
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cold atmospheric plasma-assisted deposition of ammonium functionalized glass coatings for enhanced antibacterial properties†
Venkat
Kasi‡
ab,
Md
Mahabubur Rahman‡
bc,
Akshay
Krishnakumar‡
 bc,
Ulisses Heredia
Rivera
ab,
Devendra
Sarnaik
ab,
Mithun
Sinha
d and
Rahim
Rahimi
*ab
bc,
Ulisses Heredia
Rivera
ab,
Devendra
Sarnaik
ab,
Mithun
Sinha
d and
Rahim
Rahimi
*ab
aSchool of Materials Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: rrahimi@purdue.edu
bBirck Nanotechnology Center, Purdue University, West Lafayette, IN 47907, USA
cSchool of Electrical and Computer Engineering, Purdue University, West Lafayette, IN 47907, USA
dDivision of Plastic Surgery, Department of Surgery, Indiana University School of Medicine, Indianapolis, IN 46202, USA
First published on 21st March 2025
Abstract
Implant-associated bacterial infections are a major healthcare concern, and significantly impact patient health, leading to increased morbidity, extended hospital stays, and substantial economic burden. The rise of antibiotic-resistant bacterial strains further complicates treatment protocols, as these resistance mechanisms often exhibit enhanced bacterial pathogenicity and virulence. A key strategy to address this challenge involves the development of antibacterial surface modifications for implantable devices that provide broad-spectrum efficacy against various opportunistic bacterial pathogens while preserving biocompatibility. In this context, the present work focuses on the conformal deposition of ammonium compound-infused glass ceramic coatings using cold atmospheric plasma (CAP) technology. The air plasma-based process facilitates the in situ polymerization and direct deposition of glass composite coatings on a wide range of polymeric and metallic surfaces under atmospheric conditions. To identify the optimal composition of ammonium compounds in the SiOx (AF-SiOx) matrix for effective chemical bonding while simultaneously providing antibacterial properties, a systematic study of various processing precursor compositions was conducted. A series of material characterization and chemical surface composition studies, including SEM, EDX, and XPS, were employed to determine the effective plasma process parameters, final composition, and chemical bonding characteristics of the AF-SiOx matrix. Results show that increased infusion of APS leads to enhanced covalent bonding within the matrix, resulting in stronger SiOx crosslinking in the functional coating. Additionally, among the various precursor compositions, at least 0.6 wt% APS is necessary within the SiOx matrix to provide broad-spectrum antibacterial efficacy against two common implant-associated bacterial pathogens, Escherichia coli and Staphylococcus aureus, while also ensuring biocompatibility. It is envisioned that the developed CAP technology will enable a scalable, rapid, and low-temperature deposition of antibacterial coatings for both temporary and permanent implants, aimed at reducing current complications associated with such infections.
1. Introduction
Rapidly rising population and advancements in medical technology have led to a dramatic increase in the number of surgical implant procedures, in the past few decades. With one in six people projected to be 60 years or older by 2030, according to WHO forecasts, and the rising incidence of bone fractures, the orthopedic implant market which primarily utilizes metallic surfaces is expected to reach $32.5 billion, growing at a 3.7% CAGR.1 Similarly, the cardiovascular, dental, and breast implant markets, which also rely on a combination of metallic and polymeric materials, are experiencing significant growth, driven by the rising demand for minimally invasive procedures and advancements in implant technology.2 Despite such growing demand, implant-associated infections (IAIs) remain a significant concern, affecting 33% of patients, with 50–70% of these infections being bacterially related.3 In these IAIs, opportunistic bacterial pathogens adhere to the implant surface, start proliferating, and produce an extracellular biofilm matrix that develops immune evasion and antibiotic tolerance properties. With host eucaryotic cells and bacterial pathogens racing to attach and proliferate on the implanted surface, bacteria multiply rapidly and form such complex biofilm structures that inherently disrupt the normal healing process, impeding integration and function of the implant.4 Failure to prevent such debilitating bacterial infections can lead to severe consequences, including systemic infections, prolonged hospital stays, and an increased risk of amputation. Consequently, the first line of treatment for such infections is the intravenous administration of antibiotics to target to clear the bacterial pathogens from the infected area.5 However, with the increase of antibiotic-resistant bacterial strains and limited antibiotic availability, there is a need to develop effective antibiotic alternatives to deal with such infections.6 In this regard, prevention of infection progression can be achieved through surface modification of implants with antibiotic alternatives, without compromising their functionality and biocompatibility.7Previous research groups have investigated the applicability of various organic and inorganic functional coatings to improve the antibacterial properties on implant surfaces. In particular, organic antibacterial surfaces can be broadly categorized into polymers with inherent antibacterial properties and inert polymers that are infused with antibacterial agents, such as metallic nanoparticles or antimicrobial peptides, to enhance bacterial adhesion resistance and contact-based bactericidal properties.8 Although these functional coatings demonstrate broad-spectrum antibacterial efficacy against various bacterial pathogens, most reported methods rely on wet chemical synthesis, where a polymer is dissolved in solvents to form slurries for casting or spray coating.9,10 Despite the solvent being removed during the drying process, the residual solvent can become trapped within the polymer matrix, posing a risk of long-term leaching and associated toxicity.11,12 Conversely, while UV-based polymerization approaches eliminate the use of solvents, complications from unreacted monomer residues within the structure remain, potentially leading to systemic toxicity and carcinogenicity. On the other hand, while inorganic functional coatings based on antibacterial metallic nanoparticles such as silver, copper, magnesium, and zinc have demonstrated pronounced antibacterial efficacy, long-term application of such coatings on implants can lead to gradual leaching of metallic ions, which may cause severe inflammation and progressively result in implant rejection.13
In this context, inorganic ammonium persulfate (APS) has been observed as a highly effective antimicrobial alternative for implants, due to its broad-spectrum antibacterial activity and favorable low-toxicity profile.8 APS has been previously reported to have effective antibacterial properties against Staphylococcus aureus (S. aureus) and Escherichia coli (E. coli) bacterial populations at a relatively low concentration of 150 mM.14,15 The mechanism is assumed to be based on the charge attraction between the positively charged ammonium compounds and the negatively charged phospholipid bilayer of the bacterial cell membrane triggering autolysis and resulting in cell death.16 Thus, incorporation of APS could be an effective way to improve the antibacterial properties while retaining the biocompatibility characteristics of an implant surface. However, APS is water-soluble and therefore requires encapsulation within a protective matrix to promote long-term antibacterial stability on surfaces. Hence, various efforts have been reported to incorporate antibacterial compounds into coating matrices, using polymers, ceramics, and hybrid systems to develop effective and durable antibacterial coatings. For example, in terms of polymeric structures, Gharibi et al.17 reported a solgel synthesis of ammonium compounds as antibacterial agents with silane and poly(vinyl alcohol), which exhibited promising antibacterial activity against E. coli and S. aureus bacterial populations. Despite several advantages of polymer-based matrices including flexibility and versatility, these techniques still suffer from the aforementioned limitations of solvent entrapment. On the other hand, incorporating antibacterial compounds into ceramic matrices offers numerous advantages over polymeric surfaces, including enhanced hardness, abrasion resistance, chemical resistance, and biocompatibility, while also improving antibacterial efficacy. For instance, a recent study reported by Saif et al. demonstrated the sol–gel assisted synthesis of an antibacterial compound-infused glass matrix with an enhanced antibacterial efficacy against two commonly implant-associated bacterial pathogens, E. coli and S. aureus, within 72 h of exposure.18 However, despite the unique antibacterial properties of many previously reported ceramic structures, they often utilize sol–gel technology, which is limited by its compatibility with certain metallic substrates due to the high-temperature requirements for the sol–gel calcination process. Additionally, the strong thermal mismatch between ceramic coatings and the underlying metallic substrate, combined with high-temperature treatments, can often lead to complications such as crack formation and poor adhesion of the deposited layer.
