
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeviate from the norm: ambipolar-acceptor frameworks as a new design paradigm for low bandgap conjugated polymers†
Wyatt D.
Wilcox
a,
Evan W.
Culver
a,
Nicolas C.
Nicolaidis
 b,
Spencer J.
Gilman
a,
Tomas J.
Marsh
b,
Paul C.
Dastoor
b and
Seth C.
Rasmussen
*a
b,
Spencer J.
Gilman
a,
Tomas J.
Marsh
b,
Paul C.
Dastoor
b and
Seth C.
Rasmussen
*a
aDepartment of Chemistry and Biochemistry, North Dakota State University, NDSU Dept. 2805, P.O. Box 6050, Fargo, ND 58108-6050, USA. E-mail: seth.rasmussen@ndsu.edu
bCentre for Organic Electronics, University of Newcastle, Callaghan, NSW 2308, Australia
First published on 4th March 2025
Abstract
A new design paradigm is presented for the production of low bandgap (Eg < 1.5 eV) polymers which takes advantage of the dual nature of building blocks that can simultaneously exhibit both strong donor and strong acceptor properties. By replacing the donor unit of commonly applied donor–acceptor (D–A) frameworks with such ambipolar units, the resulting ambipolar-acceptor framework maintains the classical D–A backbone, while effectively adding a second acceptor. As illustrative examples of this approach, the synthesis of a series of alternating copolymers are reported that pair the ambipolar unit thieno[3,4-b]pyrazine (TP) with various traditional acceptors, including 2,1,3-benzothiadiazole, quinoxaline, and 2H-benzotriazole, successfully producing soluble, processible materials with band gaps of 1.02–1.12 eV. Analysis of these materials reveals the tunability of this approach and provides convincing evidence that the LUMOs of the TP and traditional acceptor hybridize to give a delocalized and suitably stabilized polymer LUMO, thus contributing to the low bandgaps of the resulting polymers. To demonstrate the potential of these materials, initial application to bulk heterojunction photonic devices are also reported, revealing photoresponse out to beyond 1100 nm, with initial NIR photodetectors providing detectivity (D*) values as high as 8.9 × 1011 Jones at 815 nm.
Introduction
Although generally believed to be a modern subject, the preparation and study of conjugated organic polymers dates back to the 1830s.1 Of course, the polymeric nature of these materials was not recognized until the start of the 20th century and their electronic properties were largely overlooked until the first reports of their conductivity in the early 1960s.1–5 Since that time, the study of conjugated polymers has continued to grow, ultimately leading to the current field of organic electronics, with various demonstrated applications including electrochromics, sensors, organic field effect transistors (OFETs), organic light-emitting diodes (OLEDs), and organic photovoltaics (OPVs).6–12The successful application of organic semiconducting materials is dependent on various critical properties, including effective conjugation length, energies of the frontier molecular orbitals and the resulting bandgap, charge mobilities, solubility and processability, and overall stability.13 Of these, the material's band gap (Eg) is often deemed especially vital.13–19 Defined as the energy between the filled valence and empty conduction bands, the Eg corresponds to the HOMO–LUMO gap of the solid-state material and determines such optical properties as the onset of absorbance or the energy of any potential emission. As most conjugated polymers exhibit Eg values of 1.5–3.0 eV, these materials primarily absorb and emit light in the visible regime. However, lowering the Eg can allow more effective absorption in the red and near infra-red (NIR) wavelengths, a critical capability for both OPVs11,12,19 and NIR photodetectors.19–25 In addition, a smaller Eg allows greater thermal population of the conduction band, leading to more intrinsic charge carriers and enhanced conductivity.13–17 As reducing the Eg is typically coupled with destabilization of the HOMO energy, this can also lead to lower potentials of oxidation and stabilization of the p-doped (i.e., oxidized) state.13–15 As such, the development of design criteria for lowering the Eg of conjugated materials remains a critical factor for the production of technologically useful low (Eg < 1.5 eV)13–17 and reduced (Eg = 1.5–2.0 eV)13–16 bandgap polymers.
While a number of factors contribute to the material's Eg,13–16 there are generally only two primary design methodologies for the successful production of true low Eg polymers. The first of these was introduced in 1984 by Wudl, Heeger, and coworkers, which involved efforts to enhance the quinoidal nature of the aromatic backbone of polythiophene.26,27 For polythiophene, theoretical studies have shown the quinoidal form to have a much lower Eg than the aromatic form, and that the Eg can be decreased by increasing the quinoidal contribution to the ground state of the polymer.15–19,28 As shown for the original example of polyisothianaphthene in Fig. 1, this reduction in Eg is attributed to both a stabilization of the LUMO and destabilization of the HOMO, with the effect on the HOMO nearly twice that of the LUMO.17,28
 | ||
| Fig. 1 Minimizing Egvia enhancing the quinoidal character of the ground state. | ||
These early efforts utilized fused-ring units such as isothia-naphthene, sometimes referred to as proquinoidal units.18 That is, aromatic units without quinoidal content in the monomeric form, but able to induce quinoidal character in the resulting polymers. More recent approaches have been to move beyond proquinoidal units to units that adopt a true quinoidal con-stitution in the ground state.17,18 Although efforts to enhance the quinoidal nature of conjugated polymers continues to be an important approach to reducing Eg,14,17,18 an alternate approach has grown to become the most commonly applied design criteria for low Eg polymers.
