaction separately with 3-bromo-9H-fluoren-9-one 8 and (4-bromophenyl)(phenyl)methanone 9 to obtain luminogens SK-6 and SK-4 respectively. Knoevenagel condensation of SK-4 with malononitrile afforded the desired luminogen SK-5. All the intermediates and the targeted luminogens, SK-1–SK-6, were purified by silica gel column chromatography and were characterized using 1H, 13C NMR, FT-IR, and HRMS analysis methods.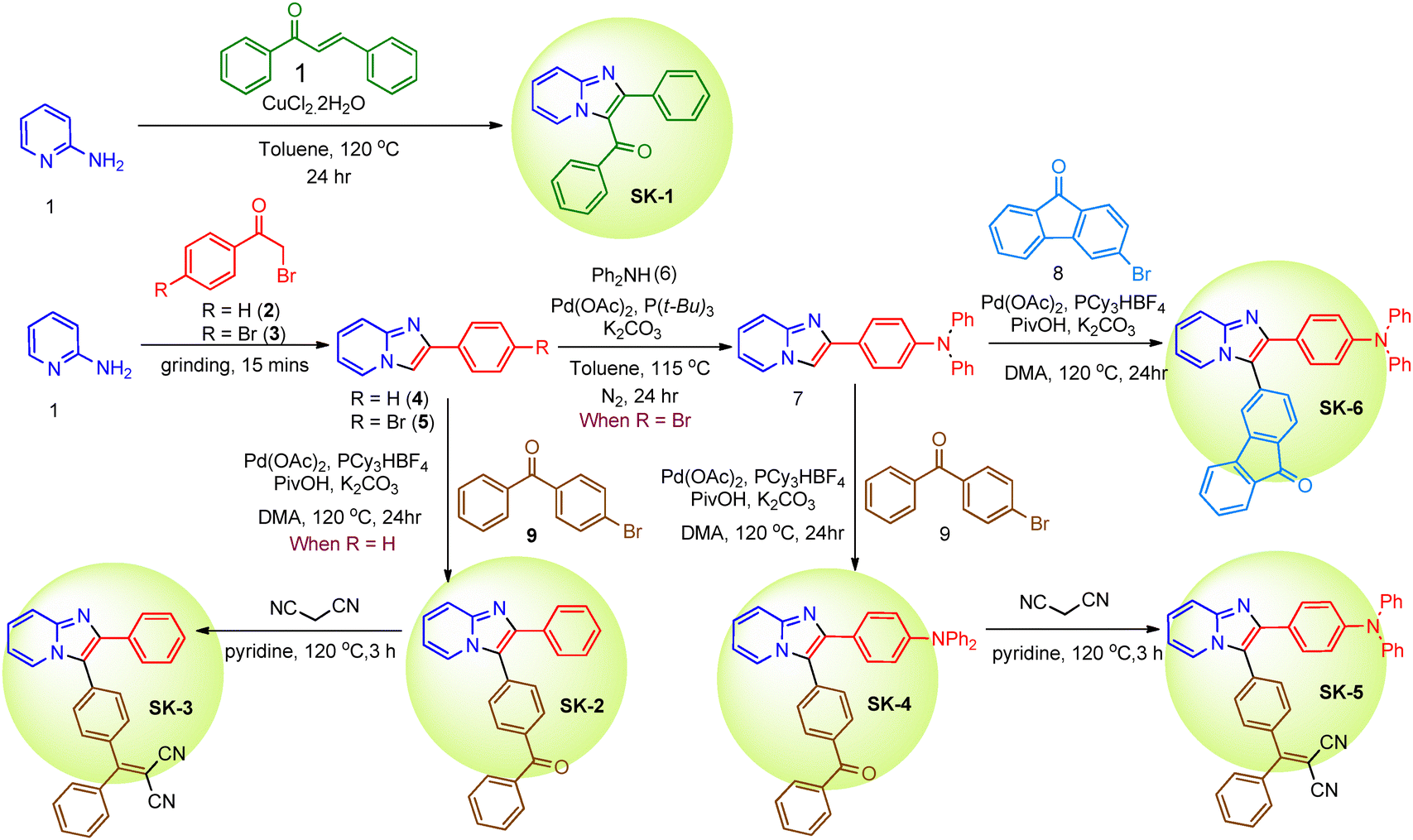 | ||
| Scheme 1 Synthetic scheme for the preparation of luminogens SK-1 to SK-6. | ||
Thermal stability is an important feature of any technological development as it ensures the durability of organic molecules to withstand high current density during fabrication. Thermal analysis (TGA) was conducted by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) to investigate the thermal stabilities of luminogens SK-1–SK-6. DSC-TGA analysis was performed from 35 to 600 °C at a scanning rate of 10 °C min−1. The TGA profiles of the synthesized luminogens SK-1–SK-6 are presented in Fig. S1 (see ESI†), which gives a brief idea of the weight loss of the compound with respect to an increase in temperature. The TGA data show that the compounds are thermally stable except for SK-4, which shows lower decomposition temperatures. SK-1 to SK-6 do not show any considerable loss up to 215 °C, 300 °C, 324 °C, 348 °C, 338 °C, and 341 °C respectively. However, beyond that temperature, a steady weight loss up to 600 °C was observed. The final weight loss % at 600 °C for compounds SK-1 to SK-6 is 100%, 100%, 85%, 100%, 60% and 80% respectively. Additionally, DSC (Fig. S2 (see ESI†)) analysis indicated that the Td of the luminogens was observed at 132 °C for SK-1, 166 °C for SK-2, 256 °C for SK-3, 200 °C for SK-4, 220 °C for SK-5 and 243 °C for SK-6, highlighting their thermal robustness suitable for optoelectronic applications (see ESI† for more details). Among them, SK-2 to SK-6 have higher Td, compared to SK-1 due to the stronger structural rigidity.
2.2. DFT and electrochemical studies
An insight into the photophysical phenomena of the luminogens was obtained using density functional theory (DFT) with the B3LYP/6-31G(d,p) basis set.55 The optimized electronic structures and frontier molecular orbital energies of compounds SK-1–SK-6 are presented in Fig. 3 (Fig. S3, details can be seen in the ESI†). Careful investigation of the depicted optimized structures revealed twisted molecular conformations in all these luminogens. The molecules adopted twisted configurations with the torsion angle ranging from 20.79° to 34.09° between the imidazo[1,2-a]pyridine core and the C-2 peripheral phenyl ring. Additionally, the imidazo[1,2-a]pyridine ring and C3 phenyl bridge exhibit twisted conformations with torsion angles ranging from 57° and 59° for SK-2 to SK-6 (Table S1, see ESI†). A similar observation was made for SK-1, where the torsion angle between the imidazo[1,2-a]pyridine core and –COPh was approximately 61°. Furthermore, the two phenyl rings connected by –CO or –C(CN)2 also exhibit substantial distortion. However, molecular tethering in fluorenone infusing planarity prevented any sort of distortion in SK-6. | ||
| Fig. 3 HOMO–LUMO band gaps of SK-1 to SK-6 and their energy-minimized structures. | ||
The twisted aromatic rings are expected to serve as a rotor to quench the fluorescence non-radiatively in the solution state as well as prevent close intermolecular packing, thereby preventing the molecules from detrimental π–π stacking in the solid state. This feature endows luminogens SK-2–SK-6 with typical aggregation-induced emission (AIE) characteristics. The HOMO and the LUMO distribution of the luminogens as depicted in Fig. 3 reveal that except for SK-1, the HOMO of the luminogens SK-2–SK-6 are mainly localized on the imidazo[1,2-a]pyridine group or slightly extended on the NPh2 unit and the LUMO is mostly localized on the –COPh or –C(CN)2Ph units. For SK-1 the depicted HOMO and LUMO electron densities are evenly localized throughout the molecules. The HOMO of SK-2 shows high electron densities at the imidazo[1,2-a]pyridine ring as compared to the HOMO orbital of SK-3. The substantial shifting of electron clouds indicates the occurrence of twisted intramolecular charge transfer in these AIEgens upon excitation, which might result in the large Stokes shift. The band gap obtained from DFT calculations shows energies as follows: SK-1 (4.05 eV), SK-2 (3.45 eV), SK-3 (3.02 eV), SK-4 (2.72 eV), SK-6 (2.34 eV), and SK-5 (2.13 eV), which correlate well with their molecular structures. The relatively smaller ΔE for compounds SK-3 to SK-6, as compared to SK-1 and SK-2, is due to the incorporation of donor (D)–acceptor (A) components within the molecular framework, which is expected to bathochromically shift their emission spectra. The reduction in the ΔE value is also evident upon installation of the dicyanovinylidene moieties in SK-3 and SK-5. Moreover, the HOMO–LUMO spatial separation as is evident in SK-4, SK-5, and SK-6 is expected to reduce the energy gap between the molecules' lowest excited singlet and triplet states (ΔEST), enabling the molecules to show desirable thermally activated delayed fluorescence (TADF) features by the reverse intersystem crossing (RISC) mechanism.
