
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Greenish yellow-emitting carbon dot-based films for luminescent solar concentrator applications†
Yunxiang
Liu
,
Yoshiki
Iso
 * and
Tetsuhiko
Isobe
*
* and
Tetsuhiko
Isobe
*
Department of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: iso@applc.keio.ac.jp; isobe@applc.keio.ac.jp; Fax: +81 45 566 1551; Tel: +81 45 566 1558 Tel: +81 45 566 1554
First published on 12th November 2024
Abstract
Currently, most carbon dots (CDs) are synthesized from carbon-based materials in autoclaves with high pressure, and then applied in various fields. This work explored the facile synthesis of CDs at atmospheric pressure, which were then purified and embedded in the ethylene-vinyl acetate (EVA) copolymer to form CDs@EVA films and applied in luminescent solar concentrator (LSC) devices. First, crude CDs were synthesized by refluxing p-phenylenediamine (p-PD) in diphenyl ether at 250 °C for 2 h under ambient air conditions, followed by purification using silica gel column chromatography to obtain purified CDs with a photoluminescence (PL) quantum yield of 56% and an average particle size of 15.1 ± 4.4 nm. The CDs@EVA films were fabricated at CD concentrations of 0.05 wt%, 0.10 wt%, and 0.15 wt%, and their thickness was varied from ∼100 μm to ∼500 μm. The yellowish-transparent CDs@EVA films exhibited greenish-yellow emission with absorption peaks at 350 nm and 470 nm, corresponding to the n–π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds and the π–π* transition between the highest occupied molecular orbital and the lowest unoccupied molecular orbital of π-conjugated electrons. The PL intensity reached its maximum at 0.05 wt% CD concentration and 408 μm thickness due to self-absorption of emission from CDs. According to the current–voltage curve measurements under AM1.5G simulated sunlight, this film exhibited the maximum short-circuit current (Isc) and power conversion efficiency (η) of the LSC device. Additionally, incident photon-to-current efficiency spectrum measurements revealed that the enhanced Isc and η in CDs@EVA films mainly resulted from emission by absorption through the n–π* and π–π* transitions.
N bonds and the π–π* transition between the highest occupied molecular orbital and the lowest unoccupied molecular orbital of π-conjugated electrons. The PL intensity reached its maximum at 0.05 wt% CD concentration and 408 μm thickness due to self-absorption of emission from CDs. According to the current–voltage curve measurements under AM1.5G simulated sunlight, this film exhibited the maximum short-circuit current (Isc) and power conversion efficiency (η) of the LSC device. Additionally, incident photon-to-current efficiency spectrum measurements revealed that the enhanced Isc and η in CDs@EVA films mainly resulted from emission by absorption through the n–π* and π–π* transitions.
1. Introduction
A luminescent solar concentrator (LSC) is a device that efficiently converts solar energy through luminescent layers, which convert high energy ultraviolet or visible light into lower energy visible or near infrared light. The absorbed sunlight was re-emitted to the edges of the device via total internal reflection, where it is collected by solar cells.1 LSCs are often integrated into building windows, skylights, and curtain walls, providing energy while maintaining the aesthetics and transparency of the building.2,3LSC devices can significantly enhance the photoelectric conversion efficiency of solar cells by expanding the spectral response range. This integration reduces loss of light through the conversion and concentration of light while protecting the solar cell materials, thus improving system stability and longevity.
Luminescent materials are pivotal in determining the performance of LSCs. As a result, the exploration of luminescent materials represents a critical research avenue for advancing LSC technology. Traditionally, organic dyes,4–7 and coordination compounds of rare-earth ions, such as Eu3+,8–11 have been widely utilized. However, there has been a growing focus on inorganic semiconductor quantum dots (QDs), such as CdSe/ZnS,12–15 and CsPbX3 (X = Cl, Br, I),16–19 due to their superior properties. Compared to other commonly employed luminescent materials, QDs offer higher photostability, tunable optical properties, enhanced photoluminescence (PL) efficiency, and multicolor emission capabilities. Therefore, using QDs as luminescent layers in LSC devices, issues in solar energy technology can effectively be addressed, improving overall efficiency and performance.20 However, the high toxicity of elements such as cadmium and lead, the complex synthesis process, environmental stability, and other issues of QDs still need to be addressed.21
Carbon dots (CDs) are nanomaterials composed of carbon, typically ranging from a few nanometers to tens of nanometers in size.22 Synthesis methods include the top-down processing of laser ablation and electrochemical etching, and the bottom-up processing of hydrothermal synthesis,23 with most current CDs produced in autoclaves under high pressure.24–27 Due to their excellent fluorescence properties, good biocompatibility, strong chemical stability, and simple, low-cost synthesis methods, CDs have broad application prospects in bioimaging, sensing, drug delivery, and optoelectronic devices.28–31
CDs can absorb ultraviolet and short-wavelength visible light from solar energy and re-emit it as long-wavelength visible light in all directions. Taking advantage of their fluorescence properties, CDs can be used in luminescent layers for application in LSC devices,32–40 as shown in Table S1 (ESI†). According to recent research, nitrogen-doped carbon dots (N-CDs) have shown effectiveness in LSC applications by enhancing light absorption, PL efficiency, and stability.36,37,41 Nitrogen doping alters the electronic structure of the CDs and introducing nitrogen-related surface states, which significantly improves absorption in the ultraviolet and visible regions, enhances PL quantum yield (PLQY), reduces non-radiative transitions, and improves light stability, leading to better energy conversion efficiency.42–44 Li et al. achieved a power conversion efficiency of 3.94% by integrating green to yellow-green-emitting N-CDs into 6.67 μm thick poly(methyl methacrylate) films, overcoming the limitations of materials such as organic dyes and QDs, which suffer from narrow absorption bands, rapid photobleaching, high toxicity, and cost.36 Wang et al. enhanced the performance of LSCs by embedding blue to yellow-green-emitting N-CDs in polyvinylpyrrolidone films, achieving a power conversion efficiency of 4.97% with an optimal configuration of 7 layers containing 0.5 wt% N-CDs.37 This way resolved compatibility issues between QDs and polymer matrices, which often lead to aggregation and PL quenching. These findings suggest that CDs provide a more environmentally friendly and cost-effective solution for LSC fabrication, enhancing the potential for practical applications such as semi-transparent photovoltaic windows.