This second approach was introduced by Havinga and co-workers in 1992, which applied a conjugated backbone of alter-nating electron-rich (donor) and electron-poor (acceptor) groups in order to reduce the polymer Eg.29,30 As illustrated in Fig. 2, lowering the Egvia donor–acceptor (D–A) frameworks is typically explained by hybridization of the frontier orbitals of the donor and acceptor, thus producing a hybrid D–A material with HOMO levels characteristic of the donor and LUMO levels characteristic of the acceptor.13–17,31,32 The HOMO–LUMO energy of the D–A unit is thus smaller than either of the respective homodimers and further chain extension of the D–A framework would continue hybridization to give a reduced polymer Eg. It should be pointed out that such pictorial representations found in the literature commonly show equivalent mixing of both the HOMO and LUMO.16,17,19 While such equivalent mixing can occur in some cases,33 this is usually not the case.20,31–34 Rather, the HOMO levels of the donor and acceptor are normally more energetically similar than the corresponding LUMO levels, thus leading to reduced mixing between the respective LUMOs.32 This is particularly true for many materials of strong acceptors, where LUMO levels are too energetically and spatially separated to see any substantial mixing in the initial D–A unit, although some additional mixing can sometimes occur in the final polymer.31,32
 | ||
| Fig. 2 Hybridization of frontier orbitals of donors (D) and acceptors (A) to give symmetric (D–D or A–A) or hybrid (D–A) dimeric units. | ||
In terms of their application of D–A frameworks, monomers are generally viewed as either electron-rich donors, electron-poor acceptors, or a neutral spacer units. This view, however, has been complicated by the realization that the common building block thieno[3,4-b]pyrazine (TP, Fig. 3)35–39 is not a simple acceptor as generally applied, but simultaneously acts as both a strong donor and strong acceptor.32,40 Consequently, the TP unit contributes strongly to both the HOMO and LUMO in D–A frameworks and it is the internal D–A interaction37 of TPs that dominates their electronic behavior. Due to the dual donor and acceptor nature of TPs, these units have been designated ambipolar units in order to differentiate them from traditional donors or acceptors.31,32,40
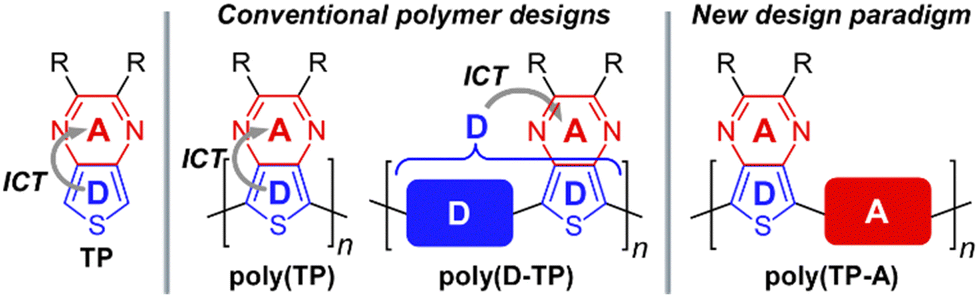 | ||
| Fig. 3 Ambipolar thieno[3,4-b]pyrazine (TP) and its corresponding polymeric materials. | ||
Historically, TP units have been used to generate homo-polymeric materials or applied as acceptors in D–A frameworks for the successful production of a wide range of low Eg polymers.35 However, as it is now understood that TPs also exhibit a high-lying HOMO, this characteristic should logically allow for a new design paradigm for low Eg materials in which ambipolar units such as TP are paired with an acceptor, rather than the traditional pairing with a donor (Fig. 3). Previous communications in 2018 presented initial examples of this approach,41,42 with the current report presenting further optimization of those efforts, as well as expansion of the scope of these materials to include a series of ambipolar-acceptor frameworks that pair TP with the traditional acceptors 2,1,3-benzothiadiazole (BTD),43,44 quinoxaline (Qx),45–47 and 2H-benzotriazole (BTA).48,49 In the process, the present discussion will outline the nature and scope of this new design paradigm, report the full optical and electronic characterization of these materials, provide a detailed comparison to related model D–A polymers, and present the initial application of these materials to bulk heterojunction devices.
Results and discussion
Ambipolar nature of TP relative to common donors and acceptors
Prior to presenting working examples of this new design paradigm, it is worth further clarifying the ambipolar nature of TP and exactly how this relates to traditional donors or acceptors. Of course, a complication here is that the terms donor and acceptor are often used throughout the literature as simply “electron-rich” or “electron-poor” units, without any firm parameters as to what exactly qualifies them as such. However, as outlined in Fig. 2, acceptors of significant strength are considered to have low-lying LUMOs, thus allowing electrochemical characterization of these energy levels as shown in Fig. 4a for TP and the accepters BTD, Qx, and BTA used in the current study. Based on the potential for the TP reduction, it represents a quite strong acceptor, falling halfway between the reductions of the traditional acceptors BTD and Qx. In comparison, the reduction of BTA occurs at significantly higher negative potentials and falls on the upper limit of what can be characterized via electrochemistry. As such, this data more than justifies the historic use of TP as an acceptor in D–A frameworks. | ||
| Fig. 4 (a) Cyclic voltammograms of TP and various acceptors; (b) cyclic voltammograms of TP and various donors; (c) plot of frontier energy levels of donors, acceptors, and ambipolar units as determined from electrochemistry. | ||
Similarly, donors of significant strength are considered to have high-lying HOMOs, again allowing electrochemical characterization as shown in Fig. 4b for TP and the donors thiophene and 3,4-ethylenedioxythiophene (EDOT). Here, TP undergoes oxidation at potentials ca. 600 mV less than thiophene and at nearly the same potential as that of EDOT. As such, TP can also be considered to be a quite strong donor, a conclusion also supported by various studies of oligomeric models.32,40
From such electrochemical studies, the relative frontier orbital energies can be estimated relative to vacuum, with the corresponding frontier orbital energies plotted in Fig. 4c. Here, we can now provide working definitions in which donors exhibit HOMO energies high enough to be characterized via electrochemistry, but with LUMO energies that fall outside normal electrochemical solvent windows. Likewise, acceptors exhibit LUMO energies low enough to be measured, but with HOMO energies that fall outside normal solvent windows. As ambipolar units act as both donors and acceptors, both frontier orbitals can be determined by normal electrochemical methods, as illustrated in Fig. 4c for TP and its extended analogue acenaphtho[1,2-b]thieno[3,4-e]pyrazine (ATP).50
Polymer synthesis
Of course, the novelty of this new ambipolar-acceptor design approach involves some synthetic complications. For example, the most common synthetic routes to D–A polymers utilize a dihalo-functionalized acceptor with either a distannyl- or diboroester-functionalized donor, thus suitable for Stille or Suzuki cross-coupling. This is the case for TP-based materials as well, with the bulk of previous TP copolymers produced via 5,7-dibromothieno[3,4-b]pyrazines.35 Thus, in order to successfully cross-couple what has been previously viewed as two acceptor units, new distannyl- or diboroester-acceptor intermediates would be needed.In order to circumvent this issue, efforts instead turned to direct arylation polymerization (DArP), which offers a number of advantages over traditional cross-coupling methods.51–56 In terms of its application to the desired TP-acceptor polymers here, the most critical aspect is that DArP facilitates C–C bond formation between aryl C–H units and aryl halides without the need for conventional organometallic species (i.e., distannyl monomers). As a result, DArP provides a viable route to the cross-coupling of unfunctionalized TPs with a corresponding dihalo-functionalized acceptor. Furthermore, as TP only possesses C–H bonds at the two thiophene α-carbons, the β-defects sometimes observed in the application of DArP are not a possibility here.