Additionally, cyclic voltammetry (CV) experiments were performed using a three-electrode system to evaluate the HOMO–LUMO energy levels and band gaps of luminogens SK-1–SK-6. The electrochemical investigation was performed with respect to a 0.1 M solution of tetra-butylammonium hexafluorophosphate (TBAP) in THF, with Ag/AgCl as the reference electrode and platinum gauze as the counter electrode. The voltammogram showed a quasi-reversible oxidation process for SK-1 to SK-6 with onset potentials of 1.52 V, 1.53 V, 1.51 V, 1.17 V, 1.18 V, and 1.14 V respectively, Fig. S4 (see ESI†).
These energy levels were calculated from the onset oxidation potential (Eonset) and the optical band gap Eg using the equations HOMO = −(4.8 + Eonset) eV and LUMO = (HOMO + Eg) eV, where Eg = 1240/λonset and λonset is the onset absorption wavelength (Table 1). Calculated energy levels of highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) commensurate well with the theoretical values determined from DFT calculations.
| Compound | λ onset (nm) | Experimental | Theoretical | |||||
|---|---|---|---|---|---|---|---|---|
| E HOMO (eV) | E LUMO (eV) | E (Onset/ox) [V] | Band gap (eV) | E HOMO (eV) | E LUMO (eV) | Band gap (eV) | ||
| SK-1 | 343 | −6.32 | −2.71 | 1.52 | 3.61 | −6.31 | −2.27 | 4.04 |
| SK-2 | 337 | −6.33 | −2.66 | 1.53 | 3.67 | −5.89 | −2.44 | 3.67 |
| SK-3 | 404 | −6.31 | −3.25 | 1.51 | 3.07 | −5.98 | −2.96 | 3.02 |
| SK-4 | 347 | −5.97 | −2.4 | 1.17 | 3.57 | −5.15 | −2.43 | 2.72 |
| SK-5 | 440 | −5.98 | −3.08 | 1.18 | 2.81 | −5.25 | −3.12 | 2.13 |
| SK-6 | 350 | −5.94 | −2.4 | 1.14 | 3.54 | −5.23 | −2.88 | 2.35 |
2.3. Photophysical properties
The photophysical properties of synthesized luminogens were investigated by measuring their absorption and emission spectra in a 1 mM solution of THF, as depicted in Fig. 4(a). The absorption maxima of luminogens SK-1 to SK-6 observed at 342, 337, 404, 347, 440, and 440 nm respectively are attributed to the π–π* electronic transitions. The keto group attached directly to the imidzo[1,2-a]pyridine moiety in SK-1 resulted in slightly bathochromically shifted absorption maxima as compared to SK-2, where the acceptor group is separated by a phenyl from the donor imidazo[1,2-a]pyridine. The introduction of dicyanovinylidene moieties in SK-3 and SK-5 resulted in bathochromically shifted absorption maxima as compared to SK-2 and SK-4 respectively. The insertion of an additional electron donating diphenylamine at the periphery in SK-4, SK-5, and SK-6 resulted in an enhancement in the electron donating ability of the luminogens, leading to increased π–π* electronic transitions and hence bathochromically shifted absorption maxima. Incidentally, the introduction of the dicyanovinylidene moiety in SK-5 as an electron acceptor and the diphenylamine group in SK-6 as an electron donating group counteracted, resulting in similar absorption maxima for both the luminogens. | ||
| Fig. 4 (a) Normalized absorbance spectra of luminogens SK-1–SK-6 in THF, (b) normalized emission spectra of luminogens SK-1–SK-6 in THF, (c)–(g) solid state emission spectra of SK-2 to SK-6, and (h) photos of solid powders of compounds SK-1 to SK-6 as observed under UV light (365 nm). | ||
Furthermore, the photoluminescence spectra of the luminogens were measured in 1 mM THF solution of the luminogens, and SK-1 and SK-2 were not found to be very emissive while SK-3 was faintly emissive, Fig. 4(b). Except for SK-1 and SK-3, all the molecules were found to be emissive in the solution state albeit with different fluorescence intensities. SK-2, SK-4, and SK-6 are found to be more emissive than SK-5. In the solution state, SK-4 and SK-5 were found to be very weakly emissive with emission maxima observed at 534 nm and 505 nm. Fluorophores SK-2, SK-4, SK-5, and SK-6 exhibit cyan, yellow, orange, and dark yellow fluorescence respectively in the solution state, Fig. S5 (see ESI†), while SK-2, SK-3, SK-4, SK-5, and SK-6 appears blue, green, cyan, yellowish-orange, and yellow, fluorescence in the solid-state when observed under 365 nm UV-lamp irradiation. Moreover, their strong emissive nature in the solid state, observed under 365 nm UV light, prompted us to investigate their solid-state photoluminescence properties. It was observed that except for SK-1, all of these luminogens exhibited intense solid-state emissions, Fig. 4(c)–(h). The emission maxima of the luminogens in the solid state are presented in Table 2 and Fig. 4. The slight red shift of the emission maxima could be due to the rigidification of the molecular structure, which limits the free intramolecular motion of the rotors. The fluorescence quantum yields (PLQYs) of all the molecules in the solution state and solid states were recorded by an absolute method using an integrated sphere and are tabulated in Table 2. Very high fluorescence quantum yields (ΦFL) of 10.62 and 32.61 (Table 2) were noted for SK-2 and SK-4, while moderate PLQYs of 0.016 and 0.662 in THF solvent were observed for SK-5 and SK-6 respectively. The moderately emissive nature of SK-6 as compared to SK-4 might be due to the greater degrees of freedom of the more planarized structure of the fluorenone ring to undergo free rotation in the excited state as compared to the two untethered phenyl rings in SK-4. Interestingly, the luminogens were found to be quite emissive in the solid state. The absolute photoluminescence quantum yields (%) of the luminogens recorded using integrated sphere methods show values of 1.35, 26.7, 28.45, 29.1, and 9.4 for SK-2, SK-3, SK-4, SK-5, and SK-6 respectively.
| SK-1 | SK2 | SK-3 | SK-4 | SK-5 | SK-6 | |
|---|---|---|---|---|---|---|
| a Calculated by the relative method using quinine sulfate as a standard. b Calculated by the absolute method using an integrating sphere. c Non-emissive. | ||||||
| Absorption (nm) | 259, 342 | 252, 338 | 312, 405 | 244, 346 | 304, 450 | 257, 350 |
| Emission (THF) (nm) | 397 | 474 | 566 | 534 | 505 | 534 |
| Emission (solid) (nm) | — | 450 | 532 | 518 | 591 | 564 |
| % ΦFL (THF)a | —c | 10.62 | —c | 32.61 | 0.016 | 0.662 |
| % ΦFL (solid)b | — | 1.35 | 26.7 | 28.45 | 29.1 | 9.4 |
2.4. Solvatochromism
To further establish the D–π–A features of the luminogens, the absorption and emission spectra of the designed luminogens were recorded in solvents with varying polarity, Table S2 and Fig. S6–S11 (see ESI†). The solvents chosen were based on their increasing order of polarity and consisted of n-hexane, toluene, chloroform, acetone, methanol, acetonitrile, DMF, and DMSO. Luminogens SK-1–SK-3 demonstrated essentially unchanged absorption spectra upon varying the solvent polarities, implying that their ground state is not affected by solvent polarity. Likewise, an insignificant 6 nm hypsochromic shift of the absorption maxima in the case of SK-4, SK-5, and SK-6 on moving from non-polar to polar solvents also ruled out any significant solvatochromic features of these molecules as well. More importantly, fluorophores SK-2, SK-3, SK-4, and SK-6 displayed moderate to intense emission in some of the organic solvents like hexane, toluene, chloroform, acetone, and DMSO, which indicated remarkable solvent compatibility. Luminogen SK-1 was found to be non-emissive in most of the common organic solvents. Interestingly, SK-5 did not show any solvatofluorochromic properties, while SK-2, SK-3, SK-4, and SK-6 displayed positive solvatofluorchromism, which may be related to a weak ICT effect and strong non-radiative transition across the donor (D)–acceptor (A) molecule. The solvatofluorochromic features of SK-2, SK-3, SK-4, and SK-6 may be attributed to the excited state stabilization by the highly polar solvents, which enhances the ICT effect.2.5. Aggregation-induced emission
The strong emission observed on the TLC plate and in the solid state hinted about the potential aggregation-induced emissive (AIE) features of these molecules. This led us to investigate their AIE properties. The AIE properties of luminogens SK-1 to SK-6 were investigated by dissolving the molecules in THF (1 mM) and recording their photoluminescence spectra with incremental addition of water into the THF solution, Fig. 5 and Fig. S12–S17 (see ESI†). Luminogen SK-1 was emissive in THF solution; however, with the gradual increase in the water fraction the emission intensity was found to decrease and it became non-emissive at an fw of 99% H2O, representing a typical attribute of an aggregation caused quenching luminogen. The emission quenching is possibly due to the initiation of the TICT process leading to emission quenching through non-radiative decay pathways. SK-2 solution was emissive, whose intensity gradually decreased with the increase in water fraction (fw) up to 80% and suddenly rose to a moderate intensity when the water fraction reached the nano-aggregated state at an fw of 99%, indicating its emissive properties in both the solution and aggregated states. Contrary to SK-2, luminogen SK-3 was moderately emissive in the solution state, and with the steady increase in the water fraction, the emission intensity of SK-3 was found to increase and became maximum when the water fraction fw reached 99%, highlighting the aggregation-induced emission enhancement behaviour due to the restricted free rotations of the phenyl rings. | ||
| Fig. 5 (a)–(f) Plot of the I/I0vs. water fractions of SK-1 to SK-6. | ||
The emission profile of SK-4 was found to be quite distinctive compared to SK-2 and SK-3. SK-4 was found to be highly emissive at a 0% water fraction whose emission intensity rapidly decreased at an fw of 10% and was almost non-emissive up to an fw of 60%. Beyond that, there was a steady rise in the emission intensity upon incremental water addition, with the highest emission intensity observed at an fw of 99%, indicating the dual-state emissive behaviour of SK-4. Luminogen SK-5 was moderately emissive in the solution state and there was a slow reduction in the emission intensity with the increase in the water fraction up to an fw of 70%. This drop in emission intensity, along with a marginal bathochromic shift in emission peaks, could be attributed to the initiation of the TICT process leading to emission quenching through non-radiative decay pathways. At 80% water fraction, there was a sudden dip in the emission intensity, which then rapidly raised to maxima at an fw of 99%, indicating the dual state emissive behaviour of the luminogen.