To address this need, the present work focuses on the conformal deposition of ammonium functionalized glass (AF-SiOx) ceramic coatings using cold atmospheric plasma (CAP) technology.19 This novel approach facilitates in situ polymerization and direct deposition of glass composite coatings with ammonium functionality on a variety of polymeric and metallic surfaces under atmospheric conditions. Ammonium persulfate (APS) is incorporated into the SiOx ceramic matrix to develop an effective antibacterial coating for implant surfaces that ensure long-term efficacy while maintaining biocompatibility. As shown in Fig. 1a, the silica precursor (hexamethyldisilazane (HMDSN)) and APS solutions are injected through two separate channels with adjustable flow rates. The silica precursor, carried by the carrier gas through the main channel, is vaporized and delivered in the form of atomized precursors into the upper region of the plasma glow, while the APS solution is introduced through a customized secondary channel into the lower plasma region in a controlled manner. Upon exposure to plasma, the precursors undergo a series of fragmentation and radical generation events that initiate a chain reaction among the activated species, leading to the polymerization of silica. During the polymerization process, fragmentation specifically occurs in the –CH3 groups of the monomer, resulting in the release of atmospheric H2 and CO2 that escape from the surface, while various elements from APS are incorporated into the silica network. Additionally, during plasma exposure, sulphate ion radicals generated from APS, which are strong oxidizing agents with high oxidation potential, facilitate a higher degree of polymerization, resulting in a more crosslinked silica network.20 As a result of the fragmentation and recombination processes in CAP deposition, the silica coating is effectively functionalized with ammonium and sulphate ions that are tightly integrated into the structural network. To determine the optimal ratio of APS within the SiOx structure for long-term antibacterial efficacy and biocompatibility, a systematic study was conducted by varying the APS composition (Fig. 1b). The morphological, functional, and elemental characteristics of the deposited AF-SiOx were analyzed using SEM, FTIR, and XPS analysis. The antibacterial efficacy of the developed AF-SiOx was tested against two common implant-associated bacterial pathogens, E. coli and S. aureus, along with its biocompatibility with human epithelial cells (HCT-8).
 | ||
| Fig. 1 Cold atmospheric plasma assisted deposition of ammonium compound infused glass substrate (AF-SiOx). (a) Schematic illustration of the silica deposition process enriched with ammonium compound functionality, (b) illustration of the antibacterial properties of the APS infused glass coating caused by disruption in the bacterial cell membrane. | ||
2. Materials and methods
2.1. CAP assisted deposition of AF-SiOx coatings
AF-SiOx coatings were deposited onto both metallic and polymeric surfaces using an open-air atmospheric pressure plasma jet system (Plasmatreat PE39) that consists of a plasma generator and a precursor delivery setup with adjustable flow rates. High-purity nitrogen (99.99%) was utilized as both plasma gas and carrier gas for precursor delivery. The plasma gas, connected directly to the plasma head, was maintained at a flow rate of 35 L min−1, with an operating frequency of 23 kHz and an applied voltage of 260 V, generating plasma at a power of 560 W. Meanwhile, the carrier gas, responsible for delivering the vaporized precursors into the plasma glow, was set at a flow rate of 10 L min−1. Additionally, the plasma head was securely mounted onto a robotic system equipped with precise controls for 3-dimensional movement, scan speed, and source-to-sample distance, ensuring consistent coating deposition.In this study, the deposition of AF-SiOx coatings was achieved by injecting precursors through two separate channels, allowing the reactions to occur within the plasma glow. The silica SiOx precursor, HMDSN (Sigma-Aldrich, St. Louis, USA), was delivered through the main channel at a flow rate of 5 g h−1, while an APS solution (Sigma-Aldrich, St. Louis, USA) was supplied through a secondary channel at a flow rate of 0.4 mL min−1. The APS concentration, delivered to the plasma glow, varied between 0.1 wt% and 1.0 wt% in the precursor mixture to systematically characterize the deposited AF-SiOx coatings. As a proof-of-concept, the AF-SiOx deposition was demonstrated on two different substrate types: (1) a polymeric substrate—commercially available polyimide (PI) sheets provided by DuPont (thickness ∼60 μm, DE, USA) and (2) a metal substrate—304 stainless steel (SS) coupons (Eoiips, 1′′ diameter discs, 19-gauge thickness). The polymer samples were prepared by cutting the PI sheets into circular pieces with a diameter of 25 mm. Prior to coating deposition, all the samples were sonicated in isopropanol and deionized water for 5 min each to remove surface contamination, followed by drying with a pure nitrogen gun. To achieve complete coverage of the substrate surface, a three-dimensional robotic system, capable of movement in all directions, was programmed to perform a single scan across the substrate at a scanning speed of 200 mm s−1, maintaining a consistent distance of 20 mm between the nozzle and the substrate surface. Given that the diameter of the plasma stream is approximately 4–5 mm, the robot was programmed to maintain a track width of 2 mm, as illustrated in Fig. S1, ESI,† which allows each successive scan to overlap with the previous one, thereby ensuring full coverage of the surface area.
2.2. Surface and chemical characterization
The surface morphology and the composition distribution of the AF-SiOx coated SS and PI surfaces with varying precursor ratios were analyzed by performing scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX), respectively, using FE-SEM (Hitachi-S 4800, Tokyo, Japan). High-resolution surface images of the AF-SiOx coated SS and PI surfaces were obtained using field emission scanning electron microscopy (FE-SEM, Hitachi-S 4800, Tokyo, Japan) at an accelerating voltage of 5 kV, a current of 15 μA, and a working distance of 15 mm. Prior to analysis, the samples were coated with Au/Pd using an SPI sputter coater (SPI Supplies, West Chester, Pennsylvania, USA) (argon sputtering at a current of 18–20 μA for 20 seconds) to minimize charge buildup during imaging. Chemical structures of the AF-SiOx coated on SS and PI surfaces with varying precursor ratios were analyzed using a high-resolution attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectrometer (Nicolet iS5 series, Thermo Scientific, Madison, Wisconsin, USA). Absorption spectra were collected over a wavenumber range of 500 to 4000 cm−1, averaging 30 scans after background noise removal. Overlapping peaks were deconvoluted using Origin Pro 2023 software to obtain individual peak intensities. The deconvolution process involved baseline correction using the second derivative points and normalization for consistency, followed by Gaussian peak fitting to resolve the overlapping bands. During deconvolution, the full width at half maximum (FWHM) was maintained consistent across all the peaks to ensure uniform comparison. Additionally, the sum of all deconvoluted peak intensities was maintained to match the original spectrum, ensuring both accuracy and reliability in spectral analysis.21–23The composition and oxidation states of the elements in the AF-SiOx coated SS and PI surfaces with different precursor ratios were determined using X-ray photoelectron spectroscopy (XPS) analysis (Kratos Axis Ultra DLD spectrometer with monochromatic Al Kα radiation, 1486.6 eV). A commercial Kratos charge neutralizer was employed to prevent non-homogeneous charging of the non-conductive material and to enhance resolution. XPS spectra were analyzed using CasaXPS software, with curve fitting performed using a Gaussian/Lorentzian shape following Shirley background subtraction to deconvolute the high-resolution narrow scan spectra. Similar to the FTIR deconvolution, the FWHM was kept consistent across peaks of the same element, and the sum of all deconvoluted peak intensities was maintained to match the original spectrum.
