
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal–organic framework with dual fluorescent emission based on 3-aminoisonicotinate: luminescence thermometry and Fe3+ sensing in hybrid membranes†
Oier
Pajuelo-Corral
 *a,
Eneko
Alkain
b,
Ricardo F.
Mendes
c,
Filipe A.
Almeida Paz
c,
Antonio
Rodríguez-Diéguez
d,
Jose Angel
García
e,
Jose M.
Seco
b and
Javier
Cepeda
*b
*a,
Eneko
Alkain
b,
Ricardo F.
Mendes
c,
Filipe A.
Almeida Paz
c,
Antonio
Rodríguez-Diéguez
d,
Jose Angel
García
e,
Jose M.
Seco
b and
Javier
Cepeda
*b
aPOLYMAT Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), Facultad de Química, 20018 Donostia-San Sebastian, Spain. E-mail: oier.pajuelo@ehu.eus
bDepartamento de Química Aplicada, Facultad de Química, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 20018 Donostia-San Sebastián, Spain. E-mail: javier.cepeda@ehu.eus
cDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal
dDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad de Granada, Av. Fuentenueva S/N, 18071 Granada, Spain
eDepartamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 48940, Leioa, Spain
First published on 22nd April 2025
Abstract
This work comprises the characterization of a 3D metal–organic framework (MOF), namely, {[Zn(μ-3isoani)2]·4H2O}n, synthesized through the coordination of 3-aminoisonicotinic acid (H3isoani) with Zn2+ ions. The assembly of the building blocks resulted in a doubly interpenetrated open 3D crystalline structure with a quartz-like topology that possessed solvent-accessible voids occupied with water molecules. The compound exhibited excellent photoluminescence properties with a dual fluorescent emission arising from the organic molecule as corroborated by calculations using the time-dependent density functional theory (TD-DFT). The independent nature of each emission band with temperature inspired us to study the compound as a ratiometric luminescent thermometer. Notably, it showed the best performance in the 200–300 K range. In addition, the MOF was dispersed in polymethylmethacrylate (PMMA) for the construction of an easy-handled membrane via the solution-casting method for detecting aqueous Fe3+ ions with a competitive KSV value of 3.7 × 103 M−1 and a limit of detection (LOD) of 2.8 × 10−5 M.
Introduction
In the last decade, metal–organic frameworks (MOFs) have become a hot topic in materials science because of their tailorable crystalline structures, which can be rationally designed by adhering to the principles of coordination chemistry and by the appropriate selection of metal ions and organic molecules, which are similar to the bricks that sculpt crystalline architectures.1,2 Historically, the main applications of these fascinating porous materials are in gas adsorption and separation of small molecules owing to their open structure, well-defined pore shape and high specific surface areas (above 7000 m2 g−1).3–5 Nonetheless, the open structure of MOFs makes them prone to interact with the environment that could modulate the properties of these materials, making them useful for sensing applications. Indeed, the number of MOFs with sensing applications has incredibly grown in the last few years owing to their advantages of high flexibility to adapt to the environment and large number of properties, such as magnetic,6,7 electrochemical8 and photoluminescence9,10 properties, to be employed as transduction signals. Importantly, the sensor material's response to stimuli must be fast, sensitive and easily detectable. Notably, photoluminescent (PL) MOFs meet all these requirements as their bright and intense emission is prone to detect noticeable changes in either intensity (which can follow turn-off or turn-on mechanisms) or wavelength of the emission bands.11 For instance, the existing dependence of PL signals of MOFs to temperature enable them to be employed in luminescent thermometry, facilitating the development of simple devices that impose a high level of temperature control in industrial processes to ensure the quality of the final product.12,13 Luminescent materials are well known for their use as thermometers since increasing the temperature usually decreases the emission intensity owing to the rise in non-radiative deactivation pathways.14,15 Nonetheless, most of the currently reported thermometers are single intensity-based ones, which are often susceptible to errors that may arise from changes in the concentration of the probe, excitation power or detection capacity.16 Additionally, although all the experimental conditions are kept constant during the measurements (counting on the excitation wavelength or power source), the variability in absorption and scatter cross-section from sample to sample may lead to a reduction in the accuracy of the method.17 To overcome these drawbacks, ratiometric luminescence thermometry makes use of two independent signals from the same material to overcome the potential problems of single signal-based thermometers. This fact removes the dependency of the concentration or the drifts of the excitation source from temperature sensing, giving rise to a new generation of self-referenced thermometers.18At the same time, controlling leakage in industrial processes is of equal importance since toxic wastes can reach rivers and streams, posing a danger to the environment and human health. PL-MOFs have proven to be effective for detecting toxic species as corroborated by the huge number of sensors explored in the literature to detect biomolecules,19 explosives20–22 and metal ions.23,24 However, they are far from being applied in sensing devices since the low solubility in common solvents and their fragility makes them difficult to handle. To tackle this issue, developing processing techniques to shape MOFs into more manageable materials, such as membranes or foams, is of vital importance to go one step beyond the sensing applications of these materials.25,26 In this sense, the dispersion of MOFs into a polymeric matrix for the creation of mixed-matrix membranes (MMMs) opens the door to manufacture advanced composites that synergistically merge the excellent sensing capacities of the crystalline materials and the standout mechanical properties of the polymers, converging in a superior hybrid material that allows shaping the MMM on demand. Indeed, different polymeric matrices such as polyethersulfone (PES),27 polydopamine (PDA),28 poly(vinylidene fluoride) (PVDF)29,30 or polydimethylsiloxane (PDMS)31,32 have been combined with MOFs for the construction of MMMs, with most of them oriented to the separation of small gas molecules. In contrast, their use as sensing devices has been less explored. It is important to point out that the polymeric matrix of photoluminescent MOFs should not cause interferences with the excitation nor with the emission of the metal–organic material, since it considerably reduces the number of available polymers. For those cases, the use of polymethylmethacrylate (PMMA)33 or Matrimid34 are preferred because they do not interact with the PL properties of the MOF and allow their use in PL sensing applications.
Continuing with our quest for synthesizing MOFs with PL properties based on 3-aminoisonicotinate (3isoani) ligand,35,36 we present the PL performance of the open 3D MOF formulated as {[Zn(μ-3isoani)2]·4H2O}n. The compound shows dual fluorescent emission, which has been exhaustively studied experimentally at variable temperature and computationally supported by TD-DFT calculations. Besides, taking advantage of the dual emission, the sensing capacity of the compound is studied for its use as a ratiometric fluorescent thermometer. Likewise, its activity as a chemical sensor has been explored for a matrix-mixed membrane based on PMMA containing the powdered MOF, creating an advanced and easy-handling device to detect Fe3+ ions in aqueous media.
