
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring core size, shell thickness, and aluminium doping of Au@ZnO core@shell nanoparticles†
Quynh
Nguyen
 a,
Attilio
Zilli
b,
Michele
Celebrano
b and
Andrea
Baldi
*a
a,
Attilio
Zilli
b,
Michele
Celebrano
b and
Andrea
Baldi
*a
aVrije Universiteit Amsterdam, Department for Physics and Astronomy, De Boelelaan 1100, 1081HV Amsterdam, The Netherlands. E-mail: a.baldi@vu.nl
bPolitecnico di Milano, Department of Physics, Piazza Leonardo da Vinci, 32, 20133 Milano, Italy
First published on 13th March 2025
Abstract
Plasmonic materials, such as gold nanoparticles (AuNPs), exhibit significant extinction and near-field enhancement across the visible and near-infrared spectrum, attributable to localized surface plasmon resonances (LSPRs). Epsilon-near-zero (ENZ) materials, such as aluminium doped zinc oxide (AZO) are known in non-linear optics for their ability to generate and manipulate light-matter interactions through processes like higher harmonic generation. Combining doped ZnO with plasmonic materials therefore holds promise for enhancing non-linear efficiencies and tuning their operational wavelengths. To date, however, only top-down structures based on plasmonically decorated thin ENZ films have been realized, and no colloidal and scalable route to obtain these hybrid materials has been reported yet. Here, we introduce a novel colloidal synthesis approach for fabricating Au@AZO core@shell nanoparticles with tunable core size, shell thickness, and dopant concentration, allowing for the spectral alignment of the LSPRs of the AuNPs with the non-linear optical properties of the AZO shells. Our method involves the colloidal synthesis of gold cores followed by an ascorbic acid-assisted process to deposit polycristalline ZnO and AZO shells, resulting in core diameters ranging from 25 to 69 nm, shell thicknesses from 16 to 47 nm, and aluminium doping levels between 0 and 4 at%. Our procedure widens the range of hybrid plasmonic nanostructures that can be colloidally synthesised, opening new possibilities for the large scale fabrication of high-performance nanomaterials for integration in photonic, photocatalytic, and sensing applications.
1. Introduction
Plasmonic materials, such as gold nanoparticles (AuNPs), are renowned for their strong light–matter interaction, including high extinction and near-field enhancement. These properties arise from localized surface plasmon resonances (LSPRs), which are the collective oscillation of the free electrons of metal nanoparticles (NPs) when exposed to light. The resonance wavelength of AuNPs can easily be tuned across the visible (VIS) and near-infra-red (NIR) range by varying their size, shape, and surrounding, opening the door for applications in nanoscale (bio)sensing, photocatalysis, and photothermal processes.1–8The near-field enhancement of LSPRs has been utilized to amplify intrinsically weak non-linear responses, such as second- (SHG) and third-harmonic generation (THG). Yet, these performances were achieved by highly engineered nanostructures, which require a demanding fabrication, the use of extremely short pulses, and are not readily scalable.9–15
To date, the integration of plasmonics with non-linear optics (NLO) has primarily focused on lithographically fabricated two-dimensional structures.16–18 However, several applications such as biomedical labelling and imaging demand scalable syntheses of large quantities of colloidal NPs. A viable approach is to synthesise plasmonic NPs coated by a NLO thin shell, where the second or third harmonics of the shell are, both spatially and spectrally, overlapping with the LSPR of the core to further enhance the intrinsically weak non-linear optical signals.
Zinc oxide (ZnO) is chemically and mechanically stable.19–26 Additionally, while the toxicity of ZnO has been well studied and documented, specific surface modifications and application methods have been developed to mitigate these effects, making them suitable for bio-imaging under controlled conditions.27–29 Additionally, ZnO displays NLO properties that can be modulated by chemical doping.10,30,31 In particular, aluminium doped zinc oxide (AZO) exhibits a vanishing real part of the permittivity in the near-infrared (near-IR) range, classifying it as an epsilon-near-zero (ENZ) material. The level of aluminium doping controls the wavelength at which the permittivity crosses zero, providing an additional dimension of tunability.32 ENZ materials have been recently suggested as promising materials for NLO, thanks to their sizeable nonlinear susceptibility that combines with the strong field enhancements at specific wavelength, typically set in the NIR region of the spectrum. This phenomenon, known as ENZ condition, occurs in transparent conducting oxides when the real part of the electric permittivity approaches zero yielding a metal-like behavior.33,34
An ideal system to combine plasmonic near-field enhancement with ENZ materials could therefore be composed of a gold core coated with an aluminium-doped ZnO shell, where the size and shape of the core, and the thickness and dopant level of the shell are tuneable at will.