This increase in water content initiated restricted internal rotation (RIR) processes and limited intermolecular charge transfer (ICT) effects, resulting in high emission in the aggregated state. Luminogen SK-6 was moderately emissive in the solution state whose emission intensity rapidly raised as the water fraction fw reached 10% and then onwards demonstrated a steady decrease in the intensity and it became almost non-emissive at an fw of 90%. However, at an fw of 99%, a marginal enhancement in the emission intensity was observed, enabling the molecule to follow the traits of aggregation-caused quenching luminogens. A weak PL intensity in pure THF solvent for SK-3 and SK-6 was attributed to the uninterrupted rotation of the peripheral phenyl groups of the TPE core via a non-irradiative decay pathway induced by a strong intermolecular charge transfer process. A 3.5 and 4-fold enhancement in the emission intensity was observed in the aggregated state for SK-3 and SK-5 while SK-4 and SK-2 being emissive in both the solution and aggregated states, the enhancement factor noted was marginal and not recorded. It was noted that imidazo[1,2-a]pyridines with a phenyl ring at the terminal in SK-2 and tethered phenyl rings in SK-6 turned out to be ACQ molecules. This observation strengthens the importance of the incorporation of the rotor and distorted acceptors to achieve DSE properties. This may be the result of achieving a delicate balance between the twisting conformation and interrupted conjugation by taking advantage of both the –COPh and (CN)2Ph groups and the TPA moiety. Further confirmation of the aggregated states was provided by particle size distribution analysis by the dynamic light scattering (DLS) experiment, as well as the Tyndall effect, Fig. S18 and Table S3 (see ESI†). The size distribution analysis of the highest intensity states confirms the nano-aggregate formation, which accounts for the emission enhancement.
2.6. Fluorescence lifetime decay
To apprehend the TADF properties of these emitters and understand the nature of phosphorescence, preliminary studies on the PL emissions in THF solution were performed for compounds SK-3 and SK-4 under oxygenated conditions and also by degassing the solution with N2 gas. As seen in Fig. S19 (see ESI†) both luminogens display a significant decrease in the emission intensity when recorded under oxygenated conditions in the inert environment, Fig. S19 (see ESI†). The reduction of emission intensity under oxygenated conditions may be attributed to the quenching of the triplet excited states, thereby facilitating the non-radiative energy transfer process. Such an observation supports the TADF features of the luminogens, which require the involvement of the triplet states, which are sensitive to oxygen. Oxygen can quench these triplet states, leading to a decrease in emission intensity.Subsequently, the low-temperature (77 K) fluorescence and phosphorescence spectra of the emitters SK-2, SK-3, and SK-4 were studied in their condensed forms to confirm their TADF properties (Fig. 6(a)–(e)). At room temperature, the solution phase produced considerable emissions, primarily from the S1 state. However, when the sample was cooled to liquid nitrogen (77 K), the enhanced stiffness of the molecular environment limited intramolecular rotations around the D–A bonds. This restriction hindered nonradiative decay from the T1 state, while thermally activating nonradiative decay channels resulted in significant phosphorescence emission from the T1 state. The analysis of room-temperature fluorescence and low-temperature (77 K) phosphorescence spectra gave the singlet and triplet energy gaps. A minor energy gap between the lowest singlet (S1) and the triplet excited states (ΔEST) allows triplet excitons to convert to singlet excitons through reverse intersystem crossing (RISC) at ambient temperature. These singlet excitons can then relax via the fluorescence channel. The calculated ΔEST values are 0.4604 eV, 0.3908 eV, 0.1014 eV, 0.0246 eV, and 0.068 eV for SK-2, SK-3, SK-4, SK-5, and SK-6 respectively. (Fig. 4(a)–(c) and Table 2). A relatively small ΔEST of the above luminogens suggests easy photon upconversion from T1 to S1 via RISC, resulting in TADF properties. Additionally, the potential for triplet harvesting was assessed through time-dependent photoluminescence (PL) spectroscopy. The transient PL decay profiles of the luminogens were recorded at both room temperature and 77 K, Fig. 6(f)–(j). Time-resolved fluorescence measurements were conducted to extract time constants from the photoluminescence (PL) decay, which revealed exponential decay across all luminogens in the solid state. For fluorescence emission decay measurements, we used the corresponding excitation and emission wavelengths specific to each luminogen. The resulting spectra are illustrated in Fig. 4, and the lifetime outcomes are summarized in Table 3. All these emitters showed double exponential decay curves consisting of prompt and delayed components. Upon lowering the temperature to 77 K there was a substantial delay in the decay time for SK-2, which showed 4879 ns delayed fluorescence as compared to 0.11 ns at room temperature. Similarly, enhanced fluorescence decay time on the order of nanoseconds was exhibited by luminogens. Such delayed fluorescence is the typical attribute of the direct fluorescence process from singlet states (S1) to ground states (S0) by reverse intersystem crossing. The lifetimes of prompt/delayed components were 0.115/4879 ns for SK-2, 4.74/13.89 ns for SK-3, 4.23/8.321 ns for SK-4, 9.17/60.3 ns for SK-5, and 1.76/24 ns for SK-6. Small ΔEST values coupled with the delayed components indicated the participation of the triplet exciton through the reverse intersystem crossing process, indicating the possible TADF features of the luminogens.