Results and discussion
Structural description
Compound 1 was previously reported by Shao et al.,37 although its crystal structure has been re-determined in this work following a greener synthetic route that replaces N,N′-dimethlyformamide (DMF) with water and ethanol as solvents (see Experimental section). The assembly of Zn(II) and 3isoani ligand under hydrothermal conditions leads to the formation of a two-fold interpenetrated open 3D framework showing channels occupied by solvent water molecules, all of which crystallizes in the P31 trigonal space group with a {[Zn(μ-3isoani)2]·4H2O}n formula (compound 1 hereafter). The central Zn(II) ion exhibits a N2O2 donor set coming from two pyridine nitrogen atoms with a Zn–N distance of 2.018(6)–2.062(7) Å and two oxygen atoms of two carboxylate groups with a Zn–O distance of 1.928(5)–1.973(6) Å (Table S2, ESI†). Meanwhile, the second carboxylic oxygen atoms are placed at a distance of 2.829(6) and 2.851(5) Å and cannot be considered within the coordination sphere, rendering a slightly distorted tetrahedral geometry (Td = 1.068) as depicted by the continuous shape measurements (see Table S1 for more details, ESI†) using SHAPE software.38 3isoani bridges the tetrahedral nodes by adopting a μ-κN:κO coordination mode and imposing a Zn⋯Zn distance of 8.8 Å with an angle between coordinating ligands in the 101–123° range (Table S3, ESI†). As a result, an open 3D framework with quartz-like qtz topology (Fig. S5, ESI†) and (64·82) point symbol is obtained, as confirmed by TOPOS software.39 The resulting architecture presents large voids that require the co-crystallization of a second equivalent framework to stabilize the resulting architecture (Fig. S6, ESI†), though there is still a remarkable porosity that corresponds to 32.1% of the unit cell according to PLATON software.40 The material's porosity comes from two different channels running along the crystallographic c axis (Fig. 1): narrow triangular-shaped channels, which do not contain guest molecules owing to their slim size; and hexagonal channels containing several lattice water molecules. These molecules are greatly disordered in such a way that they could not be located in the Fourier map, preventing further analysis of their interactions with the framework. Nevertheless, these interactions do not seem to be strong in view of their ease release from the powdered material during the sample drying stage. In fact, there is no low temperature mass release related to water molecules in the thermogravimetric measurement (Fig. S1, ESI†), meaning that lattice molecules remaining in the dried sample might be released during the equilibration process of the measurement. In any case, it can be confirmed that the squeezed electron density removed in the final refinement stage of the X-ray structure accounts for four water molecules, and this amount has been taken for the formula unit.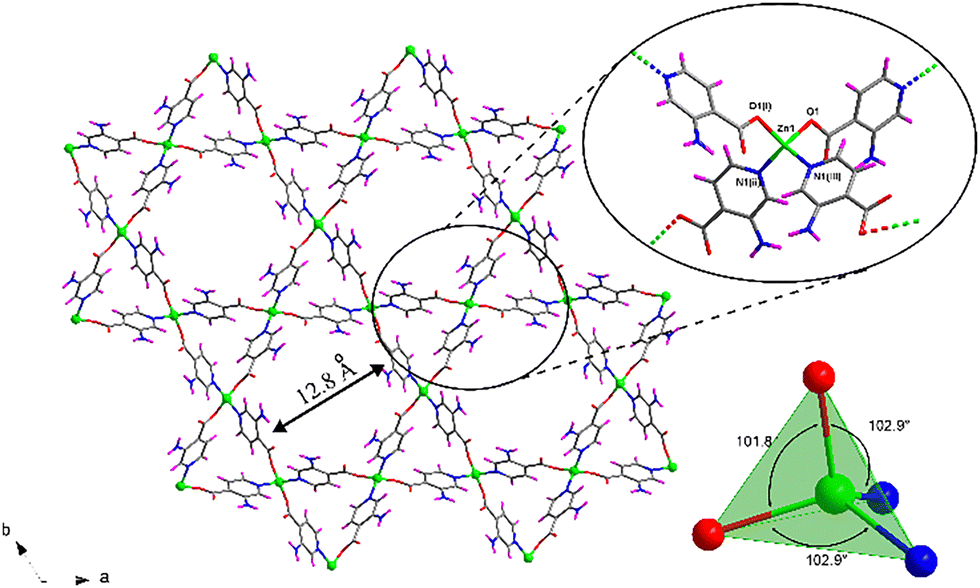 | ||
| Fig. 1 . Framework of compound 1 along the [001] direction of the unit cell showing the hexagonal and trigonal channels. The inset depicts a detailed view of the coordination of 3isoani ligand around Zn(II) ions. Colour coding: carbon (grey), hydrogen (pink), nitrogen (blue), oxygen (red) and zinc (green). Water lattice molecules are omitted. | ||
Photoluminescent measurements
Photoluminescent properties of compound 1 were first studied at ambient temperature on polycrystalline powder under a high vacuum. The recorded steady-state spectrum is composed of two well-differentiated broad bands peaking at 425 and 495 nm, with the second band showing almost twice the intensity of the first one (Fig. 2). | ||
| Fig. 2 Excitation and emission spectra of compound 1 recorded at RT under high vacuum (ca. 4 × 10−7 bar). The inset shows a photograph of a single crystal illuminated in the microscope. | ||
The separation between the two bands, of around 70 nm, exceeds the shift of vibrational levels, indicating that they originated from different electronic transitions.41 The closed-shell nature of Zn(II) precludes any ligand-to-metal (LMCT) or metal-to-ligand charge transfers (MLCT) and metal-centered emission, meaning that the organic ligand is the main substance responsible for the photoluminescent emission. This is confirmed by the steady-state emission spectrum collected for the free H3isoani ligand using the same setup, which shows a similar profile with the appreciation of two distinguishable bands at 428 and 562 nm, respectively (Fig. S7, ESI†). The shift in the band maxima between compound 1 and H3isoani, particularly for the second band, could be derived from its coordination. Back to the steady-state spectrum of 1, the presence of several bands is also known to be related to fluorescence and phosphorescence emissive processes, commonly observed in organic phosphors showing emission afterglows.42–44 However, this does not seem to be the case of the second (high-wavelength) band because the delayed emission spectrum collected under similar conditions of the steady-state reveals a null signal in the region of 560 nm, ruling out the possibility of phosphorescence in the present compound. Therefore, both bands must correspond to different fluorescent singlet-to-singlet electronic transitions. The excitation spectra recorded at the two emission maxima are similar and share a broad band covering the 250–400 nm spectral range (Fig. S8, ESI†). The similarities between these two excitation spectra suggest that a high-energy Sn excited state could be reached from the ground state (S0), and that the former could populate the other two low-lying excited states from which radiative relaxation would take place. Dual emission organic molecules are non-habitual since most of the fluorescent organic molecules suffer a rapid internal conversion from the Sn state to the S1 state and then relax to the S0 state with the emission of a photon in agreement with Kasha's rule.45 Nonetheless, dual fluorescent emissions have been reported in organic molecules from the simultaneous emissions from two different excited singlet states. In those cases, the most common case considers an equilibrium between a parent local excited state and a twisted intramolecular charge transfer,46 although the existence of dual charge transfer states,47 excimer states,48 excited state intramolecular photon transfer49 and structural modification upon exposure to UV-light50 may also derive in fluorescent dual emission. The emission capacity of compound 1 was also confirmed by the micrographs taken on single crystals under UV and visible light, where the cyan blue emission observed under λex = 365 nm is concordant with the steady-state emission spectrum (Fig. S9, ESI†). In addition, the absolute emission quantum yield (QY) measured for the high-energy emission band in the solid-state using the same experimental conditions at room temperature has a value of 2.1%. Other Zn-based MOFs have shown discrete quantum yields,51,52 which could result from the open and flexible nature of these crystalline materials that cannot hinder the vibrational non-radiative relaxation pathways.