The thickness of the AF-SiOx coatings deposited with varying concentrations of APS (0 wt%–1.0 wt%) was measured using an Alpha-Step D-600 stylus profilometer manufactured by KLA Corporation (CA, USA) at a scanning speed of 20 μm s−1. A silicon wafer was used as the substrate for these measurements to avoid the effect of substrate surface roughness. During the deposition process, a section of the sample was masked with Kapton tape, which was removed afterward to create a step profile between the coated and uncoated areas. The stylus of the profilometer was then moved across the surface to measure the height difference between the coated and uncoated regions of the samples. All measurements were repeated three times to calculate the average and standard deviation of the film thickness.
The surface wettability of the AF-SiOx coated SS and PI surfaces with different precursor ratios was analyzed by measuring the water contact angle (WCA) using a sessile drop method with the help of an optical contact angle measurement instrument (Kruss Goniometer DSA100, KRUSS, Hamburg, Germany). A 2 μL droplet of deionized water was placed on the coated surface, and the contact angles (CAs) were analyzed using the Young–Laplace contour fitting method. All contact angle measurements were performed in triplicate per condition to deduce the mean and standard deviation values.
2.3. Antibacterial efficacy
Bactericidal efficacy of the AF-SiOx coated SS and PI surfaces with varying precursor ratios was tested against a model Gram-positive Staphylococcus aureus (S. aureus, ATCC 25923) and a Gram-negative Escherichia coli (E. coli, ATCC 25922). In order to evaluate the efficacy of the AF-SiOx coatings against bacterial attachment, a surface dependent contact killing based test was performed. All bacterial populations revived from frozen stocks were initially cultured overnight in Tryptic Soy Broth (TSB, 30 g L−1) at 37 °C for 18 h. After washing the cultures three times in PBS buffer, the bacterial suspensions were adjusted to approximately 105 CFU per mL using optical density measurements (OD600nm ∼ 0.2). Next, 1 cm2 samples of the AF-SiOx coated surfaces were placed into a 12-well plate, followed by the addition of 100 μL of the diluted bacterial suspensions directly on the substrate. After 6 h of incubation at 37 °C, the adherent bacteria were detached from the sample surfaces by sonicating and vortexing in PBS buffer. The resulting solution was serially diluted in PBS buffer, plated on the TSB agar plate, and incubated for 18 h to determine the reduction in the viable bacteria upon contact with the SiOx deposited surfaces. Next, inhibition zone analysis of the SiOx deposited SS surfaces with different APS concentrations was tested to elucidate the leaching characteristics of the APS antibacterial coatings. For this test, 105 CFU per mL of both the bacterial species was uniformly spread over a standard TSB plate, followed by the addition of the SiOx-coated SS substrates with varying APS concentrations. Progressively, the plates were then incubated for 24 h at 37 °C to quantify the level of inhibition around the deposited surface.Next, to assess the stability of the CAP assisted SiOx-coating, the as prepared samples were divided into two groups: one group was incubated in simulated body fluid (SBF) for 2 h, while the other was used as prepared, without any further modification. To assess the performance of the CAP-assisted coating, the same test was performed on the SS substrate by drop-casting the SiOx precursor with increasing concentrations of APS and drying it under ambient conditions for 3 h. All contact killing analyses were conducted following the procedure described previously.
2.4. Biocompatibility assessment
In vitro safety assessment of the AF-SiOx coating deposited on stainless steel (SS), a widely used metal for biomedical implants, with varying APS concentrations, was evaluated through a biocompatibility assessment using HCT-8 epithelial cell lines obtained from ATCC CCL (VA, USA). For this test, the epithelial cells (passage #12-14) were cultured in Dulbecco modified Eagle medium (DMEM) supplied with 10% fetal bovine serum (D-10F) and 1% of penicillin–streptomycin antibiotics (all procured from Thermo Fisher Scientific, MA, USA). Initially, frozen stocks were cultured in a T-75 flask at 37 °C in a humidified atmosphere (95% humidity) with 5% CO2 for three days until 80% confluency. Next, the adherent cells were trypsinized, washed, resuspended, and diluted to a cell concentration of 5000 cells per cm2 in the culture media before performing the biocompatibility analysis. First, the change in the metabolic activity of the cells upon exposure to the AF-SiOx coated SS and PI surfaces was analyzed by performing the standard MTT cell proliferation assay. For this test, 100 μL of the diluted cell suspensions were added into a 12-well plate for 24 h to facilitate the attachment of the cells to the well plates, followed by the addition of the coated SS and PI surfaces (area = 1 mm2). After 24 h of exposure, the coated surfaces were withdrawn from the well, and an MTT cell proliferation assay was performed according to the manufacturer's instructions. Progressively, the cells were exposed to 10 μL of MTT tetrazolium salt for 3 h, then treated with 200 μL of detergent solution and incubated for an additional 1 h. Finally, the absorbance of the resulting solution was analyzed using a BMG Labtech clariostar (NC, USA) at a wavelength of 550 nm. Additionally, trypan blue staining was performed to visualize cell morphology and membrane disruption after exposure to AF-SiOx-coated SS and PI surfaces. After 24 h of cell incubation with the coated samples, the samples were withdrawn, and 10 μL of trypan blue stain was added to each well. The plates were then incubated for 5 min, imaged using a Nikon Ti2 Eclipse microscope (NY, USA) with appropriate filters under a 10× optical lens, and analyzed using NIS-Elements D software.3. Results and discussion
3.1. Morphological and chemical analysis
Initially, the optical microscopy images of AF-SiOx-coated SS and PI surfaces with varying precursor ratios were analyzed to observe changes in the visual appearance of the surfaces. The overall plasma assisted deposition of the AF-SiOx coating onto various SS and PI surfaces is shown in Fig. 2a. As observed in Fig. 2b and c, no significant changes in the colour differences were observed on the SS and PI surfaces with the SiOx coating and AF-SiOx coating with low concentrations of APS (<0.6 wt%). This can be attributed to the thin and the intrinsic transparent nature of the CAP assisted AF-SiOx coating. However, at higher concentration of APS, above 0.6 wt% in the precursor mixture, there is a slight increase in opaqueness due to the incorporation of ammonium and sulphate ions into the silica network. As such, high magnification SEM analysis was performed to analyze the change in the surface morphological characteristics of the AF-SiOx coated SS with varying precursor ratios. As shown in Fig. 3a and b, at lower APS concentrations, the AF-SiOx deposition resulted in a clear, smooth coating on the SS surface. This smooth uniform coating can be attributed to the precursor being activated in the plasma glow and then deposited onto the surface, where it subsequently polymerizes to form an APS infused SiOx matrix. Conversely, at higher APS concentrations (>0.6 wt%), APS itself acts as a strong photo initiator, leading to an increase in the radical density within the reaction zone. This results in intense generation of reactive oxygen species (ROS) such as O−, O2−, and nitrogen radicals contribute to the fragmentation of the HMDSN monomers. Meanwhile, APS absorbs plasma energy, further facilitating the generation of additional reactive species through photoexcitation, which accelerates the polymerization process. This increased radical density in the plasma glow promotes the pre-polymerization of fragmented monomers, leading to their early incorporation into the growing polymer matrix.24 These pre-polymerized monomers become embedded in the silica matrix, contributing to an increased cross-linking density, while also inducing slight localized roughness due to distribution of the polymerized species across the coating surface. | ||
