
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of bipolar charge transport of derivatives of 1-phenyl-1H-benzo[d]imidazole with horizontal molecular orientation on the performance of OLEDs based on thermally activated delayed fluorescence or phosphorescence†
Simas
Macionis
 a,
Ehsan Ullah
Rashid
a,
Jurate
Simokaitiene
a,
Rita
Butkute
a,
Oleksandr
Bezvikonnyi
ab,
Dmytro
Volyniuk
a,
Dalius
Gudeika
a,
Tien-Lung
Chiu
c,
Jiun-Haw
Lee
d,
Zi-Wen
Su
d,
Chia-Hsun
Chen
d,
Ruta
Budreckiene
e,
Mariia
Stanitska
a,
Oleksandr
Navozenko
f and
Juozas V.
Grazulevicius
*a
a,
Ehsan Ullah
Rashid
a,
Jurate
Simokaitiene
a,
Rita
Butkute
a,
Oleksandr
Bezvikonnyi
ab,
Dmytro
Volyniuk
a,
Dalius
Gudeika
a,
Tien-Lung
Chiu
c,
Jiun-Haw
Lee
d,
Zi-Wen
Su
d,
Chia-Hsun
Chen
d,
Ruta
Budreckiene
e,
Mariia
Stanitska
a,
Oleksandr
Navozenko
f and
Juozas V.
Grazulevicius
*a
aDepartment of Polymer Chemistry and Technology, Faculty of Chemical Technology, Kaunas University of Technology, K. Baršausko st. 59, LT-51423, Kaunas, Lithuania. E-mail: juozas.grazulevicius@ktu.lt
bKTU “M-Lab” Laboratory Center, Kaunas University of Technology, Studentų g. 63A, LT-51369, Kaunas, Lithuania
cDepartment of Electrical Engineering, Yuan Ze University, Chungli, Taoyuan, 32003, Taiwan. E-mail: tlchiu@saturn.yzu.edu.tw
dGraduate Institute of Photonics and Optoelectronics and Department of Electrical Engineering, National Taiwan University, Taipei, 10617, Taiwan. E-mail: jiunhawlee@ntu.edu.tw
eDepartment of Biochemistry, Lithuanian University of Health Sciences, A. Mickeviciaus st. 9, LT-44307, Kaunas, Lithuania
fDepartment of Experimental Physics, Faculty of Physics, Taras Shevchenko National University of Kyiv, Akademika Glushkova Av. 4, 03127, Kyiv, Ukraine
First published on 9th January 2025
Abstract
The synthesis and properties of two derivatives of 1-phenyl-1H-benzo[d]imidazole with differing numbers of tert-butylcarbazole electron-donating moieties are reported. The compounds exhibit high thermal stability, with 5% weight loss temperatures exceeding 341 °C and glass transition temperatures of over 149 °C. They display moderate triplet energies of 2.63 and 2.66 eV. The synthesized compounds were employed as host materials in phosphorescence and TADF-based organic light-emitting diodes (OLEDs). An investigation of the angle-dependent emission intensity of light-emitting layers containing the phosphorescent emitter Ir(ppy)2(acac) doped into the examined compounds revealed a notably high internal outcoupling efficiency in OLEDs, exceeding 30%. This efficiency is attributed to the significant horizontal molecular orientation factor, reaching up to 87%. Based on the characterization of the hosting properties of 1-phenyl-1H-benzo[d]imidazole derivatives, the most significant influence on device performance is attributed to their charge-transporting properties. An OLED with the phosphorescent emitter Ir(ppy)2(acac) and a host material exhibiting bipolar charge transport demonstrated an external quantum efficiency of 13%. Additionally, the picric acid sensitivity of one of the compounds was examined. Triplet-facilitated emission was completely quenched upon the addition of a nitroaromatic explosive as a guest in a film.
1. Introduction
The technology of organic light emitting diodes (OLEDs) is undergoing rapid development owing to the advantages of color quality, response time and lower energy consumption of OLEDs when compared to competing devices in the market of display and illumination technology.1 The singlet and triplet excitons are formed upon applying electricity in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 due to the spin statistics.2,3 As a result, prompt fluorescent emitters have a limit of internal quantum efficiency (IQE) of 25%.4 The main path to increase the efficiency of OLEDs is utilization of the triplet excited states in electroluminescence (EL), potentially increasing the IQE value to 100%. In phosphorescence OLEDs (PhOLEDs), heavy metal complexes (such as iridium or platinum complexes) which facilitate spin–orbit coupling and in turn accelerate the radiative deactivation through phosphorescence are utilized. Despite the increase in performance of the devices, the use of heavy metal atoms brings certain problems, such as high production costs and environmental concerns.5–7 PhOLEDs have been developed to date, with reported external quantum efficiency (EQE) values of over 30%.8
:3 due to the spin statistics.2,3 As a result, prompt fluorescent emitters have a limit of internal quantum efficiency (IQE) of 25%.4 The main path to increase the efficiency of OLEDs is utilization of the triplet excited states in electroluminescence (EL), potentially increasing the IQE value to 100%. In phosphorescence OLEDs (PhOLEDs), heavy metal complexes (such as iridium or platinum complexes) which facilitate spin–orbit coupling and in turn accelerate the radiative deactivation through phosphorescence are utilized. Despite the increase in performance of the devices, the use of heavy metal atoms brings certain problems, such as high production costs and environmental concerns.5–7 PhOLEDs have been developed to date, with reported external quantum efficiency (EQE) values of over 30%.8
OLEDs exploiting thermally activated delayed fluorescence (TADF) emerged as a promising alternative to PhOLEDs.9 TADF is a delayed fluorescence of up-converted triplet excitons.10 Thermal motion of atoms facilitates the reverse intersystem crossing (RISC) making delayed fluorescence possible.11 The energy splitting between the first singlet and triplet excited states has to be low for effective RISC.12 Thus, the design strategy for a TADF molecule is based on the separation of highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) on separate moieties.4 In many cases, the hybridization of locally exited (LE) and charge transfer (CT) states occurs forming new hybridized local and charge transfer (HLCT) states. The phenomenon of HLCT, as well as the hot-exciton transition, deals with the RISC at higher excited states rather than the first excited state. This leads to the emission of up-converted excitons even if the singlet–triplet energy splitting is not negligible.3 One of the most efficient and well-studied TADF emitters 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN)13 was reported to possess TADF properties operating through HLCT states.14–16 Previously, our group reported on blue OLEDs with hosts exhibiting HLCT.17 Specifically, OLEDs with the di-tert-butylcarbazole-containing TADF emitter doped into the derivative of dimethoxycarbazolyl-disubstituted dibenzofuran as a host showed an EQE value of 5.8%.17 PhOLEDs containing imidazole derivatives with di-tert-butylcarbazole or carbazole moieties as HLCT-hosts exhibited EQE of up to 7.6%.18 Bipolar or electron-transporting derivatives of carbazole and benzimidazole exhibited good performance as hosts for TADF OLEDs and PhOLEDs.19–22 Derivatives of carbazole exhibit good hole-transporting properties and excellent thermal stability as well as exciplex-forming properties and triplet emission which can be tuned by the purposeful molecular design.23–29 Attachment of the tert-butyl group to the carbazole moiety of the HLCT-TADF emitter 4CzIPN led to the improvement of the morphology of the emitting layer (EML) and enhanced OLED characteristics.30 An electron accepting benzimidazole moiety is a promising building block for the design of donor–acceptor–donor type emitters.31,32 Imidazole-based emitters33–36 including derivatives of benzimidazole37,38 are characterized by hybridization of excited states due to the twisted geometries and conformational heterogeneity. Consequently, imidazole-based compounds have been successfully applied as hosts in PhOLEDs, with relatively high EQE values of over 26%, and low efficiency roll-offs at high luminescence (Table S1, ESI†).39
The efficiencies of TADF-based and phosphorescence OLEDs can be improved by improving the internal outcoupling efficiency via molecular orientations of emitters in EMLs.40–43 Many efficient TADF and phosphorescent emitters exhibit random molecular orientations in the EMLs.44 Fortunately, their orientation can be improved using appropriate hosts.45 Thus, charge injecting and charge-transporting properties, triplet channel energy losses, triplet harvesting abilities, and vertical or horizontal molecular orientations should be taken into account when new hosts for TADF and phosphorescence OLEDs are being developed.46 However, it is difficult to obtain the best combinations of properties for a single host material. In addition, it is not clear which hosting properties have the strongest influence on the performance of OLEDs. We partly aimed to study this issue by developing compounds containing the same electron-accepting unit and the different number of the same electron-donating units.