To unravel the electronic transitions giving rise to the dual-fluorescent emission, TD-DFT calculations were performed on a suitable model of compound 1. More details about the computational methods are given in the Experimental Section. The calculated absorption spectrum reveals the occurrence of two bands at ca. 280 and 365 nm which reproduce quite well to those found in the diffuse reflectance spectrum of 1 (λabs = 255 and 365 nm, see Fig. 3a).
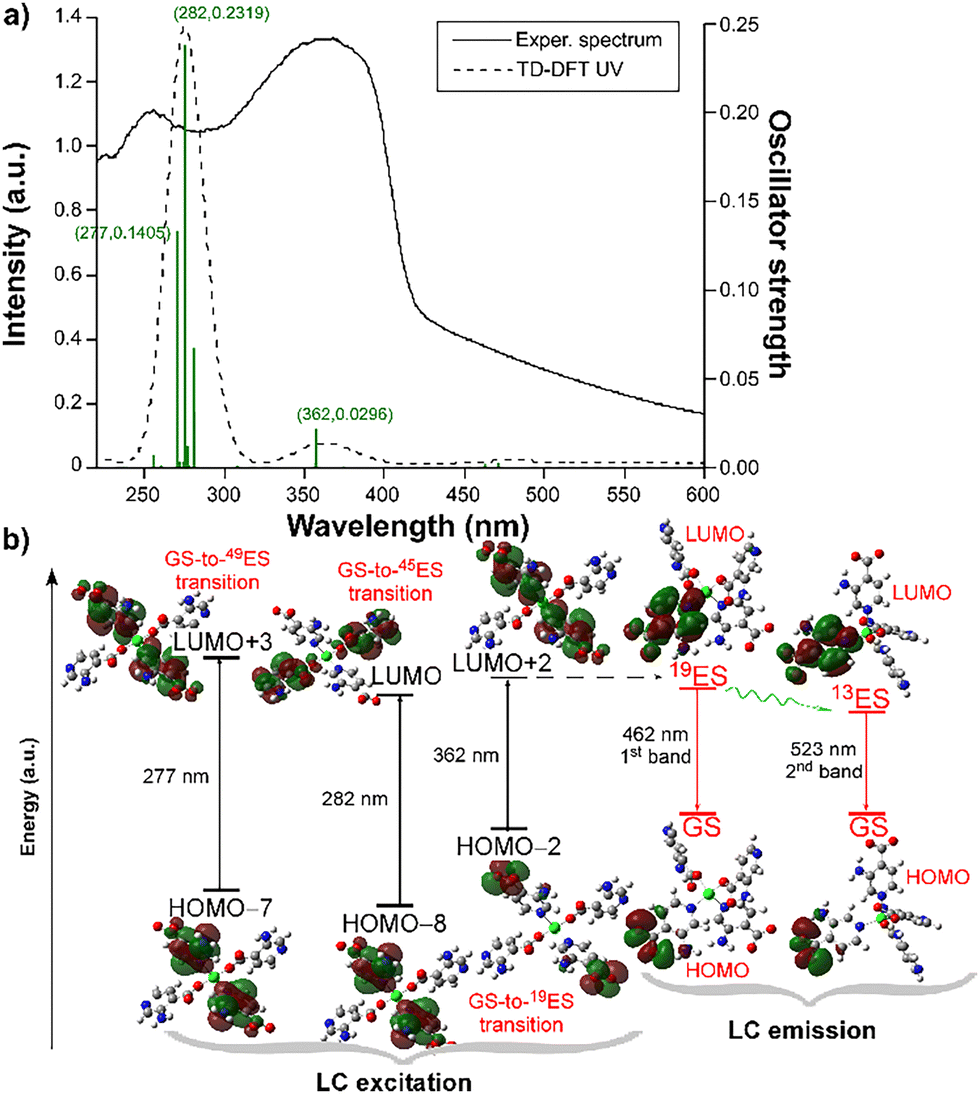 | ||
| Fig. 3 (a) Comparison of the TD-DFT and absorption spectra of compound 1, showing the most intense calculated excitations. (b) Energy diagram showing the main calculated excitations and emissions occurring at compound 1. | ||
These two bands can be described as ligand-centered (LC) excitations of a π–π* nature in view of the corresponding HOMO−n and LUMO+n orbitals involved in the electronic transitions (see Fig. 3b and Table S6 for further details, ESI†), as expected for a d10 ion in which LMCTs are not usually observed.53,54 However, the fact that the low-energy (second) band (at λabs = 365 nm) also corresponds to the main band observed in the excitation spectrum (Fig. S8, ESI†), whereas the first band (at λabs = 255 nm) is not observed in the latter spectrum, makes us assume that the luminescence process in this compound mainly involves the second band. In other words, the excitation process under laser excitation (at λex = 325 nm) is governed by the 19th excitation state (19ES), which is accessed when compound 1 is excited in the near UV region. This fact is also confirmed by the weak luminescence shown by the compound under excitation at 255 nm, where a similar double-band emission pattern is collected (Fig. S10, ESI†), meaning that 49ES and 45ES can transfer energy to 19ES, although the efficiency of the process is low because part of the excitation energy is non-radiatively dispersed in the system. The geometry optimization of the 19ES shows that the coordination sphere is not significantly modified (keeping the distorted tetrahedral environment) and that the most noticeable change corresponds to the relative rotation of mean planes of the 3isoani ligands (for instance, the angle is 54.5° in the geometry of GS whereas it increases up to 59.6° in the 19ES). Based on the latter geometry, the 19ES–GS energy difference by means of vertical excitation gives 2.68 eV (λ19EScalc,lum = 462 nm, see Fig. 3b), which is concordant with the luminescence energy of the first band (λem,1 = 428 nm), meaning that the first band derives from the radiative relaxation of the 19ES. Nonetheless, this excited state does not explain the occurrence of the second band, wherein another lower-lying excited state must be responsible. Among others, 13ES could be considered as a good candidate in view of its relatively high oscillator strength (f = 0.008) observed in the absorption calculation. A similar vertical excitation calculation using the optimized geometry of the 13ES gives a good agreement with the maximum wavelength of the second band (λ13EScalc,lum = 523 vs. λem,2 = 495 nm), meaning that the second band emission is derived from this excited state. Taking into account that both bands are also present in the free H3isoani molecule and that both share the excitation band at ca. 365 nm, it may be assumed that the 19ES → 13ES transfer is favored in the molecule.