It is possible to synthesise Au@ZnO core@shell NPs by combining the colloidal synthesis of the metallic cores to an ascorbic acid-assisted step for the formation of an oxide shell, in which ascorbic acid (AA) forms a CTAB–[AA–Zn(OH)4]2− complexing agent to facilitate the deposition of Zn2+ ions onto the Au cores. However, previous studies largely overlooked possibilities of tailoring the structural parameters of the NPs35–40 or led to the formation of a hydrated zinc hydroxide (Zn(OH)2) shell in the absence of an annealing final step.41 Additionally, no colloidal synthesis of Au@AZO core@shell NPs has been reported, likely due to the high sensitivity of the AZO syntheses to the reactant concentrations, the pH level of the solution, the ratio of surfactant to ascorbic acid, and the temperature of the reaction.42,43
In this work, we demonstrate the colloidal synthesis of Au@ZnO and Au@AZO core@shell NPs with controlled shell thickness, core size and shape, and aluminium dopant concentration. As a first step to demonstrate tunability of the synthetic process, we establish a procedure to vary the shell thickness of Au@ZnO NPs by varying the initial concentration of Au cores while keeping the concentration of the other reactants constant. We then demonstrate that ZnO shells can easily be grown on spherical Au cores of different diameters as well as on gold nanorods. Finally, we develop a novel procedure to obtain Au@AZO NPs by adding an aluminium doping step before calcination. We corroborate the synthetic results with UV-Vis spectroscopy, scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), inductively coupled plasma mass spectrometry (ICP-MS), and Mie theory calculations of the extinction cross-section spectra of the core@shell NPs.44
2. Results and discussion
The synthesis of Au@ZnO core@shell NPs has been adapted and modified from the method described by Zeto et al., and proceeds via a three-step process: (i) first the Au cores are synthesised with established colloidal procedures, (ii) then a shell of Zn(OH)2 is nucleated around the cores via a hydrothermal treatment, and (iii) finally the Au@ZnO NPs are obtained by calcination of the Au@Zn(OH)2 NPs at 500 °C in air.38 For the synthesis of Au@AZO NPs, an additional step of redispersing the Au@Zn(OH)2 NPs in a pH-neutral Al3+ solution is added before calcination. All synthetic details are given in the Experimental Section.The result of a typical Au@ZnO synthesis is shown in Fig. 1, for Au cores of 43 ± 3 nm in diameter and a ZnO shell thickness of 47 ± 4 nm. Fig. 1(a) shows the extinction spectrum of the reaction solution, containing AuNPs, L-ascorbic acid (AA), cetyltrimethylammonium bromide (CTAB), hexamethylenetetramine (HMTA), and zinc nitrate (Zn(NO3)2), prior to hydrothermal synthesis. At this stage, light extinction is solely attributed to the LSPR of the AuNPs with a peak at 534 nm. Fig. 1(b) shows the extinction spectrum of the as-synthesised Au@Zn(OH)2 dispersion after the hydrothermal reaction. The formation of a hydrated Zn(OH)2 shell with an average thickness of 59 ± 5 nm induces a ∼15 nm redshift in the LSPR wavelength to 549 nm, due to the slight increase in the refractive index of the gold surrounding from water to Zn(OH)2.45 The NP dimensions and core@shell structure are confirmed by SEM.
 | ||
| Fig. 1 Normalised extinction spectrum (top row), secondary electron and backscattered electron SEM image (middle row), and size distribution (bottom row) of (a) as-synthesised AuNPs with a diameter of 43 nm, (b) Au@Zn(OH)2 NPs after hydrothermal synthesis with an average shell thickness of 59 nm, and (c) Au@ZnO NPs after calcination with an average shell thickness of 47 nm. All scalebars are 200 nm. | ||
To convert the hydrated Zn(OH)2 shell into crystalline ZnO, we centrifuge the Au@Zn(OH)2 NPs, redisperse them in water, dry them in air at 150 °C for 30 min, and calcinate them at 500 °C in air for 2 h. The obtained powder is then let to cool down and redispersed in water. Fig. 1(c) shows the extinction spectrum of a suspension of Au@ZnO core@shell NPs, highlighting a further ∼86 nm red-shift of the LSPR to 635 nm, due to the higher refractive index of ZnO compared to Zn(OH)2. The spectrum also shows the emergence of a peak at 375 nm, corresponding to the band gap energy of ZnO (E ≈ 3.37 eV) and indicating the presence of crystalline wurtzite phase ZnO, as confirmed via X-ray diffraction (XRD) measurement (see SI 1.1, ESI†). The SEM images demonstrate a uniform particle shape and narrow size distribution, as also indicated by the narrow LSPR peaks observed in the UV-Vis extinction spectra. The inset of backscattered electron SEM image in Fig. 1(c) (middle row) shows the presence of a Au core in all the Au@ZnO NPs.
The shell thickness of the final Au@ZnO NPs can be controlled by varying the number of gold NPs in the initial reaction solution while keeping the concentration of the remaining reactants constant. The Au@ZnO NPs shown in Fig. 1(c), are obtained with an initial concentration of 3.1 × 1011 AuNPs per ml leading to a final diameter of the Au@ZnO core@shell NPs of 137 nm, corresponding to a ZnO shell thickness of 47 nm. In Fig. 2, we demonstrate the coating of the same 43 nm AuNPs with ZnO shell thicknesses of 21, 25, 30, and 34 nm, obtained from reaction solutions with initial AuNP concentration of 10.8 × 1010, 9.6 × 1010, 7.5 × 1010, and 5.6 × 1010 NPs per ml, respectively. The initial AuNP concentrations in the reaction solution are estimated via Beer–Lambert law from the OD of the solutions, using Mie theory to calculate the extinction cross section of a 43 nm AuNP (see calculations in SI 1.2, ESI†).46
 | ||
| Fig. 2 Secondary electron SEM images (top), backscattered electron SEM images (middle) and corresponding size distribution histograms (bottom) of Au@ZnO core@shell NPs with varying ZnO shell thicknesses synthesised from reaction solutions with initial AuNP concentrations of (a) 10.8 × 1010, (b) 9.6 × 1010, (c) 7.5 × 1010, and (d) 5.6 × 1010 NPs per ml. All scalebars are 200 nm. | ||
Precise knowledge of the AuNPs concentration allows us to estimate the yield of the ZnO synthesis reaction.