We have explored the facile liquid phase synthesis of CDs from p-phenylenediamine (p-PD) by refluxing in diphenyl ether (DPE) under atmospheric pressure.45–48 Our work innovatively employs p-PD as a single precursor and achieves the facile one-step synthesis of high-PLQY CDs under ambient conditions. This method not only simplifies the synthesis process but also offers the advantages of low cost, ease of operation, and environmental friendliness, further enhancing the feasibility and scalability of the technology. In this study, the purified p-PD derived CDs were embedded in the ethylene-vinyl acetate (EVA) copolymer to form CDs@EVA films, as shown in Scheme 1. In most works, common polymers such as polyvinylpyrrolidone and poly(methyl methacrylate) are used as the matrix films for LSCs.36,37 In contrast, we selected the EVA copolymer as the matrix material, because of its high transparency in the UV and visible ranges, high durability against sunlight, and its use in practical solar cells. The optical properties of the EVA film and CDs@EVA films with different thicknesses and CD concentrations were measured for comparison. Furthermore, the performance of these films in LSC devices under AM1.5G simulated sunlight was evaluated to understand the potential of p-PD derived CDs in LSC. Soda glass was chosen as a light guide plate in LSCs because it is widely used as window glass.
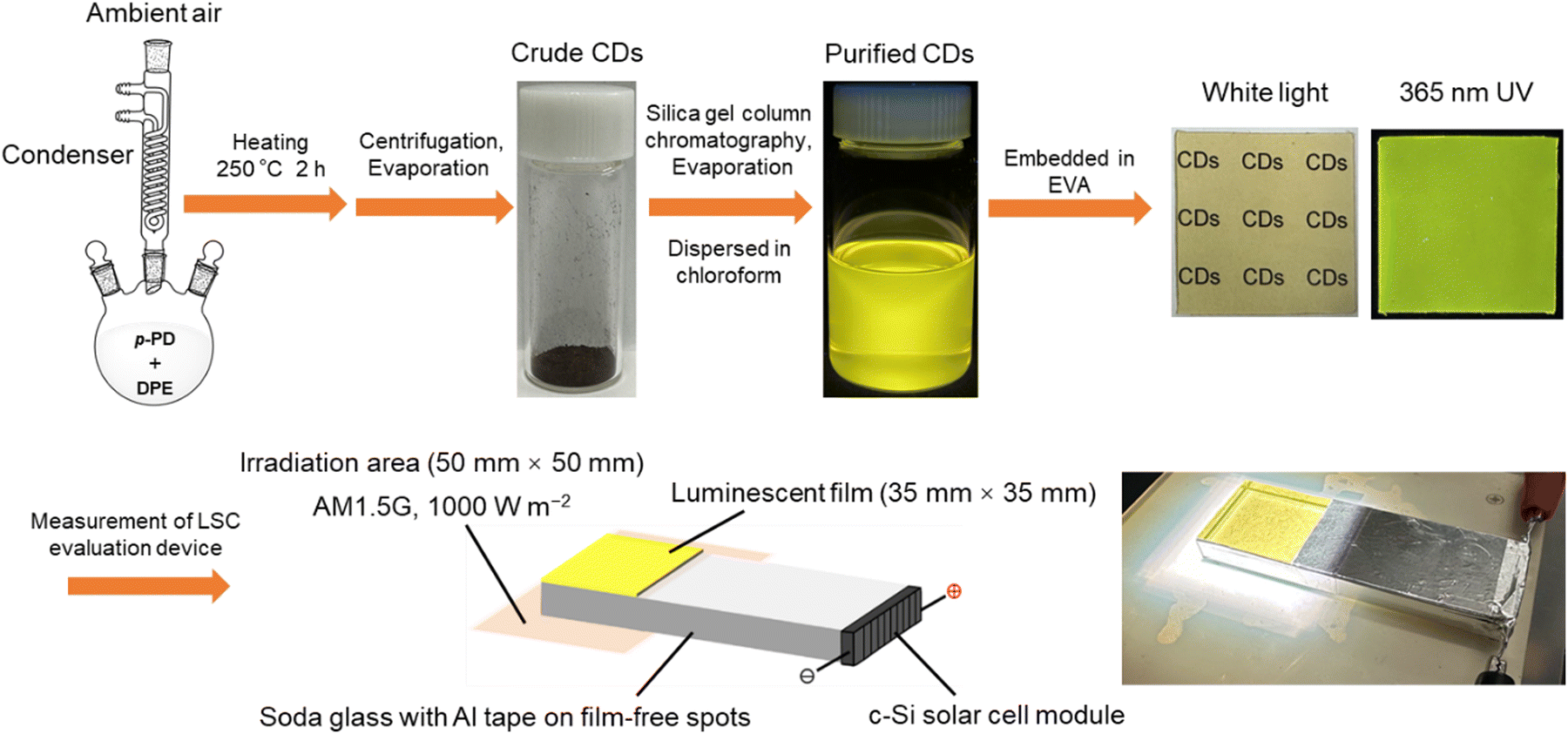 | ||
| Scheme 1 Schematic illustration on synthesis of CDs from p-PD and fabrication of fluorescent EVA films for LSC. | ||
2. Experimental section
2.1. Materials
p-PD (>98.0%) and 1-octadecene (ODE; >90.0%) were purchased from Tokyo Chemical Industry. DPE (>99.0%) was purchased from FUJIFILM Wako Pure Chemical Corporation. Chloroform (>99.0%), hexane (>96.0%), dichloromethane (99.5%), methanol (>99.8%), and silica gel (100–210 μm neutral spherical particles) were purchased from Kanto Chemical. A granular EVA copolymer (Evaflex EV150) was purchased from DuPont-Mitsui Polychemicals. All reagents were used as received without further purification.2.2. Synthesis of crude CDs
p-PD (0.20 g, 1.85 mmol) was mixed with DPE (15 mL) and heated at 250 °C for 2 h under ambient air conditions according to our previous procedure.45 After cooling to room temperature, the resulting black suspension (see Fig. S1, ESI†) was divided into three equal parts and 33 mL of hexane was added to each. The mixtures were centrifuged at ∼11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g (10 cm radius rotor at 10000 rpm) for 10 min. This step was repeated three times, and then the precipitate was dried overnight under ambient air conditions. Finally, all of the precipitates were mixed with methanol and then evaporated to obtain crude CDs (see Fig. S2, ESI†). The product yield calculated from the weight ratio of crude CDs to p-PD was 77%.