The general polymerization conditions investigated for the crosscoupling of 2,3-bis(2-ethylhexyl)thieno[3,4-b]pyrazine with 4,7-dibromo-2,1,3-benzothiadiazole are given in Table 1. THF was initially chosen as the polymerization medium as it has previously proven to be a good solvent for solubilizing TP materials. However, to allow access to higher temperatures, the reactions were carried out in a sealed microwave vial to allow superheating of the solvent under pressure. While the initial reaction conditions utilizing THF at 100 °C successfully generated the desired materials, both the solvent and the corresponding polymerization temperature are considered among the most critical variables for DArP and attempts to further optimize these conditions were thus carried out as detailed in Table 1.53 These efforts first began with the polymerization temperature, in which the temperature of the reaction in THF was increased from 100 to 120 °C. While the previous conditions at 100 °C required a full 48 h of reaction time in order to achieve sufficient molecular weights and yield,41 it was now found that the reaction time at 120 °C could be halved to 24 h with no loss in polymer yield. Furthermore, the elevated temperature also resulted in higher molecular weight samples and a slight drop in the Eg from 1.05 to 1.02 eV.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Reaction time (h) | Polymer yield | M n | PDI | λ max (CHCl3, nm) | λ max (film, nm) | E g (eV) |
| a Carried out in a sealed microwave vial to allow superheating of the solvent. | |||||||||
| 1 | THF | 100 | 24 | 74% | 697 | ||||
| 2 | THF | 100 | 48 | 99% | 8100 | 1.45 | 813 | 850 | 1.05 |
| 3 | THF | 120 | 24 | 99% | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
1.59 | 790 | 840 | 1.02 |
| 4 | xylenes | 120 | 24 | 99% | 4600 | 1.42 | 798 | 842 | 1.09 |
| 5 | xylenes | 150 | 24 | trace | |||||
| 6 | DMAc | 120 | 24 | 78% | 11500 |
2.54 | 625 | 652 | 1.28 |

Alternate solvents were then investigated, beginning with xylenes as a higher boiling solvent that could access even higher reaction temperatures. At 120 °C, xylenes gave comparable yields to THF, but with a reduced Mn and an increase in Eg. Further increasing the temperature to 150 °C did not improve these results, instead giving only trace amounts of product. Finally, N,N-dimethylacetamide (DMAc) was also investigated as it has been a popular choice for conjugated polymers via DArP.56 DMAc, however, gave lower yields and much higher Eg values. As the Mn values via DMAc are similar to those via THF, it is assumed that DMAc resulted in defects of some form that reduced the backbone conjugation.
As the combination of THF and 120 °C gave the best overall results, these then became the standard conditions for the preparation of a series of polymers, as outlined in Scheme 1. These methods gave the desired polymer products in yields of ca. 80–99%, with Mn values of 8000–14000. All of these materials exhibit good solubility, although more solubilizing 2-ethylhexyl sidechains were required on the TP when paired with BTD due to the lack of any solubilizing sidechains on the acceptor unit.
 | ||
| Scheme 1 Synthesis of thieno[3,4-b]pyrazine-acceptor frameworks. | ||
Optical and electronic characterization
The optical and electronic properties of the resulting TP-A polymers are summarized in Table 2, with the visible-NIR spectra of thin film samples given in Fig. 5. As with most D–A polymers,57 all of the TP-A materials exhibit two absorption bands, with the low energy band assigned to an intramolecular charge transfer (ICT) from the polymer backbone to one or both acceptor units. As can be seen from the thin film spectra, the absorbance onsets correspond to low Eg values of 1.02–1.12 eV, with the trend in the observed Eg values consistent with the relative strengths of the acceptor units applied (Fig. 4).| Polymer | λ max (CHCl3, nm) | λ max (film, nm) | E optg (eV) | HOMO (eV)a | LUMO (eV)b |
|---|---|---|---|---|---|
a
E
HOMO = −(E[onset,oxvs.Fc+/Fc] + 5.1) (eV), ref. 58.
b
E
LUMO = − (E[onset,red vs.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Fc+/Fc] + 5.1) (eV), ref. 58.
c Ref. 59.
d In toluene.
e Ref. 60.
f In o-dichlorobenzene. Fc+/Fc] + 5.1) (eV), ref. 58.
c Ref. 59.
d In toluene.
e Ref. 60.
f In o-dichlorobenzene.
|
|||||
| EHTP-BTD | 790 | 840 | 1.02 | −5.25 | −3.93 |
| TP-TP | 970 | 890 | 0.96 | −5.00 | −3.90 |
| TP-Qx | 765 | 828 | 1.07 | −5.25 | −3.87 |
| TP-BTA | 740 | 774 | 1.12 | −5.20 | −3.80 |
| EHTP-Ph | 620 | 640, 690 | 1.50 | −5.50 | −3.70 |
| ProDOT-BTDc | 645d | 662 | 1.53 | −5.22 | −3.70 |
| TQ1e | 612f | 629 | 1.75 | −5.35 | −3.60 |
 | ||
| Fig. 5 Visible-NIR spectra of TP-A thin films. | ||
While most low bandgap materials achieve such low Eg values by raising the HOMO energy, the TP-A materials here all exhibit relatively stabilized HOMO levels of ca. −5.20 to −5.25 eV. This difference can be easily seen in comparison to the GRIM polymerized poly(2,3-dihexylthieno[3,4-b]pyrazine) (TP-TP),61 which exhibits a relatively similar Eg (ca. 0.96 eV), although with a higher HOMO of −5.0 eV. In the case of TP-TP, the combination of ambipolar TP units results in hybridization of two strong donors, resulting in a very destabilized HOMO, as well as the hybridization of two strong acceptors, resulting in a deep LUMO. By replacing a TP unit with a traditional acceptor, the total donor content is significantly reduced, resulting in a deeper HOMO typically of many D–A frameworks. While this does raise the Eg to some extent, the deeper HOMO of the TP-A materials provides some additional environmental stability, while also offering a clear benefit in applications to technological devices.
What really sets this new class of low Eg materials apart from typical D–A frameworks is the deeper LUMO levels achieved by this new design approach. While the exact nature of the LUMO resulting from the inclusion of two acceptor units was an initial question during the development of this design approach, a combination of computational studies,32,41 analysis of oligomeric models,32 and the trends presented in Table 2, all support a single, delocalized LUMO resulting from hybridization of the LUMOs of TP and the second acceptor species. For example, the LUMO values given in Table 2 track well with the respective acceptor strengths of the TP-A pairs (Fig. 4a), with stronger acceptor combinations giving the most stabilized LUMOs. This trend includes the TP-TP combination of the GRIM polymerized homopolymer, which also had been previously concluded to exhibit a delocalized LUMO via hybridization of the neighbouring TP units.62 However, unlike the TP-A materials, the TP homopolymer lacks the traditional acceptor in order to stabilize the HOMO level, again resulting in the higher HOMO level. The trend observed here in the LUMO levels across all TP-A pairings also emphasizes the fact that this design paradigm is not limited to the use of acceptors that are stronger than TP (i.e., BTD), although again, stronger acceptors result in the most stabilized polymer LUMO and thus lower bandgaps.