We estimated the theoretical maximum yield of ZnO shell synthesis around a AuNP core. Assuming complete conversion of the zinc nitrate precursor to zinc oxide, the calculations indicate a maximum yield of ∼7.3 mg of Au@ZnO NPs.
These calculations were based on the initial amount of zinc nitrate used, and the properties of the AuNPs, including their OD and radius. The detailed derivations and the full set of assumptions made in these calculations are given in SI 1.3 (ESI†).
Fig. 3(a) shows the extinction spectra of the particles shown in Fig. 2, after the hydrothermal treatment but before the final annealing step, i.e. corresponding to Au@Zn(OH)2 core@shell NPs. As expected, suspensions with a larger initial concentration of AuNPs have higher overall extinction across the visible and a slightly blue-shifted LSPR, indicating a thinner Zn(OH)2 shell (Fig. 3(b)). Fig. 3(c) shows the total volume of ZnO in the final Au@ZnO core@shell NP suspensions of Fig. 2, calculated from the initial AuNP concentration, cAuNPs, the radius of the gold cores, rAu, and the radius of the final Au@ZnO core@shell NPs, rAu@ZnO, according to:
 | (1) |
 | ||
| Fig. 3 (a) Extinction spectra of as-synthesised Au@Zn(OH)2 core@shell NPs after hydrothermal treatment with varying initial AuNP concentrations. (b) LSPR wavelength position determined via a Lorentzian fit of the spectra in (a) as a function of the initial AuNP concentration. (c) Total volume of ZnO in the Au@ZnO core@shell NPs, calculated from the initial Au NP concentration and the ZnO shell thickness using equation (1). | ||
The horizontal dashed line in Fig. 3(c) represents the maximum obtainable ZnO volume per ml, calculated from the total amount of zinc nitrate added to the hydrothermal reaction, indicating a nearly quantitative conversion of the initial Zn(NO3)2 into ZnO shells around the Au cores.
Our synthetic procedure can be used to synthesise ZnO shells around Au particles of different diameters and shapes. In Fig. 4(a)–(c) we show SEM images of Au@ZnO core@shell NPs with spherical core diameters of 25, 43 and 69 nm. Fig. 4(d) shows the encapsulation of non-spherical gold nanorods with average width and length of 54 × 118 nm. Coating different Au NPs with ZnO shells allows us to tune their optical response across the visible (Fig. SI.2, ESI†).
 | ||
| Fig. 4 Secondary electron SEM images (top row) and backscattered electron SEM images (bottom row) of Au@ZnO NPs with (a) 25 nm, (b) 43 nm, and (c) 69 nm spherical cores and (d) 54 × 118 nm nanorod cores. All scalebars are 200 nm. | ||
As a final step in the development of our synthesis, we altered the shell composition by intercalating Al3+ ions into the ZnO lattice, using a novel approach. Zeto et al. have successfully embedded copper and rare earth metals (Ce, Er, Nd, Tm, and Yb) into the ZnO shells of Au@doped-ZnO core@shell nanostructures.38 However, the synthesis of Au@AZO core@shell NPs remains largely unexplored. Previous studies on ZnO NPs and thin films showed that aluminium doping significantly affects the morphology of the final AZO crystallites.42,47,48
As a first approach to obtain Au@AZO core@shell NPs, we followed previous experimental procedures for the synthesis of AZO49,50 and added a stoichiometric quantity of Al(NO3)3 to the initial hydrothermal reaction mixture containing Zn(NO3)2 (procedure 1 in Scheme 1). However, such an approach proved to be unsuccessful in leading to well-controlled core@shell NPs, resulting in large quantities of AZO agglomerates decorated with the AuNPs (Fig. SI.3, ESI†). Our working hypothesis to explain the difficulty in incorporating Al3+ ions in the ZnO shell structure is that the acidity of the aluminium nitrate solution (pH ∼ 3) prevents the precipitation of Zn(OH)2, which is unstable below pH 6.51,52 In fact, the significance of the structural damage to the final AZO shell increases for higher Al3+ dopant concentration.
 | ||
| Scheme 1 Overview of the two experimental approaches used to dope the ZnO shell with aluminium. Procedure 1 follows the literature syntheses of AZO NPs and Au@doped ZnO core@shell NPs, whereby the dopant is added to the other reactants at the beginning of the ZnO synthesis. In procedure 2, the aluminium nitrate solution is neutralized to a pH of 6.5 and used to redisperse the Au@Zn(OH)2 NPs obtained after hydrothermal treatment. | ||
To mitigate the impact of pH on the overall hydrothermal synthesis, we also tried a different approach whereby the Al3+ dopant solution was neutralized to a pH of 6.5 using ammonium hydroxide (NH4OH) before addition to the reaction mixture prior to hydrothermal heating. Despite the formation of Au@AZO core@shell NPs, we noticed a significant decrease in particle size and shell homogeneity with increasing concentrations of the dopant (Fig. SI.4, ESI†).