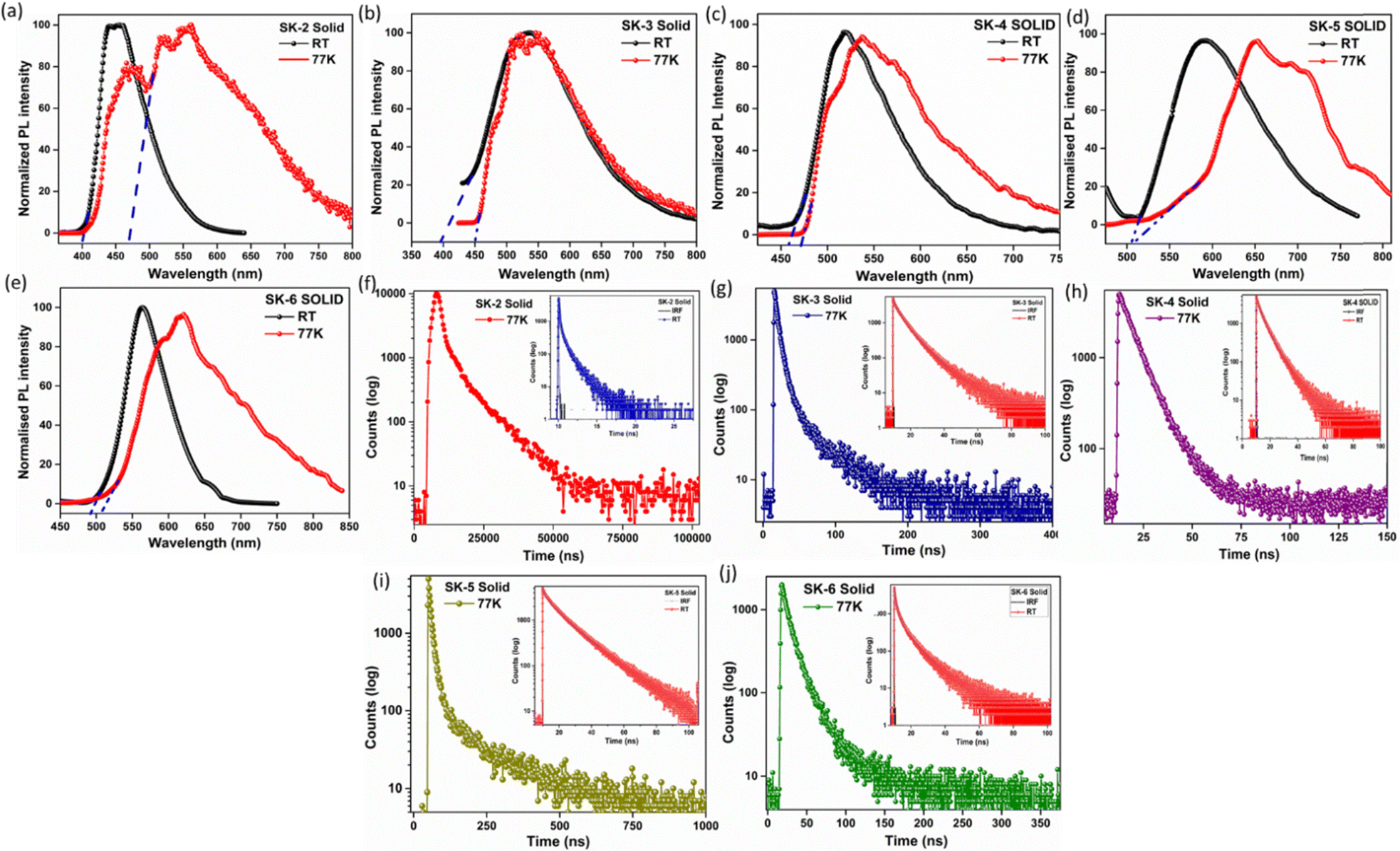 | ||
| Fig. 6 (a)–(e) Room temperature and low temperature (77 K) photoluminescence spectra of SK-2 to SK-6; (f)–(j) fluorescence decay profile of the luminogens SK-2 to SK-6 at room temperature and low temperature (77 K). | ||
| Compound | λ abs (nm) liquida | λ em (nm) liquidb | λ em (nm) solidc | FWHMd (nm) | ϕ PL (%) thin filme | ΔST (eV) Calculatedf | ΔST (eV) Experimentalg | τ p (ns) thin filmh | τ d (ns) thin filmi |
|---|---|---|---|---|---|---|---|---|---|
| a Absorbance at room temperature in THF. b Emission wavelength at room temperature in THF. c Solid-state emission at room temperature. d FWHM values from solid-state emission graphs. e Calculated by the absolute method using an integrating sphere. f Calculated by DFT calculations. g Calculated from the onset of the fluorescence and phosphorescence peaks of solid films at room temperature and 77 K respectively. h Prompt fluorescence decay profiles of compounds in thin films at room temperature. i Delayed fluorescence decay profiles of compounds in thin films at 77 K. | |||||||||
| SK-1 | 259, 342 | 397 | — | — | — | 0.840 | — | — | — |
| SK-2 | 252, 338 | 473 | 450 | 74 | 1.35 | 0.4554 | 0.4604 | 0.115 | 4879 |
| SK-3 | 312, 405 | 566 | 534 | 121 | 26.7 | 2.59 | 0.3908 | 4.74 | 13.89 |
| SK-4 | 244,346 | 535 | 518 | 90 | 28.45 | 0.110 | 0.1014 | 4.23 | 8.321 |
| SK-5 | 304, 450 | 505 | 558 | 117 | 29.1 | 0.058 | 0.0246 | 9.17 | 60.3 |
| SK-6 | 257, 350 | 535 | 560 | 69 | 9.4 | 0.090 | 0.068 | 1.76 | 24 |
X-ray crystallographic analysis by single crystal X-ray diffraction (XRD) was employed to confirm the structural integrity and determine the exact three-dimensional atomic arrangement of the synthesized crystal. To support our explanation from theoretical calculations and analyze the molecular packing in crystals to verify the strong solid-state emissions, we tried growing single crystals of all the luminogens. However, after multiple attempts, we could successfully grow the single crystals of two of the luminogens SK-2 and SK-3 (Crystal id: 2426806 for SK-2 and 2426807 for SK-3). The slow evaporation from the solvent mixture of acetone and pet-ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulted in the formation of single crystals of SK-2 while the slow evaporation from a 1:1 CHCl3/MeOH solvent mixture resulted in cubic-shaped crystals of SK-3. The crystal structures of both luminogens SK-2 and SK-3 and their corresponding single crystallographic data are summarized in Fig. 7, Fig. S20, S21 and Tables S4, S5 (see ESI†).
:1) resulted in the formation of single crystals of SK-2 while the slow evaporation from a 1:1 CHCl3/MeOH solvent mixture resulted in cubic-shaped crystals of SK-3. The crystal structures of both luminogens SK-2 and SK-3 and their corresponding single crystallographic data are summarized in Fig. 7, Fig. S20, S21 and Tables S4, S5 (see ESI†).
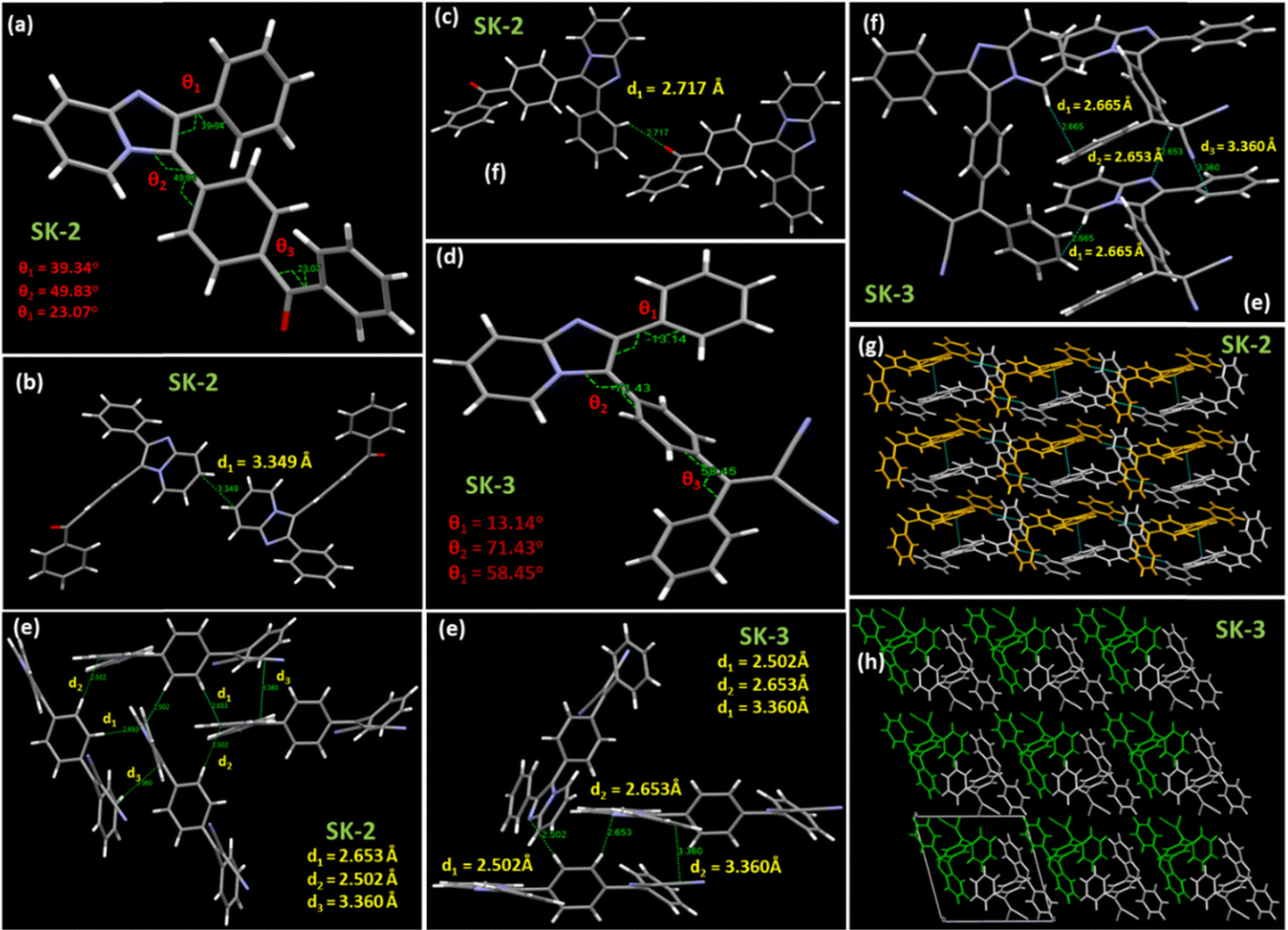 | ||
| Fig. 7 (a) and (b) Crystal structures of SK-2 and SK-3; (c) and (d) interplanar interactions in the crystal lattice of SK-2; (e) and (f) interplanar interactions in the crystal lattice of SK-3; and (g) and (h) the molecular packing modes, and intermolecular interactions in the crystal lattice of SK-2 and SK-3. | ||
The analysis of the crystal structures of both molecules revealed ordered molecular arrangements with highly twisted conformations. The solid-state emissive nature of these compounds can be attributed to the stator–rotor structures, twisted 3D conformations, and multiple short contacts within the crystal lattice. Large torsional angles between the central imidazo[1,2-a]pyridine ring and C2 peripheral phenyl rings were observed, and the distances between the two nearest imidazo[1,2-a]pyridine planes are 3.349 Å and 2.653 Å. The molecular conformation in the SK-2 molecule is twisted with the torsion angle (θ2) between the imidazo[1,2-a]pyridine ring and the adjacent phenyl ring of –COPh being 49.83°, while that for SK-3 is 71.43° (Fig. 7(a) and (d)). In addition to that, the dihedral angle between the imidazo[1,2-a]pyridine ring and the phenyl ring attached to the C2 site also showed significant variation. For SK-2, the observed dihedral angle (θ1) is 39.43° while that for SK-3 it is 13.14°. The installation of the dicyanovinylidene moiety in SK-4 has triggered the molecular distortion as can be evident from the higher torsional angle (θ3) of 58.45° in SK-3 as compared to 23.07° in SK2. Small interplanar distances in SK-3 with twisted architecture help in preventing the molecules from close packing, which is thought to be responsible for the RIMs as evident from their emissive nature in their aggregated states. SK-3 is more emissive in the aggregated state as compared to SK-2. A significant difference in the dihedral angle is observed in SK-2 and SK-3, which accounts for their differences in their emissive properties in their aggregated states as well as their photoluminescence quantum yields. Besides insignificant π–π interactions, other intermolecular short contacts are observed in the crystal structures of both luminogens SK-2 and SK-3. These weak interactions could rigidify molecular conformation and suppress the nonradiative channel, leading to the enhanced emission intensity of the two molecules in the solid state. These abundant intermolecular and intramolecular interactions could lock and rigidify the molecular conformation and restrict the molecular motions of SK-2 and SK-3, resulting in the possibility of bright fluorescence in the solid state. SK-2 exhibits high emission in the solution state (PLQY = 10.62%), while SK-3 was almost non-emissive in the solution state, Hence the solution state PLQY could not be calculated. The relatively small dihedral angle (θ2 = 49.83°) in SK-2 as compared to the dihedral angle in SK-3 (θ2 = 71.43°) allowed the phenyl group to attain a conformation within the crystal lattice where the carbonyl group attached to the Ph ring could enjoy a weak intermolecular interaction with the neighbouring Ph ring with a short distance of 2.717 Å. The short contact disrupts the free rotation to a certain extent in SK-2 while the free rotations of the phenyl rotors in SK-3 could consume the excited-state energy and result in weak emission in solution.