000g (10 cm radius rotor at 10000 rpm) for 10 min. This step was repeated three times, and then the precipitate was dried overnight under ambient air conditions. Finally, all of the precipitates were mixed with methanol and then evaporated to obtain crude CDs (see Fig. S2, ESI†). The product yield calculated from the weight ratio of crude CDs to p-PD was 77%.
2.3. Purification of crude CDs through silica gel column chromatography
The crude CDs were completely dispersed in an eluent with a volume ratio of dichloromethane to methanol of 20:1, and then purified using silica gel column chromatography (see Fig. S3, ESI†). The non-fluorescent fraction 1, mainly consisting of yellow components under white light, was removed first. The eluent ratio was then changed to 2:1, and the weakly yellow-emitting fraction 2 and the strongly red-emitting fraction 3, which consisted of red components under white light, were collected. Finally, the solvent in fraction 3 only was removed using a rotary evaporator and air-dried overnight at room temperature to obtain purified CDs (see Fig. S2, ESI†). The product yield calculated from the weight ratio of purified CDs to p-PD was 25%. Purified CDs (1 mg) were dispersed in chloroform (10 mL) under ultrasonication, and then diluted fourfold to obtain the CD chloroform dispersion for analysis. A p-PD chloroform solution was also prepared using the same procedure for comparison.
2.4. Fabrication of CDs@EVA films
Granular EVA (2.6 g), chloroform (30 mL), and ODE (1.5 mL) were mixed and stirred at room temperature for 1 h to obtain an EVA solution. To fabricate the CDs@EVA films at concentrations of 0.05 wt%, 0.10 wt%, and 0.15 wt%, the purified CDs (1.3 mg, 2.6 mg, and 3.9 mg) were each mixed with the EVA solution and dispersed ultrasonically for 1 h. After stirring for 24 h, the mixture was further dispersed ultrasonically for 2 h. The obtained CD dispersions were then purged with Ar for 5 min, followed by stirring and defoaming using a planetary centrifugal mixer (Thinky, AR-100) for 30 s each. Finally, 5 mL, 10 mL, 15 mL, and 20 mL of dispersions were cast into Petri dishes and dried under ambient conditions over 36 h to obtain various CDs@EVA films with different thicknesses and CD concentrations. An EVA film without CDs was also prepared using the same procedure. All films were cut into square pieces (35 mm × 35 mm). The thickness of the films was measured using a micrometer.2.5. Characterization studies
The particle size and morphology were imaged using a field emission transmission electron microscope (FE-TEM; FEI, Tecnai G2) operated at 200 kV. The FE-TEM sample was prepared by dropping a drop of the CD chloroform dispersion on a copper grid with an ultra-thin carbon support film (Oken Shoji, UHR-C10) and drying overnight. Fourier transform infrared (FT-IR) spectra of p-PD and the purified CDs in pressed KBr pellets were measured using a FT-IR spectrometer (JASCO, FT/IR-4200). The ultraviolet-visible (UV-vis) absorption spectrum of the CD chloroform dispersion was measured using a UV-visible spectrophotometer (V-750, JASCO). The in-line and total transmission spectra of the film samples were measured using the same UV-vis spectrophotometer equipped with a standard film holder (FLH-741, JASCO) and an integrating sphere (ISV-922, JASCO), respectively. PL and PL excitation (PLE) spectra of the CD chloroform dispersion and film samples were obtained using a fluorescence spectrometer (JASCO, FP-8500). Absolute PLQYs were measured with a quantum efficiency measurement system (QE-2000-311C, Otsuka Electronics). The device for evaluating the LSC performance is shown in Fig. S4 (ESI†). The light guide plate (100 mm × 35 mm × 10 mm) wrapped with aluminum tape and the monocrystalline silicon (c-Si) solar cell module (35 mm × 10 mm; KIS) were assembled together. The CDs@EVA films was attached to the region of the light guide plate without aluminum tape (35 mm × 35 mm) and illuminated with AM 1.5G simulated sunlight (1000 W m−2). Current–voltage (I–V) curves of this device were measured using a source meter unit (ADCMT, 6242) under AM1.5G simulated sunlight (1000 W m−2, see Fig. S5, ESI†) generated by a solar simulator (XES-40S1, San-Ei Electric). Incident photon-to-current efficiency (IPCE) measurements were performed using a source meter unit (B2901A, Keysight Technologies) under monochromatic light irradiation (MLS-1510, Asahi Spectra). The results of the I–V curve and IPCE analysis were averaged five and three measurements, respectively.3. Results and discussion
3.1. Properties of purified CDs
The FE-TEM image of the purified CDs shows that the average particle size was 15.1 ± 4.4 nm, as shown in Fig. S6 (ESI†).45,48 The FT-IR spectra of p-PD and purified CDs are shown in Fig. S7 (ESI†). The presence of amino groups in p-PD and CDs was verified by N–H stretching vibrations at 3200 cm−1, 3305 cm−1, and 3374 cm−1, and N–H bending vibrations at ∼1630 cm−1.45–47 Semicircular ring vibrations at 1520 cm−1 and 1450 cm−1 confirmed the presence of aromatic ring structures. The similarity between the FT-IR spectra of p-PD and CDs suggests that the CDs have polyaromatic macromolecular structures which were formed from p-PD by simple bond cleavage and reorganization through dehydrogenative polymerization.As shown in Fig. 1A, the CD chloroform dispersion appeared yellow under white light and exhibited yellow emission under 365 nm UV light, while the colorless transparent p-PD chloroform solution exhibited no visible emission (see Fig. S8, ESI†). According to the UV-vis spectrum, PL/PLE mapping, and three-dimensional PL/PLE spectrum of the p-PD chloroform solution (Fig. S9, ESI†), an absorption peak was observed at 317 nm, possibly due to the π–π* transition of the benzene ring, and no visible emission was observed.
