
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning thermomechanical properties of hydrogen-bonded materials by using a mixed cocrystal approach†
Gary C.
George
III
 a,
Liulei
Ma
a,
Jack R.
Gaffney
a,
Richard K.
Brooks
b,
Daniel K.
Unruh
c,
Ryan H.
Groeneman
*b and
Kristin M.
Hutchins
*a
a,
Liulei
Ma
a,
Jack R.
Gaffney
a,
Richard K.
Brooks
b,
Daniel K.
Unruh
c,
Ryan H.
Groeneman
*b and
Kristin M.
Hutchins
*a
aDepartment of Chemistry and MU Materials Science & Engineering Institute, University of Missouri, Columbia, MO 65897, USA. E-mail: kristin.hutchins@missouri.edu
bDepartment of Natural Sciences and Mathematics, Webster University, St. Louis, MO 63119, USA. E-mail: ryangroeneman19@webster.edu
cOffice of the Vice President for Research, University of Iowa, Iowa City, IA 52242, USA
First published on 2nd April 2025
Abstract
The ability to tune the thermomechanical properties of organic solids by utilizing a mixed cocrystal approach is described. The components of each solid are self-assembled through hydrogen bonds, and changing the composition of the solid at the molecular level provides control over the solid-state property. Specifically, two binary solids are prepared using the same hydrogen-bond donor molecule and an unsymmetrical, isosteric hydrogen-bond acceptor. The mixed cocrystal is realized by incorporating both acceptors into the solid material. The thermomechanical response of the mixed cocrystal lies numerically in between the two binary systems along all three principal directions of the solid. Mixed cocrystals are underexplored when compared to their binary counterparts, and this work demonstrates the tunability in solid-state material properties that can be achieved using the mixed approach.
In recent years, the power to alter the solid-state structure of an organic solid by modifying the molecular structure has been widely demonstrated.1–3 Furthermore, addition of a second molecular component allows for synthesis of supramolecular solids such as coordination polymers, cocrystals, and host–guest complexes, while offering a significant opportunity to tune both the solid-state structure and properties by controlling the identity of the second component and the corresponding self-assembly process.4–7 These solids are typically sustained by supramolecular interactions such as coordination, hydrogen, or halogen bonds, and other electrostatic or van der Waals forces.8 Incremental changes to either component of these solids can significantly influence not only the resulting structure but the properties as well.
Cocrystals are typically binary multi-component solids and have received considerable attention over the last two decades.9 Mixed cocrystals are a class of molecular solids that contain two isosteric molecules, which are interchangeable in a given crystallographic position, along with an additional molecule in the lattice (Fig. 1).10,11 Mixed cocrystals have been significantly less investigated, but exhibit promise because of the inherent ability to incorporate multiple components in the solid that impart different chemical or physical properties to the solid material.12
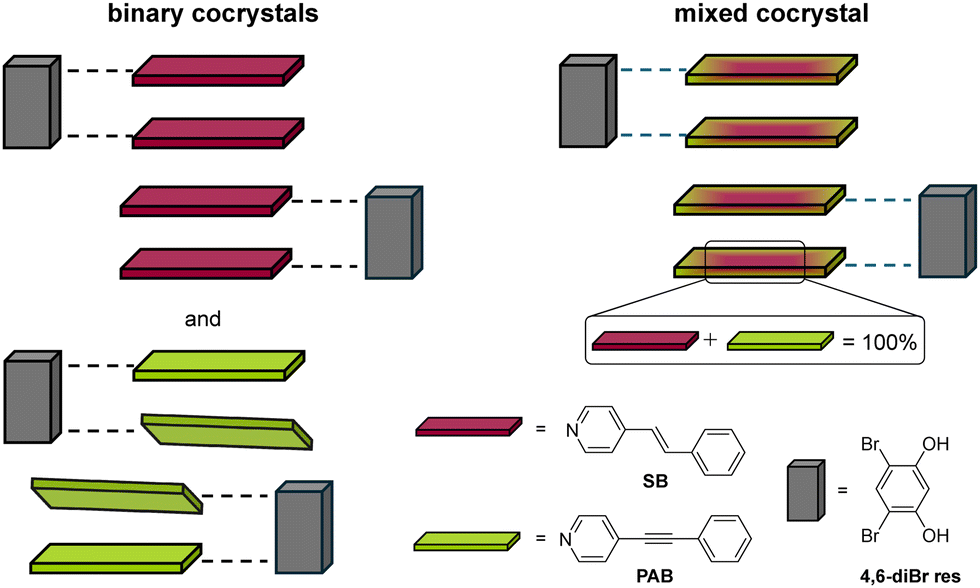 | ||
| Fig. 1 Structures of binary and mixed cocrystals with molecular components shown. Dashed lines represent hydrogen bonds. | ||
Thermomechanical behaviors of molecular crystals are challenging to predict, and recently, several groups have made efforts to control and tune these properties.13–16 Molecular crystals with unique thermomechanical behaviors can be used in sensors, actuators, or multi-functional materials.17 Our groups have recently demonstrated the property of thermal expansion (TE)18 can be fine-tuned in mixed cocrystals19,20 by using molecules that are either capable or incapable of undergoing dynamic molecular motion21 in the solid state. In particular, we have reported symmetrical molecules containing a motion-capable group such as ethylene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) yield larger TE tensors in solids where motion occurs when compared to isostructural solids containing a motion-incapable group such as acetylene (C
C) yield larger TE tensors in solids where motion occurs when compared to isostructural solids containing a motion-incapable group such as acetylene (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C).