Herein, we present the synthesis and properties of the derivatives of 1-phenyl-1H-benzo[d]imidazole and di-tert-butylcarbazole as hosts with TADF capabilities for phosphorescence and TADF-based OLEDs. Both synthesized compounds exhibit TADF. They were tested as emitters as well as hosts in PhOLEDs and TADF-based OLEDs. As the emitter, the compound with a single di-tert-butylcarbazole moiety (1tCzBzCN in Scheme 1) showed the best performance in host-free OLEDs. Additionally, as a host, compound 1tCzBzCN showed better performance in green phosphorescence OLEDs than compound 2tCzBzCN. The angle-dependent photoluminescence measurements revealed the horizontal orientation factors of 0.84 for the films containing 1tCzBzCN and 0.87 for the films containing 2tCzBzCN. This observation suggests an enhanced outcoupling factor for the emitting layers composed of green phosphorescence emitters doped in the studied compounds. Compound 1tCzBzCN exhibits bipolar charge carrier transport, while 2tCzBzCN exhibits a unipolar one. Compound 1tCzBzCN was utilized as a host for the fabrication of blue and green emitting PhOLEDs with external quantum efficiencies of 10.2 and 13%, respectively. In addition, in this study, we demonstrate the complete quenching of TADF emission of 2tCzBzCN in a solid state by picric acid. To the best of our knowledge, this is the first report on the optical sensing of nitroaromatic explosive compounds based on TADF quenching in the solid state.
 | ||
| Scheme 1 Synthesis of 1tCzBzCN and 2tCzBzCN. | ||
2. Experimental section
2.1 Materials
N-Phenyl-1,2-phenylenediamine, 2,6-difluoro-4-formylbenzonitrile, and 3-fluoro-4-formylbenzonitrile were purchased from Fluorochem and used as received. Dimethylformamide (DMF), dimethyl sulfoxide (DMSO) and cesium carbonate (Cs2CO3) were purchased from Sigma-Aldrich and used as received. 3,6-Di-tert-butylcarbazole was synthesized according to the procedure reported earlier.47 2-Fluoro-4-benzonitril-1-phenyl-1H-benzo[d]imidazole (1, yield 70%) and 2,6-difluoro-4-benzonitril-1-phenyl-1H-benzo[d]imidazole (2, yield 47%) were synthesized from N-phenyl-1,2-phenylenediamine and the corresponding 4-formylbenzonitriles were synthesized according to the procedure described earlier.48 After the initial workup, the target intermediates were recrystallized from hot methanol.3. Results and discussion
3.1 Synthetic procedures
Compounds 1tCzBzCN and 2tCzBzCN were synthesized in a simple two-step synthesis process (Scheme 1). In the first step, the corresponding fluoro-substituted 4-formylbenzonitriles were coupled with N-phenyl-1,2-phenylenediamine to form the 4-benzonitril-1-phenyl-1H-benzo[d]imidazole core and provide intermediate compounds. Following the first step, nucleophilic substitution reactions were carried out using Cs2CO3 as a base to introduce electron-donating 3,6-di-tert-butylcarbazole moieties and form the target compounds 1tCzBzCN and 2tCzBzCN.3.2 Physical properties
 | ||
| Fig. 1 Ground state optimized geometry and molecular charge distribution of 1tCzBzC and 2tCzBzC according to B3LYP/6-31G**. | ||
The molecular orbital analysis provides key insights into the electronic distribution in 1tCzBzCN and 2tCzBzCN. In the HOMO, the wavefunction is localized on the benzonitrile and tert-butylcarbazole moieties in 1tCzBzCN, while in 2tCzBzCN, it extends over the benzonitrile and both tert-butylcarbazole moieties. This broader distribution in 2tCzBzCN reflects the stronger electron-donating environment provided by the two tert-butylcarbazole units, which contributes to its slightly higher HOMO energy (−5.30 eV) compared to that of 1tCzBzCN (−5.45 eV). In the LUMO, the wavefunction is predominantly localized on the benzonitrile and benzoimidazole moieties of both the compounds, highlighting the consistent electron-accepting nature of the benzonitrile unit. However, the LUMO energy of 2tCzBzCN (−2.11 eV) is slightly lower than that that of 1tCzBzCN (−1.89 eV). This stabilization arises from the enhanced π-conjugation between the benzonitrile and benzoimidazole units in 2tCzBzCN, facilitated by its smaller dihedral angle (22°), which allows for an improved orbital overlap.
 | ||
| Fig. 2 TGA (a) and DSC (b) and (c) curves of 1tCzBzCN and 2tCzBzCN. | ||
| Compound | T −5%, °C | T m, °C | T g, °C | T cr, °C |
E
oxvs.Fconset, V |
E
redvs.Fconset, V |
IPCV, eV | EACV, eV | E bg, eV |
|---|---|---|---|---|---|---|---|---|---|
|
T
−5% – 5% mass loss temperature; Tm – melting temperature; Tg – glass transition temperature; Tcr – crystallization temperature; Eoxonset – onset oxidation potential vs. Fc measured from CV; Eredonset – onset reduction potential vs. Fc measured from CV; IPCV – ionization potential, calculated from IPCV = Eoxvs.Fconset + 4.8; EACV – electron affinity, calculated from EACV = Eredvs.Fconset + 4.8; Ebg – band gap values calculated from Ebg = IPCV − EACV. |
|||||||||
| 1tCzBzCN | 341 | 287 | 149 | — | 0.79 | −2.15 | 5.59 | 2.65 | 2.94 |
| 2tCzBzCN | 426 | 412 | 192 | 277 | 0.94 | −1.86 | 5.74 | 2.94 | 2.80 |
vs.Fconset, Eredvs.Fconset), respectively. Both the synthesized compounds exhibited single oxidation peaks owing to the resident di-tert-butylcarbazole moieties in the molecules (Fig. 3). The ionization potential (IPCV) values calculated from CV data were found to be 5.59 and 5.74 eV for compounds 1tCzBzCN and 2tCzBzCN, respectively. Compound 1tCzBzCN exhibited a lower electron affinity (EACV) of 2.65 eV than 2tCzBzCN (2.94 eV). Band gap energy values (Ebg) estimated using IPCV and EACV were found to be 2.94 eV for 1tCzBzCN and 2.8 eV for 2tCzBzCN. The summary of data obtained from CV measurements is displayed in Table 1.