When the photoluminescent measurements are repeated at a temperature of 10 K, the fluorescence emission spectrum of compound 1 preserves the two main contributions although a shoulder peaking at 515 nm is well distinguished at the second band (with the main component now slightly shifted to λmax,2 = 480 nm) in addition to a splitting of the maximum of the first band (λmax,1 = 412 and 428 nm) in contrast to RT. Moreover, there is an expected significant increase in the emission intensity at low temperatures owing to the decrease in non-radiative relaxation pathways.44,55 In any case, it deserves to be remarked that the increase in the emission intensity at lower temperature does not equally affect both emission bands, since both bands show similar intensities at 10 K (Fig. S11, ESI†). This behavior can be exploited using compound 1 as a ratiometric thermometer, for which further studies are discussed in the ratiometric temperature sensing section below.
On another level, the presence of the carboxylate decorated pyridine ring may endow the material with a long-lasting phosphorescent (LLP) behavior at low temperatures, as observed for other Zn-based coordination compounds built from positional isomers such as [Zn(μ-2ani)2]n and [Zn(μ-6ani)2]n (where 2ani = 2-aminonicotinate and 6ani = 6-aminonicotinate).56,57 Accordingly, decay curves were recorded at different emission wavelengths at a fixed excitation of 325 nm but very abrupt decays were collected, which are indicative of fluorescent processes. Fitting of the curves with the multiexponential expression [It = A0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2)] that considers different radiative pathways taking place in the emission to determine the emission lifetimes of the compounds afford short lifetimes of 4.5 ns and 11.9 ns and emission wavelengths of 420 and 515 nm, respectively (Fig. S12 and Table S5, ESI†). These short emission lifetimes discard the occurrence of LLP as observed for 2ani and 6ani-based compounds. The different behavior observed for the amino-derivatives of pyridine carboxylate ligands can be explained by resorting to the distinct supramolecular interactions occurring in this metal–organic compound. As empirically analyzed for molecular-based phosphorescent materials reported so far,58 a key point enabling LLP is derived from the restriction of the molecular motion in the crystal building by means of strong interactions in such a way that the lowest-lying triplet states are shielded from non-radiative quenching. In the case of compound 1, the open nature of its crystalline structure somewhat minimizes the supramolecular interactions. For instance, the amine group, known to be an oscillator that quenches the phosphorescent emissions,59 does not establish rigid hydrogen bonds in 1 because the only acceptors are lattice water molecules (which are easily lost from the pores, as discussed below); conversely there are strong intramolecular hydrogen bonds established in the case of the 2ani and 6ani ligands in their respective compounds.56,57 In line with this hypothesis, another previously reported 2-aminoisonicotinate (2ain)-based compound with a {[Zn(μ-2ain)2]·DMF}n formula60 that possesses a similar porous two-fold interpenetrated architecture, also shows fluorescent emissions of a similar order to that of compound 1.
exp(−t/τ1) + A2exp(−t/τ2)] that considers different radiative pathways taking place in the emission to determine the emission lifetimes of the compounds afford short lifetimes of 4.5 ns and 11.9 ns and emission wavelengths of 420 and 515 nm, respectively (Fig. S12 and Table S5, ESI†). These short emission lifetimes discard the occurrence of LLP as observed for 2ani and 6ani-based compounds. The different behavior observed for the amino-derivatives of pyridine carboxylate ligands can be explained by resorting to the distinct supramolecular interactions occurring in this metal–organic compound. As empirically analyzed for molecular-based phosphorescent materials reported so far,58 a key point enabling LLP is derived from the restriction of the molecular motion in the crystal building by means of strong interactions in such a way that the lowest-lying triplet states are shielded from non-radiative quenching. In the case of compound 1, the open nature of its crystalline structure somewhat minimizes the supramolecular interactions. For instance, the amine group, known to be an oscillator that quenches the phosphorescent emissions,59 does not establish rigid hydrogen bonds in 1 because the only acceptors are lattice water molecules (which are easily lost from the pores, as discussed below); conversely there are strong intramolecular hydrogen bonds established in the case of the 2ani and 6ani ligands in their respective compounds.56,57 In line with this hypothesis, another previously reported 2-aminoisonicotinate (2ain)-based compound with a {[Zn(μ-2ain)2]·DMF}n formula60 that possesses a similar porous two-fold interpenetrated architecture, also shows fluorescent emissions of a similar order to that of compound 1.
Ratiometric temperature sensing
As mentioned above, the distinct temperature-dependent evolution of the two bands composing the emission spectrum prompted us to use compound 1 as a luminescent thermometer (Fig. 4). In essence, both bands decrease in intensity upon increasing the temperature from 10 K onwards. However, they do not decrease proportionally, especially in the 200–300 K temperature range where the first band (high-energy band, λem,1 = 428 nm) decreases in intensity upon increasing temperature, whereas the second band (of low-energy, λem,2 = 495 nm) does not show temperature dependency and remains constant. Therefore, this unequal temperature-evolution is promising for the compound to behave as a ratiometric luminescent thermometer in the selected temperature range. Taking into account that the structure of the two main bands is strongly affected by the temperature since it splits both band maxima into two (giving a total of four components), the spectra were first converted to energy units (eV) and then properly decomposed by fitting the bands according to Gaussian functions (see more details in Fig. S13 section of the ESI†). The thermometric analysis has been conducted in terms of the relative integrated intensity (areas) of the corresponding functions obeying the relationship between both bands (Δ = I2/I1). As illustrated in the inset of Fig. 4, two distinct regimens can be found when Δ is plotted against temperature: a low temperature region covers the 50–200 K range where the Δ value barely changes with the temperature (see inset in Fig. 4), and there is a second regimen in the 200–300 K region where the change is more pronounced. Both regimens can be fitted linearly, showing the highest sensitivity between 200 and 300 K in the following equation:| Δ = 0.03606T − 3.5706 | (1) |
 | (2) |
 | (3) |
 | ||
| Fig. 4 Fluorescence emission spectra of compound 1 at variable temperatures maintaining the excitation at 325 nm. Inset: Best fit of the relationship between the ratiometric intensity of the bands. | ||
The best fitting results were of 0.14 K−1 and 3.2% K−1 at 250 K for the absolute and maximum relative sensitivity, respectively. The obtained values are comparable to other MOFs for ratiometric temperature sensing (Table 1). The thermometric activity was also examined in the high-temperature range (above 300 K), revealing a less intriguing behavior with diminished sensitivity (see Fig. S14, ESI†).
| MOF | T range (K) | S a (K−1) | S r (% K−1) | Ref. |
|---|---|---|---|---|
| ZnATZ-BTB | 30–130 | 0.09 | 5.29 (30 K) | 41 |
| [Sr(NH2-bdc)(DMF)]n | 10–320 | 0.12 | 7.5 (130 K) | 13 |
| Tb0.957Eu0.043cpda | 40–300 | 0.0037 | 16.0 (300 K) | 62 |
| Tb0.9Eu0.1(pia) | 100–300 | 0.0353 | 3.53 (300 K) | 63 |
| Tb0.99Eu0.01(BDC)1.5(H2O)2 | 290–320 | 0.0014 | 0.31 (318 K) | 64 |
| ZJU-88Ɔperylene | 293–353 | 0.0128 | 1.28 (293 K) | 65 |
| Eu@UiO-(bpydc) | 293–353 | 0.0228 | 0.31 (293 K) | 66 |
The wide variety of possible emission centers in MOFs makes these materials suitable for this purpose, for which the most common emissive-center combinations could be: (i) metal/ligand, (ii) metal/metal and (iii) metal/guest.61 It must be highlighted that, as shown in Table 1, most reported MOFs for ratiometric thermometry are built up from two emissive centers, usually by combining rare-earth Ln3+ ions such as Eu3+ with Tb3+, whereas the use of close-shell ions for constructing ratiometric luminescent thermometers has been comparatively scarcely reported.