The synthesis of dopant-controlled Au@AZO NPs was achieved by first synthesizing Au@Zn(OH)2 core@shell NPs in the absence of any Al3+ doping, centrifuging the product and redispersing it in a pH-neutral aqueous solution of aluminium nitrate (Al(NO3)3).
The Au@Zn(OH)2 core@shell NPs are left in the Al3+ solution for 30 minutes before drop casting on a silicon substrate and annealing. This process is illustrated in procedure 2 in Scheme 1. Using a non-neutralized aluminium nitrate solution results in compromised NP morphology and uniformity (Fig. SI.5, ESI†).
Fig. 5 shows SEM images of Au@AZO NPs obtained by redispersing the Au@Zn(OH)2 core@shell NPs in pH-neutralized solutions of 0 mM, 0.055 mM, 0.11 mM, and 0.23 mM Al(NO3)3, corresponding to nominal Al-doping of 0, 1, 2, and 4 at%, respectively. A uniform core@shell structure of the Au@AZO NPs is visible up until a nominal doping percentage of 2 at%, with 4 at% doping leading to some loss of homogeneity of the AZO shell To confirm the incorporation of Al3+ ions into the semiconducting shell we performed energy dispersive X-ray spectroscopy (EDX) using a line-scan across a Au@ZnO particle and a 2 at% doped Au@AZO particle (Fig. 6(a) and (b)). The blue lines corresponding to the aluminium Kα1 transition, clearly show the presence of aluminium within the doped particle. To measure the dopant percentage in the AZO shells we performed inductively coupled mass-spectrometry (ICP-MS) on washed Au@AZO NPs, as illustrated in Fig. 6(c).
 | ||
| Fig. 5 SEM images acquired using secondary electrons (top row) and backscattered electrons (bottom row) of Au@AZO core@shell NPs with nominal dopant percentages of (a) 0, (b) 1, (c) 2, and (d) 4 at% Al. All scalebars are 200 nm. The photos of the final NP suspensions are also shown. | ||
 | ||
| Fig. 6 EDX line scans across (a) a Au@ZnO core@shell NP and (b) a Au@AZO core@shell NP with nominal dopant of 2 at% Al. Intensities were measured for Zn (black lines), Au (red lines), and Al (blue lines). The insets show the SEM images of the particles and the traces of the line scans. (c) Aluminium dopant concentration measured with ICP-MS on washed Au@AZO NPs with nominal Al doping from 0 to 4 at% Al. The small residual Al content in the 0 at% sample is likely due to contamination from the acids used to digest the samples. | ||
The aluminium content measured in the blank was subtracted from the one detected in each NP sample to correct for baseline contamination. The aluminium percentage in each sample was then determined from the ratio of the corrected aluminium signal to the zinc signal. When aiming for a nominal doping of 0, 1, 2, and 4 at% of aluminium, ICP-MS measurements show that our samples contain 0.4, 0.9, 2.2 and 3.4 at%, respectively. The ICP-MS data demonstrates a quantitative incorporation of Al3+ into the ZnO shell, which we further confirm using extinction spectroscopy.
Doping zinc oxide with aluminium causes a decrease in refractive index of the surrounding medium of the Au cores,53 resulting in a blue-shift of the LSPR. The extinction spectra measured on Au@AZO NPs suspensions confirm this trend (Fig. 7(a)) as the LSPR shifts from 617 nm for Au@ZnO to as low as 575 nm for a Au@AZO core@shell NP with a nominal shell doping of 4 at%. In literature, the successful doping of ZnO with Al3+ in thin AZO films is typically monitored by the lowering of the AZO bandgap with increasing dopant concentration.54,55 However, the high scattering of the Au@AZO NPs causes a strong increase in extinction at shorter wavelengths. This high scattering background affects the shape and spectral position of the ZnO bandgap, preventing us from using it as an indicator of dopant concentration.