In order to clearly illustrate the effect of the two-acceptor combination, the model polymer EHTP-Ph was also prepared which maintains the TP-phenylene backbone of the TP-A series, but without the second traditional acceptor. The first effect observed in removing the second acceptor is a deeper HOMO for EHTP-Ph. This difference can be attributed to the fact that benzene is actually a weaker donor than the fused-ring benzo acceptors, as evidenced by their respective HOMO energies (BTD63 −7.2 eV; benzene64 −7.4 eV). More critically, however, is the fact that EHTP-Ph does not exhibit the stabilized LUMOs observed in the TP-A series, highlighting the importance of the second acceptor and its contribution via mixing between the two acceptor units. The combination of the deeper HOMO and higher-lying LUMO for EHTP-Ph thus results in an Eg of 1.50 eV, ca. 0.5 eV higher than the TP analogues containing the second acceptor.
Finally, the properties of the TP-A polymers can be com-pared and contrasted to the traditional D–A polymers, poly(3,4-propylenedioxythiophene-alt-2,1,3-benzothiadiazole) (ProDOT-BTD)59 and poly(2,3-bis(3-octyloxyphenyl)quinoxaline-alt-thio-phene) (TQ1),60 both of which can be considered analogues of TP-BTD and TP-Qx in which the TP has been replaced with either ProDOT or thiophene respectively. As ProDOT-BTD and EHTP-BTD share the same traditional acceptor and ProDOT is a donor of similar strength to TP (i.e., ProDOT is similar to EDOT, Fig. 4b), one would expect the HOMO energies of the two materials to be similar, which is exactly that shown in Table 2 (−5.25 vs. −5.22 eV). The Eg value of ProDOT-BTD, however, is ca. 0.5 eV greater than that of TP-BTD, largely due to the more stabilized LUMO of the latter. In the same way, the polymers TQ1 and TP-Qx share the same acceptor, but as TP is a stronger donor than thiophene (Fig. 4b), one would expect the HOMO of TQ1 to be more stabilized than that of TP-Qx. As shown in Table 2, this is exactly what is found (−5.35 vs. −5.25 eV). Even taking this into account, however, the Eg value of TQ1 is ca. 0.7 eV greater than TP-Qx, again due to the more stabilized LUMO of the latter.
Of course, the lack of LUMO stabilization for EHTP-Ph, ProDOT-BTD, and TQ1 illustrates that sufficient hybridization between the acceptor units relies not just on suitable energy matching of acceptor LUMOs, but also on the relative spatial distance between the acceptors. That is, the stabilization seen in the TP-A frameworks are not due to the combination of two different acceptors alone, but due to the fact that two acceptor units of suitably matched LUMO energies are also close enough to facilitate significant mixing. As such, one would expect the LUMO stabilization exhibited in the TP-A frameworks to be eliminated if spacer units were introduced into the backbone, thus spatially separating the TP and A units.
The question of quinoidal effects
Considering that TP is a well-known proquinoidal unit18 and that the new paradigm introduced via ambipolar-acceptor frameworks overturns long-held assumptions for D–A models, some readers will attempt to dismiss the low bandgaps achieved in the TP-A materials as simply the result of expected quinoidal effects, rather than any new insight into D–A frameworks. Furthermore, some authors have even questioned the effect of the D–A approach in general, suggesting that it is actually the geometrical mismatch between quinoidal and aromatic units that accounts for the lower Eg in such frameworks.65–67 As such, a discussion of any potential quinoidal effects in the current materials is warranted.To begin with, the fact that the LUMOs of the TP-A frameworks here are dependent on the presence and strength of the traditional acceptor, it is clear that these play a role in determining both the LUMO and the resultant Eg. At the same time, as the proquinoidal TP unit is held constant throughout the series, it is not really possible to account for differences in Eg due to quinoidal effects alone. While one could argue that the differences could be due to each acceptor unit impacting the TP quinoidal contribution to a greater or lesser extent, the fact that the chemical composition of the conjugated backbone is essentially held constant throughout the series makes this unlikely. Of course, none of this precludes a combination of quinoidal and D–A effects at play.
At the same time, it must be highlighted that the trends found in Table 2 are also in excellent agreement with a previous oligomer study of D–A effects, including those of TP-BTD.32 As the oligomers of that study fell below the minimum chain length expected for meaningful quinoidal effects,68 the trends in the oligomers were concluded to be solely due to D–A effects. As such, if quinoidal effects provided a significant contribution to the energies and Eg values of the polymers reported here, but not to the previous oligomers, one would expect to see some deviation between the two sets of results.
Finally, as previously discussed above, the polymers EHTP-BTD and ProDOT-BTD only differ in the replacement of TP with ProDOT, with both polymers exhibiting nearly equal HOMO energies (Table 2). Furthermore, these similar HOMO energies are consistent with the similar donor strengths of TP and ProDOT. However, while TP is a proquinoidal unit, ProDOT is not, and thus one cannot make a valid argument for quinoidal effects in the case of ProDOT-BTD. As introduced above, the enhancement of quinoidal effects lowers the Eg by destabilizing the HOMO and stabilizing the LUMO, with a greater effect observed for the HOMO energies. Thus, if quinoidal effects were contributing to EHTP-BTD, but not to ProDOT-BTD, the TP material would be expected to exhibit a HOMO of higher energy in comparison to its ProDOT analogue. The fact that they exhibit similar HOMOs would require equivalent quinoidal contributions in both cases, which is just not realistic. However, as the HOMO energies are in complete agreement with the D–A model, one must conclude that any potential quinoidal effects are minimal in the TP-A materials reported here.