Study on the origin of the dual emission
Captivated by the good thermal sensitivity offered at low temperature based on the relative intensity changes occurring on the two emission bands shown by this luminescent compound, we decided to study the origin of its dual emission in greater depth and check the possible physico-chemical factors involved. To that end, an emission spectrum was acquired on freshly ground single crystals of 1 at RT in the open atmosphere (without applying a vacuum to the sample). To our surprise, the spectrum only showed an intense band (that was previously named as high-energy band, λmax,1 = 425 nm), whereas the second band vanished to leave a large tail extending across 250 nm in its place (Fig. 5). Intrigued by the drastic change in the emission pattern, the sample was then submitted to the usual vacuum (note that all previous measurements are performed under a high-vacuum of ca. 4 × 10−7 bar). Interestingly, the emission pattern was progressively changing as the vacuum increased in the sample cryostat, with the first band (λmax,1 = 425 nm) decreasing its intensity and the second band (λmax,2 = 495 nm) showing the opposite effect (Fig. 5). It is worth highlighting that the second band exceeds the first one by a small amount in the present study, although the relative intensity of the first band does not experience the expected abrupt drop to become a minor band as observed for the first sample studied (see Fig. 2). | ||
| Fig. 5 . Evolution of the emission spectrum of compound 1 recorded at RT under variable vacuum. | ||
Being the dual emission of compound 1 a ligand-centred phenomenon in which two excited states (19ES in the first band and 13ES in the second) are involved, the observed behaviour seems to be related to structural changes occurring in the porous and flexible framework, possibly related to the release of the weakly bounded lattice water molecules. To corroborate this hypothesis, the previously analysed sample 1 was heated at 50 °C for 3 h, after which the sample was completely dehydrated as confirmed by means of a TG/DTA measurement (Fig. S2, ESI†). The emission spectrum of this sample under high-vacuum (see Fig. 5) resembles that initially shown for the compound, meaning that all previous spectroscopic data (steady-state and lifetime spectroscopy as well as the thermometry) have been taken on a dehydrated sample. As a consequence, considering that no previous treatment had been performed on that sample, we can conclude that compound 1 may be spontaneously dehydrated by releasing the lattice water molecules of the pores. This fact is in good agreement with their disordered arrangement into the pores, which involves a lack of strong interactions with the framework surface, high mobility and ease of evaporation.
Sensing activity in membranes
Based on the strong dependency of the global emission of the compound according to the presence/absence of water in the pores, a further step explored the capacity to detect cations in water. Nonetheless, given the well-known low chemical stability of MOFs in water,67 we decided to protect 1 by creating a mixed-matrix membrane (MMM) that not only enhances the stability of the MOF but further facilitates its handling. For the membrane fabrication, we followed the solvent-casting method by the dispersion of freshly prepared compound 1 into polymethylmethacrylate (PMMA) dissolved in dichloromethane. More details about the experimental procedure are given in the experimental section. PMMA was selected among other polymeric matrices to disperse the compound based on its good optical properties, aiming to avoid any interference with the MOF emission.33 For the formulation of the membrane, different MOF-to-polymer ratios were tested to achieve a balanced composite, trying to avoid an excessive MOF/polymer ratio that undermines mechanical properties of the membrane making it brittle, or low amounts of MOF resulting in weak emission intensities. The collected diffraction pattern (Fig. S19, ESI†) confirmed that the structural integrity and crystallinity of compound 1 in the membrane (1@PMMA hereafter) are well-preserved. The fact that compound 1 is well dispersed on the membrane was also confirmed by the photographs taken in the microscope (Fig. S20, ESI†) and SEM images where a homogenous distribution of crystallites of compound 1 are observed in 1@PMMA (Fig. S31, ESI†), maintaining the rod-shaped morphology and size of the as-prepared samples of 1 (see ESI†). First, the emission spectrum was collected to ensure that the photoluminescent properties remain unaltered in the membrane. As observed in Fig. S15 (ESI†), the vacuum-dependent behaviour is reproduced for 1@PMMA in such a way that a second band (λmax,2 = 480 nm, in addition to the main band at λmax,1 = 420 nm) is shown when the film is degassed under high vacuum for few hours. Moreover, this vacuum-induced effect is shown to be fully reversible even for the material embedded in the PMMA membrane, in such a way that the single-band emission is recovered by exposing the sample to open atmosphere for some time (Fig. S16, ESI†). The fact that the process is accelerated by soaking the membrane in water supports the idea that this effect is also related to the de-/rehydration of compound 1 (by loss/refilling of the pores with water molecules). The thermometric response of the membrane was analyzed by means of the ratiometric intensity of the bands (Δ = I2λ=420/I1λ=480), finding slightly weaker dependence of the signal with temperature than for the bulk compound and a lower maximum Sr of 2.7% K−1 at 250 K (Fig. S17, ESI†).On another level, the sensing activity of the membrane was studied in aqueous solutions in spite of its single-band luminescence observed in aqueous medium implying lower sensitivity. To that end, the membrane was firstly immersed in water and an emission spectrum (Fig. S22, ESI†) was collected at room temperature (λex = 310 nm) in agreement with previous experiments performed under vacuum. As previously concluded, the occurrence of the high-energy band dominating the spectrum of this compound implies the upload of pores with water molecules, which could be the case of 1@PMMA for several reasons: (i) having employed fresh sample, (ii) the presence of water in the dichloromethane employed during the preparation of the composite and (iii) the uptake of water molecules into the pores when the composite is immersed in water.