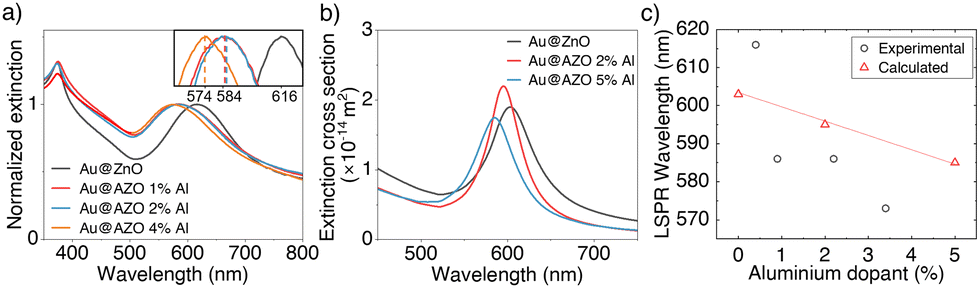 | ||
| Fig. 7 (a) Normalised extinction spectra of Au@AZO core@shell NP dispersions with nominal dopant percentage up to 4 at% Al. The Au@AZO NPs with 0, 1, and 2 at% doping have dimensions of 121 ± 8 nm (0 at% Al), 130 ± 9 nm (1 at% Al), and 127 ± 6 nm (2 at% Al), as obtained from an analysis of the SEM images in Fig. 5. Since the Au cores have an average diameter of 43 nm, these dimensions correspond to shell thicknesses of 39 nm (0 at% Al), 43.5 nm (1 at% Al), and 42 nm (2 at% Al), respectively. The inset shows a zoomed in view of the LSPR peak positions. (b) Calculated extinction cross sections of Au@AZO particles with 43 nm core-diameter, 40 nm shell thickness and dopant percentage of 0, 2 and 5 at% Al. (c) LSPR peak positions for the spectra shown in panels (a) and (b) as a function of aluminium dopant percentage.53,56,57 | ||
Using Mie theory and literature values for the refractive indices of doped ZnO, we calculated the extinction cross section of core@shell particles with 43 nm diameter cores and 40 nm thick shells with Al3+ dopant percentages of 0, 2 and 5 at% (Fig. 7(b)).46,53,56,57 The position of the LSPR shifts towards shorter wavelengths for particles with increased dopant concentration, qualitatively reproducing the trend observed in our experimental spectra (Fig. 7(c)).
3. Experimental section
3.1. Chemicals
Gold(III) chloride hydrate (HAuCl4), L-ascorbic acid (AA), cetyltrimethylammonium bromide (CTAB), cetyltrimethylammonium chloride (CTAC), hexamethylenetetramine (HMTA), zinc nitrate hexahydrate (Zn(NO3)2·6H2O), aluminium nitrate nonahydrate (Al(NO3)3·9H2O), and sodium borohydride (NaBH4) were purchased from Sigma-Aldrich and used without any further purification. Synthesis reactions were carried out using Milli-Q grade water, which had a resistivity of 18.1 MΩ cm at 25 °C.3.2. Synthesis of CTAB-capped Au nanospheres
CTAB-capped Au nanospheres with diameters between 10 nm and 79 nm are obtained via a seed-mediated synthesis reported previously and consisting of three consecutive steps: (1) the synthesis of Au nanosized clusters, (2) their growth into 10 nm Au NPs (seeds), and (3) the growth of the Au seeds into larger spheres.58To prepare Au nanoclusters, 5 ml of 0.5 mM HAuCl4 are added to 5 ml of 0.2 M CTAB in a round-bottom flask in a water bath at 27 °C under stirring. Then, 0.6 ml of 0.01 M fresh ice-cold sodium borohydride are added rapidly to the reaction mixture. The stirring is then stopped, and the reaction mixture is left undisturbed for 3 h.
To prepare the 10 nm seed solution, 20 ml of 0.2 M CTAC, 15 ml of 0.1 AA and 50 μl of the previously obtained Au nanocluster solution are added in a separate flask under stirring and kept at 27 °C. Using two 10 ml pipettes, 20 ml of 0.5 mM HAuCl4 are added via one-shot injection. The reaction is left to continue for 15 min to obtain a 10 nm AuNP seed solution with the characteristic vibrant red colour. The AuNPs are centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm (7943 × g) for 60 min. The supernatant is removed, and the pellet is redispersed in 0.02 mM CTAC. This procedure is repeated twice. Finally, the optical density (OD) at the LSPR wavelength of the seed solution is adjusted to OD = 3.06, corresponding to an estimated concentration of 1.37 × 1013 NPs per ml.
500 rpm (7943 × g) for 60 min. The supernatant is removed, and the pellet is redispersed in 0.02 mM CTAC. This procedure is repeated twice. Finally, the optical density (OD) at the LSPR wavelength of the seed solution is adjusted to OD = 3.06, corresponding to an estimated concentration of 1.37 × 1013 NPs per ml.
To obtain AuNPs with a diameter of 43 nm, 80 ml of an aqueous solution of 0.1 M CTAC are added to 4.8 ml of 0.01 M AA together with 798 μl of the 10 nm AuNP seed solution in a round-bottom flask and kept at 27 °C. Subsequently, 80 ml of 0.5 mM HAuCl4 are added drop-wise while stirring with a syringe pump at a rate of 80 ml h−1. After the HAuCl4 addition, the solution is left to stir for another 15 minutes. The solution is then centrifuged at 5000 rpm for 15 minutes and the final product was redispersed in water and diluted to an OD of 13, as confirmed using extinction spectroscopy. For AuNPs with a diameter of 43 nm, an OD of 13 corresponds to a concentration of ∼6.31 × 1011 AuNPs per ml. To synthesise 25 nm, 69 nm and 79 nm AuNPs the same procedure is being following with 4000 μl, 40 μl and 27 μl of the 10 nm AuNP seed solution, respectively.
