
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom-temperature synthesis of highly luminescent methylammonium lead bromide nanocubes encapsulated in block copolymer micelles: impact of solvent choice on crystallization and stability†
Belda Amelia
Junisu
 ,
Ya-Sen
Sun
* and
Bo-Cheng
Zhao
,
Ya-Sen
Sun
* and
Bo-Cheng
Zhao
Department of Chemical Engineering, National Cheng Kung University, Tainan 701, Taiwan. E-mail: yssun@gs.ncku.edu.tw
First published on 6th January 2025
Abstract
Herein, we demonstrate a facile, room-temperature method for synthesizing highly luminescent methylammonium lead bromide (MAPbBr3) nanocubes encapsulated within colloidal polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP) micelles in a non-polar 1,3,5-trimethylbenzene (TMB) solvent. The synthesis involves five distinct stages: micellization of the PS-b-P2VP block copolymer, dissolution and complexation of PbBr2 precursors, coordination between bromoplumbate complexes and P2VP segments within the micelle core, multiple emulsion, and confined crystallization of perovskite nanocubes. Unlike conventional ligand-assisted methods, the colloidal micelles act as soft nanoreactors by controlling the nucleation and growth of the nanocubes through multiple emulsion. Moreover, the micelles serve as colloidal templates, preserving a significant concentration of [PbBr3]− ions formed during the complexation stage. We also demonstrate that the choice of solvent for transporting MA+ and Br− ions from the TMB phase to the micelle core significantly influences the dimensions of the resulting MAPbBr3 perovskite. Utilizing methanol as the transport medium yields encapsulated cubic MAPbBr3 nanocubes with a high photoluminescence quantum yield (PLQY) of ∼77%, and these encapsulated nanocubes exhibit long-term stability with a superior PLQY of ∼88%. In contrast, employing dimethylformamide (DMF) to solvate MA+ cations mainly yields MAPbBr3 microcrystals with reduced PLQY, as the formation of encapsulated MAPbBr3 nanocubes is hindered. This hindrance is attributed to the high entropic penalty associated with the diffusion and penetration of DMF-solvated MA+ cations through the PS shells. We believe that this versatile approach can be extended to synthesize a wide range of nanomaterials with well-defined morphologies, monodisperse sizes and long-term stability.
Introduction
Perovskites have received significant attention in recent years as promising materials across various fields. Their unique and flexible compositions enable precise tuning of optoelectronic properties, making them attractive for applications such as photovoltaics,1,2 light-emitting devices,3,4 and photodetectors.5 The desirable optical and electronic properties of hybrid perovskites can be attributed to their ABX3 crystal structure, where “A” represents a monovalent cation, “B” is a bivalent cation, and “X” represents halide ions.6,7 While inorganic perovskites (e.g., CsPbBr3, CsPbI3) offer superior thermal and environmental stability along with excellent optoelectronic properties,8–11 hybrid organic–inorganic perovskites (HOIPs) (e.g., MAPbBr3, MAPbI3) provide a balance of good performance, easier processing, and greater flexibility at a lower capital cost.12,13Despite the promising properties of perovskite materials, challenges remain in their synthesis and processing. The most critical challenge is their stability under ambient conditions.14,15 In particular, the stability of perovskite nanocrystals (NCs) can be influenced by intrinsic factors such as morphology and size distribution. Numerous studies has shown the difficulty to obtain perovskite NCs with well-defined morphologies and monodisperse size distribution via solution processing.16,17 Perovskite NCs with different morphologies (such as nanocubes, nanoplatelets, and irregular shapes) and sizes, exhibit varying degrees of stability.18,19 Moreover, the size of the perovskite NCs is also crucial. Smaller nanocrystals (e.g. 4 nm) tend to be less stable compared to larger ones due to increased surface chemistry and reactivity.20,21
Many bottom-up approaches for the synthesis of perovskite NCs employ ligands to control growth kinetics and prevent agglomeration.22–24 However, these ligands (e.g., oleic acid, octylamine) mainly act as stabilizers without controlling morphology. Block copolymers (BCPs) represent a compelling alternative, capable of providing both stabilization and morphology control. In particular, BCP self-assembly can generate well-defined nanostructures that serve as templates or nano-reactors for the growth of perovskite NCs.25 The thermodynamic incompatibility of the constituent polymer blocks drives microphase separation, leading to the formation of myriad ordered nanostructures.