 | ||
| Fig. 1 (A) Photographs of purified CDs in chloroform under white light and 365 nm UV light. (B) PL/PLE spectra of purified CDs in chloroform. | ||
Fig. 1B shows the UV-vis and PL/PLE spectra of the purified CDs in chloroform. Table S2 (ESI†), summarizes the PL/PLE properties of the purified CDs synthesized three times with their average values. An absorption peak at 466 nm was observed, attributed to the π–π* transition between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the π-conjugated electron.45–47 The strongest excitation peak was observed at 470 nm with a corresponding emission peak at 550 nm. Both peaks are attributed to the π–π* transition between the HOMO and the LUMO of π-conjugated electrons. The absorption and excitation peaks at 350 nm are possibly due to the n–π* transition of CN bonds. PLQYs of the purified CDs in chloroform were measured (see Fig. S10, ESI†). According to Table S2, (ESI†), the average PLQY of the purified CDs synthesized three times was 56% under excitation at 470 nm. According to the PL/PLE mapping and three-dimensional PL/PLE spectra (Fig. S11, ESI†), the emission did not depend on the excitation wavelength.
3.2. Properties of CDs@EVA films
By embedding the purified CDs into EVA resin, CDs@EVA films with concentrations of 0.05 wt%, 0.10 wt%, and 0.15 wt% and thicknesses ranging from ∼100 μm to ∼500 μm were prepared (see Table S3, ESI†). The photographs of the EVA and CDs@EVA #3 films under white light and 365 nm UV light are shown in Fig. 2 and Fig. S12A, B (ESI†). Under white light, the EVA film was colorless and transparent, while all CDs@EVA films were yellowish transparent, with the yellow color deepening as the concentration and thickness increased. Under 365 nm UV light, the EVA film did not emit fluorescence, while all CDs@EVA films exhibited greenish-yellow emission. At the same CD concentration, the PL intensity increased with increasing the film thickness, reached the maximum, and then decreased. At similar film thickness, the films with higher CD concentration exhibited lower PL intensity. | ||
| Fig. 2 Photographs of the EVA and CDs@EVA #3 films under the white light and 365 nm UV light. | ||
The difference in the fluorescence color between the CDs@EVA films and the CD chloroform dispersion is attributed to positive fluorescence solvatochromism.45
Fig. 3A and B shows the in-line transmittance and in-line absorbance spectra, respectively, of EVA and CDs@EVA #3 films shown in Fig. 2. The results of other CDs@EVA films are shown in Fig. S13 (ESI†). The EVA film exhibited high in-line transmittance and no absorption peak. For CDs@EVA films, the absorption peak due to the π–π* transition was observed around 470 nm.45–47 This absorbance of CDs@EVA films gradually increased with increasing CD concentration and film thickness (see Fig. S14, ESI†).