Structural aspects of the current TP-A materials
An aspect of the TP-A series that may contribute to the effective mixing between the two acceptors is significant hydrogen-bonding between the aromatic C–H units of the traditional acceptors and the pyrazine nitrogen of the TP. Originally observed in oligomeric models (Fig. 6a),32 these hydrogen bonds in the EHTP-BTD polymer is evidenced by large downfield shifts of the BTD hydrogens in the polymer 1H NMR spectra (Fig. 6b). This hydrogen-bonding, coupled with complementary S⋯N interactions, results in a very flat, rigid backbone for the TP-A series, which also accounts for the reduced solubility seen across this series in comparison to other conjugated materials. Attempting to gain insight into the potential contributions of these interactions, the 5,6-difluoro-2,1,3-benzothiadiazole (F2BTD) analogue of EHTP-BTD was prepared, in which replacement of the BTD hydrogens with fluorine would prohibit the hydrogen-bonding interaction. Unfortunately, the resulting EHTP-F2BTD polymer (Fig. 6c) exhibited an extremely large blue-shift (ca. 322 nm) in the thin film absorption, resulting in an Eg of ca. 1.60 eV. This dramatic increase in Eg was thus attributed to not only removing the hydrogen-bonding effect, but considerable electronic repulsion between the fluorine and nitrogen lone pairs. | ||
| Fig. 6 Attractive and repulsive interactions in dimeric TP-BTD (a), polymeric EHTP-BTD (b), and EHTP-F2BTD (c). | ||
Bulk heterojunction devices
In order to probe the potential of the TP-A frameworks in technological devices, bulk heterojunction (BHJ) devices were fabricated from 1:1 blends of TP-BTA and phenyl-C61-butyric acid methyl ester (PCBM). The binary blends produced sufficiently smooth films of ∼100 nm thickness and the resulting J–V plot and external quantum efficiency (EQE) for a representative device is shown in Fig. 7. The EQE response exhibits a high-energy maximum at ca. 375 nm, with additional low-energy maxima at ca. 770 and 815 nm. This low-energy response matches well with the absorbance of pure TP-TBA films, while the higher energy response is attributed to contributions of PCBM.69 As can be seen, TP-BTA contributes to the device response in the NIR region down to ca. 1200–1300 nm, in good agreement with the thin-film absorption shown in Fig. 5. Moreover, the EQE response of the devices is significant across the entire 300–1100 nm wavelength range.
 | ||
| Fig. 7 External quantum efficiency (EQE) and J–V plot of a 1:1 TP-BTA:PCBM device. | ||
Although the characteristics of these unoptimized devices pale in comparison to the high-efficiency OPV devices now possible, they are noteworthy in their ability to provide photo- response in the NIR. While a fair number of low Eg polymers have been applied to photonic devices, the family of such materials capable of response below 1000 nm still encompasses less than 20 polymers.20
In order to evaluate the ability of these devices to act as NIR photodetectors, the noise current of the BHJ devices was determined so that the specific detectivity (D*(λ)) could be calculated. Such D*(λ) values reflect the ability to detect signals of weak irradiation intensity and are considered the most critical parameter for NIR photodetectors,20–23 with values >1011 Jones (or cm Hz½ W-1) considered competitive with inorganic photodetectors. The respective responsivity (R(λ)) and D*(λ) values for the TP-BTA:PCBM device are shown in Fig. 8. Within the NIR region, maximum values of 0.010 A W-1 and 2.43 × 109 Jones are found at 815 nm, with D*(λ) remaining above 109 Jones out to 1000 nm.
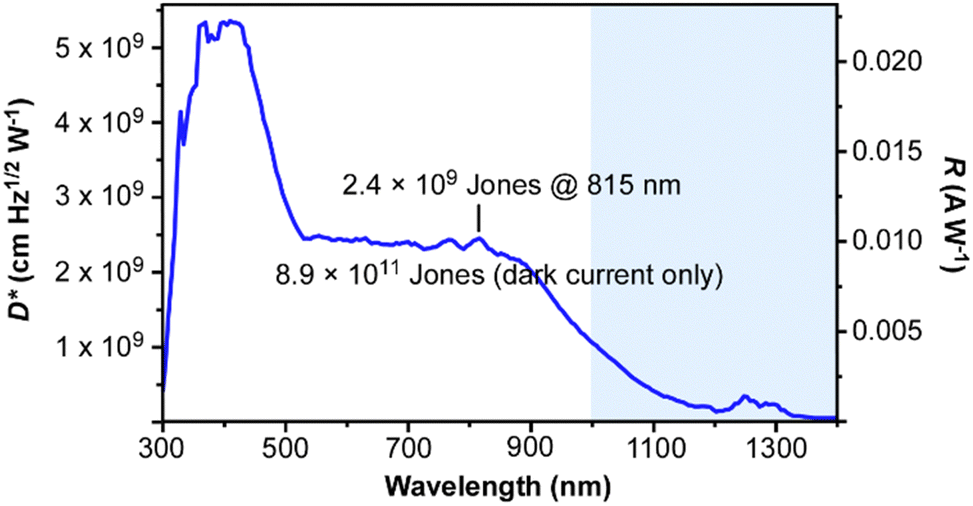 | ||
| Fig. 8 Specific detectivity (D*(λ)) and responsivity (R(λ)) for a 1:1 TP-BTA:PCBM device. | ||
Furthermore, much like with the photoresponse shown in Fig. 7, the detectivity of these NIR photodetectors extend beyond 1000 nm, with measurable values out to ca. 1300 nm.
Of course, NIR photodetectors based on low Eg polymers and small molecules capable of response below 1000 nm have now achieved D*(λ) values >1013 Jones.20–22,25 Still, it must be noted that the bulk of these values have not been determined relative to the total noise current as was done for the TP-BTA devices here. Rather, most determine D*(λ) solely from the dark current.21–24 As such, the specific detectivity is generally overestimated, as the measured noise current is usually much larger than the theoretical shot noise limit.21,23,25 In fact, some argue that the contribution of thermal noise to the total noise current is significant in organic NIR photodetectors, particularly those based on low Eg materials,22,24 with the overestimation of D*(λ) predicted to be greater than an order of magnitude when based only on dark current.24 To emphasize the effect of determining D*(λ) via dark current vs. total current, the D*(λ) value for the TP-BTA device at 815 nm was also determined using the dark current only, resulting in the considerably higher value of 8.9 × 1011 Jones. This value determined from the dark current thus compares very well with the best reported NIR photodetectors, placing it among the top 10 of such reported devices. At the same time, this also emphasizes the very real problem with determining these values by dark current only, as this significantly overestimates the real detectivity of these devices. Nevertheless, the current results of these initial non-optimized devices exhibit significant promise and reflect the potential for the future development of NIR photodetectors based on such TP-A frameworks.
Lastly, the deep HOMO and low-lying LUMO of the TP-A frameworks offer the possibility of their application as non-fullerene acceptors in OPV devices. To test this possibility, devices were fabricated from blends of poly(3-hexylthiophene) (P3HT) with either TP-BTD or TP-BTA. While 1:1 devices did produce photocurrent, the overall performance was rather poor. This behaviour was largely due to quite low short circuit currents, arising from poor film morphology, with large crystallites evident in the blend films. It is hypothesised that this morphology problem is again reflective of the rigid nature and structural arching of the molecules, resulting from the intra-molecular hydrogen-bonding shown in the Fig. 6. Still, if these limitations could be overcome, these materials could lead to a new family of NIR-absorbing, non-fullerene acceptors.