| Fig. 2 Plasma assisted deposition of the SiOx coating onto PI and SS surfaces with varying APS concentration. (a) Image of the application of the CAP system with two inlets for the silica precursor and APS delivery into the plasma jet. Photographs of (b) PI and (c) 304 SS, with zoomed areas of various samples. Images show the surfaces of the pristine substrate, and the surfaces coated with silica coatings from the precursor containing 0 to 1.0 wt% APS. | ||
 | ||
| Fig. 3 Morphological and elemental distribution analysis of glass coatings prepared with various concentrations (0, 0.2, 0.4, 0.6, and 1 wt%) of APS in the reaction mixture. Surface SEM images at (a) low (5k×) and (b) high (25k×) magnification, EDX elemental mapping of (c) silicon and (d) nitrogen on the surface of the glass coatings. | ||
Furthermore, to quantify the chemical composition of elements across the AF-SiOx-coated SS surface and observe the effect of varying APS concentrations, EDX mapping analysis was performed. As observed in Fig. 3c and d, the SiOx coating without APS infusion exhibited the presence of Si with trace levels of the N content. However, with an increase in the APS concentration up to 0.4 wt%, a gradual increase in the N content was observed without significant changes in Si levels. Upon further increasing APS concentration above 0.6 wt%, a significant increase in the N content accompanied by a gradual reduction in Si was noted. Although at 1.0 wt% APS concentration, some nonuniformity was detected on the surface, EDX mapping of high-magnification SEM images (Fig. S2, ESI†) confirmed a uniform spatial distribution of oxygen, silicon, carbon, and nitrogen, indicating that the surface shares the same compositional characteristics as the surrounding coating, identifying them as same ammonium-functionalized silica. Moreover, regardless of processing conditions, all CAP-assisted AF-SiOx coatings observed a highly uniform and homogeneous spatial distribution of the surface elements throughout the coating area.
Next, in order to evaluate the chemical structure and the level of crosslinking of the AF-SiOx coating, FTIR functional group analysis was performed. Fig. 4a shows the FTIR curves of the AF-SiOx coating for different concentrations of APS ranging from 0 wt% to 1.0 wt%. As observed in this figure, for the coated samples with varying precursor ratios, characteristic vibrational absorption bands of Si–O–Si and –OH groups within the range of 950 to 1250 cm−1 and 3200 to 3400 cm−1, respectively, were observed.25–27 In addition, for the AF-SiOx coating with APS concentration below 0.4 wt% an absorption band of a significant Si–CH3 group arising from the precursor can be identified at 1280 cm−1. However, upon an increase in the APS concentration above 0.4 wt%, a significant and noticeable reduction in the Si–CH3 group was observed due to increased monomer fragmentation.
 | ||
| Fig. 4 Functional group analysis: (a) FTIR spectra of the glass coating prepared with varying concentrations of APS in the precursor mixture from 0 to 1 wt%. (b)–(g) Deconvoluted FTIR spectra over the range of 970–1230 cm−1 and the trends of the peak ratios for (h) NH4+/Si–O–Si, (i) SO42−/Si–O–Si, and (j) Si–CH3/Si–O–Si. | ||
Interestingly, a trace amount of Si–CH3 was still observed at 1.0 wt% of APS concentration, which could be attributed to the remnant monomer and dangling bonds present in the final AF-SiOx coating. Concurrently, as the APS concentration in the reaction mixture increases, characteristic peaks corresponding to groups such as NH, NH4+, and SO42− gradually emerge in the spectra. Here, the broad peak within the range of 2900 to 3300 cm−1 corresponds to various modes of neutral NH, while the sharp peak at 1422 cm−1 is attributed to the bending vibration of ammonium (NH4+).28,29 These results clearly validate that increased APS concentration in the precursor mixture leads to greater incorporation of NH4+ and enhanced cross-linking within the silica matrix coating.
Furthermore, the degree of cross-linking, as well as the trends in peak intensities and shifts of these groups relative to the characteristic Si–O–Si peak, was analyzed to elucidate the effect of APS concentration on the incorporation of ammonium and sulfate functionalities within the AF-SiOx coatings. Since both the asymmetric Si–O–Si peak and the sulfate ion peak overlapped in the same region between 970 and 1200 cm−1, the broad peak resulting from their overlap in this region was deconvoluted to obtain the individual peak intensities (Fig. 4b–g).30
In general, SiOx coatings show three distinct asymmetric stretches of Si–O–Si bonds in the specified region, as illustrated in the deconvoluted curves in Fig. 4b. The first deconvoluted peak, observed around 1023–1035 cm−1, is attributed to silicon sub-oxidized groups (either O–Si–C or O–Si–Si). The second peak, found in the range of 1065–1078 cm−1, corresponds to a siloxane functional group network resembling SiO2. Finally, the third component, occurring between 1135–1150 cm−1, arises from cage-like entities (O–Si–H).31–37 When APS is introduced into the SiOx matrix, the peaks associated with the suboxide and network structures shift slightly to the right and left, respectively. Additionally, a new peak appears at 1044–1047 cm−1, which is attributed to the S–O bond in the SO42− group.38,39 As shown in the deconvoluted curves (Fig. 4b–g), the relative intensity of the S–O peaks increases with higher APS concentrations, while the intensity of the suboxide groups exhibits a decreasing trend. The relatively higher peak intensity of the network structures, compared to the networked suboxide groups and cage structures, indicates a greater degree of monomer fragmentation, leading to stronger crosslinking and enhanced network structures in the AF-SiOx coating. The peak intensity ratios of SO42−/Si–O–Si (network), NH4+/Si–O–Si (network), and Si–CH3/Si–O–Si (network), as shown in Fig. 4h–j, respectively, reveal a gradual increase in of SO42−/Si–O–Si (network), NH4+/Si–O–Si (network) with the increase in the APS concentration, while the Si–CH3/Si–O–Si ratio decreases. These results confirm that higher APS concentration leads to increased incorporation of ammonium and sulfate within the silica matrix coating, while also enhancing cross-linking.