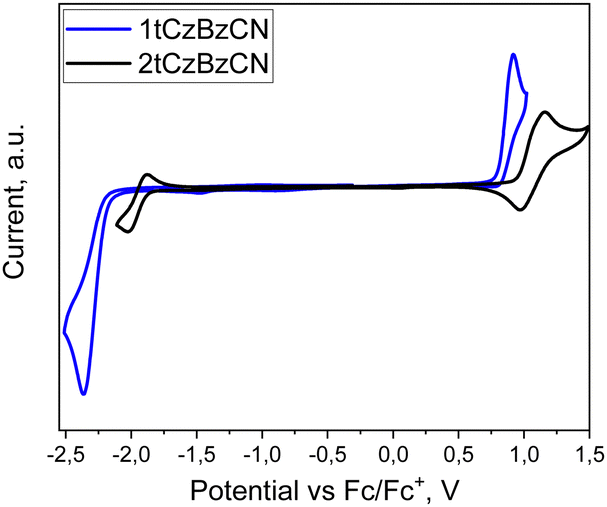 | ||
| Fig. 3 CV curves of 2tCzBzCN and 1tCzBzCN. | ||
| Compounds | PLQYtola | PLQYfilmb | PLQYdopedc | S1, eVd | T1, eVd | ΔEST, eVd | τ 1, nsae | τ 2, msbf |
|---|---|---|---|---|---|---|---|---|
| a Toluene solutions at room temperature in air. b Neat films at room temperature in air. c Films of the molecular mixtures with Ir(ppy)2(acac) (10 wt%) at room temperature in air. d Films at 77 K temperature under an argon atmosphere. e Lifetime of prompt fluorescence at room temperature, in air. f Lifetime of delayed fluorescence at room temperature, in a vacuum. | ||||||||
| 1tCzBzCN | 0.26 | 0.20 | 0.63 | 2.99 | 2.66 | 0.33 | 4.22, 10.11 | 0.58 |
| 2tCzBzCN | 0.09 | 0.39 | 0.48 | 2.68 | 2.63 | 0.05 | 1.85, 4.03 | 0.46 |
PL spectra of the dilute solutions of compounds 1tCzBzCN and 2tCzBzCN in solvents of different polarities manifested the positive solvatochromic effect highlighting the CT nature of emission (Fig. 4a). The addition of the second di-tert-butylcarbazole donor moiety resulted in a redshift of PL spectra of 2tCzBzCN when compared to 1tCzBzCN, likely due to the extension of π-conjugation. Unlike 2tCzBzCN, the solution of 1tCzBzCN exhibited a redshift of 5 nm when the non-polar toluene (0.36 D) was changed to a moderately polar THF (1.75 D). An even longer bathochromic shift of 27 nm was observed when THF was changed to a highly polar DMF (3.82 D) (Fig. 4a). The increasing redshift with the increase of polarity of the solvent leads to the switch from LE character of emission to CT as for previously reported HLCT emitters.52–55 The HLCT nature of the compounds is further substantiated by quantum chemical calculations, as elaborated later in this section. The removal of oxygen led to the enhancement of the PL only for the film of 2tCzBzCN with a 3.3-fold increase in intensity and no additional peaks (Fig. S3, ESI†). No PL intensity increase was observed for the film of 1tCzBzCN. The increase of PL intensity is attributed to the utilization of triplet excitons in the emission as the triplet energy is quenched in the presence of oxygen.2 Collisional interactions with molecular oxygen could cause the drop of PLQY values for the films as the values were estimated for the samples under ambient air conditions. Deoxygenated toluene solutions of the compounds exhibited only prompt fluorescence with lifetimes of up to 10 ns (Fig. S1, ESI†). PL decay curves of the films of the compounds recorded in a vacuum were characterized by the appearance of the long-lived components of the emission with lifetimes up to 0.5 ms (Table 2 and Fig. S2, ESI†). The energy values of the first singlet S1 and triplet T1 excited states were estimated from the onsets of fluorescence and phosphorescence (delay of 1 ms) bands, respectively, recorded under an inert atmosphere at 77 K (Fig. 4b). These values were taken for the calculation of the singlet–triplet energy splitting (ΔEST) (Table 2). The thermal activation of delayed fluorescence was confirmed as the TADF component with sub-ms lifetimes increased upon heating (Fig. 4c). The lifetime derived from the PL decay is slightly lower for 2tCzBzCN (0.46 ms) with ΔEST of 0.05 eV than that of 1tCzBzCN (0.58 ms) with ΔEST of 0.33 eV. This observation shows that more efficient TADF has a shorter lifetime (Table 2).29,56 There is experimental evidence of the HLCT nature of TADF of 1tCzBzCN.14,16 The relatively high ΔEST together with the undoubtfully TADF nature of emission suggests a hot exciton upconversion which occurs at higher energy levels than S1 and T1. The films of both compounds show no change in PL spectral behavior in the temperature range of 77 to 300 K. This indicates that the singlet excitons and the triplet excitons upconverted via RISC are deactivated radiatively from practically the same excited states with energy levels close to the triplet excited states from which phosphorescence occurs at low temperatures (Fig. S4, ESI†). The emission band with a vibronic substructure related to the phosphorescence disappears upon heating.
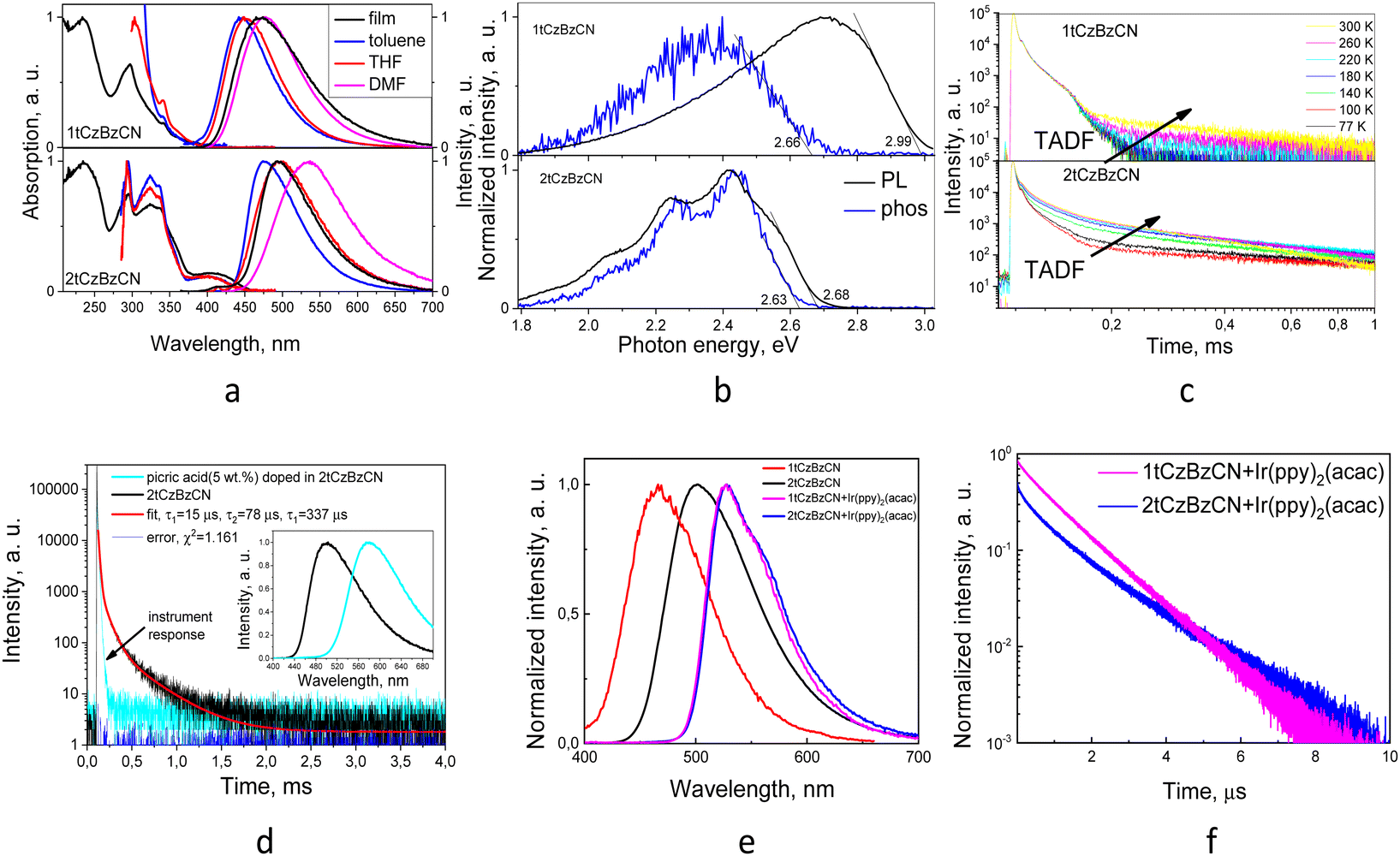 | ||
| Fig. 4 Absorption and PL spectra of the films and dilute toluene, THF and DMF solutions (a); PL and phosphorescence spectra, recorded at 77 K of the films (b); PL decay curves, recorded at different temperatures of the films (c) of compounds 1tCzBzCN and 2tCzBzCN. PL decay curves of the films of the mixtures of picric acid (5 wt%) doped in 2tCzBzCN recorded at room temperature in a vacuum. Inset: PL spectra of the films (d). PL spectra (e), PL decay curves (f), recorded at room temperature of the films of the molecular mixtures of Ir(ppy)2acac (10 wt%) and compounds 1tCzBzCN and 2tCzBzCN. | ||