Synthetic complexity
Lastly, it is worth addressing the growing issue of synthetic complexity in conjugated polymers. In the pursuit of superior materials with targeted properties, particularly those capable of providing enhanced device performance, high-performance polymers have become increasingly more structurally complex. In fact, many modern materials can be composed of three or more different monomeric units, with the total synthesis requiring multiple sequential steps to control the desired order of monomer connectivity, as well as any potential regio-chemistry involved.13 In comparison, the TP-A materials here are simple alternating copolymers of only two symmetrical monomeric units.In order to allow meaningful comparison of synthetic complexity (SC) within a group of related materials, Po and co-workers defined an SC index in 2015 to estimate the relative cost of synthesized materials in a way that balances key factors that determine the overall cost.70 Factors that determine the SC index includes the number of synthetic steps, the reciprocal synthetic yield, the number of unit operations required for isolation/purification, the number of column chromatographic purifications, and the number of hazardous chemicals involved. Applying the SC index to a group of best performing NIR photodetector polymers results in a SC index of 52.2 for TP-BTA. In comparison, the bulk of previous high-performing materials in such devices have SC index values of 75.1–92.5 (See ESI†). As such, these TP-A not only provide competitive device performance, but do so with a significant reduction in synthetic complexity.
Conclusions
A new design paradigm for low band gap conjugated polymers has been presented in which an ambipolar TP unit incorporating dual donor and acceptor qualities is paired with a traditional acceptor, including 2,1,3-benzothiadiazole (BTD), quinoxaline (Qx), and 2H-benzotriazole (BTA). These materials were prepared via direct arylation polymerization to produce processible materials with Eg values of 1.02–1.12 eV, in which the Eg is dependent on the strength of the traditional acceptor applied.Optical and electrochemical characterization of these materials, along with the model polymer EHTP-Ph, supports the view that the low Eg values of these materials are due to D–A effects in which the TP acts as the donor and both monomers act as acceptors. As a result, this produces materials with deep HOMOs and sufficiently stabilized polymer LUMOs resulting from the hybridization of the LUMOs of both acceptors. At the same time, no evidence could be found that would support the possibility that the lower Eg values could be due to any quinoidal effects in these materials. While the application of these materials to conventional BHJ OPV devices was disappointing, the application of TP-BTA to NIR photodetectors resulted in very competitive specific detectivity (D*(λ)) values and with a material of significantly reduced synthetic complexity. As such, this new design model provides significant promise for the future generation of both successful low Eg materials and their application to NIR photodetectors.
Author contributions
Conceptualization, S. C. R.; methodology, S. C. R., N. N., and P. C. D.; formal analysis, S. C. R., N. N., W. D. W., E. W. C., and S. J. G.; investigation, W. D. W., E. W. C., T. M., and S. J. G; data curation, W. D. W., E. W. C., N. N., T. M., and S. J. G.; writing – original draft preparation, S. C. R.; writing – review and editing, S. C. R., W. D. W., E. W. C., and S. J. G.; visualization, S. C. R.; supervision, S. C. R. and P. C. D.; funding acquisition, S. C. R., and P. C. D.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the National Science Foundation (CHE-2002877) and North Dakota State University for support of this research, as well as Pinjing Zhao for access to his dry box and Daniel Elkington for discussions around the experimental determination of the noise floor of the devices. Portions of this work was performed in part of the materials node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities to Australia's researchers.Notes and references
- S. C. Rasmussen, Conjugated and Conducting Organic Polymers: The First 150 Years, ChemPlusChem, 2020, 85, 1412–1429 CrossRef CAS PubMed.
- S. B. Mainthia, P. L. Kronick, H. Ur, E. F. Chapman and M. M. Labes, Electronic conductivity of complexes of poly-p-phenylene, Polym. Prepr., 1963, 4(1), 208–212 CAS.
- R. McNeill, R. Siudak, J. H. Wardlaw and D. E. Weiss, Electronic Conduction in Polymers, I. The Chemical Structure of Polypyrrole, Aust. J. Chem., 1963, 16, 1056–1075 CrossRef CAS.
- B. A. Bolto and D. E. Weiss, Electronic Conduction in Polymers, II. The Electrochemical Reduction of Polypyrrole at Controlled Potential, Aust. J. Chem., 1963, 16, 1076–1089 CrossRef CAS.
- B. A. Bolto, R. McNeill and D. E. Weiss, Electronic Conduction in Polymers, III. Electronic Properties of Polypyrrole, Aust. J. Chem., 1963, 16, 1090–1103 CrossRef CAS.
- Handbook of Conducting Polymers, ed. J. R. Reynolds, T. A. Skotheim and B. Thompson, CRC Press, Boca Raton, FL, 2019, 4th edn Search PubMed.
- Handbook of Thiophene-based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Hoboken, 2009 Search PubMed.
- C. B. Nielsen and I. McCulloch, Recent advances in transistor performance of polythiophenes, Prog. Polym. Sci., 2013, 38, 2053–2069 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Synthesis of light-emitting conjugated polymers for applications in electroluminescent devices, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Fluorescent thiophene-based materials and their outlook for emissive applications, Chem. Commun., 2015, 51, 4528–4543 RSC.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated polymer-based organic solar cells, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- M. C. Scharber and N. S. Sariciftci, Efficiency of bulk-heterojunction organic solar cells, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- S. C. Rasmussen, S. J. Gilman and W. D. Wilcox, Conjugated Polymers: Synthesis & Design, ACS in Focus, American Chemical Society, Washington DC, 2023 Search PubMed.
- S. C. Rasmussen, S. J. Gilman and W. D. Wilcox, The Eternal Quest for Practical Low Bandgap Polymers, Gen. Chem., 2023, 9, 220010 Search PubMed.
- S. C. Rasmussen, Low-Bandgap Polymers, in Encyclopedia of Polymeric Nanomaterials, ed. K. Muellen and S. Kobayashi, Springer, Heidelberg, 2015, pp. 1155–1166 Search PubMed.
- M. C. Scharber and N. S. Sariciftci, Low Band Gap Conjugated Semiconducting Polymers, Adv. Mater. Technol., 2021, 2000857 CrossRef CAS.
- T. Mikie and I. Osaka, Small-bandgap quinoid-based π-conjugated polymers, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- X. Ji and L. Fang, Quinoidal conjugated polymers with open-shell character, Polym. Chem., 2021, 12, 1347–1361 RSC.
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Low-Bandgap Near-IR Conjugated Polymers/Molecules for Organic Electronics, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- S. C. Rasmussen, S. J. Gilman, E. W. Culver and W. D. Wilcox, Organic NIR Photodetectors: Pushing Photodiodes Beyond 1000 nm, Gen. Chem., 2021, 7, 200019 Search PubMed.