The combination of the TICT effect and RIM mechanisms in the elaborately designed luminogens would endow the molecules with not only excellent AIE properties but also a large Stokes shift. We have utilized Crystal Explorer software to map the Hirshfeld surface depicted in Fig. 8, providing a clear visualization of interaction strengths.56 The interactions are color-coded universally: red for strong, white for medium, and blue for weak, Fig. S22 and S23 (see ESI† for details). Both SK-2 and SK-3 crystals exhibit a significant proportion of C–H type interactions, accounting for 33.3% in SK-2 and 32.4% in SK-3. Notably, both crystals show minimal C–C interactions, ranging from 2.3% to 3.2%. The most prominent interaction, illustrated in Fig. 4, is H⋯H, which constitutes 52.2% and 48.3% of the total crystal packing in SK-2 and SK-3, respectively. Additionally, SK-3 has a significantly higher extent of N–H interactions compared to SK-2, with 12.5% in SK-3 and 3.7% in SK-2. van der Waals interactions appear to be the primary factor in crystal packing due to their substantial contribution. In the packing diagram of SK-2 shown in Fig. 4, weak intramolecular interactions are observed with distances of around 3.349 Å. In contrast, SK-3 exhibits more non-covalent interactions with distances ranging from 2.47 to 2.88 Å. These multiple weak interactions could synergistically rigidify the molecular conformations in the solid state, suppressing non-radiative dissipation and enhancing solid-state emissivity.
 | ||
| Fig. 8 (a) and (c) View of the three-dimensional Hirshfeld surfaces and intermolecular interactions of SK-2 and SK-3 generated in Crystal Explorer; (b) and (d) bar graphs displaying possible interactions in SK-2 and SK-3 calculated through 2D finger plots from Crystal Explorer software. | ||
2.7. Biocompatibility assessment
Considering the emissive nature of the luminogens in their aggregated states, we intended to explore their cell-imaging potential. Luminogens SK-3, SK-4, and SK-6 were chosen as representative cases. Since the biocompatibility assessment is crucial for analyzing materials intended for biological purposes, mouse fibroblast cells (L929) were used to check their cellular viability. The above-mentioned luminogens were dissolved in tetrahydrofuran (THF) to make a concentrated solution (1 mg mL−1).57,58 At the time of material addition, final concentration ranges were made in complete DMEM. The biocompatibility results are shown in Fig. 9, which shows that all the luminogens beyond 1 μg mL−1 concentrations are toxic for the L929 cells. Considering the cell viability data on L929 cells, the safe concentration for further cytotoxicity evaluation against the cervical cancer cell line was found to be 1 μg mL−1. However, even at a 1 μg mL−1 concentration, SK-4 shows more toxicity to L929 cells than SK-3 and SK-6, Fig. 9. Furthermore, the luminogens displayed reasonable photostabilities, a feature that will allow for continuous observation of dynamic processes without significant signal degradation (Fig. S24 and Table S6, see ESI†). | ||
| Fig. 9 Comparative in vitro cell viability studies on L929 cells reveal the concentration-dependent cytotoxic response of SK-3, SK-4, and SK-6 (data are presented as mean ± SD of three replicates). | ||
2.8. Cytotoxicity assessment
The in vitro study conducted on SiHa cells has demonstrated that the imidazo[1,2-a]pyridine-based compounds SK-3, SK-4, and SK-6, even at low concentrations, successfully inhibit the proliferation of SiHa cells. Fig. 10 shows that all of these luminogens possess concentration-dependent anti-metastatic potential towards SiHa cells at a concentration ranging from 0.1 to 1 μg mL−1. The calculated IC50 values of SK-3, SK-4, and SK-6 are 0.58 μg mL−1, 0.52 μg mL−1, and 0.68 μg mL−1 respectively. The anti-proliferative potential of synthesized imidazo[1,2-a] pyridine-derived luminogens is presented in Fig. S25 (ESI†). The dose-dependent effect on the cells was observed in the morphology of the cells, resulting in cell shrinkage and membrane blebbing as apoptotic evidence. The anticancer potential of imidazo[1,2-a]pyridines has been documented over the past few years.59–63 As per the literature, the established mechanism of the anticancer activity of these moieties was due to the inhibition of tubulin polymerization, c-Met inhibition, and inhibition of PI3K/Akt, CDKs, IGF-IR, and CENP-E pathways.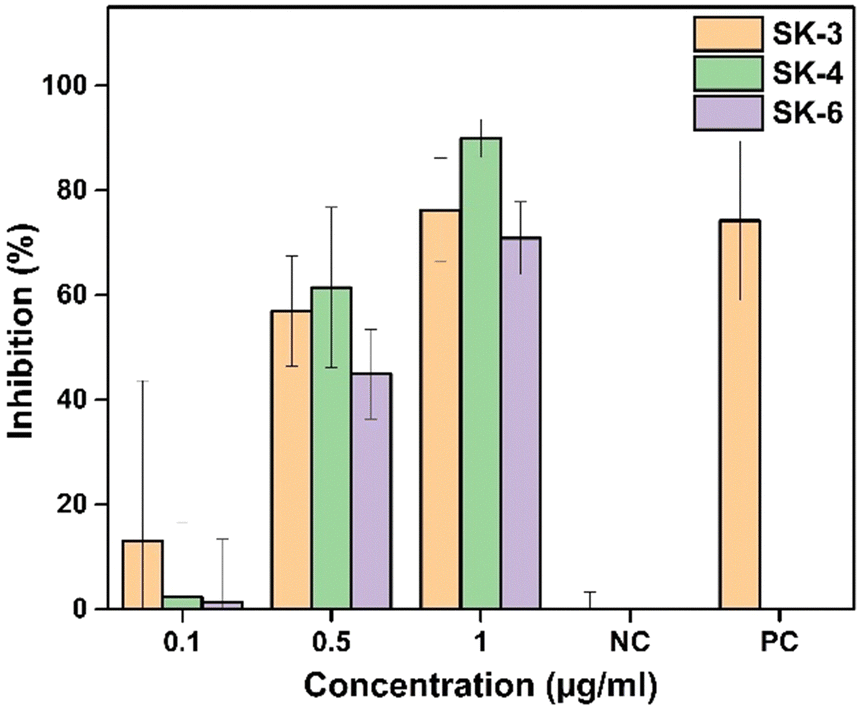 | ||
| Fig. 10 Comparative in vitro cytotoxicity assessment of the luminogens on cervical cancer SiHa cells (data are presented as mean ± SD of three replicates). | ||
Apart from that, the anti-proliferative potential of the luminogens could be due to the generation of reactive oxygen species (ROS). Therefore, the ROS generation potential of SK-3, SK-4, and SK-6 was investigated employing H2DCF-DA as a ROS indicator, which becomes strongly emissive at around 530 nm in the presence of ROS, Fig. 11 and Fig. S26 and S27 (see ESI†). The efficiency of the ROS generation was investigated by irradiating the DCFH-DA solution with different luminogens using white light as the excitation light source. In the presence of luminogens SK-3, SK-4, and SK-6, the fluorescence of H2DCF-DA was gradually enhanced with increasing irradiation time; however, this change was not observed in H2DCF-DA alone (see Fig. 11). Therefore, the antiproliferative potential of the luminogens, although it seems to be due to the incorporation of the imidazo[1,2-a]pyridin scaffold, however, can be boosted by white light irradiation to initiate the PDT for cancer cell ablation.