C).
We sought to further investigate the hypothesis that using a mixed cocrystal approach offers a pathway for tuning thermomechanical properties of organic solids. Both of our previous studies19,20 included symmetrical molecules containing an acceptor moiety on each side, and strong noncovalent bonds held this symmetrical molecule in place from both sides, limiting structural flexibility. Thus, we modified the motion-capable molecule to an unsymmetrical structure lacking an acceptor on one side, which could disrupt the solid-state structure and influence corresponding properties. Here, we show that local self-assembly is well controlled in the binary and mixed solids; however, the extended packing of the two binary systems does differ, likely due to the unsymmetrical component. However, the directions of TE across the series are analogous. Most importantly, the thermal and thermomechanical properties of the mixed cocrystal lie in between the two binary solids, demonstrating the ability to tune material properties using molecular-level control strategies.
We selected 4,6-dibromoresorcinol (4,6-diBr res) as a ditopic hydrogen-bond donor and two monotopic, unsymmetrical hydrogen-bond acceptors, 4-stilbazole (SB) and 4-(phenylethynyl)pyridine (PAB) (Fig. 1). The crystal structure of (4,6-diBr res)·2(SB) has been previously reported,22 so we knew cocrystallization was possible with one of our selected binary pairs. The donor, 4,6-diBr res, was synthesized using a modified literature method,23 while PAB24 and SB25 were synthesized as reported. The binary cocrystals, (4,6-diBr res)·2(SB) and (4,6-diBr res)·2(PAB), were prepared by combining a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of 4,6-diBr res and the acceptor, respectively, in ethanol. The mixed cocrystal (4,6-diBr res)·(SB)·(PAB) was prepared by combining a 1:1:1 molar ratio of 4,6-diBr res, SB, and PAB in ethanol. Each solution was allowed to evaporate slowly, which yielded single crystals suitable for X-ray diffraction in each case. Powder X-ray diffraction confirmed the bulk phase purity of each solid (Fig. S1–S3, ESI†). 1H NMR spectroscopy was also used to verify the composition of the mixed cocrystal (Fig. S4–S7, ESI†). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to characterize the thermal behavior of each cocrystal (Fig. S8–S13 and Table S7, ESI†).
:2 molar ratio of 4,6-diBr res and the acceptor, respectively, in ethanol. The mixed cocrystal (4,6-diBr res)·(SB)·(PAB) was prepared by combining a 1:1:1 molar ratio of 4,6-diBr res, SB, and PAB in ethanol. Each solution was allowed to evaporate slowly, which yielded single crystals suitable for X-ray diffraction in each case. Powder X-ray diffraction confirmed the bulk phase purity of each solid (Fig. S1–S3, ESI†). 1H NMR spectroscopy was also used to verify the composition of the mixed cocrystal (Fig. S4–S7, ESI†). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to characterize the thermal behavior of each cocrystal (Fig. S8–S13 and Table S7, ESI†).