- X. Liu, Y. Lin, Y. Liao, J. Wu and Y. Zheng, Recent advances in organic near-infrared photodiodes, J. Mater. Chem. C, 2018, 6, 3499–3513 RSC.
- Q. Li, Y. Guo and Y. Liu, Exploration of Near-Infrared Organic Photodetectors, Chem. Mater., 2019, 31, 6359–6379 CrossRef CAS.
- P. C. Y. Chow and T. Someya, Organic Photodetectors for Next-Generation Wearable Electronics, Adv. Mater., 2020, 32, 1902045 CrossRef CAS PubMed.
- Z. Wu, Y. Zhai, H. Kim, J. D. Azoulay and T. N. Ng, Emerging Design and Characterization Guidelines for Polymer-Based Infrared Photodetectors, Acc. Chem. Res., 2018, 51, 3144–3153 CrossRef CAS PubMed.
- Y. Chen, Y. Zheng, J. Wang, X. Zhao, G. Liu, Y. Lin, Y. Yang, L. Wang, Z. Tang, Y. Wang, Y. Fang, W. Zhang and X. Zhu, Ultranarrow-bandgap small-molecule acceptor enables sensitive SWIR detection and dynamic upconversion imaging, Sci. Adv., 2024, 10, eadm9631 CrossRef CAS PubMed.
- F. Wudl, M. Kobayashi and A. J. Heeger, Poly(isothianaphthene), J. Org. Chem., 1984, 49, 3382–3384 CrossRef CAS.
- M. Kobayashi, N. Colaneri, M. Boysel, F. Wudl and A. J. Heeger, The electronic and electrochemical properties of poly(isothianaphthene), J. Chem. Phys., 1985, 82, 5717–5723 CrossRef CAS.
- J. L. Bredas, A. J. Heeger and F. Wudl, Towards organic polymers with very small intrinsic band gaps. I. Electronic structure of polyisothianaphthene and derivatives, J. Chem. Phys., 1986, 85, 4673–4678 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, A new class of small band gap organic polymer conductors, Polym. Bull., 1992, 29, 119–126 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Alternate donor-acceptor small-band-gap semiconducting polymers: Polysquaraines and Polycroconaines, Synth. Met., 1993, 55, 299–306 CrossRef CAS.
- S. J. Evenson, M. E. Mulholland, T. E. Anderson and S. C. Rasmussen, Minimizing Polymer Band Gap via Donor-Acceptor Frameworks: Poly(dithieno[3,2-b:2′,3′-d]pyrrole-alt-thieno[3,4-b]pyrazine)s as Illustrative Examples of Challenges and Misconceptions, Asian J. Org. Chem., 2020, 9, 1333–1339 CrossRef CAS.
- T. E. Anderson, E. W. Culver, I. Badía-Domínguez, W. D. Wilcox, C. E. Buysse, M. C. R. Delgado and S. C. Rasmussen, Probing the nature of donor–acceptor effects in conjugated materials: a joint experimental and computational study of model conjugated oligomers, Phys. Chem. Chem. Phys., 2021, 23, 26534–26546 RSC.
- A. D. Thilanga Liyanage, B. Milián-Medina, B. Zhang, J. Gierschner and M. D. Watson, ¿Conjugated? Copolymers from a Pechmann Dye Derivative, Macromol. Chem. Phys., 2016, 217, 2068–2073 CrossRef CAS.
- B. P. Karsten, L. Viani, J. Gierschner, J. Cornil and R. A. J. Janssen, An Oligomer Study on Small Band Gap Polymers, J. Phys. Chem. A, 2008, 112, 10764–10773 CrossRef CAS PubMed.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Thieno[3,4-b]pyrazines and their applications to low band gap organic materials, Chem. Commun., 2011, 47, 11394–11410 RSC.
- D. D. Kenning, K. A. Mitchell, T. R. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, Thieno[3,4-b]pyrazines: Synthesis, Structure, and Reactivity, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS PubMed.
- S. C. Rasmussen, D. J. Sattler, K. A. Mitchell and J. Maxwell, Photophysical characterization of 2,3-difunctionalized thieno[3,4-b]pyrazines, J. Lumin., 2004, 190, 111–119 CrossRef.
- L. Wen, J. P. Nietfeld, C. M. Amb and S. C. Rasmussen, Synthesis and Characterization of New 2,3-Disubstituted Thieno[3,4-b]pyrazines: Tunable Building Blocks for Low Band Gap Conjugated Materials, J. Org. Chem., 2008, 73, 8529–8536 CrossRef CAS PubMed.
- S. C. Rasmussen, M. E. Mulholland, R. L. Schwiderski and C. A. Larsen, Thieno[3,4-b]pyrazines and Its Extended Analogues: Important Buildings Blocks for Conjugated Materials, J. Heterocyclic Chem., 2012, 49, 479–493 CrossRef CAS.
- L. Wen, C. L. Heth and S. C. Rasmussen, Thieno[3,4-b]pyrazine-based Oligothiophenes: Simple Models of Donor-Acceptor Polymeric Materials, Phys. Chem. Chem. Phys., 2014, 16, 7231–7240 RSC.
- E. W. Culver, T. E. Anderson, J. T. L. Navarrete, M. C. R. Delgado and S. C. Rasmussen, Poly(thieno[3,4-b]pyrazine-alt-2,1,3-benzothiadiazole)s: A New Design Paradigm in Low Band Gap Polymers, ACS Macro Lett., 2018, 7, 1215–1219 CrossRef CAS PubMed.
- T. E. Anderson, E. W. Culver, F. Almyahi, P. C. Dastoor and S. C. Rasmussen, Poly(2,3-dihexylthieno[3,4-b]pyrazine-alt-2,3-dihexylquinoxaline): Processible, Low Bandgap, Ambipolar-Acceptor Frameworks via Direct Arylation Polymerization, Synlett, 2018, 2542–2546 CAS.
- Y. Wang and T. Michinobu, Benzothiadiazole and its π-extended, heteroannulated derivatives: useful acceptor building blocks for high-performance donor-acceptor polymers in organic electronics, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- J. Du, M. C. Biewer and M. C. Stefan, Benzothiadiazole building units in solution-processable small molecules for organic photovoltaics, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- J. Yuan, J. Ouyang, V. Cimrova, M. Leclerc, A. Najarid and Y. Zou, Development of quinoxaline based polymers for photovoltaic applications, J. Mater. Chem. C, 2017, 5, 1858–1879 RSC.