Time-dependent density functional theory (TD-DFT) calculations were performed using the LC-ω*PBE functional and the 6-31G** basis set to get insightful aspects of excited state properties. The range separation parameter “ω” is tuned for the gaseous medium. The tuned ω value is 0.178 Bohr−1 for 1tCzBzCN and 0.155 Bohr−1 for 2tCzBzCN. The theoretical vertical excited-state energies were adjusted by applying a redshift of 0.4 eV for 1tCzBzCN and 0.2 eV for 2tCzBzCN (as summarized in Table S2, ESI†) to achieve closer alignment with experimental results. This adjustment effectively accounts for the influence of the molecular environment, providing a more accurate depiction of the excited-state dynamics. The TD-DFT calculations unveiled the presence of four triplet states (T1–T4) lying below the S1 in 1tCzBzCN, highlighting the potential for hot exciton RISC due to multiple intermediate triplet levels. To unravel the nature of these excitations, the natural transition orbitals (NTOs) were generated (Fig. S5, ESI†). The NTOs of S1 exhibit a combination of localization on the benzonitrile moiety and CT from the tert-butylcarbazole moiety to the benzoimidazole fragment. This observation is consistent with the HLCT nature of the S1 state. The T1 and T3 states share the similar excitation pattern, showing localization on benzonitrile and benzoimidazole units with CT from the tert-butylcarbazole moiety. In contrast, T2 is predominantly localized on the tert-butylcarbazole unit, while T4 shows a mixed localization on the benzonitrile and tert-butylcarbazole moieties. Despite the highest spin–orbit coupling (SOC) value between S1 and T1 (0.70 cm−1, Fig. S6, ESI†), the significant ΔE(S1–T1) gap (0.4 eV) renders the S1–T1 pathway thermodynamically unfavourable for efficient RISC. However, T4 emerges as a viable mediator in the hot exciton RISC. T4 exhibits the smallest energy gap with S1 (0.03 eV) and the second-highest SOC value (0.24 cm−1) for the S1–T4 pathway. Additionally, the T4–T3 energy gap (0.1 eV) is larger than the S1–T4 gap, reducing the likelihood of internal conversion and favouring RISC as the dominant process.
The NTOs for the S1 state of 2tCzBzCN reveal a characteristic HLCT nature, with electronic density localized on the benzonitrile moiety and charge transfer from the tert-butylcarbazole to the acceptor units. Similarly, the T1 state shows predominant localization on the benzonitrile moiety, minor contributions from the benzoimidazole unit, and charge transfer from the tert-butylcarbazole fragment to the acceptor moieties. In the T2 state, the NTOs depict a mixture of localization on benzonitrile and benzoimidazole units with charge transfer from one of the tert-butylcarbazole moieties. This variation in orbital character for both T1 and T2, compared to S1, enables both states to act as potential pathways for RISC. Among these, the S1–T2 pathway is characterized by a small energy gap but exhibits a low SOC value, indicating that this pathway is relatively weak for RISC. In contrast, the S1–T1 pathway, with a moderate theoretical energy gap of 0.15 eV, demonstrates the significantly higher SOC value (0.90 cm−1), marking it as the dominant and highly efficient pathway for RISC. The pronounced SOC value for the S1–T1 pathway translates to a faster RISC rate in 2tCzBzCN, facilitating efficient conversion of triplet excitons to singlet states. These dynamics contribute to the shorter PL lifetime of 2tCzBzCN, compared to that of its counterpart (1tCzBzCN).
Additionally, the sensitivity of TADF of compound 2tCzBzCN to the presence of nitroaromatic explosive compounds was examined. Expectedly, the film of picric acid was found to be non-emissive. The PL spectra and PL decay curves of the film of the molecular mixture of picric acid (5 wt%) and 2tCzBzCN are shown in Fig. 4d. For comparison, the PL spectrum of the film of neat 2tCzBzCN is shown. The peak of the PL spectrum of the molecular mixture of 2tCzBzCN and picric acid appeared at 578 nm. It was considerably redshifted with respect to the peak of the PL spectrum of the neat layer of 2tCzBzCN which was observed at 500 nm. The bathochromic shift is explained by intermolecular interactions of the two compounds. Electronic excitation energy transfer occurs between the host and the guest effectively quenching TADF. Consequently, the long-lived component of TADF with a lifetime of up to 0.34 ms was eliminated in the PL decay curve of the molecular mixture of 2tCzBzCN and picric acid. Only prompt fluorescence was observed when picric acid was added (5 wt%). At a higher concentration of picric acid of 25 wt%, the host–guest system became non-emissive demonstrating that picric acid totally quenched prompt fluorescence as well as TADF. To gain insights into the interaction mechanism between 2tCzBzCN and picric acid, quantum chemical calculations were conducted at the LC-ωPBE/6-31G** theoretical level. The optimized geometry of the complex of 2tCzBzCN and picric acid is shown in Fig. S7 (ESI†). It highlights the spatial arrangement of the interacting species. The molecular orbitals of the complex show that the HOMO is primarily localized on the electron-rich tert-butylcarbazole moieties of 2tCzBzCN, while the LUMO is concentrated on the highly electron-deficient trinitrophenol (picric acid). This clear transfer of charge density from 2tCzBzCN to picric acid signifies strong non-covalent interactions. This charge transfer mechanism plays a pivotal role in modulating the exciton dynamics, providing a theoretical basis for the observed quenching of emission of 2tCzBzCN by picric acid. The quantum chemical calculations further confirm a substantial interaction energy of −8.03 kcal mol−1, indicating that the formation of the complex is both thermodynamically favourable and spontaneous.