Single-crystal X-ray diffraction data demonstrated the cocrystal (4,6-diBr res)·2(SB) crystallizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Tables S1 and S2, ESI†). The asymmetric unit contains one molecule of 4,6-diBr res and two molecules of SB. The components are held together by O–H⋯N hydrogen bonds [O⋯N 2.689(3) and 2.690(3) (Å) at 290 K] to form a three-component assembly (Fig. 2a). The ethylene group within one SB molecule is disordered at 290 K, while the other SB molecule is fully occupied. The occupancies of this disorder were determined by free variable refinement, where the ratio of the two positions must sum to 1.00. At 290 K, the major and minor conformations are present in a 0.95/0.05 ratio. Upon cooling, the ratio of the conformations varied, which is indicative of dynamic pedal motion, with values of 0.96/0.04 at 270 K and 0.98/0.02 at 250 K. With additional cooling, the disorder of the ethylene group fully resolved and in turn the dynamic motion ceased at 230 K (Fig. 2a).
(Tables S1 and S2, ESI†). The asymmetric unit contains one molecule of 4,6-diBr res and two molecules of SB. The components are held together by O–H⋯N hydrogen bonds [O⋯N 2.689(3) and 2.690(3) (Å) at 290 K] to form a three-component assembly (Fig. 2a). The ethylene group within one SB molecule is disordered at 290 K, while the other SB molecule is fully occupied. The occupancies of this disorder were determined by free variable refinement, where the ratio of the two positions must sum to 1.00. At 290 K, the major and minor conformations are present in a 0.95/0.05 ratio. Upon cooling, the ratio of the conformations varied, which is indicative of dynamic pedal motion, with values of 0.96/0.04 at 270 K and 0.98/0.02 at 250 K. With additional cooling, the disorder of the ethylene group fully resolved and in turn the dynamic motion ceased at 230 K (Fig. 2a).
 | ||
| Fig. 2 X-ray crystal structure of (4,6-diBr res)·2(SB) at 290 K: (a) three-component hydrogen-bonded assembly at 290 and 230 K and (b) extended packing highlighting quadruple stack of SB molecules. Hydrogen bonds are shown with yellow dashed lines and other contacts shown with black dashed lines. | ||
In (4,6-diBr res)·2(SB), the aromatic rings within each SB molecule are nearly coplanar with the mean planes twisted from coplanarity by 8.07° (ordered SB) and 8.35° (disordered SB) at 290 K. The rings undergo minimal changes in planarity upon cooling. Within the hydrogen-bonded assembly, the stacked SB molecules are rotated away from coplanarity by 28.26°. Due to inversion symmetry, there is a quadruple stack of SB molecules that is capped at both ends with a pair of 4,6-diBr res molecules (Fig. 2b). Each 4,6-diBr res engages in a Br⋯π contact with a neighboring phenyl ring of SB. This stacking pattern extends approximately along the a axis and is accompanied by dimeric C–H⋯Br contacts between 4,6-di-Br molecules26 along the c axis, to constitute a sheet. The sheets are stacked parallel along the b axis and interact through C–H⋯Br and C–H⋯π contacts.
To our surprise, simple substitution of PAB for SB in cocrystallization with 4,6-diBr res yielded cocrystals that lie in a different crystal system. The components within (4,6-diBr res)·2(PAB) crystallize in the space group P21/c (Tables S3 and S4, ESI†). The asymmetric unit contains one 4,6-diBr res and two molecules of PAB, affording a similar three-component hydrogen-bonded assembly sustained by O–H⋯N hydrogen bonds [O⋯N 2.729(3) and 2.749(3) (Å) at 290 K] (Fig. 3a). Due to the rigid acetylene core within PAB, there is no possibility of molecular pedal motion. The aromatic rings within a molecule of PAB are twisted from coplanarity by 7.77° and 15.89°. Within the hydrogen-bonded assembly, the stacked PAB molecules are twisted from coplanarity by 59.47°, significantly more when compared to the SB system.
 | ||
| Fig. 3 X-ray crystal structure of (4,6-diBr res)·2(PAB) at 290 K: (a) three-component hydrogen-bonded assembly and (b) extended packing highlighting offset quadruple stack of PAB molecules. Hydrogen bonds are shown with yellow dashed lines. | ||
Although the replacement of SB with PAB in the cocrystal could be expected to cause minimal disruption to the solid-state structure, the extended packing of (4,6-diBr res)·2(PAB) does differ from (4,6-diBr res)·2(SB). Similarly, (4,6-diBr res)·2(PAB) also contains a pair of hydrogen-bonded assemblies that form a quadruple stack; however, the closest PAB molecules are displaced and stacked with little overlap of the π surfaces. The capping group on the quadruple stack is a PAB molecule, rather than 4,6-diBr res, and it interacts with the stack through an edge-to-face π-stacking interaction (Fig. 3b). The bromine atoms of 4,6-diBr res engage in C–H⋯Br contacts with the para hydrogen on the phenyl ring of PAB and Type I Br⋯Br halogen contacts27 with adjacent 4,6-diBr res molecules in the sheet. The sheets also interact through C–H⋯Br, C–H⋯O, and C–H⋯π contacts.