By controlling the BCP composition, molecular weight, and solution processing conditions, a diverse array of morphologies can be obtained and offer soft colloidal templates to mediate the synthesis of inorganic nanomaterials.26–28 Polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP) BCPs have emerged as promising templates for enhancing the stability of perovskite NCs in both colloidal solutions29–32 and thin films.33–35 The efficient coordination between the P2VP block and metal precursor species promotes selective nucleation within the P2VP cores. For instance, Hou et al.30 and Lee et al.31 successfully demonstrated the encapsulation of CsPbBr3 perovskite NCs within PS-b-P2VP core–shell micelles, achieving remarkable stability against degradation in various polar solvents. Similarly, Hui et al.29 utilized PS-b-P2VP micelles to synthesize MAPbX3 nanoparticles exhibiting enhanced thermal stability, while Nah et al.32 further corroborated the efficacy of micelle encapsulation in producing stable MAPbX3 perovskite NCs. However, a recurring limitation in these studies is the formation of micelles with less compact architectures, often accompanied by limited control over nanocrystal shape. Such non-uniformity increases the prevalence of defects and lattice distortions, ultimately leading to non-radiative recombination and reduced photoluminescence quantum yield (PLQY). Therefore, achieving monodisperse perovskite NCs with uniform shape is crucial for enhancing its PLQY.
Ligand-assisted re-precipitation (LARP) methods has been known as a more cost-effective alternative to produce perovskite NCs at room temperature.13,21 LARP leverages the principle of supersaturated recrystallization, whereby the solubility of a highly saturated precursor solution is rapidly decreased—either by cooling, solvent evaporation, or the addition of a poor solvent (i.e., an antisolvent, usually polar solvents).36 This non-equilibrium condition induces rapid precipitation and crystallization until equilibrium is re-established. BCP colloidal templates function analogously to LARP by providing physical and chemical confinement for perovskite NCs. However, unlike LARP, BCP colloidal templates do not necessitate polar solvents for nanocrystal purification, thereby circumventing potential degradation and emission quenching associated with such polar solvents.25
The growth of perovskite nanocrystals within BCPs is a multifaceted process involving several key steps: micellization of the BCP, solvation and salt complexation of precursors, coordination interactions between precursor species and BCP segments, and crystallization.29–31 Solvent quality significantly influences each of these steps. Notably, poor solvent quality can lead to collapsed or less organized BCP conformations. Furthermore, sufficient solvation is critical to ensure the formation of precursor salt complexes. For example, precursor complexes are typically produced by solvating PbX2 precursors in an octadecene (ODE)37–39 or dimethylformamide (DMF) solvent.40 In the initial stage of synthesis, precursor complexes should selectively interact with BCP active groups. For instance, coordination interactions between precursor species and pyridine groups in P2VP blocks are often driven by Lewis acid–base interactions. The lone pair of electrons on the nitrogen atoms of quarternized P2VP blocks can delocalize to the empty 6p-orbital of Pb-complex species.41 As the concentration of coordinated precursors increases, supersaturation conditions are eventually reached.42 Under such condition, rapid crystallization produces perovskite NCs encapsulated within micelle templates.29–31
Despite the potential of BCP colloidal micelles for templated synthesis of hybrid organic–inorganic perovskite NCs, a comprehensive understanding of the underlying mechanisms remains elusive. This study aims to systematically understand how soft colloidal templating of using PS-b-P2VP micelles mediated the synthesis of encapsulated methylammonium lead bromide (MAPbBr3) perovskite nanocrystals (NCs) in a non-polar solvent (1,3,5-trimethylbenzene). The synthesis of encapsulated perovskite NCs involved PS-b-P2VP micellization, formation of hierarchical emulsion, solvation and complexation of precursor salts, coordination interactions between PS-b-P2VP and salt ions, and nucleation and growth of perovskite crystals in a confined space. Furthermore, the influence of two different polar solvents (methanol and DMF) on MABr solvation and crystallization kinetics of the resulting MAPbBr3 perovskite NCs were further examined.