 | ||
| Fig. 11 (a)–(c) Relative fluorescence intensity (I/I0) of DCFH-OH, (DCFH-OH + SK-3) mixture, (DCFH-OH + SK-4) mixture and (DCFH-OH + SK-6) mixture respectively, in the presence of white light irradiation at different time intervals (I0 is the fluorescence intensity before light irradiation). | ||
2.9. In vitro cellular uptake
To evaluate the bio-imaging abilities of SK-3, SK-4, and SK-6, the intracellular uptake was assessed on SiHa cells. The cells were imaged by confocal laser scanning microscopy (CLSM) at 63× (oil). Double nucleus staining was done with DAPI and PI to assess the effectiveness of SK-3, SK-4, and SK-6 as antiproliferative agents. PI mainly stains the dead cells' nucleus, whereas DAPI stains the nucleus of both live and dead cells. SiHa cells were treated with 1 μg mL−1SK-3, SK-4, and SK-6 for 24 hours and the cells were visualized at excitation wavelengths of 405 nm (DAPI), 488 nm (luminogens), and 561 nm (PI). Among the synthesized AIEgens, SK-6 not only enters the cytosol but also enters the nucleus and evenly stains it (Fig. 12). Luminogen SK-4 was also evenly distributed in the cytosol with higher fluorescence intensity, Fig. 12. Fig. 12 indicates that SK-3 entered the cytoplasm; however, the fluorescence intensity was lower than that of SK-4. | ||
| Fig. 12 (a)–(e) Control cells without treatment ((a) DIC, (b) DAPI, (c) GFP, (d) PI, and (e) merged DIC and DAPI); (f)–(l) control cells treated with SK-3 at 1 μg mL−1 conc. ((f) DIC, (g) DAPI, (h) fluorescence from SK-3, (i) PI, (j) merged SK-3 and DAPI, (k) merged SK-3 and PI, (l) merged SK-3, DAPI and PI); (m)–(s) control cells treated with SK-4 at 1 μg mL−1 conc. ((m) DIC, (n) DAPI, (o) fluorescence from SK-4, (p) PI, (q) merged SK-4 and DAPI, (r) merged SK-4 and PI, (s) merged SK-4, DAPI and PI); (t)–(z) control cells treated with SK-6 at 1 μg mL−1 conc. ((t) DIC, (u) DAPI, (v) fluorescence from SK-6, (w) PI, (x) merged SK-6 and DAPI, (y) merged SK-6 and PI, (z) merged SK-6, DAPI and PI). | ||
The immunofluorescence data (maximum intensity projections of confocal stacks) revealed that all the luminogens entered efficiently into the cervical cancer cells, Fig. S12 (ESI). The fluorescence intensities of SK-3, SK-4, and SK-6 were calculated using the green/GFP filter under 488 nm excitation. The corrected total cell fluorescence (CTCF) was calculated and plotted after normalizing with untreated cells (i.e., CTCF of luminous-treated cells – CTCF of untreated cells). Fig. S25 (see ESI†) presents the comparative corrected total cell fluorescence of SK-3, SK-4, and SK-6.
2.10. OLED fabrication
Considering the TADF features of the designed luminogens, two OLED devices were fabricated using SK-3 and SK-4 as emissive layers (EMLs). The OLED geometries employed in this investigation were: ITO/PEDOT:PSS/NPD/CBP/SK-3/BPhen/LiF-Al and ITO/PEDOT:PSS/NPD/SK-4/TpBi/LiF-Al (see Fig. 13(a) and (b)) (note: no light emission was observed for devices of SK-3 with the first geometry). In these devices, poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) was used as the hole injecting layer (HIL), N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPD) and 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) were used as the hole transporting layers (HTLs), bathophenanthroline (BPhen) and 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TpBi) were chosen as the hole blocking layers (HBLs) and LiF/Al was used as the cathode, Fig. 13(c). Initially, an ITO coated substrate (14–16 Ω sq−1, Ossila limited) of dimensions 20 × 15 mm2 was etched to make four independent active devices using Zn powder and HCl. Subsequent substrate cleaning was done using the standard protocol of cleaning involving soap solution, distilled water, acetone, propanol, and trichloroethylene (TCE) vapors and finally drying using nitrogen gas. The cleaned substrates were UV-treated for 25 minutes after which the layer of PEDOT:PSS was spin-coated onto them. Spin coating was done at a speed of 6000 RPM for 30 s, which gave a 30 nm thick layer. The substrates were post-annealed at 160 °C for 30 minutes to obtain a hard, uniform layer of PEDOT:PSS. All subsequent layers of NPD, EMLs, TpBi, BPhen, LiF, and Al were thermally evaporated in a vacuum at a base vacuum of 3 × 10−6 mbar. | ||
| Fig. 13 (a) and (b) Energy band diagram of the OLED device with SK-3 and SK-4 as EMLs respectively, (c) the materials used to fabricate the OLED devices, (d) electroluminescence spectra of SK-3 and SK-4 OLEDs, (e) 1931 chromaticity diagram for both devices (inset shows the pictures of the glowing devices), (f) current density (J) and luminance (L) vs. voltage (V) for both SK-3 and SK-4 devices (the linear scale to the left is for J and the log scale to the right is for L), and (g) EQE vs. current density for both devices. | ||
2.11. Electroluminescence properties
The electroluminescence (EL) spectra of the devices of SK-3 and SK-4 are shown in Fig. 13. The peak emission of SK-3 was at 583 nm, while that of SK-4 was at 538 nm (Fig. 13(d)). Also, the spectra of SK-4 had an additional shoulder peak at 578 nm. From the 1931 CIE plot of both the devices shown in Fig. 13(e), we observe that the SK-4 device looked yellowish green (CIE: 0.33, 0.44) and the SK-3 device looked yellow (CIE: 0.50, 0.45) in color (this is also evident from the pictures of the glowing devices shown in the inset). From the plots of current density (J) and luminance (L) versus voltage for the devices (see Fig. 13(f)), we see that both devices show a low turn voltage, VON. It is 5.0 V for SK-3 and ∼4.5 V for SK-4. However, both the currents and luminance are lower for SK-2 when compared with SK-4. The device's external quantum efficiency (EQE) is compared for both devices in Fig. 13(g). SK-4 shows a good EQEmax of ∼10.00%, while SK-3 shows a lower, yet reasonable EQEmax of ∼6.4%. However, SK-4 showed a higher efficiency roll-off as compared to SK-3. The device performance parameters are tabulated in Table 4.| EML | V on (@ L = 1 Cd m−2) in V | L max (Cd m−2) | CE (Cd A−1) (@ J = 10 mA cm−2) | PE (lm W−1) (@ J = 10 mA cm−2) | Λ peak (nm) | CIE (x,y) | EQEmax | EQE@J = 10 mA cm−2 |
|---|---|---|---|---|---|---|---|---|
| EML – emissive layer; Von – turn on voltage; Lmax – maximum brightness; CE – current efficiency; PE – power efficiency; λpeak – emission wavelength; EQEmax – maximum external quantum efficiency; EQE – external quantum efficiency. | ||||||||
| SK-3 | 5.0 | 2121 | 10.3 | 7.4 | 583 | (0.50, 0.45) | 6.4% | 6.4% |
| SK-4 | 4.5 | 7500 | 19.26 | 19.11 | 538 | (0.33, 0.44) | 10.0% | 8.3% |
3. Conclusion
This study underscores the critical importance of balancing molecular distortion and electronic rigidity in the design of Dual State Emissive (DSE) molecules. By synthesizing six novel D–A-structured imidazo[1,2-a]pyridine–arylketone conjugates (SK-1 to SK-6), this research demonstrates how incorporating bulky aryl rotors like triphenylamine (TPA) and aryl ketones (–COPh) across the imidazo[1,2-a]pyridine can achieve twisted configurations that enhance emission performance. The findings reveal that SK-3 and SK-4 exhibit DSE effects, while SK-2 and SK-5 show AIE characteristics with good photoluminescence quantum yield (PLQY) values. The luminogens also display high thermal stability, stable electrochemical properties, and effective SiHa cell imaging capabilities with low cytotoxicity. The calculated ΔEST values and lifetimes of prompt/delayed components accentuate the luminogens as potential thermally activated delayed fluorescence (TADF) solid-state emitters. As a proof of concept, SK-3 and SK-4 were successfully used in OLED device fabrication, with SK-4 showing a higher external quantum efficiency (EQEmax) of approximately 10.00% compared to SK-3's 6.4%. This study highlights the potential of DSE molecules for multifunctional applications in optoelectronics and bioimaging, paving the way for significant future advancements in this field.4. Materials and methods