Despite the presence of a unique (high-energy) band in the emission spectrum of 1@PMMA, sensing experiments were conducted immersing the membrane in aqueous solutions containing different transition metal, alkaline and alkaline-earth ions. As shown in Fig. S23 (ESI†), the emission pattern remains unaltered but there is a variable emission intensity of the band with the following quenching percentage (QP, %) taking the intensity of the membrane immersed in water as reference: Mg2+ (5.37%) < K+ (6.66%) < Ca2+ (7.76%) < Na+ (9.38%) < Cd2+ (13.7%) < Co2+ (53.1%) < Ni2+ (56.0%) < Cu2+ (80.2%) < Cr3+ (99.0%) < Fe3+ (99.9%). Despite the similar quenching effect observed when 1@PMMA is in the presence of Cu2+, Cr3+ and Fe3+, we selected Fe3+ as the target analyte since it presents the highest quenching percentage among the studied ions. Accordingly, the concentration of the target ion was gradually increased starting from a very low concentration, leading to a progressive quench of the emission intensity (Fig. 6 and Fig. S24, ESI†). The data were fitted to the linear Stern–Volmer expression (eqn (4)) to analyze the sensing capacity of the material toward Fe3+.
 | (4) |
 | ||
| Fig. 6 Emission spectra of 1@PMMA with varying Fe3+ concentration (λex = 310 nm). The inset shows the Stern–Volmer plot for the titration experiment. | ||
We evaluated possible interferences of other ions in the sensing selectivity of 1@PMMA. Fig. S28 (ESI†) shows the potential interference that may be caused by different ions for Fe3+ sensing, with the main interfering effects promoted by Cr3+ and Cu2+ ions. In the view that alkaline and alkaline earths ions do not interfere with the sensing capacity of 1@PMMA toward Fe3+, we repeated titration experiments using tap water instead, which mainly contains alkaline and alkaline-earth ions. The observed trend was consistent with that seen using distilled water, showing a gradual decrease in fluorescent emission as the quencher concentration increased (Fig. S29, ESI†). Fitting the results to the S–V equation affords a KSV value of 1.7 × 103 M−1, with a LOD of 4.7 × 10−5 M in the same concentration range (2 × 10−5–4 × 10−4 M), obtaining comparable results to that obtained from distilled water.
Conclusions
In this work, the combination of Zn(II) with 3-aminoisonicotinate ligand under hydrothermal conditions leads to the formation of a doubly interpenetrated 3D open network with triangular and hexagonal channels, and the latter is filled with disordered water molecules. The photoluminescent properties of the MOF were first studied at room temperature, showing a dual fluorescent emission originating from the organic part confirmed by TD-DFT calculations. The emission is highly influenced by the lattice water molecules since their presence in the pores quenches the low-energy emission band, probably due to the resulting rearrangement of the ligand in the flexible framework. The fact that the intensity of the bands does not follow the same trend when lowering the temperature provides the compound with a maximum Sr of 3.2% K−1 at 250 K according to a ratiometric analysis, which may be considered as a promising result among the ligand-centred MOF-based luminescent thermometers reported so far. Additionally, to obtain a more manageable material, a membrane of compound 1 was prepared by dispersing it into PMMA. The membrane shows a promising detection of Fe3+ in water with high sensitivity (KSV of 3.7 × 103 M−1) and a low limit of detection (2.8 × 10−5 M).Experimental section
Chemicals
All chemicals were of reagent grade and used as commercially obtained without any further purification.Synthesis of [Zn(μ-3isoani)2]n·4H2O (1)
2 mL of a water solution containing 0.0297 g of Zn(NO3)2·6H2O (0.1 mmol) was added dropwise into 15 mL of a water/ethanol solution (2:1) containing 0.0276 g of H3isoani (0.2 mmol), which was previously basified with Et3N until the complete dissolution of the ligand. Then, the vial was placed in an oven for 3 hours at 80 °C and slowly cooled down to room temperature. Light-brown needle-shaped single crystals were grown, which were filtered and washed with water and ethanol several times. Yield: 60% based on the metal. Anal. calcd for C12H18N4O8Zn (%): C, 35.01; H, 4.41; N, 13.61. Found: C, 34.45; H, 3.72; N, 13.90.
Preparation of 1@PMMA
For the membrane fabrication, the solvent-casting method was followed testing different MOF/polymer formulations to achieve a balance between the mechanical properties and good photoluminescent emission. The optimum MOF/polymer ratio was established to be 35 mg of compound 1 and 225 mg of PMMA, which were mixed in 4 mL of CH2Cl2. Then, the mixture was sonicated for 15 minutes, casted in a glass Petri dish, and left unperturbed for 1 day at room temperature until the complete evaporation of the solvent to finally form the membrane. The membrane was cut into rectangular-shaped pieces of 2 × 3 cm for the sensing experiments.Chemical characterization
Elemental analyses, FTIR spectra, diffuse reflectance measurements and thermal analyses were performed on powdered samples of compound 1 as detailed in the ESI† (see S1 section).X-ray diffraction data collection and structure determination
The single crystal X-ray diffraction (SCXD) data used for structure determination was recorded at 100(2) K on a suitable single crystal of 1 on a Bruker VENTURE diffractometer (see further details in the ESI† and in Table 2). The structure was refined with SHELX program75 including all reflections and full-matrix least-squares on F2 employing the Olex2 software.76 Apart from the usual refining strategy (described in the ESI†), it must be noticed that lattice water molecules could not be correctly modelled. Therefore, their electron density was subtracted from the Fourier map in the final refinement cycles by means of a Squeeze routine,77 which accounts for a total of four molecules per formula unit. All crystallographic details are provided as a cif file and can be found in Cambridge Crystallographic Data Center as a supplementary publication with CCDC no. 2412031.†| 1 | |
|---|---|
| a S = [∑w(F02 − Fc2)2/(Nobs − Nparam)]1/2. b R 1 = ∑||F0| − |Fc||/∑|F0| c wR2 = [∑w(F02 − Fc2)2/∑wF02]1/2; w = 1/[σ2(F02) + (aP)2 + bP] where P = (max(F02,0) + 2Fc2)/3 with a = 0.0424. | |
| Empirical formula | C12H18N4O8Zn |
| Formula weight | 411.68 |
| Crystal system | Trigonal |
| Space group | P31 |
| a (Å) | 15.5877(10) |
| b (Å) | 15.5877(10) |
| c (Å) | 6.1552(5) |
| α (°) | 90 |
| β (°) | 90 |
| γ (°) | 120 |
| V (Å3) | 1295.2(2) |
| Reflections collected | 18941 |
| Unique data/parameters | 1798/192 |
| R int | 0.1432 |
| GoF (S)a | 1.086 |
| Flack parameter | 0.095(9) |
|
R
1
/wR2c [I > 2σ(I)] |
0.0309/0.0399 |
|
R
1
/wR2c [all] |
0.0754/0.0769 |
The X-ray powder diffraction (XRD) patterns were acquired on a Philips X’PERT powder diffractometer as detailed in the ESI.†
Photoluminescence measurements
Photoluminescent measurements were recorded in the 10–310 K range on polycrystalline samples in a closed-cycle helium cryostat enclosed in an Edinburgh Instruments FLS920 spectrometer using several excitation sources as detailed in the ESI.† Micro-luminescence photographs were taken at room temperature in an Olympus optical microscope equipped with a Hg lamp. Quantum yield measurements were performed in a Horiba Quanta-φ integrating sphere using an Oriel Instruments MS257 lamp as excitation source and an iHR550 spectrometer from Horiba to analyze the emission.Sensing experiments in the membrane
The sensing experiments were carried out by immersing 1@PMMA into the metal-containing aqueous solution. 1 × 10−3 M solutions were prepared by dissolving the nitrate salt of each metal in water. For the Fe3+ titration experiment, the membrane was initially immersed in water and the concentration of the quencher was gradually increased. The membrane was not taken out from the cuvette during the whole experiment. After each addition of the quencher, the cuvette was maintained under continuous stirring for 5 minutes to ensure a complete homogeneity of the solution before the collection of the emission spectrum.Emission spectra were recorded on a JASCO fluorescent spectrum-8300 at room temperature excited at 310 nm. A complete emission spectrum was recorded at each concentration in the 320–600 nm spectral range using a Xe lamp with data acquisition every 0.5 nm and setting the bandwidth at 5 nm for the excitation and the emission. The LOD is calculated following the IUPAC recommendation of the 3σ/slope, where σ stands for the standard deviation of ten repeated measurements of blank samples.78
Computational details
TD-DFT calculations were performed over a suitable model of compound 1 (see ESI†) using the Gaussian 16 package.79 Further details about computation strategy and model chemistry are detailed in S1 of the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Ministerio de Ciencia, Innovación y Universidades (MCIN/AEI/https://doi.org/10.13039/501100011033) and the Resilience Funds Next Generation of the European Union (TED2021-132440B-I00), Red Guipuzcoana de Ciencia, Tecnología e Innovación (FA385/2023, DG23/16), Gobierno Vasco/Eusko Jaurlaritza (IT1755-22 and IT1500-22), and Junta de Andalucía (FQM-394, B-FQM-734-UGR2 and P21_00386). The authors thank the technical and human support provided by SGIker (UPV/EHU/ERDF, EU). This work was also within the scope of the project CICECO-Aveiro Institute of Materials, (Refs. UIDB/50011/2020, DOI 10.54499/UIDB/50011/2020; UIDP/50011/2020, DOI 10.54499/UIDP/50011/2020; LA/P/0006/2020, DOI 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). RFM gratefully acknowledges FCT for the Junior Research Position (CEECIND/00553/2017).Notes and references
- N. A. Khan, Z. Hasan and S. H. Jhung, Coord. Chem. Rev., 2018, 376, 20–45 CrossRef CAS.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- H. Li, L. Li, R. B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef PubMed.