Experimental section
Materials
Two polystyrene-block-poly(2-vinylpyridine) block copolymers, PS48.5-b-P2VP70 (Mn,PS = 48.5 kg mol−1, Mn,P2VP = 70 kg mol−1, Mw/Mn = 1.13) and PS49-b-P2VP75 (Mn,PS = 49 kg mol−1, Mn,P2VP = 75 kg mol−1, Mw/Mn = 1.03), were purchased from Polymer Source Inc. PS49-b-P2VP75 was used as ligands for Fig. 1–7 and Fig. S2–S9 (ESI†). PS48.5-b-P2VP70 was used as ligands for Fig. S1 (ESI†). These two block copolymers have similar molecular weights and compositions. Therefore, they have the same micellization behavior in TMB. Lead bromide (PbBr2) and methylammonium bromide (MABr) precursor were purchased from Sigma Aldrich and used as received. 1,3,5-Trimethylbenzene (TMB, C6H3(CH3)3) was purchased from Thermo Scientific. Methanol (CH3OH) was purchased from Honeywell. Dimethyl formamide (DMF) was purchased from Sigma Aldrich. | ||
| Fig. 1 (A)–(C) TEM images and corresponding (D) SAXS (symbols: experimental data and cyan lines: fitted data), (E) WAXD, and (F) absorbance profiles of BCP, t-LHP and c-LHP solutions. In (D), red lines at low-q and high-q regions that represent slope are plotted as a visual guide. Micelle size distributions from images (A)–(C) are shown in the upper panels. Inset in images (A)–(C) show photographs of the BCP, t-LHP and c-LHP solutions, respectively. | ||
 | ||
| Fig. 2 Photos of ((A) and (B)) t-PER and ((C) and (D)) c-PER solutions. The solutions were prepared by adding different aliquots [(i, iv) 5 μL, (ii, v) 10 μL, and (iii, vi) 20 μL] of methanol-solvated (left panel) or DMF-solvated (right panel) MABr solutions into t-LHP solutions. Photos in (B) and (D) were taken under UV light exposure at λ = 365 nm. | ||
 | ||
Fig. 3 UV-vis absorbance (solid lines) and PL (dashed lines) spectra of (A) c-PERMx and (B) c-PERDx solutions (x: 5, 10, and 20). (C) PLQY of the corresponding solutions described in (A) and (B). For UV-vis and PL measurements, the original solutions were diluted to a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. For PLQY measurements, the dilution factor was 1:30. :10. For PLQY measurements, the dilution factor was 1:30. | ||
 | ||
| Fig. 4 (A) WAXD and (B) SAXS (symbols: experimental data; lines: fits) profiles of the c-PERMx and c-PERDx solutions (x: 10 and 20). Stick pattern in (A): cubic MAPbBr3 crystals (ICSD#26785). The WAXD and SAXS curves were vertically shifted for better visualization. | ||
 | ||
| Fig. 5 TEM images collected in a dried state for the (A) c-PERM5, (B) c-PERM10, (C) c-PERM20, (E) c-PERD5, (F) c-PERD10 and (G) c-PERD20 solutions, respectively. Images (D) and (H) are the HR-TEM images of selected nanocube in c-PERM10 and nanocrystal in c-PERD20, respectively. Yellow dashed lines in (D) indicate the periphery of micelles. | ||
 | ||
| Fig. 6 ((A) and (D)) HR-TEM and ((B) and (C), (E) and (F)) EDS elemental analysis of dried c-PERM10 and c-PERD20 solutions. EDS spectra were selectively collected on two different positions of each specimen by scanning N, Pb, and Br elements. The positions are labelled with 1 and 2 in images (A) and (D), respectively. | ||
 | ||
| Fig. 7 ((A) and (B)) Photos and ((C) and (D)) WAXD profiles of dried precipitates separated from the (i) t-PERM10, (ii) t-PERM20, (iii) t-PERD10, and (iv) t-PERD20 solutions after centrifugation at 7000 rpm for 10 min. Photos in (B) were taken under UV light exposure at λ = 365 nm. In (C) and (D), red and black arrows indicate the diffraction from the glass substrate. Stick patterns represent standard WAXD profiles of orthorhombic PbBr2 crystals (PDF#031-0679) and cubic MAPbBr3 crystals (ICSD#268785). | ||