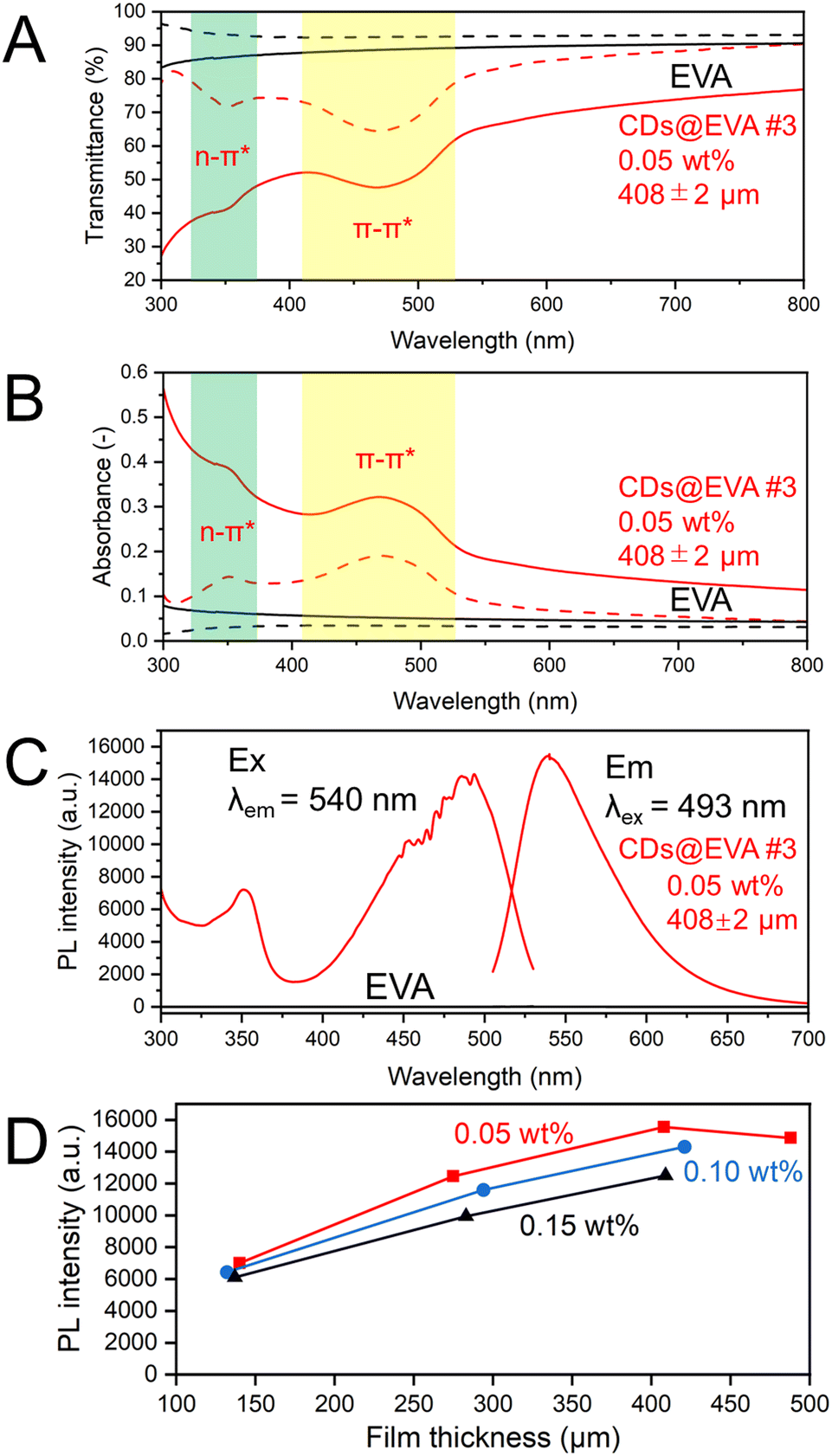 | ||
| Fig. 3 (A) In-line transmittance (solid lines) and total transmittance (dashed lines) spectra and (B) in-line absorbance (solid lines) and total absorbance (dashed lines) spectra of EVA and CDs@EVA #3 films, (C) PL/PLE spectra of CDs@EVA #3, and (D) change in the PL intensity of CD@EVA films of each CD concentration with film thickness. | ||
Fig. 3A and B also shows the total transmittance and total absorbance spectra, respectively, of EVA and CDs@EVA #3 films shown in Fig. 2. The results of other CDs@EVA films are also shown in Fig. S13, (ESI†). The measurement of total transmittance with an integrating sphere could avoid losses due to light scattering, resulting in a prominent absorption peak around 350 nm and the merging of the curves in the 600–800 nm region. The absorption peaks of the films at 350 nm and 470 nm are consistent with those of the CD chloroform dispersion. The absorbance around 470 nm for CDs@EVA films gradually increased with increasing CD concentration and film thickness (see Fig. S15, ESI†). The difference between in-line transmittance and total transmittance confirms the light scattering possibly from partially aggregated CDs.
Fig. 3C shows the PL and PLE spectra of EVA and CDs@EVA #3 films shown in Fig. 2. The results of other CDs@EVA films are shown in Fig. S16 (ESI†). All films exhibited the emission peak wavelength of 540 nm and the excitation peak wavelengths of 350 nm and 490 nm, similar to those of the CD chloroform dispersion. Respective Stokes shifts are 1.25 eV and 0.23 eV. Fig. 3D shows the changes in PL intensity with film thickness. At 0.05 wt%, the PL intensity first increased with increasing the thickness and then decreased. At the same CD concentration, the number of CDs in the film increased with increasing the film thickness, leading to an increase in PL intensity. However, when the film thickness exceeded a certain value, i.e., when the light path length of the emission from CDs exceeded a certain value, the self-absorption of CDs in the film became more pronounced, because the emission peak at 540 nm and the excitation peak at 490 nm (the absorption peak at 470 nm) were overlapped due to the small Stokes shift, which caused the PL intensity to decrease. For similar film thicknesses, the PL intensity was lower at higher CD concentrations. This can be attributed to a higher probability of self-absorption of the emission from CDs, known as concentration quenching. Accordingly, the highest PL intensity was observed for the CDs@EVA #3 film with 0.05 wt% CD concentration and 408 μm thickness.
By measuring the PLQYs of the CDs@EVA films (see Fig. S17, ESI†), it was found that with increasing the CD concentration and film thickness, the PLQY gradually decreased due to the increased self-absorption, as shown in Fig. S18 and Table S3 (ESI†). The highest PLQY of 56% was observed for the CDs@EVA #1 film with 0.05 wt% CD concentration and 140 μm thickness, which was the same as the average PLQY of the CD chloroform dispersion. Notably, CDs@EVA #1 film exhibited the lowest PL intensity at 0.05 wt% CD concentration in Fig. 3D, while it possessed the highest PLQY. This results from the PL intensity being a relative quantity that represents the number of photons emitted at a specific wavelength. Therefore, although this film has a lower PL intensity at a specific wavelength (e.g. 493 nm), it does not necessarily imply lower overall emission efficiency. In contrast, PLQY is a global parameter that is the ratio of the total number of photons emitted as fluorescence to the total number of photons absorbed by the sample. As the film thickness increased, the self-absorption of CDs in the film increased, thereby reducing the light energy conversion efficiency.