- X. Lu, Y. Tang, G. Yang and Y. Y. Wang, CrystEngComm, 2023, 25, 896–908 RSC.
- X. Zhang, V. Vieru, X. Feng, J. L. Liu, Z. Zhang, B. Na, W. Shi, B. W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef CAS PubMed.
- Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. I. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211–18220 CrossRef CAS PubMed.
- S. Zheng, D. Wu, L. Huang, M. Zhang, X. Ma, Z. Zhang and S. Xiang, J. Appl. Electrochem., 2019, 49, 563–574 CrossRef CAS.
- X. Guo, N. Zhu, Y. Lou, S. Ren, S. Pang, Y. He, X. B. Chen, Z. Shi and S. Feng, Chem. Commun., 2020, 56, 5389–5392 RSC.
- C. Jia, T. He and G. M. Wang, Coord. Chem. Rev., 2023, 476, 214930 CrossRef CAS.
- T. Wu, X. Gao, F. Ge and H. Zheng, CrystEngComm, 2022, 24, 7881–7901 RSC.
- L. Marciniak, K. Kniec, K. Elżbieciak-Piecka, K. Trejgis, J. Stefanska and M. Dramićanin, Coord. Chem. Rev., 2022, 469, 214671 CrossRef CAS.
- P. Leo, D. Briones, J. A. Garciá, J. Cepeda, G. Orcajo, G. Calleja, A. Rodríguez-Diéguez and F. Martínez, Inorg. Chem., 2020, 59, 18432–18443 CrossRef CAS PubMed.
- Q. Zhu, T. Sheng, C. Tan, S. Hu, R. Fu and X. Wu, Inorg. Chem., 2011, 50, 7618–7624 CrossRef CAS PubMed.
- D. Ma, B. Li, X. Zhou, Q. Zhou, K. Liu, G. Zeng, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8964–8966 RSC.
- M. D. Dramićanin, J. Appl. Phys., 2020, 128, 40902 CrossRef.
- C. D. S. Brites, S. Balabhadra and L. D. Carlos, Adv. Opt. Mater., 2019, 7, 1801239 CrossRef.
- Y. Cui, F. Zhu, B. Chen and G. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC.
- Y. Zhao, H. Zeng, X. W. Zhu, W. Lu and D. Li, Chem. Soc. Rev., 2021, 50, 4484–4513 RSC.
- T. Rasheed and F. Nabeel, Coord. Chem. Rev., 2019, 401, 213065 CrossRef CAS.
- A. Dutta, A. Singh, X. Wang, A. Kumar and J. Liu, CrystEngComm, 2020, 22, 7736–7781 RSC.
- O. Pajuelo-Corral, L. Razquin-Bobillo, S. Rojas, J. A. García, D. Choquesillo-Lazarte, A. Salinas-Castillo, R. Hernández, A. Rodríguez-Diéguez and J. Cepeda, Nanomaterials, 2022, 12, 3977 CrossRef CAS PubMed.
- W. Xie, M. Y. Xu, W. Jiang, G. J. Xu, S. R. Zhang, Y. H. Xu and Z. M. Su, Inorg. Chim. Acta, 2021, 528, 120635 CrossRef CAS.
- L. Liu, Y. Ran, J. Du, Z. Wang, M. Liu and Y. Mu, RSC Adv., 2021, 11, 11266–11272 RSC.
- X. Ma, Y. Chai, P. Li and B. Wang, Acc. Chem. Res., 2019, 52, 1461–1470 CrossRef CAS PubMed.
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef PubMed.
- W. W. Lestari, R. Al Adawiyah, M. A. Khafidhin, R. Wijiyanti, N. Widiastuti and D. S. Handayani, Open Chem., 2021, 19, 307–321 CrossRef CAS.
- L. Cao, F. Lv, Y. Liu, W. Wang, Y. Huo, X. Fu, R. Sun and Z. Lu, Chem. Commun., 2015, 51, 4364–4367 RSC.
- M. S. Denny, M. Kalaj, K. C. Bentz and S. M. Cohen, Chem. Sci., 2018, 9, 8842–8849 RSC.
- E. A. Feijani, H. Mahdavi and A. Tavasoli, Chem. Eng. Res. Des., 2015, 96, 87–102 CrossRef CAS.
- T. Zhu, X. Yu, M. Yi and Y. Wang, ACS Sustainable Chem. Eng., 2020, 8, 12664–12676 CrossRef CAS.
- H. Wang, S. Tang, Y. Ni, C. Zhang, X. Zhu and Q. Zhao, J. Membr. Sci., 2020, 598, 117791 CrossRef CAS.
- E. Echenique-Errandonea, R. F. Mendes, F. Figueira, P. Barbosa, S. Rojas, D. Choquesillo-Lazarte, J. Cepeda, D. Ananias, F. Figueiredo, F. A. Almeida Paz, A. Rodríguez-Diéguez and J. M. Seco, Nanomaterials, 2022, 12, 4380 CrossRef CAS PubMed.
- N. Prasetya, A. A. Teck and B. P. Ladewig, Sci. Rep., 2018, 8, 2944 CrossRef PubMed.