3.3. Synthesis of Au nanorods
Gold Nanorods (AuNRs) are synthesised via a previously reported seed-mediated synthesis.593.4. Synthesis of Au@ZnO core@shell nanoparticles
To synthesise Au@ZnO core@shell NPs, a 20 ml aqueous reaction mixture are prepared in a 100 ml Teflon-liner of a stainless steel hydrothermal reactor under stirring. This mixture includes 1 ml of 43 nm AuNPs solution, 2.74 mM CTAB, 0.55 mM L-ascorbic acid, 2.75 mM zinc nitrate, and 2.75 mM HMTA. Zinc nitrate serves as the Zn2+ source, while hexamethylenetetramine provides alkaline hydroxide ions through its decomposition under thermal conditions. Cetyltrimethylammonium bromide, a widely utilized surfactant in colloidal chemistry, is employed to ensure the spherical morphology of the NPs and prevent aggregation. The addition of L-ascorbic acid likely leads to the formation of a disordered CTAB–[AA–Zn(OH)4]2− complex that promotes the selective deposition of Zn2+ ions on the CTAB-capped Au NPs, effectively templating the growth of conformal ZnO shells.38,41 The reactor is placed in an aluminium heating mantle on a heating plate and the mixture is kept at 87.5 °C for 8 h. After the heating process, the solution is collected and centrifuged at 5000 rpm (2739 × g) for 15 min, the supernatant is removed, and the product is redispersed in an equal volume of water to remove unreacted components and surfactant. Prior to annealing, it is crucial to thoroughly remove any remaining reactants in the centrifugation step. Fig. SI.6 (ESI†) depicts SEM images of samples wherein different concentrations of CTAB remained in this step prior to annealing.The product, consisting of Au@Zn(OH)2 NPs, is drop-casted onto a flat Si wafer and dried in air at 150 °C for 30 min until dry, before calcination at 500 °C for 2 h in a furnace with a heating ramping time of 30 min. The product is then left to cool to room temperature. The obtained powder consisting of Au@ZnO NPs with a shell thickness of 47 nm, is then collected by scraping it off the Si wafer with a plastic spatula and stored in an airtight container for further characterization.
Au@ZnO NPs with shell thicknesses of 21, 25, 30, and 34 nm are obtained by adding 3.23, 2.77, 2.08, and 1.64 ml of the 43 nm AuNP solution in the initial step, respectively.
Au@ZnO NPs with core diameters of 25, 43, and 69 nm are obtained by replacing the initial AuNPs solution in the hydrothermal step with AuNPs with different diameter in the same NP concentration.
Au@ZnO NPs with Au nanorods as a core are obtained by adding 3 ml of a 118 × 54 nm AuNRs solution with an OD = 30 at the longitudinal LSPR to the reaction mixture.
3.5. Synthesis of Au@AZO core@shell nanoparticles
Au@AZO core@shell NPs are obtained via a similar hydrothermal route for the synthesis of Au@ZnO core@shell NPs. After the hydrothermal synthesis, however, the 20 ml of the product solution are centrifuged and the Au@Zn(OH)2 NPs are redispersed in an equal volume of an aqueous aluminium nitrate solution, neutralized to a pH of 6.5 with NH4OH. The aluminium nitrate concentration is adjusted to correspond to the intended doping at%, assuming a quantitative inclusion of Al3+ ions in the Zn(OH)2 shells. The Au@Zn(OH)2 NPs are left in the Al(NO3)3 solution for 30 min. The sample is then dried at 150 °C for 30 min before calcination in air at 500 °C for 2 h.3.6. ICP-MS sample preparation and measurement
To prepare the samples for ICP-MS measurements, 0.15 mg of the Au@AZO samples are dissolved overnight in 150 μl of 65% (w/w) nitric acid and 1.5 μl of freshly prepared aqua regia. On the following day, 2.5 μl of the sample are further diluted 4000 times with aqueous 1% (w/w) nitric acid to reach an approximate Zn2+ concentration below 100 ppb. To take any possible contamination into account, a blank sample was prepared following the same procedure without any NPs. The contamination found in the acids used to digest the NPs is assumed consistent across all samples, and is therefore subtracted from the detected analyte amounts. The ICP-MS measurements were conducted using a NexIOR 2000 (PerkinElmer). The calibration of the ICP-MS setup was performed using standard solutions at concentrations of 1, 5, 20, and 100 ppb for both aluminium and zinc.4. Conclusions
In conclusion, we successfully developed a robust colloidal synthesis of Au@AZO core@shell NPs, demonstrating precise control over core size and shape, as well as the polycrystalline shell thickness and composition. The distinct core@shell structure, along with its relatively narrow size distribution, was verified through SEM. Additionally, the optical properties of these nanomaterials were characterized using extinction spectroscopy, revealing strong absorption in the UV region due to the ZnO band gap and within the visible region owing to the LSPR of gold. Notably, the LSPR position is significantly modulated by core size, ZnO shell thickness and aluminium doping, demonstrating a high degree of tunability in these plasmonic/ENZ hybrid materials. Our research advances our understanding of the role of pH and surfactant concentration in the synthesis of aluminium-doped zinc oxide and provides a synthetic strategy to produce highly uniform and tuneable Au@AZO NPs, opening new avenues for their use in photonic devices.Future studies will focus on characterizing their non-linear optical properties and integrating them into real-world settings to test their performance under different environmental conditions and irradiation profiles.