Given that the three-component hydrogen-bonded assembly was persistent across (4,6-diBr res)·2(SB) and (4,6-diBr res)·2(PAB), we investigated the possibility of forming a mixed cocrystal. We expected that the three-component assembly would persist; however, the extended packing could match either one of the binary forms. A mixed cocrystal approach was realized when a 1:1 ratio of SB and PAB along with an equimolar amount of 4,6-diBr res were combined and allowed to slowly evaporate. The components of the mixed cocrystal crystallize in the space group, Pī with a formula of (4,6-diBr res)·(SB)·(PAB), which is isostructural to (4,6-diBr res)·2(SB) (Tables S5 and S6, ESI†).
Single-crystal X-ray diffraction demonstrated that the solid is indeed a mixed cocrystal rather than a ternary cocrystal, because the two acceptors occupy an equivalent crystallographic position. In a ternary cocrystal, each molecule would occupy a crystallographically unique (independent) position.10,11,28 Importantly, both hydrogen-bond acceptor sites exhibit molecular disorder such that both pyridyl sites are readily replaced with either acceptor, SB or PAB (Fig. 4). The three-component assembly is persistent, and the components self-assemble via O–H⋯N hydrogen bonds [O⋯N 2.696(3) and 2.699(3) (Å) at 290 K]. The ethylene and acetylene groups were readily located in both acceptor molecules; however, modeling the disorder of the corresponding aromatic rings was more nuanced and full details are described in the ESI.†
 | ||
| Fig. 4 X-ray crystal structure of (4,6-diBr res)·(SB)·(PAB) illustrating the three-component hydrogen-bonded assembly at 290 K. Hydrogen bonds are shown with yellow dashed lines. The ethylene moiety is shown in burgundy, and the acetylene moiety is shown in green. | ||
The mixed cocrystal was synthetically prepared using a 1:1 molar ratio of SB and PAB. Based on the single-crystal X-ray data at 290 K, SB occupies ca. 60% of the assembly and PAB occupies ca. 40% of the assembly. 1H NMR spectroscopy collected using the bulk crystalline solid demonstrated that the ratio of SB to PAB is 54%:46%. Overall, both the NMR and X-ray data show good agreement with each other, as well as the initial synthetic feed ratio. Due to the isostructurality, (4,6-diBr res)·(SB)·(PAB) has an identical extended packing as (4,6-diBr res)·2(SB). Notably, we attempted to prepare mixed cocrystals using unequal ratios of SB and PAB, but only isolated binary cocrystals in these cases. Specifics for the crystallizations are given in the ESI.†
To further understand the ability to isolate mixed cocrystals when the binary solids are not isostructural, the molecular electrostatic potential energy surfaces for 4,6-diBr res, SB, and PAB were calculated with the Spartan’20 molecular modeling program, using density functional theory (DFT) at the B3LYP/6-311++G** level.29 For 4,6-diBr res, two regions of positive potential are localized on the syn–syn hydroxyl groups, with an energy value of 316.4 kJ mol−1, demonstrating the strong potential for hydrogen-bond formation at both sites (Fig. S15, ESI†). Regions of negative potential lie on the opposite side of the oxygen atoms, adjacent to the bromine groups. Notably, the energy value on the bromine atoms is 73.6 kJ mol−1. Thus, 4,6-diBr res is a much weaker halogen-bond donor than hydrogen-bond donor, which explains why there is not competition from halogen bonding in the cocrystals. For SB and PAB, regions of negative potential lie on the pyridyl nitrogen atoms, demonstrating their hydrogen-bond-accepting ability. The energy value for SB is −197.8 kJ mol−1 and the energy value for PAB is −189.6 kJ mol−1 (Fig. S15, ESI†). The lower value for SB indicates that in competition with PAB, SB would serve as a slightly stronger hydrogen-bond acceptor for 4,6-diBr res. These calculations support the successful preparation of the mixed cocrystals with a slight bias in stoichiometry toward SB. The difference in energy may also support our observation of binary cocrystal formation when the ratios of the acceptors are unequal.