- O. Pajuelo-Corral, S. Pérez-Yáñez, I. J. Vitorica-Yrezabal, G. Beobide, A. Zabala-Lekuona, A. Rodríguez-Diéguez, J. M. Seco and J. Cepeda, Mater. Today Chem., 2022, 24, 100794 CrossRef CAS.
- O. Pajuelo-Corral, I. J. Vitorica-Yrezabal, A. Rodríguez-Diéguez, J. M. Seco and J. Cepeda, Inorg. Chem. Commun., 2024, 160, 111872 CrossRef CAS.
- Z. Shao, C. Yu, Q. Xie, Q. Wu, Y. Zhao and H. Hou, Chem. Commun., 2019, 55, 13382–13385 RSC.
- D. Llunell, M. Casanova, D. Cirera, J. Bofill, J. M. Alemany, P. Alvarez, S. Pinsky and M. Avnir, SHAPE, v1.1b, Barcelona, Spain, 2005 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- H. Zhang, C. Lin, T. Sheng, S. Hu, C. Zhuo, R. Fu, Y. Wen, H. Li, S. Su and X. Wu, Chem. – Eur. J., 2016, 22, 4460–4468 CrossRef CAS PubMed.
- S. Kuno, T. Kanamori, Z. Yijing, H. Ohtani and H. Yuasa, ChemPhotoChem, 2017, 1, 102–106 CrossRef CAS.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1–50 Search PubMed.
- S. V. Eliseeva, D. N. Pleshkov, K. A. Lyssenko, L. S. Lepnev, J. C. G. Bünzli and N. P. Kuzmina, Inorg. Chem., 2010, 49, 9300–9311 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- Z. Zhang, Y. Gao, H. Liu, Q. Bai, J. Li, L. Liu, C. Wu, B. Yang, K. Wang, B. Zou, Y. Wang and P. Lu, Dyes Pigm., 2017, 145, 294–300 CrossRef CAS.
- N. A. Kukhta, A. S. Batsanov, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 28564–28575 CrossRef CAS.
- Rohini, M. Baral and B. K. Kanungo, RSC Adv., 2016, 6, 108017 RSC.
- C. Azarias, Š. Budzák, A. D. Laurent, G. Ulrich and D. Jacquemin, Chem. Sci., 2016, 7, 3763–3774 RSC.
- Z. Xu, Q. T. Liu, X. Wang, Q. Liu, D. Hean, K. C. Chou and M. O. Wolf, Chem. Sci., 2020, 11, 2729–2734 RSC.
- T. S. Sukhikh, E. Y. Filatov, A. A. Ryadun, K. A. Kovalenko and A. S. Potapov, Molecules, 2022, 27 Search PubMed.
- P. I. Scheurle, A. Mähringer, A. C. Jakowetz, P. Hosseini, A. F. Richter, G. Wittstock, D. D. Medina and T. Bein, Nanoscale, 2019, 11, 20949–20955 RSC.
- S. Majumder, L. Mandal and S. Mohanta, Inorg. Chem., 2012, 51, 8739–8749 CrossRef CAS PubMed.
- A. K. Gupta, K. Tomar and P. K. Bharadwaj, New J. Chem., 2017, 41, 14505–14515 RSC.
- A. De Bettencourt-Dias, P. S. Barber, S. Viswanathan, D. T. De Lill, A. Rollett, G. Ling and S. Altun, Inorg. Chem., 2010, 49, 8848–8861 CrossRef CAS PubMed.
- J. Cepeda, E. S. Sebastian, D. Padro, A. Rodríguez-Diéguez, J. A. García, J. M. Ugalde and J. M. Seco, Chem. Commun., 2016, 52, 8671–8674 RSC.
- O. Pajuelo-Corral, A. Rodríguez-Diéguez, J. A. García, E. San Sebastián, J. M. Seco and J. Cepeda, Dalton Trans., 2018, 47, 8746–8754 RSC.
- E. San Sebastian, A. Rodríguez-Diéguez, J. M. Seco and J. Cepeda, Eur. J. Inorg. Chem., 2018, 2155–2174 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. De Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc. Perkin Trans. 2, 1999, 493–503 RSC.
- A. A. García-Valdivia, S. Pérez-Yáñez, J. A. García, B. Fernández, J. Cepeda and A. Rodríguez-Diéguez, Sci. Rep., 2020, 10, 8843 CrossRef PubMed.
- J. Rocha, C. D. S. Brites and L. D. Carlos, Chem. – Eur. J., 2016, 22, 14782–14795 CrossRef CAS PubMed.
- Y. Cui, W. Zou, R. Song, J. Yu, W. Zhang, Y. Yang and G. Qian, Chem. Commun., 2014, 50, 719–721 RSC.
- X. Rao, T. Song, J. Gao, Y. Cui, Y. Yang, C. Wu, B. Chen and G. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef CAS PubMed.
- A. Cadiau, C. D. S. Brites, P. M. F. J. Costa, R. A. S. Ferreira, J. Rocha and L. D. Carlos, ACS Nano, 2013, 7, 7213–7218 CrossRef CAS PubMed.
- Y. Cui, R. Song, J. Yu, M. Liu, Z. Wang, C. Wu, Y. Yang, Z. Wang, B. Chen and G. Qian, Adv. Mater., 2015, 27, 1420–1425 CrossRef CAS PubMed.
- Y. Zhou and B. Yan, J. Mater. Chem. C, 2015, 3, 9353–9358 RSC.
- Y. An, X. Lv, W. Jiang, L. Wang, Y. Shi, X. Hang and H. Pang, Green Chem. Eng., 2024, 5, 187–204 CrossRef.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 CrossRef CAS.
- D. Wang, D. Zhang, S. De Han, J. Pan, Z. Z. Xue, J. H. Li and G. M. Wang, Dalton Trans., 2019, 48, 602–608 RSC.
- S. K. Panda, S. Mishra and A. K. Singh, Dalton Trans., 2021, 50, 7139–7155 RSC.
- X. Liu, W. X. Guo, X. L. Hu, Y. Y. Wang, Q. Yue and E. Q. Gao, J. Solid State Chem., 2019, 273, 53–61 CrossRef CAS.
- X. D. Fang, J. Yao, R. Fan, X. F. Bai, Y. E. Liu, C. F. Hou, Q. Q. Xu, A. X. Zhu and B. Huang, J. Solid State Chem., 2021, 294, 121854 CrossRef CAS.
- B. B. Rath and J. J. Vittal, Inorg. Chem., 2020, 59, 8818–8826 CrossRef CAS PubMed.
- J. Li, Y. X. Zhao, Q. Wu, H. Yang, J. Lu, H. Y. Ma, S. N. Wang and Y. W. Li, CrystEngComm, 2021, 23, 8392–8403 RSC.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357–361 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- L. Li, S. Shen, R. Lin, Y. Bai and H. Liu, Chem. Commun., 2017, 53, 9986–9989 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revis. C.01, Gaussian Inc., Wallingford, UK, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2412031. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc05392c |
| This journal is © The Royal Society of Chemistry 2025 |