Potential applications of such versatile hybrid material include high-speed optical modulators, advanced photonic circuits, ultra-sensitive biosensors, as well as non-linear bio labels, contributing towards more efficient, faster, and miniaturized technologies.
Author contributions
A. Zilli, M. Celebrano, A. Baldi conceptualized the project, Q. Nguyen and A. Baldi designed the experiments, Q. Nguyen performed all the experiments and analysed the data, Q. Nguyen and A. Baldi wrote the first draft of the manuscript, all authors provided feedback to the manuscript.Data availability
The data supporting the findings in this article are available as follows: Data type: Extinction spectra, scanning electron microscopy images, ICP-MS measurements, and X-ray diffraction measurements. Calculated extinction spectra are obtained using Mie theory MATLAB scripts freely available at https://github.com/andrea-baldi/Mie_Scattering_and_Absorption_Sphere and https://github.com/andrea-baldi/Mie_Core_ShellFile formats: Data files are available in .csv and .tif formats. The data are not publicly available, but are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Sven Askes (Vrije Universiteit Amsterdam) and Rachmat Adhi Wibowo (Austrian Institute of Technology) for many fruitful and insightful discussions. Additionally, we acknowledge Sipeng Zheng and Sylvestre Bonnet (Leiden University) for the ICP-MS measurements. We also acknowledge Dr. Agostino Di Francescantonio (Politecnico di Milano) for fruitful discussions on the nonlinear optical properties of our samples.References
- A. Lalisse, G. Tessier, J. Plain and G. Baffou, J. Phys. Chem. C, 2015, 119, 25518–25528 Search PubMed.
- R. Bukasov, T. A. Ali, P. Nordlander and J. S. Shumaker-Parry, ACS Nano, 2010, 4, 6639–6650 CrossRef CAS PubMed.
- G. Baffou, I. Bordacchini, A. Baldi and R. Quidant, Light: Sci. Appl., 2020, 9, 108 CrossRef PubMed.
- B. Sepúlveda, P. C. Angelomé, L. M. Lechuga and L. M. Liz-Marzán, Nano Today, 2009, 4, 244–251 CrossRef.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CAS.
- D. Devasia, A. J. Wilson, J. Heo, V. Mohan and P. K. Jain, Nat. Commun., 2021, 12, 2612 CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- L. Bonacina, P.-F. Brevet, M. Finazzi and M. Celebrano, J. Appl. Phys., 2020, 127, 230901 CAS.
- Z. Sofiani, B. Sahraoui, M. Addou, R. Adhiri, M. A. Lamrani, L. Dghoughi, N. Fellahi, B. Derkowska and W. Bala, J. Appl. Phys., 2007, 101, 063104 Search PubMed.
- P.-Y. Chen, C. Argyropoulos and A. Alù, Nanophotonics, 2012, 1, 221–233 Search PubMed.
- R. W. Boyd, A. L. Gaeta and E. Giese, in Springer Handbook of Atomic, Molecular, and Optical Physics, ed. G. W. F. Drake, Springer Nature, 2023, pp. 1097–1110 Search PubMed.
- T. Hanke, G. Krauss, D. Träutlein, B. Wild, R. Bratschitsch and A. Leitenstorfer, Phys. Rev. Lett., 2009, 103, 257404 CAS.
- M. Celebrano, X. Wu, M. Baselli, S. Großmann, P. Biagioni, A. Locatelli, C. De Angelis, G. Cerullo, R. Osellame, B. Hecht, L. Duò, F. Ciccacci and M. Finazzi, Nat. Nanotechnol., 2015, 10, 412–417 CAS.
- B. Metzger, M. Hentschel, T. Schumacher, M. Lippitz, X. Ye, C. B. Murray, B. Knabe, K. Buse and H. Giessen, Nano Lett., 2014, 14, 2867–2872 CAS.
- S. Choudhary, S. Iqbal, M. Karimi, O. Reshef, M. Z. Alam and R. W. Boyd, ACS Photonics, 2023, 10, 162–169 CAS.
- H. Khamh, E. Sachet, K. Kelly, J.-P. Maria and S. Franzen, J. Mater. Chem. C, 2018, 6, 8326–8342 CAS.
- J. Park, J.-H. Kang, X. Liu and M. L. Brongersma, Sci. Rep., 2015, 5, 15754 CrossRef CAS PubMed.
- C. F. Klingshirn, A. Waag, A. Hoffmann and J. Geurts, Zinc Oxide: From Fundamental Properties Towards Novel Applications, Springer Nature, 2010 Search PubMed.
- D. Li and H. Haneda, Chemosphere, 2003, 51, 129–137 CrossRef CAS PubMed.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- T. Pauporté and D. Lincot, Electrochim. Acta, 2000, 45, 3345–3353 CrossRef.
- W. Y. Liang and A. D. Yoffe, Phys. Rev. Lett., 1968, 20, 59–62 CrossRef CAS.
- J. Xu, Q. Pan, Y. Shun and Z. Tian, Sens. Actuators, B, 2000, 66, 277–279 CrossRef CAS.
- M. A. Franco, P. P. Conti, R. S. Andre and D. S. Correa, Sens. Actuators Rep., 2022, 4, 100100 CrossRef.