TGA demonstrated a single decomposition event for each cocrystal that began at ca. 150 °C for (4,6-diBr res)·2(PAB) and (4,6-diBr res)·(SB)·(PAB), and ca. 175 °C for (4,6-diBr res)·2(SB) (Fig. S8–S10, ESI†). DSC characterization for each solid showed a single endothermic signal upon heating, corresponding to melting. All solids also exhibited a single exothermic signal upon cooling from the melt, corresponding to crystallization. The melting point ranges for the solids are 142–144 °C ((4,6-diBr res)·2(SB)), 116–117 °C ((4,6-diBr res)·2(PAB)), and 119–126 °C ((4,6-diBr res)·(SB)·(PAB)). Notably, the melting and crystallization temperatures of the mixed cocrystal, (4,6-diBr res)·(SB)·(PAB), lie in between the two binary solids, demonstrating the influence of the mixed environment on thermal properties (Fig. S11–S13 and Table S7, ESI†).
To investigate the impact of molecular-level mixing and the motion propensity that accompanies it on the thermomechanical behavior of each solid, a variable-temperature single-crystal X-ray diffraction experiment was conducted for all three cocrystals over the temperature range of 290 to 190 K in 20 K increments (Tables S1–S6, ESI†). The TE coefficients and principal axis directions for each cocrystal were calculated with the software PASCal30,31 by using the unit cell parameters obtained from the variable-temperature experiments (Table 1 and Fig. S16–S18, ESI†). Even though the packing of the binary cocrystal with PAB differs from the other two solids, the directions of the principal axes of TE were similar for all three cocrystals, where X1 is in the direction of the hydrogen bonds, X2 is between the sheets, and X3 is in the π-stacked direction (Fig. S19–S21, ESI†).
| Crystal | α X 1 (MK−1) [axis] | α X 2 (MK−1) [axis] | α X 3 (MK−1) [axis] | α X v (MK−1) |
|---|---|---|---|---|
| (4,6-diBr res)·2(SB) | −20(1) [0 1 1] | 70(1) [−1 3 −3] | 145(3) [1 0 0] | 196(5) |
| (4,6-diBr res)·2(PAB) | −4(1) [4 0 −3] | 106(1) [0 1 0] | 120(1) [1 0 2] | 223(1) |
| (4,6-diBr res)·(SB)·(PAB) | −14(1) [0 1 1] | 76(1) [0 4 −3] | 130(3) [7 0 1] | 194(3) |
The thermomechanical response of most solids to cooling is contraction or positive TE. However, in this system, along X1, all three cocrystals exhibited negative TE, i.e., an increase in dimension upon cooling.32 Approximately 30% of known organic crystals exhibit uniaxial negative TE.33,34 Negative TE for organic solids has been attributed to transverse vibrational motions,35–37 has been seen in frameworks that exhibit scissor-style motion,38 and has been observed perpendicular to the direction of molecular motion,19 although, in many cases, the behavior is discovered anomalously.
The X1 axis encompasses the direction of the O–H⋯N hydrogen bonds, which are the primary noncovalent bonds responsible for the self-assembly of the components (Fig. 5). These hydrogen bonds in each cocrystal exhibit minimal changes in length over the temperature range, and the angle of each hydrogen bond increases upon cooling to become closer to ideal, 180° (Table S9, ESI†). While all three crystallographic axes in each cocrystal decrease in length upon cooling (positive TE, Fig. S22–S24, ESI†), the negative TE arises from a small increase in crystallographic angle dimension(s) upon cooling. In (4,6-diBr res)·2(SB), the γ angle increases by 0.14° and in (4,6-diBr res)·2(PAB) the β angle increases by 0.64°. In the mixed cocrystal, (4,6-diBr res)·(SB)·(PAB), the β and γ angles both increase by 0.13°, demonstrating the influence of multiple acceptors in the mixed environment. The angles that exhibit negative TE behavior include contribution from the changing hydrogen bond angles. The dimeric C–H⋯Br interactions involving 4,6-diBr res molecules also lie along this direction.
