
Perylene bisimide J-aggregates in a polymer matrix: controlling self-assembly and fluorescence properties†‡
Rikuto
Kanno
 ac,
Matthias
Stolte
ab,
Vladmir
Stepanenko
ab,
Ann-Christin
Pöppler
ab,
Takaya
Terashima
c and
Frank
Würthner
*ab
ac,
Matthias
Stolte
ab,
Vladmir
Stepanenko
ab,
Ann-Christin
Pöppler
ab,
Takaya
Terashima
c and
Frank
Würthner
*ab
aCenter for Nanosystems Chemistry (CNC) and Bavarian Polymer Institute (BPI), Universität Würzburg, Theodor-Boveri-Weg, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bInstitut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto, 615-8510, Japan
First published on 18th February 2025
Abstract
Perylene bisimide J-aggregates are among the most emissive dye aggregates. However, their fluorescence quantum yield severely decreases if cast as pristine materials in solid films. Here, we present an approach to prepare solid-state materials containing fluorescent perylene bisimide (PBI) J-aggregates with fluorescence quantum yields up to 28%, which is significantly higher than in their single-crystalline state. In our research, we investigated the impact of different polymer matrices, i.e. polystyrene, poly(methyl methacrylate) and a polystyrene–polybutadiene–polystyrene triblock copolymer, on the self-assembly into J-aggregates and the fluorescence quantum yield of the dye-matrix materials for spin-coated films. The degree of self-assembly of PBIs could be controlled by the PBI concentration, the type of polymer matrix, and an annealing process after spin-coating. Notably, the J-aggregates in a polystyrene matrix showed significant differences in their fluorescence quantum yield, which we attribute to transitions between individual one-dimensional fibers and the formation of larger three-dimensional crystallites.
Research on dye aggregates has often been pursued either in solution (“self-assembly”) or in thin films for their bulk materials. In contrast, studies on the formation of dye aggregates in polymer matrices are less common with the exception of a few very popular fields such as dye-polymer blends applied in organic photovoltaics, where in recent years in particular the groups of Köhler and Stingelin provided important insights with temperature-dependent optical spectroscopies.1 However, it is not clarified if the same principles established for self-assembly for dyes in solution (concentration, temperature, solvent polarity2) also drive the formation of dye aggregates in polymer matrices. In this regard, we would first like to refer to the inspiring review written by Ringsdorf and co-workers in 19883 on “Molecular Architectures and Functions of Polymeric Oriented Systems”, where they discussed the utility of supramolecular systems as models for gaining fundamental understanding as well as for applications in both materials and life sciences. Second, we would like to refer to J-aggregates, which are among the first examples of self-assembled supramolecular matter discovered by Scheibe and Jelley in the late 1930s.4,5 J-aggregates describe a self-assembled state of organic chromophores where the molecules are arranged in a slip-stacked manner through non-covalent interactions. Initial examples were formed by less directional π–π stacking, and dispersion interaction but in the last two decades also examples for supramolecular engineering by hydrogen bonding were described.4–9 The specific slip-stacked aggregation mode leads to strong exciton coupling between neighboring chromophores, resulting in characteristic properties such as narrow and bathochromically shifted absorption bands compared to their monomeric states, small Stokes shift for a narrow fluorescence band, efficient exciton transport along the aggregate, as well as often high fluorescence quantum yields.10 In nature, some of these properties are also found in light-harvesting antennas of green sulfur bacteria, where J-aggregates of bacteriochlorophyll c effectively transport energy captured from sunlight to the reaction centre.11 Accordingly, J-aggregates have received significant attention as promising candidates for applications in organic optoelectronic devices.12–15
In our previous research, we have reported that perylene bisimide (PBI) dyes having hydrogen atoms at the imide position and four bulky phenoxy substituents in the bay area self-assemble into helical fiber-like J-aggregates driven by slipped π–π stacking and complementary hydrogen bonding both in solution and the solid state.6,16–22 While our initial design required solubilizing alkyl side chains to render PBIs soluble in conventional organic solvents, introducing o-methyl phenoxy substituents at the bay area also endowed the otherwise rather insoluble PBI pigments with notable solubility even in the absence of long alkyl residues.18 Thus, the light-weight PBI derivative 1 (Fig. 1a) showed excellent solubility at elevated temperature but self-assembled into nanofibers with high photoluminescence quantum yields (ΦPL) in its J-aggregate state in solution, reaching values as high as 80%. Elimination of bulky side chains enabled us to elucidate the packing of 1 in single crystals by X-ray diffraction and to characterize 1 as double-stranded J-aggregates (Fig. 1a). However, to our disappointment, ΦPL of the J-aggregates decreases significantly in its crystalline state to only a few percent.21 As a consequence, J-aggregates of PBI 1 were so far not yet considered for a variety of applications, such as for tuning light–matter interaction in optical microcavities and polariton lasing that we currently aim for in our collaborative research.23–25 If the poor ΦPL value could be improved for an active layer, many future applications of J-aggregates of PBI 1 could be imagined, as recently demonstrated for cyanine dye-based J-aggregates in polymer matrices.26–28