Furthermore, to determine the elemental composition and oxidation states of materials, XPS analysis of the SiOx coatings with varying APS concentrations (0 wt%, 0.4 wt%, 0.8 wt%) was performed. Fig. 5a–c shows the XPS survey scan spectra for the SiOx coatings prepared with various APS concentrations. In the survey scan spectrum of the CAP assisted SiOx coating without APS, the elements Si 2p, Si 2s, C 1s, and O 1s were identified at binding energies of 101.6 eV, 151.2 eV, 283.4 eV, and 532.6 eV, respectively,40,41 with no significant presence of nitrogen (Fig. 5a). However, upon APS introduction in the precursor mixture, a clear emergence of the N 1s and S 2p peaks was observed at 399 eV and 166 eV, respectively (Fig. 5b and c). Additionally, the intensities of these peaks (N 1s and S 2p) tend to increase with increase in APS concentration in the precursor mixture from 0.4 wt% to 0.8 wt%, indicating a higher incorporation of nitrogen and sulfur within the silica matrix. The atomic percentages of the surface elements (C, O, N, Si, and S) calculated though the XPS spectra are summarized in Table S1, ESI.† These results indicate a clear increase in the nitrogen content from 0.4 at% to 4.66 at% and the sulfur content from 0 at% to 5.1 at% upon increase in the APS concentration to 0.8 wt% in the precursor mixture.
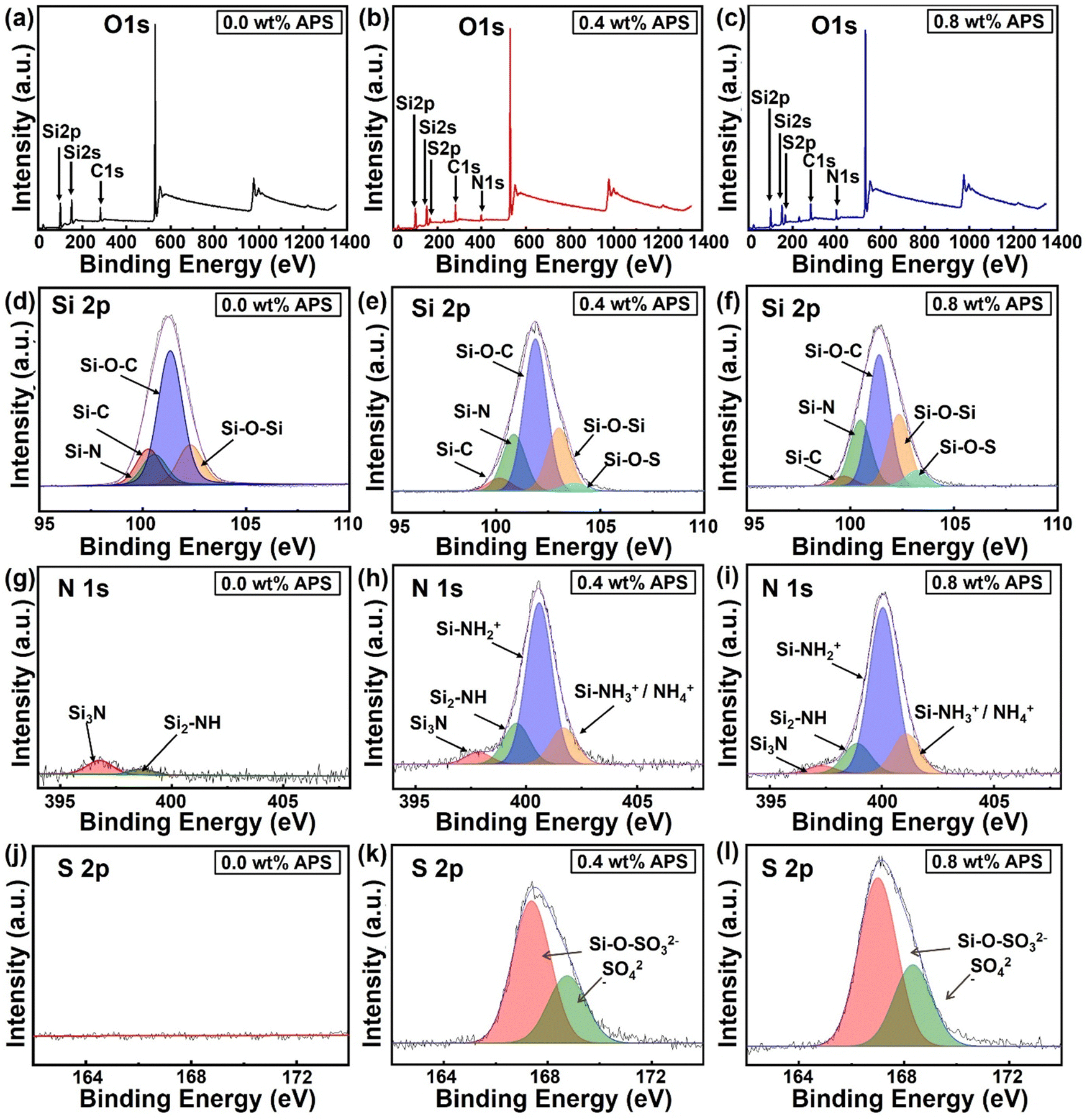 | ||
| Fig. 5 XPS analysis of coatings prepared with varying APS concentrations in the precursor mixture. (a)–(c) Survey scan spectra identifying characteristic peaks for C 1s, O 1s, N 1s, Si 2s, Si 2p, and S 2p for the coatings prepared with 0 wt%, 0.4 wt%, and 0.8 wt% APS concentration in the precursor mixture. High-resolution narrow scan spectra and their deconvoluted species of (d)–(f) Si 2p, (g)–(i) N 1s, and (j)–(l)) S 2p at the same APS concentrations, analyzing the chemical states and bonding configurations with increasing APS content. | ||
To better understand the changes in the oxidation states of the coating surface elements with varying APS concentrations, high-resolution narrow-scan spectra for Si 2p, N 1s, and S 2p were obtained and deconvoluted (Fig. 5d–l). As evidenced by the Si 2p deconvoluted spectrum (Fig. 5d–f), the coating without APS exhibits distinct peaks corresponding to Si–C, Si–N, Si–O–C, and Si–O–Si at binding energies of 99.56 eV, 100.35 eV, 101.23 eV, and 102.36 eV, respectively. The presence of these bonds in plasma-polymerized coatings derived from HMDSN can be attributed to the inherent organosilicon nature of the monomer, which contains a silazane (–Si–N–Si–) backbone and trimethylsilyl (–Si(CH3)3) groups. During the plasma-induced polymerization process, incomplete oxidation and recombination of the monomer promote the retention of Si–C, Si–O–C, and Si–N bonds within the polymerized matrix, thereby impeding the formation of a pure silica phase. Conversely, the development of a silica-like matrix is driven by partial oxidation and the crosslinking of silicon-containing species, facilitating the formation of Si–O–Si bonds. As a result, such incomplete removal of carbon and nitrogen prevents the formation of a stoichiometric silica structure (SiO2), yielding an organosilicon structure of the deposited SiOx coatings. On the other hand, introduction of APS into the precursor mixture leads to the emergence of an additional peak at 103.5 eV, corresponding to Si–O–S bonds with a significant increase in the Si–N peak (Fig. 5e and f).42,43
To further analyze how the APS concentration influences the binding states of elements during deposition, the area under the deconvoluted curves was quantified, as illustrated in Fig. S3a, ESI.† In the SiOx coating without APS, the representative area percentages for Si–C, Si–O–C, and Si–O–Si are 10.43%, 65.56%, and 15.48%, respectively. However, with the introduction of 0.4 wt% APS in the precursor mixture, a noticeable decrease in the Si–C and Si–O–C fractions to 4.41% and 51.95%, respectively, was observed, accompanied by an increase in the Si–O–Si content to 21.57%. With a further increase in the APS concentration to 0.8 wt%, Si–C and Si–O–C continued to decrease to 3.30% and 44.71%, respectively, while the Si–O–Si content further increased to 24.36%. Concurrently, the Si–N peak exhibited an upward trend, rising from 9.87% at 0 wt% APS to 19.38% at 0.4 wt% and reaching 22.41% at 0.8 wt%, while the Si–O–S peak, emerging with APS incorporation, increased from 2.69% at 0.4 wt% to 5.21% at 0.8 wt% in the silica matrix.