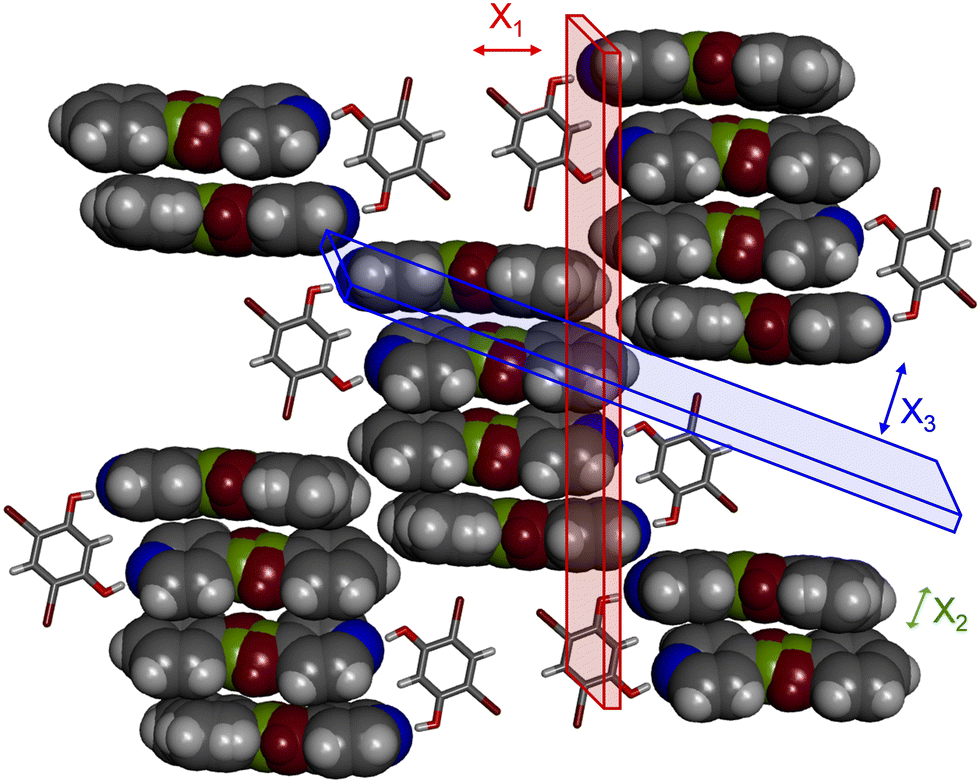 | ||
| Fig. 5 X-ray crystal structure of (4,6-diBr res)·(SB)·(PAB) with the principal axes of TE denoted by planes and accompanying arrows. The direction of X2 goes into the page. The ethylene group is in burgundy and the acetylene group is green. | ||
The TE values for X2 range from moderately positive (SB and mixed) to colossal39 (PAB). The direction of X2 encompasses the direction between stacked sheets in each cocrystal. This direction includes several weak intermolecular interactions, such as C–H⋯Br, C–H⋯O, and C–H⋯π, interactions. Within (4,6-diBr res)·2(SB), the C–H⋯Br bond distances decrease upon cooling by ca. 0.037 Å (average), the C–H⋯O interactions shorten by 0.012 Å, and the C–H⋯π distances shorten by 0.047 Å over the range. The mixed cocrystal (4,6-diBr res)·(SB)·(PAB) behaves very similarly to the binary cocrystal with SB. The C–H⋯Br distances shorten by ca. 0.038 Å (average), the C–H⋯O interactions shorten by 0.025 Å, and the C–H⋯π distances shorten by 0.042 Å over the range. Overall, this results in very similar TE coefficients along X2.
Within (4,6-diBr res)·2(PAB), the interactions that give rise to the colossal expansion along X2 are C–H⋯Br, C–H⋯O, and long/weak dispersive C–H⋯π contacts. The C–H⋯Br distances shorten by ca. 0.039 Å (average), the C–H⋯O interactions shorten by 0.047 Å, and the C–H contacts shorten by ca. 0.047 Å over the range. In addition to larger changes in distances with temperature, the difference in extended packing of (4,6-diBr res)·2(PAB) affects X2. Between sheets, acceptor molecules are primarily stacked adjacent to other acceptors or π systems, but the arrangement is offset with minimal overlap in π systems. Ultimately, this results in relatively weak van der Waals interactions, which are more thermomechanically compliant, undergoing larger changes in response to temperature.18 In the binary cocrystal with SB and the mixed cocrystal, the acceptor molecules are stacked in an edge-to-face geometry with 4,6-diBr res molecules, providing more attractive/stabilizing interactions.