 | ||
| Fig. 1 Fabrication of PBI J-aggregates in rigid polymer matrices by spin-coating from CHCl3 solution. (a) The chemical structure of PBI 1 and the structure of the J-aggregates formed by 1 in single crystals.21 (b) Chemical structures of polymers employed in the present study (PMMA, PSt, SBS triblock copolymer). (c) UV-Vis absorption spectra of thin films on glass substrates obtained by spin-coating a CHCl3 solution of PMMA (c0[Polymer] = 60 mg mL−1) with different amounts of 1 (c0[1] = 0.2–1.5 mg mL−1). Scaled absorption spectra of PBI 1 in solution as a monomer (red dashed line; CH2Cl2) and J-aggregate (blue dashed line, methyl-cyclohexane) are provided as a reference. Insets: Plots of absorbance of 1 at λabs = 643 nm in PMMA matrices scaled by c0[1] as well as the photo scans of the respective layers. | ||
In this article we focus our efforts on using polymer matrices to exploit J-aggregates of 1 in solid state hybrid materials as an alternative to their pristine solid-state materials. Thus, instead of enwrapping PBI J-aggregates with multiple solubilizing alkyl chains, we embedded 1 in polymer matrices by the conventional spin-coating technique and investigated their self-assembly behavior as well as the resultant optical properties. As we will show in the following, we could not only control the degree of J-aggregate formation by the PBI content, the choice of the respective polymer matrix and an annealing process, but also improve ΦPL of those polymer-embedded J-aggregates up to 28% compared to 2% for the pristine single crystal material.21 Although a variety of dye/polymer hybrid materials have already been reported aiming at organic laser devices,29–34 this is the first report on PBI-based J-aggregates embedded in rigid polymer matrices along with their photophysical properties.
Thin layers composed of a polymer matrix containing PBI 1 were prepared as follows: PBI 1 was dissolved in chloroform (CHCl3) combined with different amounts of one of the three polymer species poly(methyl methacrylate) (PMMA, Mw = 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), polystyrene (PSt, Mw = 35000 g mol−1), or a polystyrene–polybutadiene–polystyrene triblock copolymer (SBS, Mw = 140000 g mol−1, polystyrene block: 30 wt%). PMMA and PSt were chosen as glassy polymers with relatively high glass-transition temperatures (Tg, ∼100 °C), while SBS as a thermoplastic elastomer. All chemical structures are depicted in Fig. 1a and b, while Fig. 1a also shows the supramolecular structure of 1 known from our previous single crystal X-ray analysis.21 While the reddish PBI/polymer solutions in CHCl3 are predominantly composed of monomers and only contain small amounts of J-aggregates at the highest c0[1] of 1.5 mg mL−1 at room temperature (Fig. S1, ESI‡), spin-coating on glass substrates strongly induced the formation of blue J-aggregates in varying amounts depending on the composition of the sample and polymer species. Although spin-coating is a fast process, the steep concentration increase during thin-film formation can drive PBI 1 towards self-assembly, pending on c0[1] and the surrounding matrix material. For instance, the 1/PMMA mixtures resulted in J-aggregates only at higher PBI concentration (c0[1] > 0.6 mg mL−1), while those at lower PBI amount (c0[1] < 0.6 mg mL−1) remained in the monomeric state. Such a trend was clearly confirmed by concentration-dependent solid-state UV-Vis absorption spectra: the main absorption maximum (λabs) of the layers exhibited a bathochromic-shift of about 100 nm (2000 cm−1) from 569 nm (0.4 mg mL−1) up to 643 nm (1.5 mg mL−1) in accordance with the PBI concentration, which is typical for J-aggregate formation of 1 (Fig. 1c). Assuming in first approximation a constant layer thickness (volume) for a fixed c0[PMMA], the absorption spectra scaled by c0[1] showed a quasi isosbestic point around λiso = 580 nm. Additionally, the evolution of the maximum absorption intensity at λabs = 643 nm showed a non-sigmoidal curve when plotted against the PBI concentration (Fig. 1c inset, Fig. S1b, ESI‡). These results imply that 1 forms J-aggregates in PMMA matrices in a nucleation-elongation model as it does in solution:181 may form the dimer nuclei by intermolecular interaction among contorted π-surfaces, and then elongate into J-aggregate fibers through hydrogen bonding at the imide position.
000 g mol−1), polystyrene (PSt, Mw = 35000 g mol−1), or a polystyrene–polybutadiene–polystyrene triblock copolymer (SBS, Mw = 140000 g mol−1, polystyrene block: 30 wt%). PMMA and PSt were chosen as glassy polymers with relatively high glass-transition temperatures (Tg, ∼100 °C), while SBS as a thermoplastic elastomer. All chemical structures are depicted in Fig. 1a and b, while Fig. 1a also shows the supramolecular structure of 1 known from our previous single crystal X-ray analysis.21 While the reddish PBI/polymer solutions in CHCl3 are predominantly composed of monomers and only contain small amounts of J-aggregates at the highest c0[1] of 1.5 mg mL−1 at room temperature (Fig. S1, ESI‡), spin-coating on glass substrates strongly induced the formation of blue J-aggregates in varying amounts depending on the composition of the sample and polymer species. Although spin-coating is a fast process, the steep concentration increase during thin-film formation can drive PBI 1 towards self-assembly, pending on c0[1] and the surrounding matrix material. For instance, the 1/PMMA mixtures resulted in J-aggregates only at higher PBI concentration (c0[1] > 0.6 mg mL−1), while those at lower PBI amount (c0[1] < 0.6 mg mL−1) remained in the monomeric state. Such a trend was clearly confirmed by concentration-dependent solid-state UV-Vis absorption spectra: the main absorption maximum (λabs) of the layers exhibited a bathochromic-shift of about 100 nm (2000 cm−1) from 569 nm (0.4 mg mL−1) up to 643 nm (1.5 mg mL−1) in accordance with the PBI concentration, which is typical for J-aggregate formation of 1 (Fig. 1c). Assuming in first approximation a constant layer thickness (volume) for a fixed c0[PMMA], the absorption spectra scaled by c0[1] showed a quasi isosbestic point around λiso = 580 nm. Additionally, the evolution of the maximum absorption intensity at λabs = 643 nm showed a non-sigmoidal curve when plotted against the PBI concentration (Fig. 1c inset, Fig. S1b, ESI‡). These results imply that 1 forms J-aggregates in PMMA matrices in a nucleation-elongation model as it does in solution:181 may form the dimer nuclei by intermolecular interaction among contorted π-surfaces, and then elongate into J-aggregate fibers through hydrogen bonding at the imide position.
In contrast to PMMA, instant J-aggregate formation was induced even at lower PBI concentrations in PSt and SBS matrices as becomes evident from the lack of any monomer absorption at 569 nm (Fig. S1c and d, ESI‡). This tendency was also confirmed when we reduced the polymer concentration in CHCl3 solutions before spin-coating from 60 to 15 mg mL−1 with respect to c0[1] at 1.5 mg mL−1: while for PMMA the J-aggregate formation is enhanced upon decreasing c0[PMMA], 1 fully self-assembles into J-aggregates in PSt and SBS independent of polymer concentration (Fig. S2a–c, ESI‡). These results might be attributed to the carbonyl-rich structure of PMMA: The ester moieties on its repeating units can compete as H-bonding acceptors for the imide NH groups of 1 required to form J-aggregates. Surprisingly, the optical density of a spin-coated 1/SBS layer was significantly higher (0.3) than those of PMMA or PSt ones (0.13), probably because SBS solutions before spin-coating were more viscous than the other two polymers and resulted in thicker layers: the layer thickness of the SBS sample was 2300 nm, while those of the PMMA and PSt samples were around 800 nm (Fig. S2d, ESI‡). The effects of polymer molecular weight were also investigated: as the molecular weight of PMMA increased, the optical density of 1 J-aggregates in PMMA matrices got higher (Fig. S3a and c, ESI‡). Since the normalized absorbance spectra showed almost the same band shape, polymer molecular weight seems to have no effects on the degree of J-aggregate formation (Fig. S3b, ESI‡).
Different from absorption properties, the investigated polymer matrices significantly affected the emission properties of the PBI J-aggregates. Fig. 2a and b show the emission and excitation spectra of 1 in PMMA, PSt, and SBS matrices at the lowest and highest concentration. In PMMA matrices, monomer-like emission was observed at a lower PBI concentration (c0[1] = 0.4 mg mL−1), while increasing c0[1] to 1.5 mg mL−1 and decreasing c0[PMMA] to 15 mg mL−1 afforded almost complete J-aggregate emission at λem = 669 nm. The full-width at half maximum (FWHM) values got narrower from 2250 cm−1 of the monomer emission band down to 1250 cm−1 for the J-aggregate, which is a typical value for 1.18,21 In contrast, the PSt and SBS samples showed predominantly or even solely J-aggregate emission independent of c0[1], which agrees with the results of the absorption spectra. The major component of the fluorescence lifetime of the monomer 1 in PMMA (5.1 ns) was as expected from solution about 4.5 ns, while that of J-aggregates in the SBS matrix got significantly shorter to 1.2 ns (Fig. 2b inset). Interestingly, these fluorescence lifetimes of monomeric as well as J-aggregated species of PBI 1 were almost independent of the difference of the polymer matrices. The details of emission properties are summarized in Table 1. For the J-aggregates in different polymer matrices, absolute photoluminescence quantum yields (ΦPL) were determined with an integrated sphere setup (Fig. S4, ESI‡). In particular, the ΦPL value of J-aggregates in a PSt matrix was close to 30%, which is much higher than that of the respective PMMA layer (9%) and the solid crystalline state (2%) of 1.21 Notably, the reference PBI with benzylated imide groups which does not form J-aggregates showed almost identical fluorescence spectra and quantum yields in the three polymer matrices (Fig. S5 and Table S1, ESI‡).
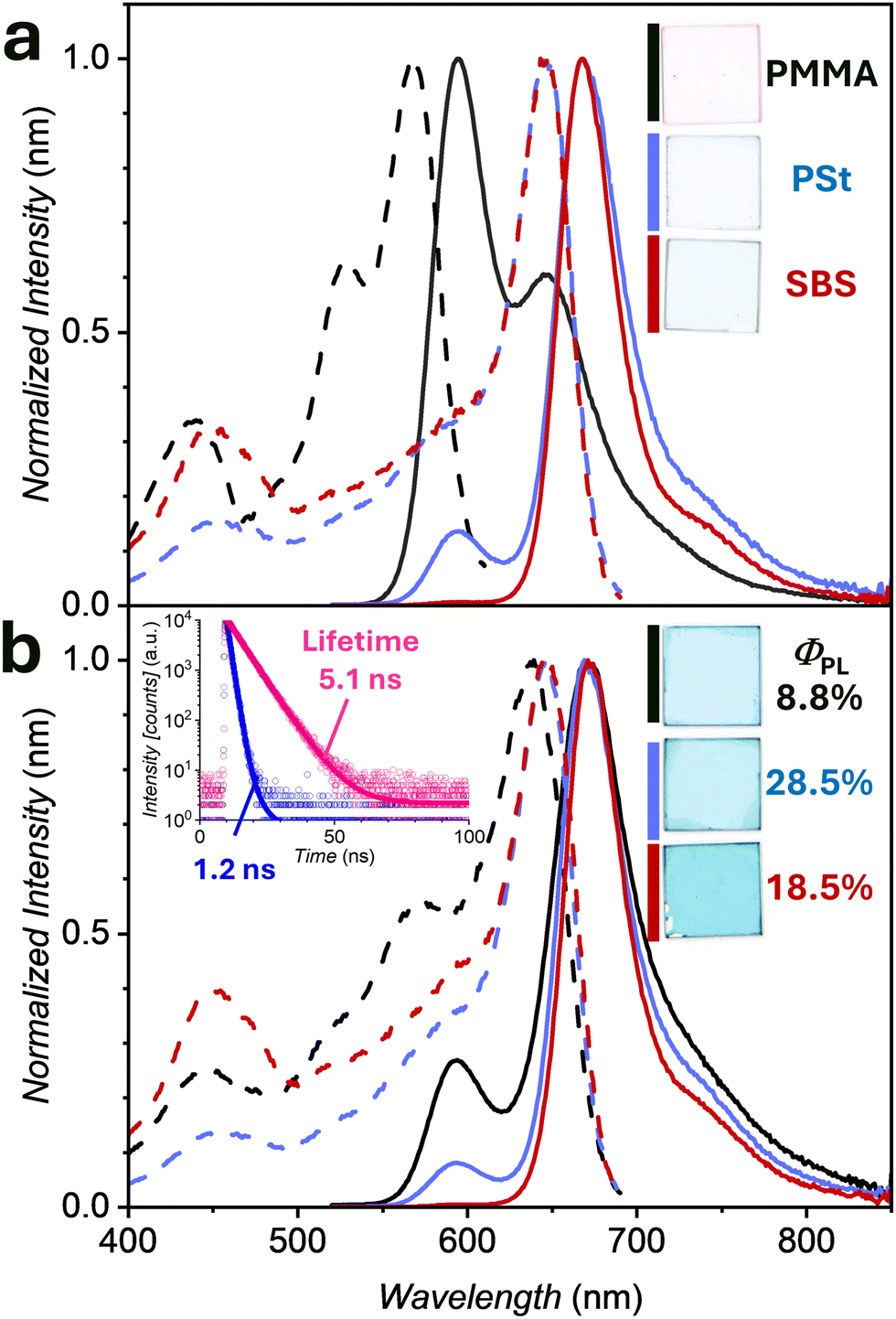 | ||
| Fig. 2 Normalized thin-film excitation (dashed lines) as well as respective emission spectra (solid lines) of spin-coated polymer matrices PMMA (black), PSt (blue) and SBS (red) containing different amounts of 1 (a/b: c0[1] = 0.4/1.5 mg mL−1) on glass substrates (c0[Polymer] = 60 mg mL−1, except PMMA in (b) where it is 15 mg mL−1). The insets in (a) and (b) display the photo scans (partially with ΦPL) of the respective layers as well as the fluorescence decay curves (open symbols) of monomeric (red) as well as J-aggregates (blue) 1 in PMMA (c0[1] = 0.4 mg mL−1) or SBS (c0[1] = 1.5 mg mL−1) under excitation at λex = 479.7 nm along with the respective fits (solid line). | ||
| Polymer | c 0[1]a [mg mL−1] | c 0[Polymer] [mg mL−1] | λ abs [nm] | λ em [nm] | FWHM [cm−1] | Φ PL [%] |
|---|---|---|---|---|---|---|
| a Concentration of 1 and polymers in the CHCl3 solutions for spin-coating. b λ ex = 460 nm. c Absolute quantum yields measured with λex = 445 nm without correction for reabsorption, which were averaged over at least three independent samples. d Cited from ref. 21. | ||||||
| PMMA | 1.5 | 15 | 641 | 669 | 1250 | 8.8 ± 0.4 |
| 1.5 | 60 | 573, 643 | 593, 666 | 1300 | — | |
| 0.4 | 541 | 595 | 2250 | 39.3 ± 2.3 | ||
| PSt | 1.5 | 60 | 647 | 669 | 1030 | 28.5 ± 0.6 |
| 0.4 | 647 | 667 | 1040 | 25.9 ± 0.5 | ||
| SBS | 1.5 | 60 | 650 | 671 | 900 | 18.5 ± 0.3 |
| 0.4 | 648 | 668 | 910 | 34.2 ± 2.4 | ||
| — | Single crystald | — | 641 | 685 | — | 2.0 |
Given that the ΦPL dependence on polymer matrix could be either attributed to the structural difference of the J-aggregate or the environment provided by the different polymers, we next analyzed the structural features of 1 J-aggregates in the polymer matrices by atomic force microscopy (AFM) and (photoluminescence) polarized optical microscopy ((PL-)POM) measurements. The close-up AFM height images show fiber-like morphologies in all three polymer-based layers containing J-aggregates of PBI 1 (Fig. 3, right). The height of those fibers permeating out of the smooth polymer layers is around 2 nm, which is a little bit larger than that of isolated aggregates deposited from solution (∼1.4 nm).18 This increase originates probably from the fact that J-aggregate fibers are partially covered by polymer chains close to the surface of the matrix layer. However, large surface area AFM as well as POM measurements exhibited a clear difference of J-aggregate structure depending on the respective polymer species. While PSt samples did not show any indication for phase separation between the matrix and PBI 1, micrometre-sized needle-like crystallites can be identified in the PMMA as well as SBS layers (Fig. 3 and Fig. S6, ESI‡). These needles are quite similar in shape to the ones of the single crystals of 1 confirmed in our previous report.18,21 These results offer a hypothesis to explain the structures of 1 J-aggregates in different polymer matrices as follows: Since PSt has good interaction with π-surfaces of 1 but does not disrupt hydrogen bonding, this polymer allows 1 to form well-solubilized one-dimensional double-stranded fibers without three-dimensional crystallites (compare structures shown in Fig. 1a). In contrast, PMMA and SBS may additionally induce further crystallization of these J-aggregate fibers because they exhibit less interactions with π-surfaces than PSt for solubilization. Given that the single crystals of 1 showed quite a low ΦPL (∼2%), the lower value of 1 in the PMMA and SBS layers than that of the PSt ones could be attributed to the small crystal-like three-dimensional structures of J-aggregates in these two matrices. In the solid state the fast exciton diffusion in such J-type coupled chromophores may lead to non-radiative relaxation on unavoidable trap sites in addition to other excited-state relaxation pathways (e.g. symmetry-breaking charge separation, singlet fission).
 | ||
| Fig. 3 Structural analysis on J-aggregates of PBI 1 in different polymer matrices. AFM height images of J-aggregates of 1 that emerge from (a) PSt, (b) PMMA, and (c) SBS matrices (c0[1] = 1.5 mg mL−1; c0[Polymer] = 60 mg mL−1). The white dotted circles highlight representative micrometre-sized crystalline domains, while the white arrows indicate the permeation of J-aggregates of 1 through the polymer layers. | ||
While the PBI concentration as well as the choice of polymer matrices affected the degree of J-aggregate formation, we were also able to control the self-assembly of 1 by annealing the polymer matrix. When we conducted solvent vapor annealing (SVA) by CHCl3 at RT for 5 min on the PMMA sample containing monomeric 1, we could observe a color change from red to blue (Fig. 4a, inset). Both the absorption and emission spectra showed the bathochromic-shifted peak, clearly demonstrating J-aggregate formation (Fig. 4a). AFM pictures also confirmed the appearance of the fiber-like morphology after SVA (Fig. 4c). Time-dependent annealing experiments revealed that five-minute SVA is enough to complete the monomer-to-aggregate transition, and further annealing did not make any difference (Fig. S7b and c, ESI‡). Neither the layer thickness nor the weight of the PMMA matrix changed before and after SVA, indicating that no CHCl3 molecules were adsorbed in the sample after softening the matrix (Fig. S7d, ESI‡). Thus, we conclude that the originally observed embedment of monomeric PBIs within PMMA is not due to the thermodynamically disfavored formation of J-aggregates but due to a kinetic trapping of monomers by being hydrogen-bonded to PMMA. As shown in the model in Fig. 4e, we can indeed relate this inhibition of supramolecular polymerization to related ones recently studied by us and others for a variety of organogelator molecules where the supramolecular polymerization could be controlled by intramolecular hydrogen bonds as depicted in Fig. 4f.35–37
 | ||
| Fig. 4 Effects of solvent vapor annealing (SVA) and thermal annealing (ΔT) on J-aggregate formation of 1 in different polymer matrices. (a) and (b) UV-Vis absorption (dashed) and scaled emission (solid, λex = 460 nm) spectra of thin films along with the respective photo scans of 1 in (a) PMMA or (b) SBS matrices before (red) and after (blue) annealing (SVA: CHCl3, 5 min; 20 °C. ΔT: 150 °C, 30 min). (c) and (d) Respective AFM height images before (left) and after (right) the annealing process. (e) Suggested structural model for the kinetic trapping of a 1 monomer by hydrogen bonding to a PMMA matrix based on the comparison with (f) a kinetically trapped PBI monomer studied for seed-induced living supramolecular polymerizations.35 | ||
In contrast, when the J-aggregates in the SBS matrix were thermally annealed (ΔT) at 150 °C (above the Tg of the polystyrene block, ∼100 °C) for 30 min, the color changed from blue to red, and absorption and emission bands were blue-shifted to the monomeric state (Fig. 4b). Concomitantly, fiber-like morphologies completely disappeared after ΔT on the AFM images (Fig. 4d). These results corroborate the disassembly of the PBI J-aggregates in the SBS matrix by thermal treatment, suggesting monomeric dyes to be the thermodynamically favored species in this environment. At this stage, taking into consideration that 1 J-aggregates did not disappear in the PSt matrix after the same thermal treatment (Fig. S9, ESI‡), we cannot provide a proper explanation for this peculiar behavior in the block copolymer matrix. What we can note is the formation of round-shaped domains (Fig. 4d) that are attributed to the phase-separated structure of the SBS block copolymer.38 Thus, disassembly of J-aggregates seems to be induced by the phase separation of the SBS block copolymer, rather than simply by the heat stimuli.
Conclusions
In conclusion, we successfully fabricated polymer layers containing J-aggregates of PBI 1 just by spin-coating a blend of PBI/polymer from solutions on glass substrates. Different polymer matrices affected the self-assembled states and the emission properties of 1 as follows: The PSt matrix strongly supported J-aggregate formation, leading to almost complete conversion from monomers to aggregates even at low c0[1] and with excellent photoluminescence quantum yields of 28%, which is much higher than that of the same PBI in its neat single-crystalline state. Based on our AFM studies, this result is attributed to the presence of (mostly) individual dual-stranded PBI chains. For the other polymer matrices, the situation was more complex as either kinetically trapped monomers or larger three-dimensional crystallites with a more quenched fluorescence were observed. Solvent vapor annealing on PMMA matrix samples revealed a transition from kinetically trapped 1 into J-aggregates while thermal annealing for PBI J-aggregates dissolved in a SBS matrix induced their disassembly into monomers. Thus, this study provides fundamental insights into the formation of PBI J-aggregates in polymeric matrices and the impact of the respective matrix on their fluorescence properties. These results are of considerable importance for the use of these and other dye aggregates in optoelectronic applications.Author contributions
Frank Würthner suggested the project, involving Ann-Christin Pöppler, Matthias Stolte, Takaya Terashima and Rikuto Kanno in the conceptualization of the executed research. Rikuto Kanno (most studies) and Vladimir Stepanenko (AFM studies) conducted all experimental studies under the supervision of Matthias Stolte and Frank Würthner. All authors contributed to the writing and editing of the manuscript and agreed on the final version.Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the framework of IRTG2991 “Photoluminescence in Supramolecular Matrices”, Project number 517122340. R. K. appreciates “JSPS Overseas Challenge Program for Young Researchers” for the financial support.Notes and references
- F. Panzer, M. J. Dyson, H. Bakr, S. Wedler, K. Schötz, M. Chauhan, P. N. Stavrinou, A. Köhler and N. Stingelin, Adv. Funct. Mater., 2024, 34, 2314729 CrossRef CAS.
- Y. Vonhausen and F. Würthner, Chem. – Eur. J., 2023, 29, e202300359 CrossRef CAS PubMed.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed. Engl., 1988, 27, 113–158 CrossRef.
- E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS.
- G. Scheibe, Angew. Chem., 1937, 50, 212–219 CrossRef CAS.
- M. Hecht and F. Würthner, Acc. Chem. Res., 2021, 54, 642–653 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- S. Yagai, Bull. Chem. Soc. Jpn., 2015, 88, 28–58 CrossRef.
- Z. Chen and Z. Chen, Org. Chem. Front., 2023, 10, 2581–2602 RSC.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- X. Hu, A. Damjanović, T. Ritz and K. Schulten, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5935–5941 CrossRef CAS PubMed.
- J. H. Kim, T. Schembri, D. Bialas, M. Stolte and F. Würthner, Adv. Mater., 2022, 34, e2104678 CrossRef PubMed.
- G. Chen, H. Sasabe, W. Lu, X.-F. Wang, J. Kido, Z. Hong and Y. Yang, J. Mater. Chem. C, 2013, 1, 6547–6552 RSC.
- G. Chen, H. Sasabe, T. Igarashi, Z. Hong and J. Kido, J. Mater. Chem. A, 2015, 3, 14517–14534 RSC.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- H. Lin, R. Camacho, Y. Tian, T. E. Kaiser, F. Würthner and I. G. Scheblykin, Nano Lett., 2010, 10, 620–626 CrossRef CAS PubMed.
- Z. Xie, V. Stepanenko, B. Fimmel and F. Würthner, Mater. Horiz., 2014, 1, 355–359 RSC.
- Z. Xie, B. Xiao, Z. He, W. Zhang, X. Wu, H. Wu, F. Würthner, C. Wang, F. Xie, L. Liu, Y. Ma, W.-Y. Wong and Y. Cao, Mater. Horiz., 2015, 2, 514–518 RSC.
- S. Herbst, B. Soberats, P. Leowanawat, M. Stolte, M. Lehmann and F. Würthner, Nat. Commun., 2018, 9, 2646 CrossRef PubMed.
- M. Stolte, R. Hecht, Z. Xie, L. Liu, C. Kaufmann, A. Kudzus, D. Schmidt and F. Würthner, Adv. Opt. Mater., 2020, 8, 2000926 CrossRef CAS.
- C. Rehhagen, M. Stolte, S. Herbst, M. Hecht, S. Lochbrunner, F. Würthner and F. Fennel, J. Phys. Chem. Lett., 2020, 11, 6612–6617 CrossRef CAS PubMed.
- P. Lova, V. Grande, G. Manfredi, M. Patrini, S. Herbst, F. Würthner and D. Comoretto, Adv. Opt. Mater., 2017, 5, 1700523 CrossRef.
- D. Horneber, J. Düreth, T. Schembri, S. Betzold, M. Stolte, S. Höfling, F. Würthner and S. Klembt, Adv. Opt. Mater., 2025 DOI:10.1002/adom.202402617.
- M. Rödel, L. N. Philipp, J. H. Kim, M. Lehmann, M. Stolte, R. Mitric, F. Würthner and J. Pflaum, ACS Photonics, 2025, 12, 107–117 CrossRef.
- G. G. Paschos, N. Somaschi, S. I. Tsintzos, D. Coles, J. L. Bricks, Z. Hatzopoulos, D. G. Lidzey, P. G. Lagoudakis and P. G. Savvidis, Sci. Rep., 2017, 7, 11377 CrossRef CAS PubMed.
- N. Polat, O. Yakar, S. K. Özdemir and S. Balci, ACS Photonics, 2024, 11, 1804–1809 CrossRef CAS.
- M. Marangi, Y. Wang, M. Wu, F. Tjiptoharsono, A. I. Kuznetsov, G. Adamo and C. Soci, Nanophotonics, 2024, 13(18), 3519–3526 CrossRef CAS PubMed.
- A. Miasojedovas, K. Kazlauskas, G. Armonaite, V. Sivamurugan, S. Valiyaveettil, J. V. Grazulevicius and S. Jursenas, Dyes Pigm., 2012, 92, 1285–1291 CrossRef CAS.
- M. G. Ramírez, M. Morales-Vidal, V. Navarro-Fuster, P. G. Boj, J. A. Quintana, J. M. Villalvilla, A. Retolaza, S. Merino and M. A. Díaz-García, J. Mater. Chem. C, 2013, 1, 1182–1191 RSC.
- R. Muñoz-Mármol, N. Zink-Lorre, J. M. Villalvilla, P. G. Boj, J. A. Quintana, C. Vázquez, A. Anderson, M. J. Gordon, A. Sastre-Santos, F. Fernández-Lázaro and M. A. Díaz-García, J. Phys. Chem. C, 2018, 122, 24896–24906 CrossRef.
- R. Yang, Z. Hu, Y. Li, J. Xia, J. Ma and J. Yang, RSC Adv., 2020, 10, 2437–2447 RSC.
- A. A. Ischenko, Pure Appl. Chem., 2008, 80, 1525–1538 CrossRef.
- W. Lin, Y. Cui, J. Yu, Y. Yang and G. Qian, Dyes Pigm., 2017, 136, 791–797 CrossRef CAS.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- H. Kar and S. Ghosh, Chem. Commun., 2016, 52, 8818–8821 RSC.
- H. Wang, Y. Zhang, Y. Chen, H. Pan, X. Ren and Z. Chen, Angew. Chem., Int. Ed., 2020, 59, 5185–5192 CrossRef CAS PubMed.
- G. Holden, E. T. Bishop and N. R. Legge, J. Polym. Sci., 1969, 26, 37–57 Search PubMed.
Footnotes |
| † In memory of Professor Helmut Ringsdorf. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00409h |
| This journal is © The Royal Society of Chemistry 2025 |