The solid films of emitters 1tCzBzCN and 2tCzBzCN at room temperature in air demonstrated relatively low photoluminescence quantum yields of 0.2 and 0.39. To pursue efficient OLED performance, it is necessary to add efficient emitters to enhance the formation of excitons. Before device fabrication, the energy transfer between the host and the dopant was analyzed using PL spectra and PL decay curves of vacuum-deposited layers of 1tCzBzCN and 2tCzBzCN, and their molecular mixtures with 10 wt% bis[2-(2-pyridinyl-N)phenyl-C](acetylacetonato)iridium(III) (Ir(ppy)2acac) (S1 ∼ 2.47 eV and T1 ∼ 2.45 eV). The PL spectra and PL decay curves are shown in Fig. 4e and f and Fig. S2 (ESI†). The whole PL spectra of the molecular mixtures are attributed to the emission of Ir(ppy)2acac, illustrating the efficient energy transfer from the hosts (1tCzBzCN or 2tCzBzCN) to the guest (Ir(ppy)2acac). The yields are 0.63 for the mixture containing 1tCzBzCN and 0.48 for the mixture containing 2tCzBzCN (Fig. S8, ESI†). TPL decay curves of the layers of 1tCzBzCN and 2tCzBzCN shown in Fig. S2 (ESI†) exhibit long lived components with the lifetimes of over 4 ms, which evidence the possibility of ISC or RISC. After mixing the synthesized compounds with Ir(ppy)2acac, long lived components disappeared in the PL decay curves (Fig. 4f). They revealed emission lifetimes of less than 10 μs. This observation leads to a conclusion that Ir(ppy)2acac facilitates exciton emission, corresponding to the increased PLQY. The PL decay curve of the molecular mixture of 1tCzBzCN and Ir(ppy)2acac is characterized by the single exponential function, indicating efficient energy transfer from the host to the dopant, leading to the enhancement of PLQY up to 0.63. This may result from very fast and efficient Förster energy transfer (FRET) and Dexter energy transfer (DET) between the host and the guest. On the other hand, for the adequate representation of the PL decay curve of the molecular mixture of 2tCzBzCN and Ir(ppy)2acac, the double exponential fit was required. This observation can be attributed to the ISC process, implying a relatively slow or inefficient FRET between the host and the dopant. This resulted in a lower PLQY of 0.48.
 | ||
| Fig. 5 TOF transients of vacuum-deposited films of compounds 1tCzBzCN (a) and (b) and 2tCzBzCN (c) under applied positive voltages for holes and negative voltages for electrons at an optically transparent electrode ITO and the corresponding charge carrier mobilities (d) as the Poole–Frenkel function of electric fields. Inset: A fragment of data of thickness measurements of compound 1tCzBzCN using a profilometer Profilm3D. | ||
3.3 Performance of organic light-emitting diodes
The series of OLEDs with the structure ITO/TAPC (50 nm)/mCP (10 nm)/EML (30 nm)/DPPS (55 nm)/LiF/Al were fabricated. ITO and Al were anode and cathode electrodes, respectively. The layers of 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC) and diphenyl-bis[4-(pyridin-3-yl)phenyl]silan (DPPS) were selected as hole and electron transport layers, respectively. N,N-Dicarbazolyl-3,5-benzene (mCP) was utilized for the deposition of the exciton blocking layer. LiF was utilized as the electron injection layer. As the light-emitting layer (EML), the neat layers of 1tCzBzCN (devices A), 2tCzBzCN (devices C) or the layers of 1tCzBzCN (devices B, E and F) and 2tCzBzCN (device D) co-deposited with the different guests were used. Green phosphorescent emitter Ir(ppy)2(acac) (10 wt%) was used as a guest for the fabrication of OLEDs B and D. Sky-blue phosphorescent emitter bis[2-(4,6-difluorophenyl)pyridinato-C2,N] (picolinato)iridium(III) (FIrpic) (9 wt%) was used as a guest in device E. The green TADF emitter 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) was used as a guest in device F. The heterostructure of OLEDs and the molecular structures of the functional compounds are presented in Fig. 6a and b.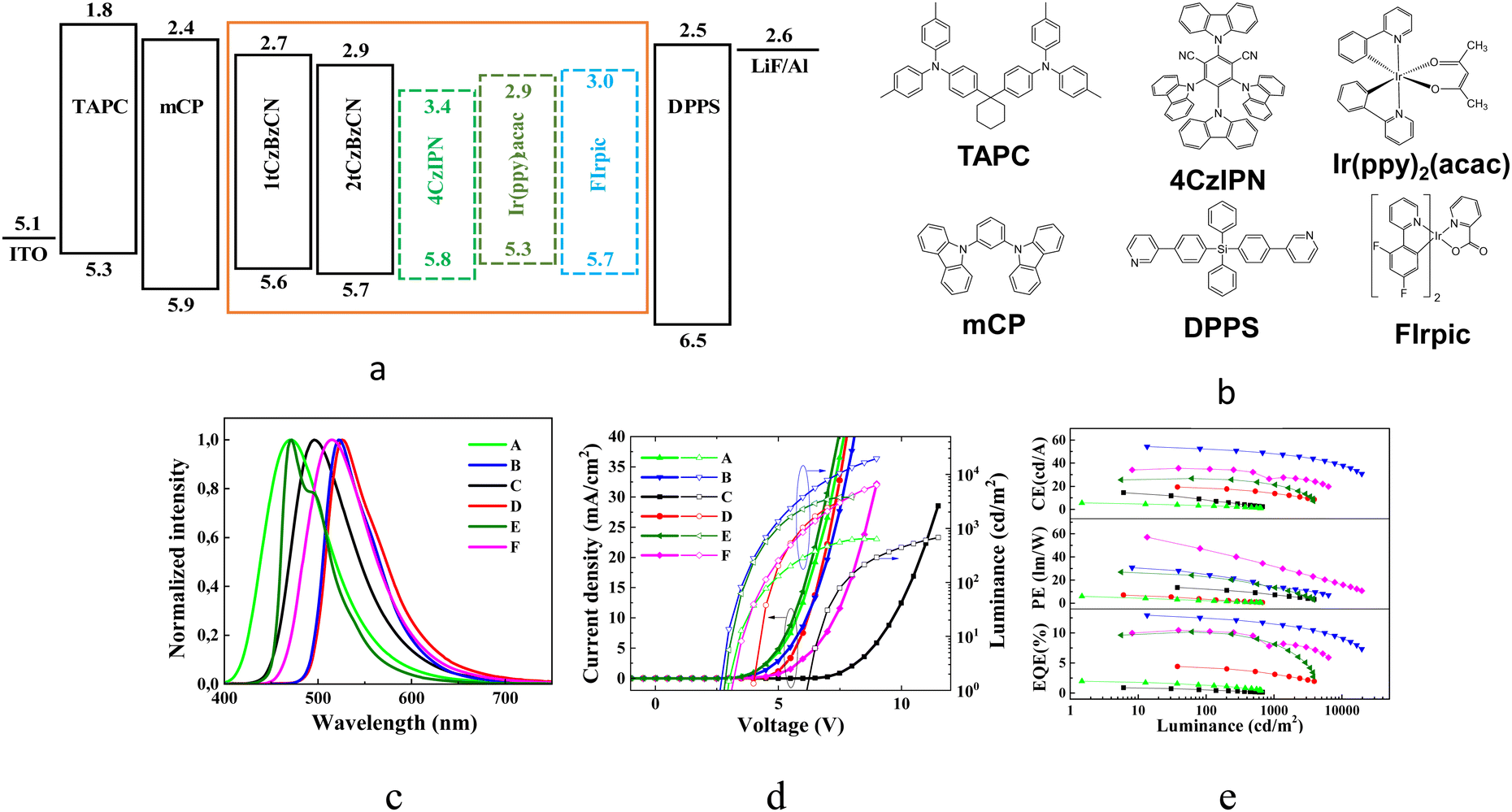 | ||
| Fig. 6 The general structure of the fabricated OLEDs (a). The chemical structures of the functional compounds (b). EL spectra (c), current density–luminance–voltage (d) and CE–PE–EQE–luminance plots (e). | ||