3.3. Properties of the LSC evaluation device with CDs@EVA films
The EVA and CDs@EVA films were attached to the LSC evaluation device, as shown in Fig. S4 (ESI†), and the I–V curves were measured under AM1.5G simulated sunlight as shown in Fig. 4 and Fig. S19 (ESI†). Table S4 (ESI†), summarizes the short-circuit current (Isc), open-circuit voltage (Voc), fill factor (FF), and power conversion efficiency (η) for the LSC device with no film, EVA film, and CDs@EVA films. As shown in Fig. 4, when EVA and CDs@EVA #3 film were attached to the LSC device, Isc increased from 0.733 mA to 0.774 mA and 0.855 mA by a factor of 1.06 and 1.17, respectively. As a result, η increased from 0.0190% to 0.0202% and 0.0226% by a factor of 1.06 and 1.19, respectively. At the 0.05 wt% CD concentration, the Isc and η first increased and then decreased with increasing film thickness, as shown in Fig. S20 and S21 (ESI†). The former results from the PL enhancement due to the increase in the number of CDs in the film, and the latter from the enhanced self-absorption due to the increase in the light path length of the emission from CDs. At CD concentrations of 0.10 wt% and 0.15 wt%, both the Isc and η decreased with increasing film thickness. This is attributed to concentration quenching due to increased self-absorption of the emission from CDs. Consequently, at similar film thicknesses, higher CD concentration caused a decrease in Isc and η. The maximum Isc and η values were achieved for CDs@EVA #3 film with 0.05 wt% CD concentration and the 408 μm thickness. This is due to the increased self-absorption of CDs by the small Stokes shift (large spectral overlap between the emission peak at 540 nm and the excitation peak at 490 nm (the absorption peak at 470 nm)) at higher CD concentrations and thicknesses.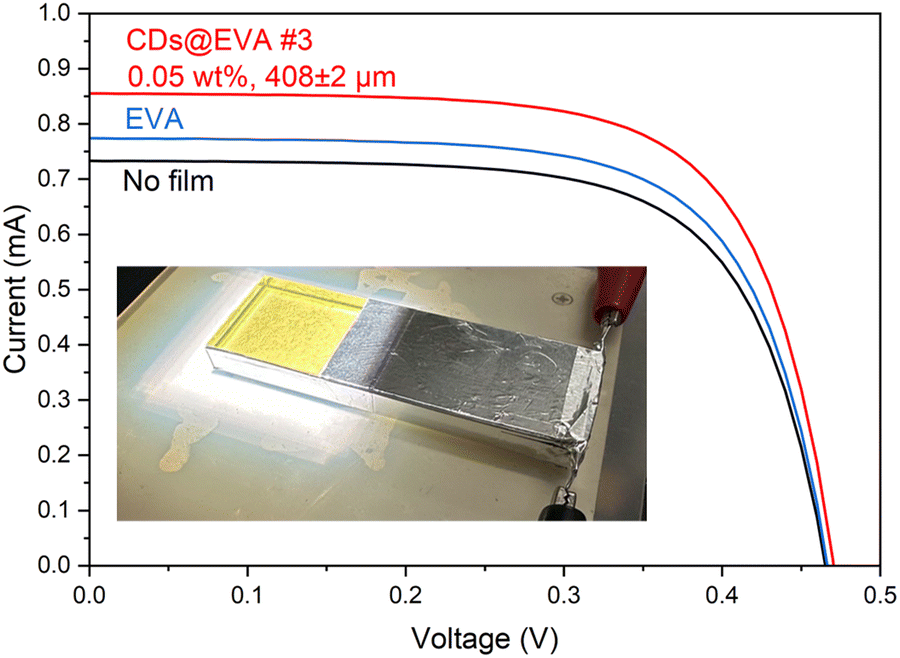 | ||
| Fig. 4 I–V curves of LSC with no film, EVA film, and CDs@EVA #3 film. The inset shows the experimental setup with the LSC with CDs@EVA #3 film under illumination. | ||
To understand the reasons for the increase in Isc and η, IPCE spectra were measured on the LSC device for the LSC device with no film, EVA film, and CDs@EVA films and shown in Fig. 5 and Fig. S22 (ESI†). The IPCE of the LSC device with no film decreased in the wavelength range above 500 nm, even though the c-Si solar cell module used can utilize light up to ∼1100 nm to generate electricity. This is attributed to the light absorption by the soda glass plate in the LSC device, as shown in Fig. S23 (ESI†). The IPCE of the LSC with the EVA film was slighter higher than that of the LSC with no film, possibly because of the lower reflection of incident light and the increase in light entering the LSC.