The X3 direction lies along the stacking direction within the sheet and is mainly comprised of π–π stacking, as well as the direction affected by dynamic molecular motion. In particular, the highest expansion within (4,6-diBr res)·2(SB) and (4,6-diBr res)·(SB)·(PAB) occur along the face-to-face π–π stacking direction (Fig. 5). In (4,6-diBr res)·2(SB), these separations decrease by ca. 0.45 Å for the stacked pair within the assembly and 0.034 Å between assemblies in the quadruple stack. In (4,6-diBr res)·(SB)·(PAB), the separation between the stacked pair in the assembly and between assemblies in the quadruple stack both decrease by ca. 0.40 Å. The stacking direction also includes interactions involving the 4,6-diBr res molecules, namely Br⋯π and C–H(pyr)⋯O. The Br⋯π contacts decrease by ca. 0.70 Å in both solids, and the C–H(pyr)⋯O separations decrease by 0.60 and 0.80 Å for the SB and mixed solids, respectively. The binary cocrystal, (4,6-diBr res)·2(SB), exhibits a small amount of dynamic motion over the temperature range, which can weaken the strength of the π-stacking interactions and contribute to increased TE along the X3 direction.
In the case of (4,6-diBr res)·2(PAB), the aromatic rings interact via edge-to-face π–π stacking interactions and res molecules engage in Br⋯Br and C–H(pyr)⋯O interactions, which all contribute to X3. Interestingly, within the quadruple stack, the distance between the centroids of PAB molecules within each assembly increases upon cooling and the distance between the assemblies stays constant. This is partially compensated for by a decrease in separation between PAB molecules at the edges of the quadruple stack and the rotated PAB ‘capping‘ molecules. The behavior of the binary PAB cocrystal contrasts the solid with SB and the mixed solid. In the latter cases, all the separations between acceptor molecules in the quadruple stack decrease upon cooling. Overall the lack of motion ability and difference in interaction behavior on cooling leads to less TE along X3 in the binary PAB cocrystal.
Our groups have previously described the first two examples of using mixed cocrystals as a novel strategy for tuning TE behavior in materials.19,20 The ability to control the composition of materials at the molecular level leads to incredible power in controlling properties. The mixed cocrystal strategy offers an additional handle for tuning structure as a way to fine-tune properties. In the examples discussed here, the TE coefficients for the mixed cocrystal lie between the binary systems along all three principal axes. Even though the mixed solid is isostructural with the binary SB solid, the thermomechanical response is clearly impacted by the inclusion of both SB and PAB at the molecular level. Moreover, the melting point of the mixed cocrystal also lies in between the two binary solids. We expect that the approach of using mixed cocrystals to influence properties is likely broader than thermomechanical behavior alone. By using differences at the molecular level, tuning material properties could be expanded to optical, electronic, and other solid-state behaviors.
Conclusions
In this contribution, we expanded the application of a mixed cocrystal approach to tune the thermal and thermomechanical properties within hydrogen-bonded solids. All three thermal expansion parameters for the mixed cocrystal lie between the two binary cocrystals. This work demonstrates that the thermomechanical properties of molecular solids can be altered even when their binary ‘parent’ cocrystals may not be isostructural, which opens the door for greater molecular diversity in controlling properties within organic solids.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† CCDC deposition numbers 2420438–2420455 contain the supplementary crystallographic data for this paper.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
K. M. H. gratefully acknowledges financial support from the National Science Foundation DMR-2411677. R. H. G. gratefully acknowledges financial support from Webster University in the form of various Faculty Research Grants. J. R. G. gratefully acknowledges financial support from the MizzouForward Undergraduate Research Training Grant. The authors acknowledge Prof. Eric Bosch and Dr. Jesus Daniel Loya for assistance with the calculations.Notes and references
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426 CrossRef CAS.
- R. Banerjee, R. Mondal, J. A. K. Howard and G. R. Desiraju, Cryst. Growth Des., 2006, 6, 999 CrossRef CAS.
- Y. Ducharme and J. D. Wuest, J. Org. Chem., 1988, 53, 5787 CrossRef CAS.
- A. C. Eaby, S. Darwish, S.-Q. Wang, A. A. Bezrukov, D. Sensharma, A. Shipman, C. J. Solanilla, B. Space, S. Mukherjee and M. J. Zaworotko, J. Am. Chem. Soc., 2025, 147, 1813 CrossRef CAS PubMed.
- D. Barman, M. Annadhasan, A. P. Bidkar, P. Rajamalli, D. Barman, S. S. Ghosh, R. Chandrasekar and P. Krishnan Iyer, Nat. Commun., 2023, 14, 6648 CrossRef CAS PubMed.
- N. G. Petrov, P. Chartier, T. Maris and J. D. Wuest, J. Am. Chem. Soc., 2022, 144, 556 CrossRef CAS PubMed.
- B. F. Abrahams, P. A. Jackson and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 2656 CrossRef CAS PubMed.
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047 RSC.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha and M. L. Cheney, et al. , Cryst. Growth Des., 2012, 12, 2147 CrossRef CAS.