All chemicals were purchased from available commercial sources like Sigma-Aldrich, Spectrochem, and S. D. Fine Chemicals and used without further purification. Organic solvents were dried and distilled before use. Silica gel-coated aluminum sheets (ACME, 254F) were used for the Thin Layer Chromatography (TLC) analysis using EtOAc and petroleum ether as the eluents to monitor the reaction progress. Melting points of all the compounds were recorded using AnalabThermoCal melting point apparatus in the open capillary tube. Fourier transform infrared (FTIR) (ATR-IR) spectra were obtained using the Alpha-II/Bruker instrument. 1H Nuclear Magnetic Resonance (1H NMR) spectroscopy was carried out on a Bruker 400 spectrometer, whereas 13C NMR was carried out on a 100 MHz spectrometer using CDCl3 as a solvent. Chemical shifts are reported in parts per million (ppm) downfield from TMS, and the spin multiplicities are described as s (singlet), d (doublet), t (triplet), and multiplet (m). Coupling constant (J) values are reported in hertz (Hz). Thermogravimetric analysis (TGA) was conducted on a PerkinElmer Diamond TG/DTA instrument at a heating rate of 10 °C min−1 under a nitrogen atmosphere with a flow rate of 150 mL min−1. UV-visible absorption spectra were obtained on a FP-8200/Jasco spectrophotometer. Cyclic voltammetry (CV) measurements were performed in an electrolyte solution of tetrabutylammonium hexafluorophosphate (tBu4NPF6) in acetonitrile (0.1 M), using platinum gauze and Ag/AgCl as the counter and reference electrodes respectively. A scan rate of 25 mV s−1 was used during the CV measurements.4.1. Biocompatibility and cytotoxicity assay
The colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was carried out to check the cell viability/toxicity. L929 (mouse fibroblast cell lines) and SiHa cells were used to analyze the cell viability and cytotoxicity assay, respectively. The cells were cultured in complete DMEM and were harvested by trypsinization for seeding in the well plate. Briefly, the cellular suspension of ∼12000–14000 cells per well was seeded in a 96-well plate. Then, the cells were incubated for 24 hours to facilitate the cellular attachment on the surface of the well at 37 °C with 5% CO2. After one wash with sterile phosphate buffer, different concentrations (0.1 to 100 μg mL−1) of AIE luminogens like SK-3, SK-4, and SK-6 were added into the wells in a triplicate manner. For positive control, 1% Triton X-100, and for growth/negative control, complete DMEM media were used. After 24 hours of treatment with the luminogens, the samples were removed, and two gentle PBS washes were given to the cells to remove the test samples properly. MTT solution (50 μg per well) was added to each well and further incubated for 2 hours (facilitates the conversion of MTT to formazan). Afterward, DMSO was added to each well to dissolve formazan. The formazan's optical density (OD) was then measured at 570 nm using a plate reader (Tecan Infinite M200 Pro). Cellular viability was calculated for the wells treated with AIEgens depending on the OD data. The percentage cell viability was calculated using the following equation:| % Cell viability = {(Absorbance of the test sample − Absorbance of media control)/(Absorbance of growth control − Absorbance of media control)} × 100 |
4.2. In vitro cellular uptake
SiHa cells were used to analyze the fluorescence behavior of the compound inside the cells. The cells were seeded on the sterile coverslips in a 12-well plate at a cell density of ∼50000 cells per well and incubated for 24 hours. Then luminogens (SK-3, SK-4, and SK-6) were added to the wells at 1 μg mL−1 concentration. The cells were incubated again for 24 hours. In growth control, the cells were treated with the complete media. The next day, two PBS washes were given to the cells, and they were then incubated for 15 minutes with 4% formaldehyde to fix the cells. Afterward, three PBS washes were given to remove formaldehyde. The nuclear staining was done using 2 μg mL−1 DAPI followed by 1 μg mL−1 PI. Three consecutive PBS washes were given after each nuclear stain. Coverslips were further fixed on a glass slide, and the cells were then viewed by confocal laser scanning microscopy (CLSM).
Author contributions
Chinmay Thakkar: synthesis, characterization, and data curation; Seemantini Kale: synthesis, characterization, and data curation; Mohammad Amir Ahemad: OLED device fabrication and data curation; Monalisa Debnath: cell imaging studies; Anjali Tripathi: DFT calculations; Purav Badani: DFT calculations; Rohit Srivastava: cell imaging studies; Sangita Bose: OLED device fabrication and data curation, writing and editing; and Satyajit Saha: project conceptualization and planning, project design, project coordination and implementation, manuscript writing and editing, funding acquisition.Data availiability
The data supporting this article have been included as part of the ESI.† The crystallographic data for SK-3 and SK-4 have been deposited at the CCDC.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
CT is grateful to DST-PURSE for his fellowship. SB and MAA would like to thank SERB-CRG/2021/001120 for financial assistance. SS acknowledges SERB-CRG/2022/000579, DST-PURSE/2020/8, DBT-BUILDER (BT/INF/22/SP47618/2023) and BRNS (53/14/01/2024), India for financial assistance. SS also acknowledges DST-FIST (SR/FST/ET-I/2018/156) for the instrumental support and SAMat Research Facilities, JNCASR, Bengaluru, for low-temperature lifetime measurement facilities.References
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Mater. Horiz., 2021, 8, 2387–2419 RSC.
- C. Anichini, W. Czepa, D. Pakulski, A. Aliprandi, A. Ciesielski and P. Samori, Chem. Soc. Rev., 2018, 47, 4860–4908 RSC.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- Y. Zhang, H. Li, M. Yang, W. Dai, J. Shi, B. Tong, Z. Cai, Z. Wang, Y. Dong and X. Yu, Chem. Commun., 2023, 59, 5329–5342 RSC.
- S. Samanta, K. Lai, F. Wu, Y. Liu, S. Cai, X. Yang, J. Qu and Z. Yang, Chem. Soc. Rev., 2023, 52, 7197–7261 RSC.
- M. H. Chua, K. L. Chin, X. J. Loh, Q. Zhu and J. Xu, ACS Nano, 2023, 17(3), 1845–1878 CrossRef CAS PubMed.
- Y. Zhang, A. Chen, M. W. Kim, A. Alaei and S. S. Lee, Chem. Soc. Rev., 2021, 50, 9375–9390 RSC.
- R. Boddula and S. P. Singh, J. Mater. Chem. C, 2021, 9, 12462–12488 RSC.
- F. Khan and R. Misra, J. Mater. Chem. C, 2023, 11, 2786–2825 RSC.
- F. Y. Zhu, L. J. Mei, R. Tian, C. Li, Y. L. Wang, S. L. Xiang, M. Q. Zhu and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 3350–3383 RSC.
- G. R. Suman, M. Pandey and A. S. Chakravarthy, Mater. Chem. Front., 2021, 5, 1541–1584 RSC.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC.