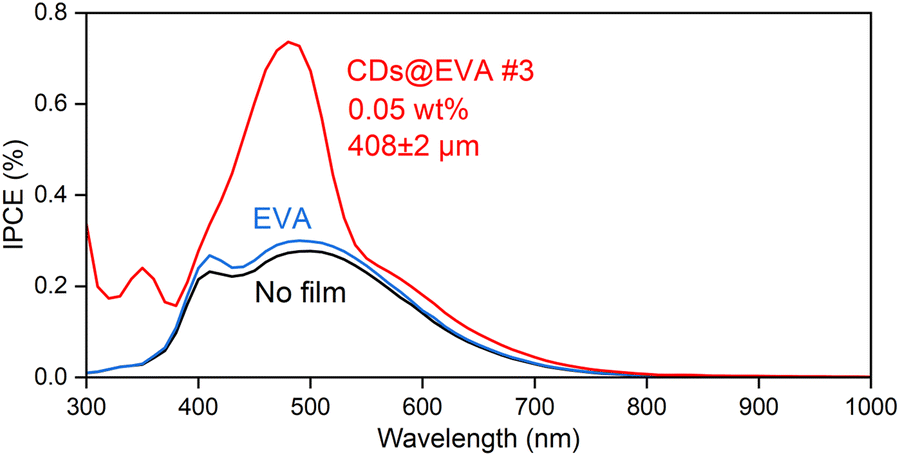 | ||
| Fig. 5 IPCE spectra of LSC with no film, EVA film, and CDs@EVA #3 film. | ||
The IPCE of the LSC with CDs@EVA films was higher than those of the LSC with no film and EVA film. One reason for this is the emission from CDs through their absorption (excitation) by the n–π* and π–π* transitions in both the UV and visible regions; therefore, the peak wavelength of IPCE at 350 nm and 493 nm corresponds to those of PLE, and the trend in maximum IPCE (see Fig. S24, ESI†) by CDs@EVA films corresponds to the trend in PL intensity shown in Fig. 3D. Another reason is the light scattering from partially aggregated CDs, because the small increase in IPCE was recognized in the region above ∼530 nm where the excitation of CDs did not occur. Light scattering intensity is inversely proportional to the fourth power of wavelength. Finally, CDs@EVA #3 film exhibited the highest IPCE in the range from 300 nm to 800 nm, resulting in the highest Isc and η values.
4. Conclusions
p-PD derived CDs synthesized in diphenyl ether at 250 °C and atmospheric pressure achieved the PLQY of 56% after centrifugation and purification. Additionally, dispersing CDs into EVA solution formed CDs@EVA films, which were applied in LSC devices to enhance the efficiency of solar cells. Due to the optical properties of the CDs, the CDs@EVA films absorbed UV light and short wavelength visible light through the n–π* and π–π* transitions, respectively, and converted them into longer wavelength visible light suitable for absorption by the c-Si solar cell module. The CDs@EVA films, with varying CD concentrations and film thicknesses, exhibited the highest Isc and η in the CDs@EVA #3 film due to self-absorption of CDs. When both EVA film and CDs@EVA #3 film were attached to the LSC device, Isc increased by factors of 1.06 and 1.17, respectively, and η increased by factors of 1.06 and 1.19, respectively. Based on IPCE spectrum measurements, the main contribution to the higher enhancement of Isc and η by CDs@EVA #3 film is the emission from CDs. These results provided valuable insights regarding the development of efficient LSC devices using luminescent CD-based materials. The PL intensity of CD chloroform dispersion and CDs@EVA #3 gradually decreased over the 5 h period (see Fig. S25, ESI†), which was attributed to the degradation possibly due to photo-oxidation reactions of the CDs under light exposure. Embedding the CDs in a high gas barrier film can significantly reduce photo-oxidation reactions of the CDs by oxygen in air, thereby greatly improving the photostability, which represents the key direction for future research.Author contributions
Y. L.: formal analysis, investigation, methodology, writing – original draft; Y. I.: supervision, writing – review and editing; T. I.: conceptualization, project administration, supervision, writing – review and editing. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.Data availability
The authors declare that all data supporting the results reported in this study are available within the paper and the ESI.† Additional data used for the study are available from the corresponding authors upon reasonable request. Y. L. thanks KLL Research Grant for PhD program at Keio University.Conflicts of interest
There are no conflicts to declare.References
- H. Hernandez-Noyola, D. H. Potterveld, R. J. Holt and S. B. Darling, Energy Environ. Sci., 2012, 5, 5798–5802 RSC.
- A. R. AbouElhamd, K. A. Al-Sallal and A. Hassan, Energies, 2019, 12, 1058 CrossRef CAS.
- M. G. Debije and P. P. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS.
- C. Liu and B. Li, J. Opt., 2015, 17, 025901 CrossRef CAS.
- K. Jo, S. Lee, G. S. Choi, B. H. Woo, Y. C. Jun, H. E. Song, T. Y. Jeon, H. H. Lee and H. J. Kim, J. Polym. Sci., 2021, 59, 59–69 CrossRef CAS.
- M. Peng, B. Dong, X. Cai, W. Wang, X. Jiang, Y. Wang, Y. Yang and D. Zou, Sol. Energy, 2017, 150, 161–165 CrossRef CAS.
- J. L. Banal, B. Zhang, D. J. Jones, K. P. Ghiggino and W. W. Wong, Acc. Chem. Res., 2017, 50, 49–57 CrossRef CAS.
- A. D’aléo, A. Picot, A. Beeby, J. Gareth Williams, B. Le Guennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258–10268 CrossRef.
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC.
- A. R. Frias, M. A. Cardoso, A. R. Bastos, S. F. Correia, P. S. André, L. D. Carlos, V. de Zea Bermudez and R. A. Ferreira, Energies, 2019, 12, 451 CrossRef CAS.
- T. Wang, J. Zhang, W. Ma, Y. Luo, L. Wang, Z. Hu, W. Wu, X. Wang, G. Zou and Q. Zhang, Sol. Energy, 2011, 85, 2571–2579 CrossRef CAS.
- X. Cao, Z. Zheng, Y. Zhang, G. Gu, J. Miao, R. Huang, D. Hou, Y. Tian and X. Zhang, J. Lumin., 2022, 252, 119368 CrossRef CAS.
- M. Grabolle, J. Ziegler, A. Merkulov, T. Nann and U. Resch-Genger, Ann. N. Y. Acad. Sci., 2008, 1130, 235–241 CrossRef CAS PubMed.