The EL spectra of devices A and C employing EMLs of neat compounds 1tCzBzCN and 2tCzBzCN correspond well with the PL spectra of the films of the respective compounds. The higher PLQY was observed for the film of 2tCzBzCN than for the film of 1tCzBzCN (Table 3). OLED A with the EML of 1tCzBzCN demonstrated more than twice as high external quantum efficiency compared to that of the 2tCzBzCN-based device (C). Thus, usage of 1tCzBzCN containing one electron-donating substituent allowed reaching the considerably higher external quantum efficiency of OLEDs compared to that of the device with 2tCzBzCN containing two di-tert-butylcarbazole moieties. This is due to the charge imbalance caused by 2tCzBzCN which also explains significantly higher turn-on and lit-on voltages of OLED C when compared to those of OLED A (Fig. 5d and 6d). Together with the PL data of guest–host systems, this observation points to 1tCzBzCN being a more efficient host than 2tCzBzCN. Among OLEDs based on Ir(ppy)2(acac), the device with 1tCzBzCN has superior values of current efficiency (CE), power efficiency (PE) and EQE relative to OLEDs with 2tCzBzCN. For the estimation of the performance of 1tCzBzCN as a host, OLEDs E and F utilizing FIrpic and 4CzIPN as emitters, were fabricated and characterized. OLEDs with EMLs based on guest–host systems showed EL spectra representing the emission of the guests without the EL contribution of the host.
| Device | EML | V on , V | CEb, cd A−1 |
PEb, lm W−1 | EQEb, % | λ, nm | 1931 CIEx,y |
|---|---|---|---|---|---|---|---|
| a Turn-on voltage at 1 mA cm−2 and the lit-on voltage 1 cd m−2. b Maximum value/at 100 cd m−2/at 1000 cd m−2. | |||||||
| A | 1tCzBzCN | 4.0/3.0 | 5.6/2.1/— | 5.9/3.2/— | 2.0/1.3/— | 472 | (0.19, 0.31) |
| B | 1tCzBzCN – Ir(ppy)2(acac) | 4.3/2.6 | 54.3/53.8/48.1 | 57/46.3/32.1 | 13.0/12.4/11.47 | 522 | (0.31, 0.64) |
| C | 2tCzBzCN | 7.6/6.2 | 14.5/8.23/— | 7.0/3.3/— | 0.9/0.5/— | 496 | (0.22, 0.48) |
| D | 2tCzBzCN – Ir(ppy)2(acac) | 4.9/4.0 | 19.4/18.8/13.8 | 13.6/12.7/7.3 | 4.4/4.3/3.2 | 526 | (0.35, 0.62) |
| E | 1tCzBzCN – FIrpic | 4.0/2.8 | 25.6/25.8/21.4 | 26.8/22.5/13.4 | 10.2/10.0/7.9 | 472 | (0.15, 0.32) |
| F | 1tCzBzCN – 4CzIPN | 5.2/3.1 | 35.6/34.1/26.7 | 30.7/24.7/13.7 | 10.4/10.2/7.88 | 514 | (0.28, 0.58) |
Time-resolved electroluminescence (TREL) spectra of devices A and C based on EMLs of the neat compounds have characteristic spikes due to imperfect charge transport at recombination sites. The inset of Fig. 7 a is an enlarged view of the spikes, which reflect a great deal of trap states inside the layer of 2tCzBzCN which impede charge carrier transport, prevent charge carrier recombination, and degrade the device performance. While device B has a long lifetime of emission, the TREL signal is shortened in comparison to that of OLED D, highlighting more efficient energy transfer. FIrpic and 1tCzBzCN have similar wavelengths of the EL peaks which are conventionally viewed as unfavorable for efficient host–guest interactions. Nevertheless, the long radiative lifetime of emission of OLED E (Fig. 7b) makes it possible to facilitate FRET even with a small spectral overlap of emission spectrum of the host and the absorption band of the guest due to vibronic coupling.58 Thus, OLED F based on green emitting 4CzIPN, and E based on sky-blue emitting FIrpic showed close values of maximum EQE of ca. 10%. For further investigation of the output coupling effect of devices B and D, the angle-dependent PL measurements were performed. They allow studying the optical transition dipole of the systems of Ir(ppy)2(acac) doped in 1tCzBzCN and 2tCzBzCNvia a laser (excitation at 325 nm) and collecting p-polarized fluorescence intensities at different emission angles as shown in Fig. 7. Two ideal cases, such as the molecular arrangement in a perfect horizontal orientation factor (Θ of 1) and an isotropic dipole orientation factor (Θ of 0.66), were also simulated to be the baseline in comparison to the experimental angle-dependent PL profiles. After fitting the two main peaks of the experimental angle-dependent PL spectra, high Θ values of 0.84 and 0.87 were obtained for OLEDs B and D, respectively. This indicates that the EMLs of devices B and D provide potentially high outcoupling efficiency of more than 30%. Device B showed a maximum EQE of 13%. This value correlates with the PLQY of the film of the corresponding guest–host system.