Examining the deconvoluted N 1s spectrum, as shown in Fig. 5g–i, the coatings deposited without APS exhibit two peaks at 396.82 eV and 398.74 eV, corresponding to Si3–N and Si2–NH, respectively. These peaks indicate nitrogen bonded to silicon groups, suggesting the presence of residual nitrogen on the coating surface due to the unfragmented monomer. With the introduction of APS into the precursor mixture, two additional peaks appear at 400.5 eV (Si–NH2+) and 401.7 eV (Si–NH3+/NH4+), which are attributed to APS-derived species, indicating increased nitrogen incorporation into the silica network. The representative percentages of these deconvoluted peaks, as compared in Fig. S3b, ESI,† reflect the changes in nitrogen species within the coating structure. The data show that Si–NH3+/NH4+ and Si–NH2+ increase from 14.46% and 64.51% at 0.4 wt% APS to 16.31% and 68.11% at 0.8 wt%, respectively. Conversely, Si3–N and Si2–NH exhibit a slight decrease, with their area percentages reducing from 4.72% and 16.30% at 0.4 wt% APS to 3.23% and 12.35% at 0.8 wt%. These results clearly indicate the increasing level of nitrogen/ammonium ion incorporation within the silica matrix with an increase in the APS concentration in the precursor mixture.
Finally, the high-resolution S 2p spectra, deconvoluted in Fig. 5j–l, reveal the sulfur incorporation trends within the coatings. As expected, the SiOx coating prepared without APS exhibits no detectable S 2p peak, confirming the absence of sulfur-containing compounds in the reaction mixture. With the introduction of APS, two characteristic sulfur oxidation states emerge, as shown in Fig. 5k and l. A smaller peak at a higher binding energy of 168.4 eV is attributed to sulfate groups that are ionically bonded, whereas a larger peak at 167.2 eV is most likely due to sulfate ions chemically integrated within the silica network.30,44 The representative area percentages of these peaks indicate a slight increase in the sulfur content with rising APS concentration (Fig. S3c, ESI†). These findings collectively suggest that carbon-containing residual groups from the monomer remain attached to the silica network in the absence of APS in the precursor mixture. However, as APS is introduced into the precursor mixture, these carbon-containing groups are progressively replaced by nitrogen- and sulfur-containing species, further modifying the chemical composition of the resulting coating.
Then the effect of variation of the APS concentration on the coating thickness was analyzed. Fig. S4a–c of the ESI† displays the transverse scanning line, the direction of the stylus, and representative thickness measurements obtained from the stylus scans across the edge of the deposited coatings. The measured thickness distribution (Fig. S4c, ESI†) shows that the average height of the SiOx coating, without any APS in the precursor mixture, is 756 nm. In comparison, when varying amounts of APS, ranging from 0.1 wt% to 1.0 wt%, are introduced to the plasma stream, the thickness of the AF-SiOx coatings increases slightly, ranging from 790 nm to 860 nm. However, at 1 wt% APS content, the AF-SiOx film exhibits a higher standard deviation, indicating greater surface roughness. This increase in roughness is attributed to the higher concentration of APS in the precursor mixture, which leads to excessive nucleation in the reaction area that disrupts the formation of uniform film making the surface rougher.
Next, the surface wettability characteristics of the SiOx coated SS and PI surfaces with varying APS concentrations were analyzed by performing a WCA analysis. Fig. 6a presents the WCA droplet images obtained for the coatings deposited on PI, with different concentrations of APS in the reaction mixture. As observed, the SiOx coating with no APS concentration exhibits pronounced hydrophobic characteristics with the highest WCA values of 102.82° ± 1.72°. This hydrophobic nature can be attributed to the high prevalence of Si–C bonds from the monomer on the surface of the pristine SiOx coating, as confirmed by FTIR analysis.45 However, as the APS concentration in the reactant mixture increased from 0.1 wt% to 1.0 wt%, a decreasing trend in the contact angle was observed, dropping from 88.79° ± 0.66° to 51.07° ± 2.65°. This change can be attributed to the increasing dominance of polar and ionic groups, such as NH and NH4+, relative to Si–O–Si and Si–C in the films, as indicated by the FTIR and XPS results, leading to enhanced surface wettability. Furthermore, WCA measurements were recorded at different time intervals, spanning from day-1 to day-7 post-deposition and summarized in Fig. 6b. As observed, the SiOx-coated PI surface at APS concentrations below 0.6 wt% exhibited a 5% increase in WCA values after 7 days of measurement. This increase can be attributed to molecular rearrangements on the surface and the removal of hydroxyl groups from the SiOx matrix in response to ambient environmental conditions. In contrast, at APS concentrations above 0.8 wt%, no statistically significant change in the WCA was observed for the PI surface with SiOx coating. This stability in WCA can be explained by the presence of a more stable NH4+ peak compared to the hydroxyl group, which helps preserve its chemical wettability characteristics. Additionally, WCA measurements performed on the AF-SiOx coatings on the SS substrates were obtained and presented in Fig. 6c and d. Overall, hydrophobic characteristics with a contact angle of ∼102° ± 0.86° were observed for the pristine SiOx coating on the SS substrate, similar to those on the PI. As expected, a similar decreasing trend in the WCA from 92° to 63° was observed with increasing APS content in the precursor for AF-SiOx coatings deposited on SS substrates. Interestingly, no significant change (<2%) in WCA values was observed for the AF-SiOx-coated SS substrates across all APS concentrations in the precursor. This is because, unlike polymers, metallic surfaces do not experience significant change in the chemical characteristics of the SiOx coating over time. However, regardless of substrate type, the ammonium compound's functionality within the SiOx matrix ensures long-term hydrophilic properties, which are essential for implant surfaces.
 | ||
| Fig. 6 Surface wettability analysis. (a) Optical images showing the WCA of the glass coatings prepared with varying concentrations of APS deposited on PI, (b) summary of the change in WCA values of the glass coatings over 7 days, (c) optical images showing the WCA of the glass coatings prepared with varying concentrations of APS deposited on SS, and (d) changes in WCA values of the coatings over 7 days. | ||
Next, the antibacterial efficacy of the AF-SiOx coating on SS and PI substrates was tested using a standard contact-killing test with a bacterial suspension of ∼6 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CFU per mL. The antibacterial potency of these coatings with varying APS concentrations in the reactant mixture is shown in Fig. 7. The pristine SiOx surface without APS showed similar bacterial concentrations to those of the control samples on both SS and PI substrates. However, with increasing APS concentration on the PI surface, a gradual decrease in bacterial population by ∼1 logCFU per mL at 0.2 wt% APS and ∼2 logCFU per mL at 0.4 wt% APS was observed, as shown in Fig. 7a. On the other hand, beyond 0.6 wt% APS, a complete reduction to untraceable levels (∼5.5 logCFU per mL) was observed for both the E. coli and S. aureus bacterial populations. A similar trend in the bactericidal properties was observed for the CAP-assisted AF-SiOx coating on the SS substrate with complete reduction to untraceable levels observed after 0.6 wt% of APS concentration (Fig. 7b). These results clearly indicate a minimum of 0.6 wt% of APS is required in the CAP assisted SiOx to achieve broad-spectrum antibacterial efficacy against both Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. Next, in order to evaluate the level of entrapment of the APS molecule within the silica matrix, inhibition zone analysis was performed. Surprisingly, no visible inhibition zone was observed around the substrate across all the deposited samples with varying APS concentration as shown in Fig. S5, ESI.† However, a region of bacterial growth inhibition beneath the substrate was observed, suggesting a contact-assisted killing of the active antimicrobial agent entrapped within the silica matrix below the surface.