- M. Lusi, CrystEngComm, 2018, 20, 7042 RSC.
- M. Lusi, Cryst. Growth Des., 2018, 18, 3704 CrossRef CAS.
- D.-K. Bučar, A. Sen, S. V. S. Mariappan and L. R. MacGillivray, Chem. Commun., 2012, 48, 1790 RSC.
- B. K. Saha and R. V. P. Veluthaparambath, Cryst. Growth Des., 2024, 24, 3467 CrossRef CAS.
- P. Harshaa and D. Das, Chem. Commun., 2024, 60, 14105 RSC.
- L. O. Alimi, P. Lama, V. J. Smith and L. J. Barbour, CrystEngComm, 2018, 20, 631 RSC.
- M. K. Panda, R. Centore, M. Causà, A. Tuzi, F. Borbone and P. Naumov, Sci. Rep., 2016, 6, 29610 CrossRef CAS PubMed.
- A. Khalil, D. P. Karothu and P. Naumov, J. Am. Chem. Soc., 2019, 141, 3371 CrossRef CAS PubMed.
- A. I. Kitaigorodsky, Molecular Crystals and Molecules; Physical Chemistry Series, Academic Press, New York, 1973 Search PubMed.
- X. Ding, D. K. Unruh, R. H. Groeneman and K. M. Hutchins, Chem. Sci., 2020, 11, 7701 RSC.
- N. Juneja, N. M. Shapiro, D. K. Unruh, E. Bosch, R. H. Groeneman and K. M. Hutchins, Angew. Chem., Int. Ed., 2022, 61, e202202708 CrossRef CAS PubMed.
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244 RSC.
- A. L. Grobelny, N. P. Rath and R. H. Groeneman, CrystEngComm, 2018, 20, 3951 RSC.
- K. Jin, B. Yue, L. Yan, R. Qiao, H. Zhao and J. Zhang, Chem. – Asian J., 2022, 17, e202200109 CrossRef CAS PubMed.
- G. C. George III, D. K. Unruh, R. H. Groeneman and K. M. Hutchins, Cryst. Growth Des., 2022, 22, 4538 CrossRef CAS.
- K. M. Hutchins, T. P. Rupasinghe, L. R. Ditzler, D. C. Swenson, J. R. G. Sander, J. Baltrusaitis, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2014, 136, 6778 CrossRef CAS PubMed.
- T. P. Rupasinghe, K. M. Hutchins, B. S. Bandaranayake, S. Ghorai, C. Karunatilake, D.-K. Bučar, D. C. Swenson, M. A. Arnold, L. R. MacGillivray and A. V. Tivanski, J. Am. Chem. Soc., 2015, 137, 12768 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrichspartn, Angew. Chem., Int. Ed., 2001, 40, 3240 CrossRef.
- Spartan 20, Version 1.0.1; Wavefunction, Inc.: Irvine, CA, USA, 2020.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321 CrossRef CAS.
- M. Lertkiattrakul, M. L. Evans and M. J. Cliff, J. Open Source Softw., 2023, 8, 5556 CrossRef.
- J. P. Attfield, Front. Chem., 2018, 6, 371 Search PubMed.
- A. D. Bond, Acta Cryst., 2021, B77, 357 Search PubMed.
- A. van der Lee and D. G. Dumitrescu, Chem. Sci., 2021, 12, 8537 CAS.
- S. Bhattacharya, V. G. Saraswatula and B. K. Saha, Cryst. Growth Des., 2013, 13, 3651 CrossRef CAS.
- I. de Pedro, A. García-Saiz, J. Dupont, P. Migowski, O. Vallcorba, J. Junquera, J. Rius and J. Rodríguez Fernández, Cryst. Growth Des., 2015, 15, 5207 CrossRef CAS.
- L. Negi, S. Kumar, B. Dwivedi and D. Das, Cryst. Growth Des., 2021, 21, 1428 CrossRef CAS.
- B. Dwivedi, A. Shrivastava, L. Negi and D. Das, Cryst. Growth Des., 2019, 19, 2519 CrossRef CAS.
- A. L. Goodwin, M. Calleja, M. J. Conterio, M. T. Dove, J. S. O. Evans, D. A. Keen, L. Peters and M. G. Tucker, Science, 2008, 319, 794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray data, 1H NMR spectra, thermal expansion analysis. CCDC 2420438–2420455. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00576k |
| This journal is © The Royal Society of Chemistry 2025 |