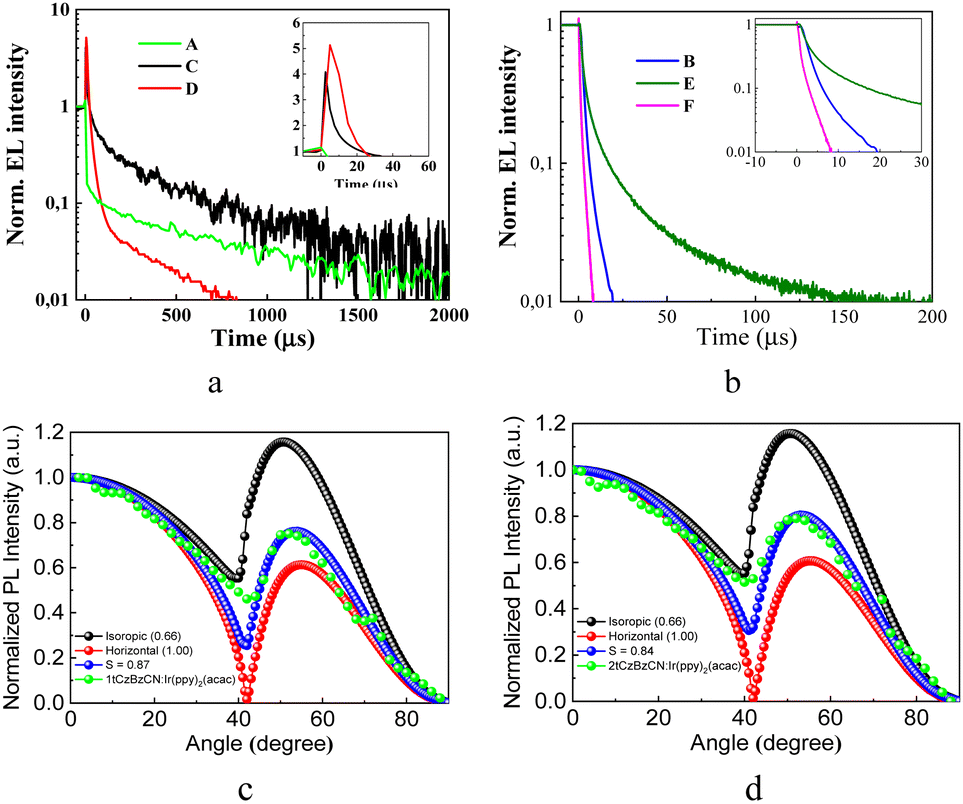 | ||
| Fig. 7 TREL signals of OLEDs A, C, and D (a) and B, E, and F (b) Inset: an enlarged view of the spikes. Simulated (Θ of 1, 0.66), experimental, and fitting (Θ = 0.92) angle-dependent PL intensity profiles of the films of the molecular mixtures of Ir(ppy)2acac (10 wt%) and compounds 1tCzBzCN (c) and 2tCzBzCN (d). | ||
4. Conclusions
Two derivatives with 1-phenyl-1H-benzo[d]imidazole and di-tert-butylcarbazole groups were synthesized and analyzed for their potential as hosts for OLEDs with TADF capabilities. TADF of 2tCzBzCN was completely quenched by the addition of picric acid due to electronic excitation energy transfer. This demonstrates the great potential of TADF for optical sensing of nitroaromatic explosives. TADF of the synthesized emitters originates from the hybridization of local excited and charge transfer states. The solid films of the compounds show moderate values of photoluminescence quantum yields of up to 39%. The derivative of 1-phenyl-1H-benzo[d]imidazole with the single tert-butylcarbazolyl group performs better in organic light emitting diodes than the derivative with two tert-butylcarbazolyl groups. The effective electronic excitation energy transfer from the host to the guest is achieved due to vibronic coupling. The angle-dependent photoluminescence measurements performed for the films of the molecular mixtures of the compounds with the green phosphorescent emitter revealed the horizontal orientation factors of 0.84 and 0.87, suggesting enhanced out-coupling factors for the emitting layers. The performance of the compounds in OLEDs is primarily influenced by their charge-transport characteristics. The derivative of phenyl-1H-benzo[d]imidazole with the single tert-butylcarbazolyl group with hole and electron mobilities of 2.34 × 10−6 and 3.58 × 10−6 cm2 V−1 s−1, respectively, at an electric field of 8.7 × 105 V cm−1 exhibited the best performance. OLEDs with this compound as a host showed an external quantum efficiency of 13%. The results of this study show that the search for hosts with high and balanced hole and electron mobilities, together with high internal outcoupling efficiency and triplet harvesting ability can result in the development of OLEDs with high external quantum efficiency.Author contributions
Conceptualization: Simas Macionis, Jurate Simokaitiene, and Dalius Gudeika; methodology and investigation: Simas Macionis, Oleksandr Bezvikonnyi, Ehsan Ullah Rashid, Jiun-Haw Lee, Zi-Wen Su, Chia-Hsun Chen, Mariia Stanitska, and Oleksandr Navozenko; validation and data curation: Rita Butkute, Ruta Budreckiene, and Dmytro Volyniuk; formal analysis and writing of original draft: Simas Macionis, Jurate Simokaitiene, Oleksandr Bezvikonnyi, Dmytro Volyniuk, and Ehsan Ullah Rashid; writing – review and editing: Tien-Lung Chiu and Juozas. V. Grazulevicius; supervision and funding acquisition: Oleksandr Navozenko and Juozas. V. Grazulevicius.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Research Council of Lithuania (LMTLT), project “Exploitation of the solid-state enhanced long-lived emission of organic emitters for the detection of nitroaromatic explosive compounds”, agreement no. S-LU-24-6. J. H. Lee acknowledges the support and funding from the NSTC, grants 113-2221-E-002-102-MY3 and 112-2221-E-002-216-MY3. T. L. Chiu acknowledges the support and funding from the NSTC, grants 113-2622-E-155-002 and 112-2221-E-155-028-MY3.References
- E. Tankelevičiūtė, I. D. W. Samuel and E. Zysman-Colman, J. Phys. Chem. Lett., 2024, 15, 1034–1047 CrossRef
.
- T. Serevičius, R. Skaisgiris, G. Kreiza, J. Dodonova, K. Kazlauskas, E. Orentas, S. Tumkevičius and S. Juršenas, J. Phys. Chem. A, 2021, 125, 1637–1641 CrossRef
- S. W. Park, D. Kim and Y. M. Rhee, Int. J. Mol. Sci., 2023, 24, 12362 CrossRef CAS
- Y. Im, M. Kim, Y. J. Cho, J. A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS
-
H. Yersin and W. J. Finkenzeller, Triplet Emitters for Organic Light-Emitting Diodes: Basic Properties, John Wiley & Sons, Ltd, 2008 Search PubMed
-
H. Yersin, Triplet Emitters for OLED Applications. Mechanisms of Exciton Trapping and Control of Emission Properties, Springer, Berlin, Heidelberg, 2012 Search PubMed
- A. Jana, V. G. Sree, Q. Ba, S. C. Cho, S. U. Lee, S. Cho, Y. Jo, A. Meena, H. Kim and H. Im, J. Mater. Chem. C, 2021, 9, 11314–11323 RSC
- J. Jayabharathi, V. Thanikachalam and S. Thilagavathy, Coord. Chem. Rev., 2023, 483, 215100 CrossRef CAS
-
J. Cameron, A. Klimash, E. J. Hussien, F. Hacivelioğlu and P. J. Skabara, Sustainable Strategies in Organic Electronics, 2022, pp. 229–280 Search PubMed
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef
- B. Zhou, D. Yan, B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef
- M. Chapran, P. Pander, M. Vasylieva, G. Wiosna-Salyga, J. Ulanski, F. B. Dias and P. Data, ACS Appl. Mater. Interfaces, 2019, 11, 13460–13471 CrossRef CAS PubMed
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 CrossRef CAS
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef
- H. Noda, X. K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed
- O. Bezvikonnyi, D. Gudeika, D. Volyniuk, A. Bucinskas and J. V. Grazulevicius, Mater. Sci. Eng., B, 2021, 273, 115441 CrossRef CAS