- J. Fujihara and N. Nishimoto, Biol. Trace Elem. Res., 2024, 202, 9–23 CrossRef CAS PubMed.
- S. L. Chia and D. T. Leong, Heliyon, 2016, 2, e00177 Search PubMed.
- T. Xia, Y. Zhao, T. Sager, S. George, S. Pokhrel, N. Li, D. Schoenfeld, H. Meng, S. Lin, X. Wang, M. Wang, Z. Ji, J. I. Zink, L. Mädler, V. Castranova, S. Lin and A. E. Nel, ACS Nano, 2011, 5, 1223–1235 CAS.
- I. Jellal, K. Nouneh, J. Jedryka, D. Chaumont and J. Naja, Opt. Laser Technol., 2020, 130, 106348 CAS.
- D. B. Ingram, P. Christopher, J. L. Bauer and S. Linic, ACS Catal., 2011, 1, 1441–1447 CAS.
- S. Gurung, A. Anopchenko, S. Bej, J. Joyner, J. D. Myers, J. Frantz and H. W. H. Lee, Adv. Mater. Interfaces, 2020, 7, 2000844 CAS.
- N. Kinsey, C. DeVault, J. Kim, M. Ferrera, V. M. Shalaev and A. Boltasseva, Optica, 2015, 2, 616–622 CAS.
- O. Reshef, I. De Leon, M. Z. Alam and R. W. Boyd, Nat. Rev. Mater., 2019, 4, 535–551 CAS.
- M. Kauranen and A. V. Zayats, Nat. Photonics, 2012, 6, 737–748 CAS.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274–5309 CAS.
- X. Shao, B. Li, B. Zhang, L. Shao and Y. Wu, Inorg. Chem. Front., 2016, 3, 934–943 CAS.
- R. Zeto, D. Cummins, A. Gallegos, M. Shao and A. M. Armani, J. Mater. Res., 2019, 34, 3877–3886 CAS.
- S. M. Majhi, P. Rai and Y.-T. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 9462–9468 CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 CAS.
- Y. Yang, S. Han, G. Zhou, L. Zhang, X. Li, C. Zou and S. Huang, Nanoscale, 2013, 5, 11808 CAS.
- N. Izu, K. Shimada, T. Akamatsu, T. Itoh, W. Shin, K. Shiraishi and T. Usui, Ceram. Int., 2014, 40, 8775–8781 CAS.
- V. V. Shinde, D. S. Dalavi, S. S. Mali, C. K. Hong, J. H. Kim and P. S. Patil, Appl. Surf. Sci., 2014, 307, 495–502 CAS.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, John Wiley & Sons, 2008 Search PubMed.
- V. Kumar, R. K. Mishra, P. Kumar and J. S. Gwag, Luminescence, 2023, 38, 1307–1318 CrossRef CAS PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- D. J. Edison, W. Nirmala, K. D. A. Kumar, S. Valanarasu, V. Ganesh, M. Shkir and S. AlFaify, Phys. B, 2017, 523, 31–38 CrossRef CAS.
- A. Vanaja, G. V. Ramaraju and S. Rao, Indian J. Sci. Technol., 2016, 9, 1–6 Search PubMed.
- M. Carofiglio, S. Barui, V. Cauda and M. Laurenti, Appl. Sci., 2020, 10, 5194 CrossRef CAS PubMed.
- E. Burunkaya, N. Kiraz, Ö. Kesmez, H. Erdem Çamurlu, M. Asiltürk and E. Arpaç, J. Sol-Gel Sci. Technol., 2010, 55, 171–176 CrossRef CAS.
- Heavy Metals in Soils: Trace Metals and Metalloids in Soils and their Bioavailability, ed. B. J. Alloway, Springer Nature, 2013, vol. 22 Search PubMed.
- R. A. Reichle, K. G. McCurdy and L. G. Hepler, Can. J. Chem., 1975, 53, 3841–3845 CrossRef CAS.
- G. Marinov, K. Lovchinov, V. Madjarova, V. Strijkova, M. Vasileva, N. Malinowski and T. Babeva, Opt. Mater., 2019, 89, 390–395 CrossRef.
- G. B. Murdoch, S. Hinds, E. H. Sargent, S. W. Tsang, L. Mordoukhovski and Z. H. Lu, Appl. Phys. Lett., 2009, 94, 213301 CrossRef.
- Y. Wu, P. M. Hermkens, B. W. H. van de Loo, H. C. M. Knoops, S. E. Potts, M. A. Verheijen, F. Roozeboom and W. M. M. Kessels, J. Appl. Phys., 2013, 114, 024308 CrossRef.
- E. Mammadov, N. Naghavi, Z. Jehl, G. Renou, T. Tiwald, N. Mamedov, D. Lincot and J.-F. Guillemoles, Thin Solid Films, 2014, 571, 593–596 CrossRef CAS.
- A. C. Gâlcă, M. Secu, A. Vlad and J. D. Pedarnig, Thin Solid Films, 2010, 518, 4603–4606 CrossRef.
- Y. Zheng, X. Zhong, Z. Li and Y. Xia, Part. Part. Syst. Charact., 2014, 31, 266–273 CrossRef CAS.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04644g |
| This journal is © The Royal Society of Chemistry 2025 |