- D. Gedefaw, M. Prosa, M. Bolognesi, M. Seri and M. R. Andersson, Recent Development of Quinoxaline Based Polymers/Small Molecules for Organic Photovoltaics, Adv. Energy Mater., 2017, 7, 1700575 CrossRef.
- M. Liu, Y. Gao, Y. Zhang, Z. Liu and L. Zhao, Quinoxaline-based conjugated polymers for polymer solar cells, Polym. Chem., 2017, 8, 4613–4636 RSC.
- I. Torres-Moya, R. Vázquez-Guilló, S. Fernández-Palacios, J. R. Carrillo, Á. Díaz-Ortiz, J. T. L. Navarrete, R. P. Ortiz, M. C. R. Delgado, R. Mallavia and P. Prieto, Fluorene-Based Donor-Acceptor Copolymers Containing Functionalized Benzotriazole Units: Tunable Emission and their Electrical Properties, Polymers, 2020, 12, 256 CrossRef CAS PubMed.
- J. Hu, X. Wang, F. Chen, B. Xiao, A. Tang and E. Zhou, Medium Bandgap D-A Type Photovoltaic Polymers Based on an Asymmetric Dithienopyran Donor and a Benzotriazole Acceptor, Polymers, 2017, 9, 516 CrossRef PubMed.
- J. P. Nietfeld, R. L. Schwiderski, T. P. Gonnella and S. C. Rasmussen, Structural Effects on the Electronic Properties of Extended Fused-ring Thieno[3,4-b]pyrazine Analogues, J. Org. Chem., 2011, 76, 6383–6388 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Direct (Hetero)Arylation: A New Tool for Polymer Chemists, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupre and M. Leclerc, Direct (Hetero)arylation Polymerization: Simplicity for Conjugated Polymer Synthesis, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- T. Bura, J. T. Blaskovits and M. Leclerc, Direct (Hetero)arylation Polymerization: Trends and Perspectives, J. Am. Chem. Soc., 2016, 138, 10056–10071 CrossRef CAS PubMed.
- P.-O. Morin, T. Bura and M. Leclerc, Realizing the full potential of conjugated polymers: innovation in polymer synthesis, Mater. Horiz., 2016, 3, 11–20 RSC.
- T. Bura, S. Beaupré, M.-A. Légaré, J. Quinn, E. Rochette, J. T. Blaskovits, F.-G. Fontaine, A. Pron, Y. Li and M. Leclerc, Direct heteroarylation polymerization: guidelines for defect-free conjugated polymers, Chem. Sci., 2017, 8, 3913–3925 RSC.
- L. Ye and B. C. Thompson, Improving the efficiency and sustainability of catalysts for direct arylation polymerization (DArP), J. Polym. Sci., 2022, 60, 393–428 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Spectral Engineering in π-Conjugated Polymers with Intramolecular Donor−Acceptor Interactions, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. David Stockdale and G. C. Bazan, Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- C. M. Amb, P. M. Beaujuge and J. R. Reynolds, Spray-Processable Blue-to-Highly Transmissive Switching Polymer Electrochromes via the Donor–Acceptor Approach, Adv. Mater., 2010, 22, 724–728 CrossRef CAS PubMed.
- R. Kroon, R. Gehlhaar, T. T. Steckler, P. Henriksson, C. Müller, J. Bergqvist, A. Hadipour, P. Heremans and M. R. Andersson, New quinoxaline and pyridopyrazine-based polymers for solution-processable photovoltaics, Sol. Energy Mater. Sol. Cells, 2012, 105, 280–286 CrossRef CAS.
- L. Wen, B. C. Duck, P. C. Dastoor and S. C. Rasmussen, Poly(2,3-dihexylthieno[3,4-b]pyrazine) via GRIM Polymeriza-tion: The Simple Preparation of a Solution Processable, Low Band Gap Conjugated Polymer, Macromolecules, 2008, 41, 4576–4578 CrossRef CAS.
- B. C. Duck, B. Vaughan, L. Wen, C. L. Heth, S. C. Rasmussen, X. Zhou, W. J. Belcher and P. C. Dastoor, Probing the structure–function relationship in pC6TP:PCBM based organic photonic devices, Sol. Energy Mater. Sol. Cells, 2013, 110, 8–14 CrossRef CAS.
- N. V. Vasilieva, I. G. Irtegovaa, N. P. Gritsan, A. V. Lonchakov, A. Y. Makarova, L. A. Shundrina and A. V. Zibarev, Redox properties and radical anions of fluorinated 2,1,3-benzothia(selena)diazoles and related compounds, J. Phys. Org. Chem., 2010, 23, 536–543 CrossRef CAS.
- P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, Accurate Oxidation Potentials of Benzene and Biphenyl Derivatives via Electron-Transfer Equilibria and Transient Kinetics, J. Org. Chem., 2009, 74, 5163–5173 CrossRef CAS PubMed.
- U. Salzner, O. Karalti and S. Durdagi, Does the donor-acceptor concept work for designing synthetic metals? III. Theoretical investigation of copolymers between quinoid acceptors and aromatic donors, J. Mol. Model., 2006, 12, 687–701 CrossRef CAS PubMed.
- M. Kertesz, S. Yang and Y. Tian, Energy gaps and their control in thiophene-based polymers and oligomers, in Handbook of Thiophene-Based Materials, ed. I. F. Perepichka and D. F. Perepichka, John Wiley & Sons, Chichester, UK, 2009, vol. 1, pp. 341–364 Search PubMed.
- P. Ou, W. Shen, R. He, X. Xie, C. Zeng and M. Li, Molecular design and density functional theory investigation of novel low-band-gap copolymers between quinoid acceptors and aromatic donors, Polym. Int., 2011, 60, 1408–1418 CrossRef CAS.
- J. Kurti and P. R. Surjan, Quinoid vs aromatic structure of polyisothianaphthene, J. Chem. Phys., 1990, 92, 3247–3248 CrossRef CAS.
- P. C. Dastoor, C. R. McNeill, H. Frohne, C. J. Foster, B. Dean, C. J. Fell, W. J. Belcher, W. M. Campbell, D. L. Officer, I. M. Blake, P. Thordarson, M. J. Crossley, N. S. Hush and J. R. Reimers, Understanding and Improving Solid-State Polymer/C60-Fullerene Bulk-Heterojunction Solar Cells Using Ternary Porphyrin Blends, J. Phys. Chem. C, 2007, 111, 15415–15426 CrossRef CAS.
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, “All that glisters is not gold”: An analysis of the synthetic complexity of efficient polymer donors for polymer solar cells, Macromolecules, 2015, 48, 453–461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic methods, device fabrication and characterization, and synthetic complexity comparisons. See DOI: https://doi.org/10.1039/d5tc00519a |
| This journal is © The Royal Society of Chemistry 2025 |