- O. Bezvikonnyi, R. S. Bernard, V. Andruleviciene, D. Volyniuk, R. Keruckiene, K. Vaiciulaityte, L. Labanauskas and J. V. Grazulevicius, Materials, 2022, 15, 8495 CrossRef CAS PubMed
- Z. J. Gao, T. H. Yeh, J. J. Xu, C. C. Lee, A. Chowdhury, B. C. Wang, S. W. Liu and C. H. Chen, ACS Omega, 2020, 5, 10553–10561 CrossRef CAS PubMed
- J. J. Huang, Y. H. Hung, P. L. Ting, Y. N. Tsai, H. J. Gao, T. L. Chiu, J. H. Lee, C. L. Chen, P. T. Chou and M. K. Leung, Org. Lett., 2016, 18, 672–675 CrossRef CAS
- S. Y. Chang, G. T. Lin, Y. C. Cheng, J. J. Huang, C. L. Chang, C. F. Lin, J. H. Lee, T. L. Chiu and M. K. Leung, ACS Appl. Mater. Interfaces, 2018, 10, 42723–42732 CrossRef CAS
- Y. M. Chen, W. Y. Hung, H. W. You, A. Chaskar, H. C. Ting, H. F. Chen, K. T. Wong and Y. H. Liu, J. Mater. Chem., 2011, 21, 14971–14978 RSC
- C. Li, C. Duan, C. Han, H. Xu, C. Li, C. Duan, C. Han and H. Xu, Adv. Mater., 2018, 30, 1804228 CrossRef PubMed
- C.-Y. Chan, L.-S. Cui, J. Uk Kim, H. Nakanotani, C. Adachi, C. Chan, L. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Adv. Funct. Mater., 2018, 28, 1706023 CrossRef
- S. M. Bonesi, M. Mesaros, F. M. Cabrerizo, M. A. Ponce, G. M. Bilmes and R. Erra-Balsells, Chem. Phys. Lett., 2007, 446, 49–55 CrossRef CAS
- H. Ikebe, K. Nakao, E. Hisamura, M. Furukori, Y. Nakayama, T. Hosokai, M. Yang, G. Liu, T. Yasuda and K. Albrecht, Aggregate, 2024, 5, e405 CrossRef CAS
- P. Ledwon, P. Zassowski, T. Jarosz, M. Lapkowski, P. Wagner, V. Cherpak and P. Stakhira, J. Mater. Chem. C, 2016, 4, 2219–2227 RSC
- A. Kadashchuk, Y. Skryshevski, A. Vakhnin, S. Toliautas, J. Sulskus, R. Augulis, V. Gulbinas, S. Nespurek, J. Genoe and L. Valkunas, J. Phys. Chem. C, 2014, 118, 22923–22934 CrossRef CAS
- Y. A. Skryshevski and A. Y. Vakhnin, Ukr. J. Phys., 2019, 64, 406 CrossRef
- Y. Joo Cho, K. Soo Yook, J. Yeob Lee, Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 6642–6646 CrossRef
- E. G. Cansu-Ergun, Polym. Rev., 2018, 58, 42–62 CrossRef CAS
- C. Tardío, J. Álvarez Conde, A. M. Rodríguez, P. Prieto, A. de la Hoz, J. Cabanillas-González and I. Torres-Moya, Molecules, 2023, 28, 4631 CrossRef
- J. Jayabharathi, G. Goperundevi, V. Thanikachalam and S. Panimozhi, ACS Omega, 2019, 4, 15030–15042 CrossRef CAS PubMed
- W. C. Chen, Z. L. Zhu and C. S. Lee, Adv. Opt. Mater., 2018, 6, 1800258 CrossRef
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam and S. Sivaraj, RSC Adv., 2020, 10, 8866–8879 RSC
- J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 6, 10138–10173 RSC
- Y. Liu, T. Tao, H. C. Hu, H. Li and X. Ouyang, Dyes Pigm., 2021, 188, 109191 CrossRef CAS
- Y. H. Gao, C. Chen, Q. Tang, B. Su, G. Zhang, B. X. Bo and W. L. Jiang, J. Mater. Sci.: Mater. Electron., 2017, 28, 7204–7211 CrossRef CAS
- C. Liu, T. Li, M. Sun, M. Xie, Y. Zhou, W. Feng, Q. Sun, S.-T. Zhang, S. Xue, W. Yang, C. Liu, T. Li, M. Sun, M. Xie, Y. Zhou, W. Feng, Q. Sun, S. Xue, W. Yang and S.-T. Zhang, Adv. Funct. Mater., 2023, 33, 2215066 CrossRef CAS
- T. Hu, Y. Guo, G. Han and Y. Yi, J. Phys. Chem. C, 2024, 128, 3148–3154 CrossRef CAS
- C. Mayr and W. Brütting, Chem. Mater., 2015, 27, 2759–2762 CrossRef CAS
- S. Sohn, M. J. Kim, S. Jung, T. J. Shin, H. K. Lee and Y. H. Kim, Org. Electron., 2015, 24, 234–240 CrossRef CAS
- D. Yokoyama, J. Mater. Chem., 2011, 21, 19187–19202 RSC
- C. K. Moon, K. H. Kim, J. W. Lee and J. J. Kim, Chem. Mater., 2015, 27, 2767–2769 CrossRef CAS
- T. Marcato and C. J. Shih, Helv. Chim. Acta, 2019, 102, e1900048 CrossRef
- T. Chatterjee and K. T. Wong, Adv. Opt. Mater., 2019, 7, 1800565 CrossRef
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS
- S. Macionis, D. Gudeika, D. Volyniuk, O. Bezvikonnyi, V. Andruleviciene, J. Haw Lee, B.-A. Fan, C.-H. Chen, B.-Y. Lin, T.-L. Chiu and J. V. Grazulevicius, Adv. Opt. Mater., 2021, 9, 2002227 CrossRef CAS
- S. H. Cheng, W. Y. Hung, M. H. Cheng, H. F. Chen, A. Chaskar, G. H. Lee, S. H. Chou and K. T. Wong, J. Mater. Chem. C, 2014, 2, 8554–8563 RSC
- S. Y. Chang, G. T. Lin, Y. C. Cheng, J. J. Huang, C. L. Chang, C. F. Lin, J. H. Lee, T. L. Chiu and M. K. Leung, ACS Appl. Mater. Interfaces, 2018, 10, 42723–42732 CrossRef CAS PubMed
- Y. M. Huang, T. Y. Chen, D. G. Chen, H. C. Liang, C. H. Wu, M. M. Lee, T. L. Chiu, J. H. Lee, Y. C. Chiu, P. T. Chou and M. K. Leung, J. Mater. Chem. C, 2021, 9, 2381–2391 RSC
- A. Kundu, S. Karthikeyan, Y. Sagara, D. Moon and S. P. Anthony, ACS Omega, 2019, 4, 5147–5154 CrossRef CAS
- J. Jayabharathi, R. Ramya, V. Thanikachalam, P. Jeeva and E. Sarojpurani, RSC Adv., 2019, 9, 2948–2966 RSC
- J. Jayabharathi, V. Thanikachalam, R. Ramya and S. Panimozhi, RSC Adv., 2019, 9, 33693–33709 RSC
- S. Arunlimsawat, P. Funchien, P. Chasing, A. Saenubol, T. Sudyoadsuk and V. Promarak, Beilstein J. Org. Chem., 2023, 19, 1664–1676 CrossRef CAS PubMed
- T. Serevičius, R. Skaisgiris, J. Dodonova, L. Jagintavičius, D. Banevičius, K. Kazlauskas, S. Tumkevičius and S. Juršėnas, ACS Appl. Mater. Interfaces, 2020, 12, 10727–10736 CrossRef
- M. Mamada, H. Katagiri, C. Y. Chan, Y. T. Lee, K. Goushi, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Funct. Mater., 2022, 32, 2204352 CrossRef CAS
- K. Stavrou, L. G. Franca, A. Danos and A. P. Monkman, Nat. Photonics, 2024, 18, 554–561 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04802d |
| This journal is © The Royal Society of Chemistry 2025 |