CFU per mL. The antibacterial potency of these coatings with varying APS concentrations in the reactant mixture is shown in Fig. 7. The pristine SiOx surface without APS showed similar bacterial concentrations to those of the control samples on both SS and PI substrates. However, with increasing APS concentration on the PI surface, a gradual decrease in bacterial population by ∼1 logCFU per mL at 0.2 wt% APS and ∼2 logCFU per mL at 0.4 wt% APS was observed, as shown in Fig. 7a. On the other hand, beyond 0.6 wt% APS, a complete reduction to untraceable levels (∼5.5 logCFU per mL) was observed for both the E. coli and S. aureus bacterial populations. A similar trend in the bactericidal properties was observed for the CAP-assisted AF-SiOx coating on the SS substrate with complete reduction to untraceable levels observed after 0.6 wt% of APS concentration (Fig. 7b). These results clearly indicate a minimum of 0.6 wt% of APS is required in the CAP assisted SiOx to achieve broad-spectrum antibacterial efficacy against both Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. Next, in order to evaluate the level of entrapment of the APS molecule within the silica matrix, inhibition zone analysis was performed. Surprisingly, no visible inhibition zone was observed around the substrate across all the deposited samples with varying APS concentration as shown in Fig. S5, ESI.† However, a region of bacterial growth inhibition beneath the substrate was observed, suggesting a contact-assisted killing of the active antimicrobial agent entrapped within the silica matrix below the surface.
 | ||
| Fig. 7 Antibacterial efficacy of the glass coatings with varying concentrations of APS in the reaction mixture deposited on (a) PI and (b) SS, against model Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. | ||
Next, in order to assess stability of the CAP assisted SiOx coating, the as prepared substrates were divided into two groups: one group was incubated in simulated body fluid (SBF) for 2 h, while the other was used as prepared, without any further modification. To assess the performance of the CAP-assisted coating, the same test was performed on the SS substrate by drop-casting the SiOx precursor with increasing concentrations of APS and drying it under ambient conditions for 3 h. Progressively, the same contact killing test was performed for all the as prepared and SBF incubated samples, as described previously.
As observed in Fig. S6a and b, ESI,† both the as-prepared CAP-assisted SiOx coating and the drop-cast coating exhibited similar antibacterial characteristics, with complete eradication of both E. coli and S. aureus bacterial population (5.5 logCFU per mL) at APS concentrations above 0.6 wt%. However, a significant alteration in the antibacterial properties was observed between the CAP-assisted and drop-cast coatings following surface washing with SBF. While the CAP-assisted coating exhibited a complete reduction in bacterial population (above 0.6 wt% of APS) after washing, the drop-cast coating showed a complete loss of antibacterial activity following the process (Fig. S6c and d, ESI†). These results clearly elucidate the influence of the CAP polymerization process on the immobilization and entrapment of APS onto the silica matrix, and its impact on the retention of antibacterial activity upon exposure to fluid.
Finally, the biocompatibility and safety of the SiOx-coated substrates with varying APS concentrations was assessed using a cell proliferation MTT assay. In this assay, a membrane-permeable tetrazolium salt turns purple upon interaction with viable cell mitochondria, which can be directly correlated with the percentage of cell viability. As shown in Fig. 8a, the optical absorbance values of cells exposed to the SiOx-coated SS and PI surfaces compared to control wells showed an enhanced cell viability above 86%, demonstrating the biocompatible nature of the SiOx coatings with different APS concentrations in the precursor mixture. Additionally, cells exposed to the AF-SiOx coated SS surfaces were stained with trypan blue to visualize and compare viable cells under an inverted microscope. As shown in Fig. 8b, no significant reduction in cell viability compared to the control wells was observed for the SiOx coated SS substrates with varying APS concentrations. These results validate the biocompatible characteristics of the CAP assisted SiOx coatings with varying APS concentration on both PI and SS surfaces. While these results validate the in vitro safety of the developed coating, further studies should focus on evaluating its safety in vivo animal models before consideration for human testing.
 | ||
| Fig. 8 Biocompatibility characteristics of the glass coatings with varying APS concentrations of the reactant mixture. (a) MTT based cell viability assay for SS and PI surfaces, and (b) the trypan blue staining analysis of the SS surface. | ||
4. Conclusions
In this work, the CAP-assisted deposition of ammonium functionalized glass coatings (AF-SiOx) with enhanced antibacterial properties was successfully demonstrated. A systematic investigation was conducted on coatings with varying concentrations of APS, assessing surface morphology, chemical composition, surface characteristics, antibacterial efficacy, and biocompatibility. The results indicate that higher concentrations of APS in the reactant mixture not only functionalize the surface with nitrogen-rich groups but also initiate polymerization, leading to a higher degree of polymerization within the glass coating matrix. Among the various precursor ratios tested, at least 0.6 wt% APS is required within the glass coatings to provide broad-spectrum antibacterial efficacy, while maintaining biocompatibility. This study highlights the effectiveness of the CAP-assisted coating technology as a scalable, rapid, and low-temperature method for producing antibacterial coatings on a variety of polymeric and metallic implant surfaces. The findings suggest that this approach offers a promising solution to the growing concern of implant-associated infections.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the staff of the Birck Nanotechnology Center for their valuable support throughout this project. Funding for this work was provided by the Center for Surface Engineering and Enhancement (CSEE) at Purdue University, as well as the 2024 Engineering in Medicine Pilot Project Program, jointly supported by the Indiana University School of Medicine and the Purdue University College of Engineering. R. R. and V. K. acknowledge the support from the School of Materials Engineering at Purdue University.References
- J. Lario Femenia, R. Poler Escoto and V. Amigó Borras, Metals (Basel), 2023, 13, 372 CrossRef CAS.
- R. Davis, A. Singh, M. J. Jackson, R. T. Coelho, D. Prakash, C. P. Charalambous, W. Ahmed, L. R. R. da Silva and A. A. Lawrence, Int. J. Adv. Manuf. Technol., 2022, 120, 1473–1530 CrossRef PubMed.
- C. Kreis, F. K. Aschenbrenner, D. Günther, N. Tholema-Hans, J. Koeppe, S. B. Rosslenbroich, M. J. Raschke and T. Fuchs, Sci. Rep., 2022, 12, 3979 CrossRef CAS PubMed.