- M. G. Hyldahl, S. T. Bailey and B. P. Wittmershaus, Sol. Energy, 2009, 83, 566–573 CrossRef CAS.
- B. Suo, X. Su, J. Wu, D. Chen, A. Wang and Z. Guo, Mater. Chem. Phys., 2010, 119, 237–242 CrossRef CAS.
- L. Jin, G. S. Selopal, X. Liu, D. Benetti and F. Rosei, Adv. Funct. Mater., 2024, 2405653 CrossRef CAS.
- Y. Zhang, W. Zhang, Y. Ye, K. Li, X. Gong and C. Liu, Sol. Energy Mater. Sol. Cells, 2022, 238, 111619 CrossRef CAS.
- F. Li, Y. Liu, H. Wang, Q. Zhan, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 8546–8554 CrossRef CAS.
- N. Zhou, D. Wang, Y. Bao, R. Zhu, P. Yang and L. Song, Adv. Opt. Mater., 2023, 11, 2202681 CrossRef CAS.
- S. Hase, Y. Iso and T. Isobe, J. Mater. Chem. C, 2022, 10, 3523–3530 RSC.
- J. G. Bruno, Application of Quantum Dots in Biology and Medicine: Recent Advances, 2022, pp. 91–102 Search PubMed.
- C. Xia, S. Zhu, T. Feng, M. Yang and B. Yang, Adv. Sci., 2019, 6, 1901316 CrossRef CAS.
- H. Liu, X. Zhong, Q. Pan, Y. Zhang, W. Deng, G. Zou, H. Hou and X. Ji, Coord. Chem. Rev., 2024, 498, 215468 CrossRef CAS.
- Z. Ye, G. Li, J. Lei, M. Liu, Y. Jin and B. Li, ACS Appl. Bio Mater., 2020, 3, 7095–7102 CrossRef CAS.
- X. Wang, J. Cheng, H. Yu and J. Yu, Dalton Trans., 2017, 46, 6417–6424 RSC.
- J. Yue, L. Li, L. Cao, M. Zan, D. Yang, Z. Wang, Z. Chang, Q. Mei, P. Miao and W.-F. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 44566–44572 CrossRef CAS.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS.
- Z. Kang and S.-T. Lee, Nanoscale, 2019, 11, 19214–19224 RSC.
- D. Xu, Q. Lin and H. T. Chang, Small Methods, 2020, 4, 1900387 CrossRef CAS.
- G. Ge, L. Li, D. Wang, M. Chen, Z. Zeng, W. Xiong, X. Wu and C. Guo, J. Mater. Chem. B, 2021, 9, 6553–6575 RSC.
- J. C. E. da Silva and H. M. Gonçalves, TrAC, Trends Anal. Chem., 2011, 30, 1327–1336 CrossRef.
- Y. Zhou, D. Benetti, X. Tong, L. Jin, Z. M. Wang, D. Ma, H. Zhao and F. Rosei, Nano Energy, 2018, 44, 378–387 CrossRef CAS.
- J. Li, H. Zhao, X. Zhao and X. Gong, Nanoscale, 2021, 13, 9561–9569 RSC.
- Y. Wu, Y. Zhan, W. Xin, W. Cao, J. Li, M. Chen, X. Jiang, J. Wang and Z. Sun, ACS Appl. Energy Mater., 2022, 5, 1781–1792 CrossRef CAS.
- G. Liu, M. Zavelani-Rossi, G. Han, H. Zhao and A. Vomiero, J. Mater. Chem. A, 2023, 11, 8950–8960 RSC.
- Y. Li, P. Miao, W. Zhou, X. Gong and X. Zhao, J. Mater. Chem. A, 2017, 5, 21452–21459 RSC.
- Z. Wang, X. Zhao, Z. Guo, P. Miao and X. Gong, Org. Electron., 2018, 62, 284–289 CrossRef CAS.
- W. Li, X. Wang, J. Lin, X. Meng, L. Wang, M. Wang, Q. Jing, Y. Song, A. Vomiero and H. Zhao, Nano Energy, 2024, 122, 109289 CrossRef CAS.
- J. Lin, L. Wang, Q. Jing and H. Zhao, Chem. Eng. J., 2024, 481, 148441 CrossRef CAS.
- H. Zhao, G. Liu, S. You, F. V. Camargo, M. Zavelani-Rossi, X. Wang, C. Sun, B. Liu, Y. Zhang and G. Han, Energy Environ. Sci., 2021, 14, 396–406 RSC.
- X. Gong, W. Ma, Y. Li, L. Zhong, W. Li and X. Zhao, Org. Electron., 2018, 63, 237–243 CrossRef CAS.
- D. Chen, W. Wu, Y. Yuan, Y. Zhou, Z. Wan and P. Huang, J. Mater. Chem. C, 2016, 4, 9027–9035 RSC.
- J. Liu, X. Liu, H. Luo and Y. Gao, RSC Adv., 2014, 4, 7648–7654 RSC.
- S. Do, W. Kwon and S.-W. Rhee, J. Mater. Chem. C, 2014, 2, 4221–4226 RSC.
- R. Sato, Y. Iso and T. Isobe, Langmuir, 2019, 35, 15257–15266 CrossRef CAS.
- K. Sato, R. Katakami, Y. Iso and T. Isobe, ACS Appl. Nano Mater., 2022, 5, 7664–7669 CrossRef CAS.
- K. Washio, K. Sato, Y. Iso and T. Isobe, ECS J. Solid State Sci. Technol., 2023, 12, 076001 CrossRef CAS.
- K. Sato, R. Sato, Y. Iso and T. Isobe, Chem. Commun., 2020, 56, 2174–2177 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04133j |
| This journal is © The Royal Society of Chemistry 2025 |