- Z. Li, X. Ji, H. Xie and B. Z. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS.
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 16–18 CrossRef.
- W. Du, X. Liu, L. Liu, J. W. Lam and B. Z. Tang, ACS Appl. Polym. Mater., 2021, 3(5), 2290–2309 CrossRef CAS.
- Y. Duo, L. Han, Y. Yang, Z. Wang, L. Wang, J. Chen, Z. Xiang, J. Yoon, G. Luo and B. Z. Tang, Chem. Rev., 2024, 124(20), 11242–11347 CrossRef CAS.
- P. Gopikrishna and P. K. Iyer, J. Phys. Chem. C, 2016, 120(46), 26556–26568 CrossRef CAS.
- T. Stoerkler, G. Ullrich, A. D. Laurent, D. Jacuemin and J. Massue, J. Org. Chem., 2023, 88(13), 9225–9236 CrossRef CAS.
- Y. Zhan, L. Lin, M. Chen and L. Wu, ACS Appl. Mater. Interfaces, 2018, 10(39), 33390–33398 CrossRef CAS.
- X. Hu, B. Fang, P. Li, J. Wang, J. Li, Y. Dai and M. Yin, Adv. Funct. Mater., 2025, 2423793 CrossRef.
- P. Chettri, A. Singhania, S. Kalita, H. Nakao, B. Bora, P. Saikia, S. Dutta, A. Bandyopadhyay and S. Ghosh, Adv. Opt. Mater., 2024, 12, 2400650 CrossRef CAS.
- N. A. Kukhta and M. R. Bryce, Mater. Horiz., 2021, 8, 33–55 RSC.
- M. D. Maciag, G. Ulrich, D. Jacquemin, J. Mysliwiec and J. Massue, Phys. Chem. Chem. Phys., 2023, 25, 15085–15098 RSC.
- F. Li, M. Wang, S. Liu and Q. Zhao, Chem. Sci., 2022, 13, 2184–2201 RSC.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24(8), 1518–1528 CrossRef CAS.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496 CrossRef CAS.
- D. Gobel, D. Duvinage, T. Stauch and B. J. Nachtsheim, J. Mater. Chem. C, 2020, 8, 9213–9225 RSC.
- Z. Huang, F. Tang, F. He, L. Kong, J. Huang, J. Yang and A. Ding, Org. Chem. Front., 2022, 9, 5118–5124 RSC.
- C. Zeng, H. Wang, B. Q. Wang, P. Hu, K. Q. Zhao and B. Donnio, Chem. Mater., 2024, 36, 7306–7316 CrossRef CAS.
- J. K. Vazquez, Y. A. Sanchez, L. A. Cortes and B. R. Molina, Chem. Mater., 2021, 33(18), 7160–7184 CrossRef.
- G. Xia, L. Si and H. Wang, Mater. Today Chem., 2023, 30, 101596 CrossRef CAS.
- A. R. Nair, G. Mohan, C. Raksha, A. Sreekumar, P. Manoj, Y. C. Sunil and A. Sivam, Sci. Rep., 2024, 14, 29795 CrossRef CAS PubMed.
- Y. Zhu, K. Liao, Y. Li, W. Zhang, B. Song, X. Q. Hao and Z. Zhu, Dyes Pigm., 2024, 224, 112004 CrossRef CAS.
- W. Xi, J. Yu, M. Wei, Q. Qiu, P. Xu, Z. Qian and H. Feng, Chem. – Eur. J., 2020, 26, 3733–3737 CrossRef CAS PubMed.
- Y. Li, J. Dai, S. Yang, N. Sui, C. Wang, X. Meng, K. Wang, H. Zhang, X. Liu, G. Lu and Y. Wang, Adv. Opt. Mater., 2024, 12, 2301475 CrossRef CAS.
- A. Cesaretti, Z. Cai, J. Kim, H. Kim, Y. Lei and B. Carlotti, ChemPhotoChem, 2023, 7, e202300040 CrossRef CAS.
- Y. Xu, L. Ren, D. Dang, Y. Zhi, X. Wang and L. Meng, Chem. – Eur. J., 2018, 24, 10383–10389 CrossRef CAS.
- Y. Ni, L. Yang, L. Kong, C. Wang, Q. Zhang and J. Yang, Mater. Chem. Front., 2022, 6, 3522–3530 RSC.
- T. C. Lin, S. T. Cho, C. L. Wu, N. E. Setyatama, P. H. Tung, B. Y. Hung, J. H. Lin, P. E. Jan, P. H. Tsai and H. W. Lin, Chem. Commun., 2024, 60, 5948–5951 RSC.
- Y. Zheng, Y. Zhou, S. Jiang, X. Xie, G. Du, X. Shen, X. Zhao and Z. Yu, Org. Chem. Front., 2023, 10, 1495–1504 RSC.
- Q. Shao, K. Liang, Y. Wang, Z. Yan, G. Xia and H. Wang, J. Mater. Chem. C, 2020, 8, 4549–4556 RSC.
- S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, RSC Med. Chem., 2023, 14, 644–657 RSC.
- X. H. Han, P. Zhao, M. K. Tang, L. Yang, Q. Wang and S. S. Zhang, Sens. Diag., 2024, 3, 1062–1067 RSC.
- X. H. Zheng, J. W. Zhao, X. Chen, R. Cai, G. X. Yang, J. J. Zhu, S. S. Tang, Z. H. Lin, S. L. Tao and Q. X. Tong, Chem. – Eur. J., 2020, 26, 8588–8596 CrossRef CAS.
- V. Kurteva, ACS Omega, 2021, 6(51), 35173–35185 CrossRef CAS PubMed.
- A. Thakur and A. Sharma, Spectrochim. Acta, Part A, 2022, 282, 121633 CrossRef CAS PubMed.
- Y. Wu, W. Yuan, H. Ji, Y. Qin, J. Zhang, H. Li, Y. Li, Y. Wang and Y. Sun, Dyes Pigm., 2017, 142, 330–339 CrossRef CAS.
- W. Song, Q. Xu, J. Zhu, Y. Chen, H. Mu, J. Huang and J. Su, ACS Appl. Mater. Interfaces, 2020, 12(17), 19701–19709 CrossRef CAS PubMed.
- G. Renno, F. Cardano, G. Volpi, C. Barolo, G. Viscardi and A. Fin, Moecules, 2022, 27(12), 3856 CrossRef CAS.
- T. Z. Liu, Y. C. Yuan and B. X. Zhao, Spectrochim. Acta, Part A, 2022, 282, 121694 CrossRef CAS PubMed.
- Y. Li, S. Ren, G. Shi, C. Yuan, G. Duan and Y. Ge, J. Lumin., 2023, 255, 119547 CrossRef CAS.
- G. Yashwantrao, V. Naik, P. Badani and S. Saha, Chem. – Eur. J., 2025, 31, e202500047 CrossRef CAS.
- G. Yashwantrao, P. Gosavi, V. Naik, M. Debnath, S. Seth, P. Badani, R. Srivastava and S. Saha, J. Mater. Chem. C, 2025, 13, 3955–3968 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A. 03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17 (2017). University of Western Australia.
- M. K. Kumawat, M. Thakur, R. B. Gurung and R. Srivastava, ACS Sustainable Chem. Eng., 2017, 3, 1382–1391 CrossRef.
- D. S. Chauhan, B. P. Reddy, S. K. Mishra, R. Prasad, M. Dhanka, M. Vats, G. Ravichandran, D. Poojari, O. Mhatre, A. De and R. Srivastava, Langmuir, 2019, 35, 7805–7815 CrossRef CAS PubMed.
- D. K. Soumyashree, D. S. Reddy, H. Nagarajaiah, L. Naik, H. M. Savanur, C. D. Shilpa, M. S. Kumari, H. Shanavaz and B. Padmashali, Arch. Pharm., 2023, 356, 2300106 CrossRef CAS PubMed.
- D. Güçlü, B. Kuzu, I. Tozlu, F. Taspinar, H. Canpinar, M. Taspinar and N. Menges, Bioorg. Med. Chem. Lett., 2018, 28, 2647–2651 CrossRef PubMed.
- A. Kamal, G. B. Kumar, V. L. Nayak, V. S. Reddy, A. B. Shaik, Rajender and M. K. Reddy, Med. Chem. Commun., 2015, 6, 606–612 RSC.
- Z. Bakherad, M. Safavi and S. Sepehri, Res. Chem. Intermed., 2019, 45, 5261–5290 CrossRef CAS.
- T. Damghani, F. Moosavi, M. Khoshneviszadeh, M. Mortazavi, S. Pirhadi, Z. Kayani, L. Saso and N. Edraki, Sci. Rep., 2021, 11, 1–20 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental details and characterization data. CCDC 2426806 and 2426807. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc01128k |
| This journal is © The Royal Society of Chemistry 2025 |