- S. Sedaghat, A. Krishnakumar, V. Selvamani, J. P. Barnard, S. Nejati, H. Wang, D. A. Detwiler, M. N. Seleem and R. Rahimi, J. Mater. Chem. B, 2024, 12, 4489–4501 RSC.
- B. Le Vavasseur and V. Zeller, Antibiotics (Basel), 2022, 11, 486 CrossRef CAS PubMed.
- H. Brüssow, Microb. Biotechnol., 2024, 17, e14510 CrossRef PubMed.
- C. Cui, Y. Zhao, Z. Bai, J. Yan, D. Qin, H. Peng, Y. Liu, J. Tong, L. Sun, X. Wu and B. Li, ACS Biomater. Sci. Eng., 2024, 10, 4093–4113 CrossRef CAS PubMed.
- X. Chen, J. Zhou, Y. Qian and L. Zhao, Mater Today Bio., 2023, 19, 100586 CrossRef CAS PubMed.
- M. A. Waqar, N. Mubarak, A. M. Khan, R. Khan, I. N. Khan, M. Riaz, A. Ahsan and M. Munir, Taylor and Francis Ltd., 2024, preprint DOI:10.1080/25740881.2024.2346331.
- H. T. Imam, K. Hill, A. Reid, S. Mix, P. C. Marr and A. C. Marr, ACS Sustainable Chem. Eng., 2023, 11, 6829–6837 CrossRef CAS PubMed.
- S. Guchhait and S. Roy, J. Sol-Gel Sci. Technol., 2019, 89, 852–865 CrossRef CAS.
- P. Bragta, R. K. Sidhu, K. Jyoti, A. Baldi, U. K. Jain, R. Chandra and J. Madan, Colloids Surf., B, 2018, 166, 339–348 CrossRef CAS PubMed.
- S. Zhang, L. Lin, X. Huang, Y.-G. Lu, D.-L. Zheng and Y. Feng, J. Nanomater., 2022, 2063265 CrossRef CAS.
- S.-R. Lee, S. Jo, S. Kim, Y. Oh and D.-K. Kim, Int. J. Food Microbiol., 2024, 419, 110749 CrossRef CAS PubMed.
- S.-R. Lee and D.-K. Kim, Food Biosci., 2024, 62, 105406 CrossRef CAS.
- K. Vasilev, S. S. Griesser and H. J. Griesser, Plasma Processes Polym., 2011, 8, 1010–1023 CrossRef CAS.
- R. Gharibi, H. Yeganeh and S. Kazemi, Mater. Sci. Eng., C, 2019, 99, 887–899 CrossRef CAS PubMed.
- M. J. Saif, J. Anwar and M. A. Munawar, Langmuir, 2009, 25, 377–379 CrossRef CAS PubMed.
- U. Heredia-Rivera, A. Krishnakumar, V. Kasi, M. M. Rana, S. Gopalakrishnan, S. Nejati, G. Gundala, J. P. Barnard, H. Wang and R. Rahimi, J. Mater. Chem. C, 2024, 12, 11861–11876 RSC.
- H. Guo, S. Pan, Z. Hu, Y. Wang, W. Jiang, Y. Yang, Y. Wang, J. Han, Y. Wu and T. Wang, Chem. Eng. J., 2023, 470, 144094 CrossRef CAS.
- M. N. Chai and M. I. N. Isa, Sci. Rep., 2016, 6, 27328 CrossRef CAS PubMed.
- A. K. Arof, S. Amirudin, S. Z. Yusof and I. M. Noor, Phys. Chem. Chem. Phys., 2014, 16, 1856–1867 RSC.
- Y. Ogihara, H. Yano, M. Watanabe, A. Iiyama and H. Uchida, Catalysts, 2016, 6, 139 CrossRef.
- M. Bertin, E. M. Leitao, S. Bickerton and C. J. R. Verbeek, Plasma Processes Polym., 2024, 21, 2300208 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- S. Barus, M. Zanetti, M. Lazzari and L. Costa, Polymer, 2009, 50, 2595–2600 CrossRef CAS.
- L. Cui, A. N. Ranade, M. A. Matos, G. Dubois and R. H. Dauskardt, ACS Appl. Mater. Interfaces, 2013, 5, 8495–8504 CrossRef CAS PubMed.
- I. Yudovin-Farber, N. Beyth, E. I. Weiss and A. J. Domb, J. Nanopart. Res., 2010, 12, 591–603 CrossRef CAS.
- S. Shankar, R. Gadi, S. K. Sharma and T. K. Mandal, Mapan – J. Metrol. Soc. India, 2022, 37, 529–544 Search PubMed.
- N. M. Bobkova and E. E. Trusova, Glass Ceram., 2017, 74, 153–157 CrossRef CAS.
- Z. J. Ding, Y. P. Wang, W. J. Liu, S. J. Ding, M. R. Baklanov and D. W. Zhang, J. Phys. D:Appl. Phys., 2018, 51, 115103 CrossRef.
- P. Okoczuk, M. Łapiński, T. Miruszewski, P. Kupracz and L. Wicikowski, Materials, 2021, 14, 2158 CrossRef CAS PubMed.
- A. Hamdi, J. Chalon, P. Laurent, B. Dodin, E. Dogheche and P. Champagne, J. Coat. Technol. Res., 2021, 18, 807–818 CrossRef CAS.
- C. C. Chang, Y. T. Wu and L. P. Cheng, J. Coat. Technol. Res., 2016, 13, 999–1007 CrossRef CAS.
- R. E. Saputra, Y. Astuti and A. Darmawan, Spectrochim. Acta, Part A, 2018, 199, 12–20 CrossRef CAS PubMed.
- C. Y. Lee, C. Y. Yan and Y. L. Cheng, Coatings, 2021, 11, 314 CrossRef CAS.
- A. Grill, J. Appl. Phys., 2003, 93, 1785–1790 CrossRef CAS.
- V. Goel, S. K. Mishra, C. Sharma, B. Sarangi, S. G. Aggarwal, R. Agnihotri and R. K. Kotnala, Mapan, 2018, 33, 209–215 CrossRef.
- A. V. Radha, L. Lander, G. Rousse, J. M. Tarascon and A. Navrotsky, J. Mater. Chem. A, 2015, 3, 2601–2608 RSC.
- A. E. Kaloyeros, Y. Pan, J. Goff and B. Arkles, ECS J. Solid State Sci. Technol., 2020, 9, 063006 CrossRef CAS.
- K. Awsiuk, A. Psarouli, P. Petrou, A. Budkowski, S. Kakabakos, A. Bernasik, J. Rysz and I. Raptis, Procedia Eng., 2011, 25, 334–337 CrossRef CAS.
- H. Szymanowski, K. Olesko, J. Kowalski, M. Fijalkowski, M. Gazicki-Lipman and A. Sobczyk-Guzenda, Coatings, 2020, 10, 794 CrossRef CAS.
- Q. H. Trinh, M. M. Hossain, S. H. Kim and Y. S. Mok, Heliyon, 2018, 4, e00522 CrossRef PubMed.
- Y. He, Z. Shan, T. Tan, Z. Chen and Y. Zhang, Polymers, 2018, 10, 930 CrossRef PubMed.
- R. P. Socha, K. Laajalehto and P. Nowak, Influence of the surface properties of silicon carbide on the process of SiC particles codeposition with nickel, Colloids Surf., A, 2002, 208, 267–275 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00520e |
| ‡ Authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |