
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(dibenzothiophenylene sulfide)s: sulfur-rich annulated frameworks with a wide-range ultrahigh refractive index†
Seigo
Watanabe
 a,
Zexin
An
b,
Hiromichi
Nishio
b,
Yoshino
Tsunekawa
b and
Kenichi
Oyaizu
*ab
a,
Zexin
An
b,
Hiromichi
Nishio
b,
Yoshino
Tsunekawa
b and
Kenichi
Oyaizu
*ab
aResearch Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: oyaizu@waseda.jp
bDepartment of Applied Chemistry, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 22nd March 2025
Abstract
Poly(phenylene sulfide) (PPS) derivatives exhibit remarkable high-refractive properties due to their elevated sulfur content. However, their refractive indices (RIs) have typically been limited to nD ∼ 1.7–1.8, primarily due to the sterically hindered side chains required for desirable amorphous properties. In this study, we demonstrate the “PPS-annulation” strategy, which involves the partial fused system of PPS, as a rational molecular design to achieve wide-wavelength ultrahigh RI. In short, poly(dibenzothiophene sulfide) (PDBTS), an annulated analog of PPS, is precisely synthesized through oxidative polymerization. PDBTS demonstrates an ultrahigh RI of nD = 1.85, excellent thermostability (Tg = 205 °C), and a completely amorphous nature owing to its aromatic/sulfur-rich and rigid backbone. Furthermore, PDBTS is further functionalized through copolymerization with other PPS derivatives (e.g., hydroxy- or methylthio-substituted PPS) to optimize the balance between their ultrahigh RI (nD over 1.82) and enhanced visible–near-infrared transparency. Overall, the “PPS-annulation” strategy offers a rational design for polymeric materials with enhanced RI, high thermostability, amorphous characteristics, and exceptional optical properties.
Introduction
Lighting devices, typified by organic light-emitting diodes (OLEDs), are crucial for sustainable energy-to-light conversion systems, requiring high light extraction efficiency alongside additional features such as lightweight and flexibility.1,2 High-refractive-index polymers (HRIPs) are key components in various optical elements, such as OLED encapsulants and waveguides, contributing to reducing optical loss.3–5 HRIPs typically exhibit refractive indices (RIs) exceeding 1.7 and offer high transparency in the near-ultraviolet (UV), visible, or near-infrared (NIR) spectra, depending on the specific application.4A typical HRIP design follows the Lorentz–Lorenz equation, involving either (1) increasing the molar refraction [R] and/or (2) decreasing the molecular volume V (i.e., the van der Waals volume of a unit or/and free volume) to enhance the RI of a polymer in the bulk state.3,4,6–9 Chalcogenide atoms, typically sulfur, and aromatic rings are essential for balancing the RI and other desired properties, such as thermostability and processability, in optical materials.4,10–12 In this context, previous studies have mainly focused on poly(phenylene sulfide) (PPS) as a promising HRIP backbone due to its excellent RI, thermal, and mechanical properties. While intrinsic PPS is semicrystalline, polymers incorporating oligomeric PPS units with various linkers (imide,13–16 triazine,17,18 and others19–21) or network structures22–24 provide amorphous HRIPs with high visible or NIR transparency. In contrast, we have demonstrated PPS derivatives4 (Fig. 1, left) with functional side groups displaying maximum PPS unit density, high solubility, and complete amorphous properties to achieve satisfactory visible transparency and a high RI (nD ∼ 1.7–1.8). In particular, hydrogen bonding (H-bonding)25,26 and sulfur-containing27 skeletons achieved ultrahigh RI (nD = 1.80–1.85), which was attributed to their maximized density and polarizability.
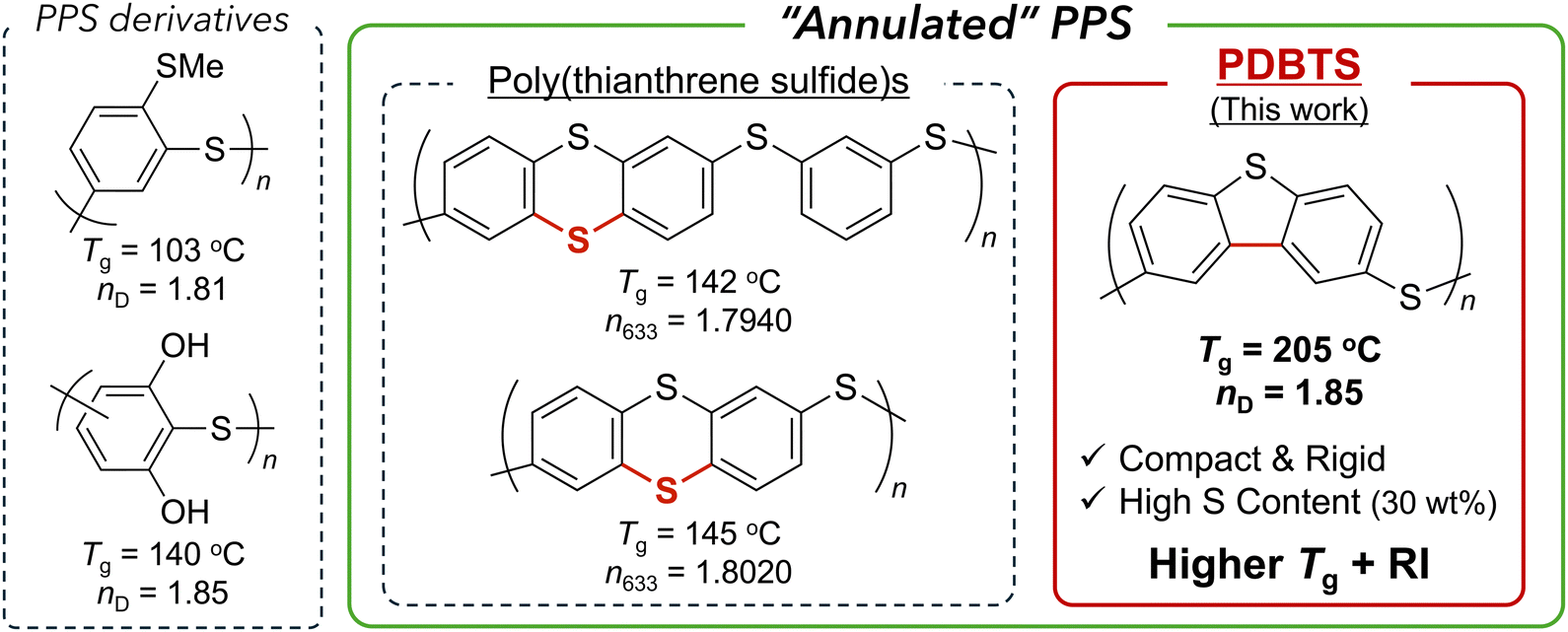 | ||
| Fig. 1 Ultrahigh-RI and amorphous PPS family: non-annulated PPS derivatives26,27 (left), poly(thianthrene sulfide)s28 (middle), and PDBTS (this work) (right). | ||
As another RI enhancement strategy, the annulation (e.g., cyclization) of aromatic rings contributes to reducing molar volume, maximizing polarizability, and promoting amorphous properties due to the rigid skeleton preventing tight π–π interactions. For example, poly(thianthrene sulfide), a sulfide-bridged analog of PPS, exhibits transparency, solution processability, and a significantly high RI (n633 = 1.8020 at maximum) (Fig. 1, middle).28 In contrast, poly(dibenzothiophene sulfide) (PDBTS) (Fig. 1, right), a direct o-phenylene-coupled analog of PPS, could be a better candidate owing to its more compact structure for achieving an even higher RI and enhanced thermostability. While PDBTS has been recognized as an electron-conducting polymer after chemical doping,29 its optical properties remain unexplored, and in particular, dibenzothiophene (DBT) has seldom been incorporated into previous HRIPs. To our knowledge, the only reported example includes DBT-containing polyimides30 exhibiting a high RI (n633 = 1.7578) and glass transition temperature (Tg = 201 °C). In short, DBT-based HRIPs could potentially achieve elevated RI levels due to their “annulated PPS” backbones with high [R] and compactness.
Herein, we introduce PDBTS and its derivatives as a novel ultrahigh-RI PPS family displaying exceptional thermal and optical properties. PDBTS is precisely synthesized through the site-selective oxidative polymerization of the DBT-containing disulfide, resulting in an amorphous, thermostable (Tg = 205 °C), and ultrahigh RI (nD = 1.85) polymer. These outstanding properties stem from its rigid, sulfur-rich, and all-aromatic backbone. Moreover, its optical and thermal properties are further enhanced using the copolymerization approach that combines PDBTS with other PPS components to modulate polarizability (specifically sulfur content) and intermolecular H-bond strength. The resulting copolymers exhibited enhanced visible light transparency while maintaining an ultrahigh RI of above 1.8. Overall, the present “annulated PPS” design based on DBT provides a straightforward strategy to enhance the thermal and RI properties of various HRIPs.
Experimental
Materials
All reagents were purchased from Tokyo Chemical Industry Co., Kanto Chemical Co., Junsei Chemical Co., and Kokusan Chemical Co. (for details, refer to the ESI†). Bis(2-methoxyphenyl) disulfide (OMeDPS) and bis(2-methylthiophenyl) disulfide (SMeDPS) were prepared following our previous reports.27,31Measurements
1H (500 MHz) and 13C (including DEPT) (125 MHz) nuclear magnetic resonance (NMR) spectra were recorded using a JEOL ECX-500 NMR spectrometer. Fast atom bombardment mass (FAB-MS) spectra were obtained using JMS-GCMATE II. Fourier-transform infrared spectra were recorded on a JASCO FT/IR 6100 spectrometer with KBr pellets. Cyclic voltammetry (CV) was performed with a BAS ALS 660D using a three-electrode system, where the working, counter, and reference electrodes were a Pt electrode (φ = 1.6 mm), a Pt wire, and an Ag/AgCl electrode, respectively. In the CV measurement, the potential was measured against an external standard (ferrocene (Fc)/ferrocenium (Fc+) redox couple, E1/2 = 0.45 V vs. Ag/AgCl). Ultraviolet-visible (UV-vis) spectra were recorded using a JASCO V-670 spectrometer at a scan rate of 100 nm min−1. Size exclusion chromatography (SEC) was performed using a SHIMAZU CBM-20A and SPD-20MA system equipped with a TOSOH TSKgel SuperHM-N column (detectors: UV and RI) and chloroform as the eluent (flow rate: 0.3 mL min−1), and molecular weight was calibrated with polystyrene standards. Powder X-ray diffraction (XRD) profiles were collected with a Rigaku RINT-Ultima III using a Cu target X-ray source (CuKα: 1.54 Å). Thermogravimetric analyses (TGA) were conducted with Rigaku TG8120 under a nitrogen flow from room temperature to 500 °C at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was performed using TA Instruments Q200 in the temperature range of 40–250 °C at a scan rate of 20 °C min−1. The thickness of a polymer film was determined by using a stylus profiler (KLA Tencor P-6).Optical property measurements
The RI spectra (n, k) were analyzed through spectroscopic ellipsometry in the film state using a Horiba UVISEL ERAGMS iHR320 spectrometer. The Abbe numbers in the visible (νD) and NIR (νNIR) ranges were calculated based on the definitions in eqn (1) and (2):15 | (1) |
 | (2) |
Synthesis of PDBTS
A typical procedure is outlined as follows (entry 3 in Table 1). To a 10 mL flask were added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.23 g, 1.0 mmol) and trifluoroacetic acid (TFA) (25 μL, 0.32 mmol) in 1,2-dichloroethane (DCE) (2 mL). Subsequently, DBTDPS (0.43 g, 1.0 mmol) was added to the solution and stirred at room temperature for 20 h. The resulting mixture was diluted by further adding DCE (5 mL) and was then precipitated in methanol with 5 vol% hydrochloric acid (300 mL in total). The precipitate was collected by filtration, was washed successively with methanol, aqueous potassium hydroxide, and water, and was then dried in vacuo. The crude product was dissolved in chloroform and was reprecipitated with acetone with 5 vol% hydrochloric acid (300 mL in total). The precipitate was collected through filtration, was successively washed with acetone, potassium hydroxide aqueous, and water, and was then dried in vacuo to yield PDBTS as a white powder (yield: 0.21 g, 49%).Synthesis of PDBTS–OMePPS copolymer (CP1)
A typical procedure is outlined as follows (run 2 in Table 2). To a 10 mL flask was added DDQ (0.46 g, 2.0 mmol) with DCE (2 mL) and TFA (50 μL, 0.66 mmol). The monomer mixture, consisting of DBTDPS (0.43 g, 1.0 mmol) and OMeDPS (0.28 g, 1.0 mmol), was added into the solution and stirred at room temperature for 20 h. After diluting with an additional DCE (6 mL), the solution was precipitated in methanol containing 5 vol% hydrochloric acid (300 mL in total). The resulting precipitate was collected by filtration, was washed successively with methanol, aqueous potassium hydroxide, and water, and was then dried in vacuo. The product was reprecipitated with chloroform/acetone with 5 vol% hydrochloric acid. The resulting precipitate was washed with acetone, aqueous potassium hydroxide, and water, and was dried in vacuo to obtain CP1 as a white powder (yield: 0.27 g, 39%).| Run | Polymer | Yield (%) | Feed ratio of [DBTDPS] (—) | Unit ratio xb (—) | M n (×103) | M w (×103) | M w/Mnc (—) |
|---|---|---|---|---|---|---|---|
| a Entry 3 in Table 1. b Determined by 1H NMR. c Determined by SEC (chloroform). | |||||||
| — | PDBTS | 49 | 1.00 | 1.00 | 0.9 | 1.3 | 1.4 |
| 1 | CP1 | 29 | 0.75 | 0.65 | 1.5 | 1.9 | 1.3 |
| 2 | CP1 | 36 | 0.50 | 0.46 | 1.8 | 2.6 | 1.5 |
| 3 | CP1 | 42 | 0.25 | 0.25 | 3.1 | 3.9 | 1.3 |
| 4 | CP2 | 25 | 0.75 | 0.75 | 0.8 | 1.2 | 1.5 |
| 5 | CP2 | 42 | 0.50 | 0.48 | 1.0 | 1.6 | 1.6 |
| 6 | CP2 | 53 | 0.25 | 0.24 | 1.2 | 1.6 | 1.3 |
Synthesis of PDBTS–SMePPS copolymer (CP2)
A typical procedure is outlined as follows (run 5 in Table 2). To a 10 mL flask was added DDQ (0.46 g, 2.0 mmol) with DCE (1 mL) and TFA (50 μL, 0.66 mmol). Once the solution became homogeneous, the monomer mixture (DBTDPS (0.43 g, 1.0 mmol) and SMeDPS (0.31 g, 1.0 mmol)) dissolved in DCE (1 mL) was added and was stirred at room temperature for 20 h. After diluting with additional DCE (6 mL), the solution was precipitated in methanol with 5 vol% hydrochloric acid (300 mL in total). The resulting precipitate was collected by filtration, was washed successively with methanol, aqueous potassium hydroxide, and water, and was then dried in vacuo. The product was reprecipitated using chloroform/acetone with 5 vol% hydrochloric acid. The precipitate was washed with acetone, aqueous potassium hydroxide, and water, and was dried in vacuo to obtain CP2 as a white powder (yield: 0.32 g, 42%).Synthesis of PDBTS–OHPPS copolymer (CP3)
A typical procedure is outlined as follows (run 8 in Table S1, ESI†). To a 30 mL flask was added CP1 (0.20 g, 0.22 mmol of –OMe) and was dissolved in dichloromethane (6.12 mL) under an Ar atmosphere. After cooling to 0 °C, a 1 M BBr3 solution in dichloromethane (3.68 mL, 3.68 mmol) was added under inert conditions. The mixture was stirred at 0 °C for 20 min and then at room temperature for 20 h. The reaction mixture was then quenched with water (6 mL) and the solvent was evaporated using a rotary evaporator. The crude product was dissolved in 6 mL of N,N-dimethylformamide (DMF) and precipitated in water with 5 vol% hydrochloric acid (200 mL). The resulting precipitate was collected by filtration, was washed with water, and was dried in vacuo. The product was redissolved in DMF and was reprecipitated in water containing 5 vol% hydrochloric acid (20-fold excess). After purifying and drying following a similar protocol, CP3 was obtained as a white powder (yield: 0.15 g, 75%).Film preparation for optical measurements
Thin films of the polymers were fabricated via drop-casting or spin-coating methods, as described in our previous report.27Results and discussion
Synthesis of PDBTS
The target PDBTS was synthesized by the oxidative polymerization of bis(3-dibenzothiophenyl) disulfide (DBTDPS). The DBTDPS monomer was prepared through a two-step synthesis: the copper-assisted thiolation of 3-bromodibenzothiophene followed by the thiol oxidation, as reported previously27,32 (refer to the ESI† for details). DBTDPS was characterized using NMR and FAB-MS measurements (Fig. S1–S5, ESI†). The electrochemical properties of DBTDPS were verified by CV, revealing an irreversible oxidation peak at 1.73 V vs. Ag/AgNO3, a higher potential than that of another typical DPS family26,27,31 (Fig. S6, ESI†). To elucidate the rationale, the optimized geometries of DBTDPS were estimated by the density functional theory (DFT) calculations (Fig. S7, ESI†). The highest occupied molecular orbital (HOMO) of DBTDPS was less distributed around the disulfide bonds due to the delocalized π-electrons resulting from the annulated DBT rings, indicating its lower oxidation ability (Fig. S7b, ESI†). The reduced nucleophilicity of DBTDPS caused by the annulated structures also lowers its reactivity towards electrophilic substitution reactions, which might lead to lower polymerization degree.Despite such less reactive nature of DBTDPS, its oxidative polymerization proceeded smoothly in a halogenated solvent with a low donor number, specifically DCE, under acidified conditions (Fig. 2a and Table 1). Since the oxidative polymerization of aromatic disulfides proceeds via the chain-growth polycondensation mechanism,33 higher monomer concentration and longer reaction time generally increase molecular weight and polymer yield.31 Starting from the benchmark condition (entry 1: [monomer] = 0.5 M, time: 20 h), longer reaction time (entry 2: 40 h) or higher monomer concentration (entry 3: 1 M) resulted in higher yield and molecular weight, with the latter optimization giving the most efficient results. Still, the resulting PDBTS exhibited an oligomeric molecular weight up to Mw = 1.3 × 103 (entry 3) with a low yield (<50%) due to the lower reactivity of the monomer compared to other typical DPS derivatives.25,27,31 The 1H NMR spectrum of DBTDPS exhibited broadened and multimodal aromatic proton signals attributed to strong π–π interchain interactions between the DBT units (Fig. S8, ESI†). In contrast, the 13C NMR spectrum revealed six distinct signals for the main-chain aromatic carbons, three of which corresponded to the C–H carbons also observed in the DEPT spectrum (Fig. 2b). Due to the low molecular weight of PDBTS, several terminal aromatic carbons (labeled * in Fig. 2b(ii) and (iii)) were also detected in the chemical shifts similar to the most peripheral carbons in DBTDPS (labeled h–k in Fig. 2b(i)). The IR spectrum showed two emerging bands for the out-of-plane vibrations (δC-H: 856 and 876 cm−1) for PDBTS (Fig. S11, ESI†), indicating the presence of adjacent and isolated aromatic C–H protons. The stretching of the thiophene rings (1410 cm−1)34 further confirmed the successful polymerization without structural defects. Considering the single-substitution pattern of aromatics with a highly symmetrical skeleton and the aforementioned characterization results, the oxidative polymerization of DBTDPS selectively produced PDBTS with a single 2,8-substituted DBT skeleton.
 | ||
| Fig. 2 Oxidative polymerization of DBTDPS. (a) Scheme. (b) Expanded 13C NMR spectra of (i) DBTDPS, (ii) PDBTS, and (iii) PDBTS (DEPT135) in chloroform-d (*: terminal aromatic carbons). The overall spectra are depicted in Fig. S4, S9 and S10 (ESI†), respectively. | ||
To gain insight into the reactivity and regioselectivity of this polymerization system, we revisited the electron distribution of DBTDPS estimated by the DFT calculations (Fig. S7, ESI,†vide supra). The 6- and 8-position carbons of the disulfide exhibit the highest Mulliken charges, indicating enhanced reactivity with active sulfonium electrophiles (Fig. S7a, ESI†). Furthermore, the HOMO is primarily located at the 8-position, while it is delocalized at the 6-position over the adjacent benzothiophene ring. This suggests a preferential reactivity towards yielding a 2,8-substituted PDBTS skeleton (Fig. S7b and c, ESI†). The resulting PDBTS was in low yield (up to 49%) and low molecular weight (Mw ∼ 1.3 × 103 (entry 3 in Table 1)) owing to the high oxidation potential of disulfides. This led to increased delocalization of π-electrons upon the polymerization progress, resulting in reduced Friedel–Crafts reactivity with another aromatic counterpart. Enhancing the reactivity could be achieved by incorporating electron-donating groups into DBTDPS to lower its oxidation potential, thereby facilitating polymerization rate and increasing the molecular weight.
Thermal and optical properties of PDBTS
The DSC thermogram of PDBTS exhibited the highest glass transition temperature (Tg) of 205 °C among all the PPS families, without any melting behaviors despite the low molecular weight (Fig. S12, ESI†). The presence of the rigid DBT backbone led to a higher rotational barrier compared to the previously studied side-chain4 or thianthrene-containing28 PPS derivatives. The XRD profiles also displayed its amorphous nature characterized by a broad halo peak (Fig. S12 inset, ESI†). This was supported presumably due to the bulky aromatic DBT backbone with a bent substitution pattern, resulting in reduced π–π interchain packing compared to the previous phenylene-containing derivatives. This behavior was in line with the findings in the previous PDBTS report,29 where bent 2,8-substitution led to an amorphous phase, while linear-like 3,7-substitution resulted in a semicrystalline phase. Furthermore, the TGA trace revealed a high degradation temperature of Td5 = 383 °C (Fig. S13, ESI†), consistent with the excellent thermal stability conferred by the all-aromatic DBT backbone.Owing to its high solubility, PDBTS was fabricated as a thin film through a solution process (see Fig. 3a, inset, left). The drop-cast film exhibited homogeneity and transparency, although it was slightly brownish due to low near-UV transmittance (%T = 82 at 400 nm, for 1 μm thickness) (Fig. 3a). This transparency was comparable to poly(thianthrene sulfide) with a similar backbone (%T = 84 at 400 nm, for 1 μm thickness).28 Furthermore, PDBTS maintained high transmittance from the long-wavelength visible range (>500 nm) to the NIR region (%T ≧ 97 above 500 nm) (see Fig. S14, ESI†). At the molecular level, the solution UV-vis spectrum (Fig. 3a, inset, right) of PDBTS exhibited a shoulder absorption peak near the visible region (∼360 nm), which was absent in the spectra of non-annulated PPS derivatives.25,35 This was attributed to the delocalized π-electrons on the annulated DBT rings, inducing the red-shift of the UV-absorption bands. The PDBTS solution was also used for spin-coating, and the resulting thin film exhibited an ultrahigh RI (nD = 1.85) and a moderate Abbe number (νD = 15) (Fig. 3b), surpassing the empirical nD–νD trade-off limit.27 These high-RI characteristics extended to longer wavelengths, with a reasonable NIR Abbe number (νNIR = 12) and a high infinite-wavelength RI of n∞ = 1.74 (Fig. S15, ESI†), indicating its potential for NIR optical applications. The extinction coefficient (k) of PDBTS (Fig. 3b, red) exhibited a long cutoff resonance wavelength (∼400 nm) resulting in anomalous RI dispersion, which is in line with the shoulder absorption observed in the solution UV-vis spectrum. In conclusion, PDBTS demonstrates ultrahigh RIs across a wide wavelength range (visible–NIR region) due to its high sulfur/aromatic content and annulated skeleton, particularly excelling in optical properties in the NIR region comparable to the previous NIR-transparent PIs (n633 < 1.8, %T > 90).15,16,36
 | ||
| Fig. 3 Optical properties of PDBTS. (a) Normalized UV-vis spectrum of the PDBTS film with a 1 μm thickness (inset: photograph of the 3.0 μm-thick PDBTS film and the UV-vis absorbance spectrum of a 0.1 mM PDBTS solution in chloroform). The original spectrum is displayed in Fig. S14 (ESI†). (b) RI spectra of PDBTS: n (black) and k (red) (inset: list of the nD and Abbe numbers). | ||
Copolymerization strategy: PDBTS with side-chain-substituted PPS
To balance the superior RI properties of PDBTS with sufficient visible transparency, we extended the design concept to PDBTS-based copolymers by incorporating another functionalized PPS to enhance the overall optical properties (Fig. 4). To improve visible transparency while maintaining high RI, the counterparts were selected according to the following criteria: side-substituted PPS with polar methoxy groups (OMePPS: CP1), polarizable methylthio groups (SMePPS: CP2), and H-bondable hydroxy groups (OHPPS: CP3). First, CP1 and CP2 were prepared through one-step synthesis involving the oxidative polymerization of the corresponding disulfide monomers (Fig. 4a and Fig. S16. ESI†). | ||
| Fig. 4 Synthesis and molecular design of the PPS copolymers containing the PDBTS skeleton. (a) Oxidative copolymerization producing the copolymer with either methoxy- (CP1) or methylthio-substituted (CP2) PPS counterparts. (b) Demethylation of CP1 to synthesize the H-bondable PPS copolymers (CP3). | ||
Based on the higher solubility and lower oxidation potential of the counterpart DPS monomer,27,31 the copolymers were obtained with a higher yield and improved solubility. Particularly, better results were achieved at a lower PDBTS content (x) (Table 2). The molecular weights increased up to Mw = 3.9 × 103 and Mw = 1.6 × 103 for CP1 (run 3) and CP2 (run 6), respectively, indicating that more polar and electron-donating skeletons promote polymerization and enhance solubility. The DOSY-NMR spectra confirmed the presence of copolymers with similar diffusion coefficients for the aromatic and methyl protons (Fig. S17, ESI†). The relative reactivity of each monomer was elucidated by monitoring the time course of copolymerization (Fig. S18–S20, ESI†). Regardless of the counterpart species (OMeDPS or SMeDPS), the x values were significantly below the feed ratio (xf) in the initial polymerization stage but gradually approached xf as the reaction progressed (Fig. S19a and S20a, ESI†). This trend indicated a higher selectivity for DPS with lower oxidation potential, leading to a larger composition, consistent with previous reports on oxidative copolymerization.27,31 The IR spectra revealed consistent changes in the peak intensity of the stretching bands of the thiophene ring (1400 cm−1) and C–X (X: S or O) bonds (νC–O: 1240 cm−1, νC–S: 1100 cm−1) in accordance with the overall composition (Fig. S21, ESI†).
The XRD profiles of CP1 and CP2 reflect their amorphous features with no specific diffraction of the crystalline phase, following a similar trend to that of the homopolymers (Fig. 5a). Their randomized sequence upon copolymerization also contributes to disordered microstructures with less interchain packing. The TGA and DSC results demonstrated that the copolymers exhibited lower Td5 and Tg values at smaller x (Fig. S22, ESI† and Fig. 5b). The DSC thermograms also displayed the thermal behavior of amorphous nature, with a single glass transition for all the copolymers. Furthermore, a series of CP1 with compact methoxy side chains achieved a higher Tg (above 147 °C) than the side-chain-flexible CP2 (Tg above 120 °C) while showing superior thermostability to typical side-chain-substituted PPS derivatives,27 based on the high rigidity of the PDBTS backbones.
 | ||
| Fig. 5 Thermal and optical properties of (i) CP1 and (ii) CP2: (a) XRD profiles, (b) DSC thermograms (scanning rate: 20 °C min−1, second heating), (c) normalized UV-vis spectra including their corresponding homopolymers (thickness: 1 μm), and (d) RI. Data for the optical properties of OMePPS and SMePPS were adopted from our previous report (ref. 31 (Copyright © 2020 The Chemical Society of Japan) and ref. 27 (Adopted under the CC-BY-NC-ND 4.0 license), respectively). | ||
Owing to the enhanced solubility of the copolymers, CP1 and CP2 were solution-processable to fabricate homogeneous thin films (Fig. S23, ESI†). These copolymers exhibited higher visible transparency compared to PDBTS (Fig. 5c and Table 3), and particularly CP1 resulted in better transmittance (over 90%T for 1-μm thick films) attributed to the less-polarizable methoxy groups. The solution UV-vis spectra followed the same trend, suggesting the absence of specific aggregation or charge transfer in the film states (Fig. S24, ESI†). The RI varied systematically depending on the unit ratio, with CP2 maintaining an ultrahigh RI (nD = 1.83–1.82) across all compositions, while CP1 exhibited a lower RI of nD = 1.80–1.76 (Fig. 5d and Table 3). These differences were primarily attributed to the unit polarizability, resulting in high RIs and low transparency for copolymers with high-[R] skeletons. Considered together with the UV-vis spectra (Fig. 5c: vide supra), the RI and transparency of CP1 and CP2 changed within the empirical RI-transparency trade-off relationship,3 without exceeding the overall optical properties of the corresponding homopolymers. In summary, the amount of polarizable units emerged as the key determinant of the optical properties of CP1 and CP2, aligning with the case of a previous OMePPS–SMePPS copolymer system.27
| Run | Polymer | Unit ratio xb (—) | ε , (103 M−1 cm−1) at 360 nm | %Tc,e at 400 nm | n D (—) | ν D (—) |
|---|---|---|---|---|---|---|
| a Entry 3 in Table 1. b Determined by 1H NMR. c Determined by UV-vis spectroscopy. d Values for chloroform solution (measured concentration: 0.1 mM). e Normalized values with 1 μm thickness. f Determined by spectroscopic ellipsometry. g Data from ref. 31. h Determined from the original spectrum data in ref. 31. i Data from ref. 27. | ||||||
| — | PDBTS | 1.00 | 3.4 | 82 | 1.85 | 15 |
| 1 | CP1 | 0.65 | 2.0 | 90 | 1.80 | 16 |
| 2 | CP1 | 0.46 | 1.9 | 93 | 1.78 | 18 |
| 3 | CP1 | 0.25 | 1.6 | 97 | 1.76 | 16 |
| — | OMePPS | 0.00 | 0.6g,h | 96g | 1.73g | 22g |
| 4 | CP2 | 0.75 | 3.3 | 87 | 1.83 | 18 |
| 5 | CP2 | 0.48 | 2.7 | 93 | 1.82 | 19 |
| 6 | CP2 | 0.24 | 2.5 | 94 | 1.82 | 20 |
| — | SMePPS | 0.00 | 1.1i | 94i | 1.81i | 19i |
PDBTS copolymers with H-bonding units
Breaking through the latent trade-off boundaries among various HRIP properties (e.g., dual tuning of thermostability, RI, and transparency), we expanded the target copolymer to CP3, which incorporates highly polarizable PDBTS and H-bondable OHPPS units (Fig. 4b and Table S1, ESI†). CP3 was synthesized by demethylating the methoxy groups of CP1 using BBr3,25,26 leading to complete hydroxylation without any residual methoxy groups, as verified by the 1H NMR spectra (Fig. S25, ESI†). The IR spectra further confirmed the reaction progress, showing H-bonded O–H stretching (νO–H: 3400 cm−1) and reduced C–O stretching bands (νC–O: 1240 cm−1) (Fig. S26, ESI†).In contrast to the strong H-bond interactions, CP3 was amorphous, showing only broad halos in its XRD profile (Fig. S27, ESI†). This was attributed to the randomly distributed rigid PDBTS segments in the OHPPS-rich unit sequence, which effectively hindered ordered assembly. From the DSC thermograms, Tg of each CP3 was slightly lowered through the demethylation of the corresponding CP2 (Fig. S28, ESI†). Owing to the high rigidity of the PDBTS units, the reduced rotational barrier upon demethylation would have a more significant effect than that of intermolecular H-bonds,25,26 resulting in the lower Tg of CP3. The TGA traces displayed the decrease in the 5% degradation temperature (Td5) upon demethylation caused by the presence of reactive hydroxy groups, resulting in earlier pyrolysis (Fig. S29, ESI†).
While intermolecular H-bonds contribute less to the thermal properties, they enhance the optical properties due to their free volume reduction effect without affecting near-UV and visible absorption.25,26Fig. 6a and Table S2 (ESI†) summarize the key optical properties of CP3. The UV-vis spectra showed similar film transparencies before and after demethylation (%T = 90–97 for CP1, 93–96 for CP3), with lower absorption in the solution spectra for CP3 than the corresponding CP1 (Fig. 6b and Fig. S30, S31(ii), ESI†). Spectroscopic ellipsometry revealed a smaller RI of CP3 for higher-x compositions (nD = 1.79, run 7 and nD = 1.77, run 8) owing to their low-[R] unit contributions. However, an anomalously higher RI was observed for the small-x composition (nD = 1.82, run 9), surpassing that of OHPPS (Fig. 6c).
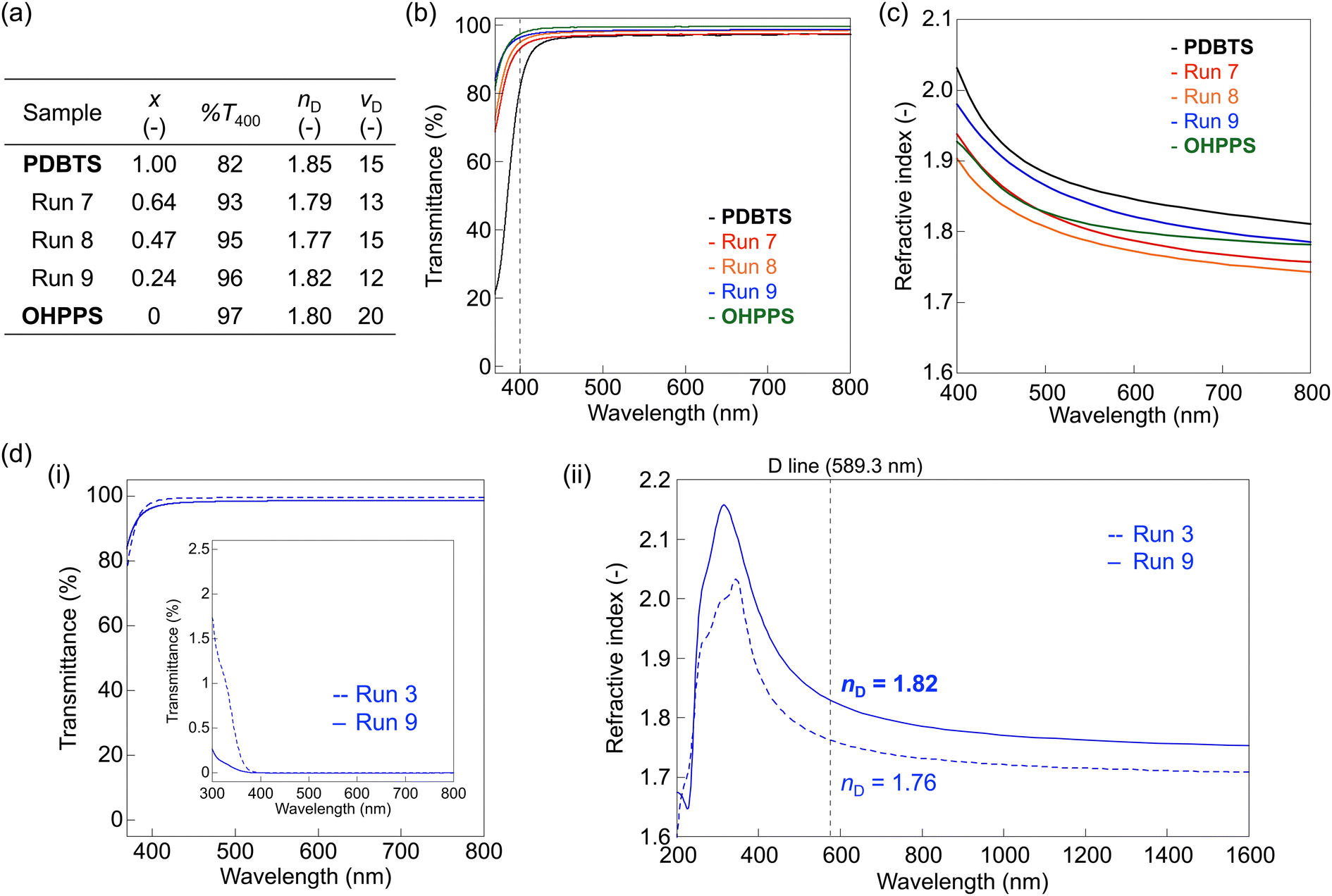 | ||
| Fig. 6 Optical properties of the demethylated copolymer (CP3). Data for OHPPS were adopted from ref. 25 (Copyright © 2022 American Chemical Society): (a) figures-of-merit, (b) normalized UV-vis spectra of CP3 and the corresponding homopolymers (thickness: 1 μm). (c) RI of CP3 and the corresponding homopolymers. (d) Extracted optical properties before and after demethylation from CP1 (run 3, x = 0.25, dotted line) to CP3 (run 9, x = 0.24, solid line): (i) normalized film UV-vis spectra (thickness: 1 μm) of the films (inset: UV-vis spectrum of a 0.1 mM solution in DMF). (ii) RI in the near-UV-visible-NIR range. | ||
To investigate this unique behavior, we excerpted the optical properties of runs 3 and 9 as depicted in Fig. 6d. The low-xCP1 and CP3 exhibited a simultaneous increase in the RI and transparency upon demethylation, unlike the medium-to-large-x copolymers (run 1/run 7 or run 2/run 8) with similar RI values (Fig. S31 and S32, ESI†). This exceptional trend was attributed to the enhanced intermolecular interactions in the low-x (OHPPS-rich) CP3, resulting in a reduction of the free volume via H-bonding without compromising transparency. In short, the synergistic combination of polarizable PDBTS units and OHPPS resulted in higher overall [R]/V than the OHPPS homopolymer, enabling CP3 (run 9) to maintain similar transparency while enhancing RI properties. This observation aligns with our previous report on the OHPPS–SMePPS copolymer system,27 supporting the importance of good balance between the highly polarizable groups and H-bonding groups to maximize RIs and transparency simultaneously. In summary, the excessive polarizability of PDBTS can be mitigated by co-polymerizing with other PPS derivatives, particularly those with sulfur-containing or H-bonding backbones. This strategy significantly enhances transparency while preserving exceptionally high RIs suitable for visible light applications. Towards even better-balanced RIs and transparency, PDBTS-based copolymers with even denser H-bonds (e.g. dihydroxy-substituted PPS)26 and PDBTS–SMePPS–OHPPS terpolymers would be the promising targets to improve chain packing while preserving the moderately high polarizability.
Conclusions
In this study, we propose PDBTS as a family of ultrahigh-RI polymers. They are characterized by their “PPS-annulation” design, which achieves amorphous properties, high polarizability, and good transparency in the broad visible–NIR range. Initially, the PDBTS homopolymer exhibited an ultrahigh RI (nD = 1.85) and excellent thermal properties (Tg = 205 °C) compared to the prior-art PPS derivatives. However, its visible transparency was limited (over 82%T for a 1 μm-thick film) due to an excessive amount of polarizable and annulated skeletons. Therefore, a copolymerization approach with other high-[R] or H-bonding PPS counterparts achieved a better balance of optical properties. In particular, the copolymer CP2 incorporating SMePPS leading to enhanced visible–NIR transparency (over 87%T) while maintaining an ultrahigh RI (nD over 1.82). Furthermore, the copolymer CP3 bearing OHPPS units displayed an exceptional RI trend with increasing hydroxy content, finally reached a reasonable Tg (143 °C), ultrahigh RI (nD = 1.82), and higher visible transparency (over 96%T). This trend was attributed to the maximized [R]/V effect through the synergistic contribution of PDBTS and H-bonding OHPPS units. While PDBTS-based polymers are difficult to handle in free-standing states due to their low molecular weight and high brittleness, their film-formability and well-balanced thermal/optical properties enable the thin-layer/coating applications in various lighting devices, such as OLEDs,37,38 light-emitting electrochemical cells (LECs),39 and waveguides5,40 toward a higher device efficiency, which will be our continuous research. These results highlight the potential of DBT backbones as an HRIP framework for enhancing thermal and optical properties beyond the empirical limit, allowing their precise tuning from the visible to NIR regions by combining DBT with versatile structural components.Author contributions
S. W. and K. O. conceived the project and designed the experiments. Z. A. performed most of the experiments with the assistance of S. W., H. N., and Y. T. S. W. conducted the computational calculations. S. W. and K. O. prepared the manuscript. K. O. supervised the project. All authors analyzed the data, discussed the results, and approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by Grants-in-Aid for Scientific Research (No. 21H04695 and 22K18335) from MEXT, Japan. S. W. acknowledges Grants-in-Aid from the Satomi Scholarship Foundation and the Nanotechnology Forum Award (Research Organization for Nano and Life Innovation, Waseda University).Notes and references
- X. Huang, Y. Qu, D. Fan, J. Kim and S. R. Forrest, Org. Electron., 2019, 69, 297–300 CrossRef CAS.
- Y.-F. Liu, J. Feng, Y.-G. Bi, D. Yin and H.-B. Sun, Adv. Mater. Technol., 2019, 4, 1800371 CrossRef.
- T. Higashihara and M. Ueda, Macromolecules, 2015, 48, 1915–1929 CrossRef CAS.
- S. Watanabe and K. Oyaizu, Bull. Chem. Soc. Jpn., 2023, 96, 1108–1128 CrossRef CAS.
- A. Nishant, K.-J. Kim, S. A. Showghi, R. Himmelhuber, T. S. Kleine, T. Lee, J. Pyun and R. A. Norwood, Adv. Opt. Mater., 2022, 10, 2200176 CrossRef CAS.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef CAS PubMed.
- D. H. Kim, W. Jang, K. Choi, J. S. Choi, J. Pyun, J. Lim, K. Char and S. G. Im, Sci. Adv., 2020, 6, eabb5320 CrossRef CAS PubMed.
- Y. Zhou, Z. Zhu, K. Zhang and B. Yang, Macromol. Rapid Commun., 2023, 44, e2300411 CrossRef PubMed.
- S. Wang, X. Li and Y. Tu, Mater. Horiz., 2025, 12, 15–19 RSC.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- L. E. Anderson, T. S. Kleine, Y. Zhang, D. D. Phan, S. Namnabat, E. A. LaVilla, K. M. Konopka, L. Ruiz Diaz, M. S. Manchester, J. Schwiegerling, R. S. Glass, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2017, 6, 500–504 CrossRef CAS PubMed.
- J. Pyun and R. A. Norwood, Prog. Polym. Sci., 2024, 156, 101865 CrossRef CAS.
- N.-H. You, T. Higashihara, S. Ando and M. Ueda, J. Polym. Sci., Part A:Polym. Chem., 2010, 48, 656–662 CrossRef CAS.
- Y. Guan, W. Dong, C. Wang and D. Shang, Polym. Int., 2017, 66, 1044–1054 CrossRef CAS.
- H. Kim, B.-C. Ku, M. Goh, H. C. Ko, S. Ando and N.-H. You, Macromolecules, 2019, 52, 827–834 CrossRef CAS.
- S. Xue, X. Lei, Y. Xiao, Y. Liu, Y. Zhang and Q. Zhang, ACS Appl. Polym. Mater., 2024, 6, 2315–2326 CrossRef CAS.
- N.-H. You, T. Higashihara, Y. Oishi, S. Ando and M. Ueda, Macromolecules, 2010, 43, 4613–4615 CrossRef CAS.
- M.-C. Fu, M. Ueda, S. Ando and T. Higashihara, ACS Omega, 2020, 5, 5134–5141 CrossRef CAS PubMed.
- K. Nakabayashi, T. Imai, M.-C. Fu, S. Ando, T. Higashihara and M. Ueda, Macromolecules, 2016, 49, 5849–5856 CrossRef CAS.
- R. Hifumi and I. Tomita, Polym. J., 2018, 50, 467–471 CAS.
- S. Watanabe, T. Yano, Z. An and K. Oyaizu, ChemSusChem, 2025, 18, e202401609 CAS.
- L. Fang, J. Sun, X. Chen, Y. Tao, J. Zhou, C. Wang and Q. Fang, Macromolecules, 2020, 53, 125–131 CAS.
- L. Fang, C. Wang, M. Dai, G. Huang, J. Sun and Q. Fang, Mater. Chem. Front., 2021, 5, 5826–5832 RSC.
- Z. Dou, J. Sun and Q. Fang, Biomacromolecules, 2024, 25, 6155–6163 CrossRef CAS PubMed.
- S. Watanabe and K. Oyaizu, Macromolecules, 2022, 55, 2252–2259 CrossRef CAS.
- S. Watanabe, H. Nishio, T. Takayama and K. Oyaizu, ACS Appl. Polym. Mater., 2023, 5, 2307–2311 CrossRef CAS.
- S. Watanabe, T. Takayama and K. Oyaizu, ACS Polym. Au, 2022, 2, 458–466 CrossRef CAS PubMed.
- Y. Suzuki, K. Murakami, S. Ando, T. Higashihara and M. Ueda, J. Mater. Chem., 2011, 21, 15727–15731 RSC.
- R. L. Elsenbaumer and L. W. Shacklette, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 1781–1787 CrossRef CAS.
- J.-G. Liu, Y. Nakamura, C. A. Terraza, Y. Shibasaki, S. Ando and M. Ueda, Macromol. Chem. Phys., 2008, 209, 195–203 CrossRef CAS.
- S. Watanabe and K. Oyaizu, Bull. Chem. Soc. Jpn., 2020, 93, 1287–1292 CrossRef CAS.
- Y. Liu, J. Kim, H. Seo, S. Park and J. Chae, Adv. Synth. Catal., 2015, 357, 2205–2212 CrossRef CAS.
- F. Aida, Y. Takatori, D. Kiyokawa, K. Nagamatsu, H. Nishide and K. Oyaizu, Chem. Lett., 2015, 44, 767–769 CrossRef CAS.
- C. L. Garcia and J. A. Lercher, J. Phys. Chem., 1992, 96, 2669–2675 CrossRef CAS.
- S. Watanabe, Y. Tsunekawa, T. Takayama and K. Oyaizu, Macromolecules, 2024, 57, 2897–2904 CrossRef CAS.
- K.-H. Nam, A. Lee, S.-K. Lee, K. Hur and H. Han, J. Mater. Chem. C, 2019, 7, 10574–10580 CAS.
- Q. Wei, R. Pötzsch, X. Liu, H. Komber, A. Kiriy, B. Voit, P.-A. Will, S. Lenk and S. Reineke, Adv. Funct. Mater., 2016, 26, 2545–2553 CrossRef CAS.
- D. W. Kim, J. W. Han, K. T. Lim and Y. H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 985–991 CAS.
- S. Watanabe, L. M. Cavinato, V. Calvi, R. van Rijn, R. D. Costa and K. Oyaizu, Adv. Funct. Mater., 2024, 34, 2404433 CrossRef CAS.
- J. Zhang, T. Bai, W. Liu, M. Li, Q. Zang, C. Ye, J. Z. Sun, Y. Shi, J. Ling, A. Qin and B. Z. Tang, Nat. Commun., 2023, 14, 3524 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer synthesis; computational studies; additional NMR and IR spectra of polymers; copolymerization and demethylation studies; and additional thermal/optical properties of the copolymers. See DOI: https://doi.org/10.1039/d4tc05458j |
| This journal is © The Royal Society of Chemistry 2025 |