Preparation of precursor solutions
The experimental procedures used in this study are depicted in Scheme 1. Perovskite NCs within BCP micelles were synthesized through three steps: micellization, precursor complexation, and confined crystallization. First, 0.5 wt% BCP colloidal solutions were prepared by dissolving 4.3 mg of PS-b-P2VP powder in 1 mL of 1,3,5-trimethylbenzene (TMB). The solutions were sonicated at 70 °C for 30 minutes and then cooled to room temperature. To ensure complete micellization, the BCP colloidal solutions were aged overnight. Next, precursor solutions were prepared by dissolving 10 mg of PbBr2 in the block copolymer solutions. These precursor solutions were stirred at 800 rpm for 24 hours using a magnetic stirrer. The resulting turbid lead-halide precursor solutions were labeled as t-LHP solutions (Scheme 1B). | ||
| Scheme 1 Procedures of colloidal templated synthesis of perovskite nanoparticles in a solution added with PS-b-P2VP micelles. First set of samples were divided by four stages: (A) BCP solutions, (B) t-LHP solutions, (C) t-PER solutions and (D) c-PER solutions. Second set of samples were divided by five stages: (A′) BCP solutions, (B′) t-LHP7 solutions, (C′) c-LHP7 solutions, (D′) t-PERc7 solutions, and (E′) c-PERc7 solutions. Note that white objects in (B), (C), (B′), and (D′) represent the magnetic stirrer. | ||
Preparation of MABr solutions
Due to MABr low solubility in TMB, MABr solutions were separately prepared in either methanol or DMF. These two solvents were selected for two reasons. First, MABr is highly soluble in methanol and DMF, ensuring complete ionization to produce MA+ and Br− ions. Second, the high polarity index of both solvents (methanol = 5.1, DMF = 6.4)43 facilitates the effective transport of ions from the polar solvent (methanol or DMF) to the P2VP core through the non-polar TMB phase. Specifically, 60 mg of MABr powder was dissolved in 200 μL of either methanol or DMF to produce 300 mg mL−1 MABr solutions. The mixtures were then stirred for 30 minutes at 50 °C to ensure complete dissolution.Synthesis of perovskite NCs
The final step involved the confined crystallization of MAPbBr3 perovskite NCs within the BCP micelles. To initiate crystallization, aliquots (5–20 μL) of the MABr solutions were added dropwise to the t-LHP solutions. The mixtures were stirred continuously at 800 rpm for 30 minutes for the nucleation and growth of MAPbBr3 nanocubes within the P2VP cores. The formation of MAPbBr3 NCs was evidenced by a color change from white in the precursor solutions to yellow in the perovskite solutions (Scheme 1C). The resulting perovskite solutions were then centrifuged at 7000 rpm to separate the encapsulated MAPbBr3 nanocubes from microstructures. The encapsulated MAPbBr3 nanocubes were dispersed in supernatants (c-PER, Scheme 1D), while the microparticles existed as precipitates. The clear supernatants (briefly denoted as c-PERM5, c-PERM10 and c-PERM20) and precipitates were collected separately for further structural characterization. The same procedures were performed on c-PERD5, c-PERD10 and c-PERD20 solutions that were prepared by adding 5, 10 or 20 μL of MABr aliquots in DMF to the t-LHP solutions, followed by centrifugation. The subscripts denote the used solvents (M: methanol; D: DMF) and the amount of added MABr aliquots. All solutions were prepared at room temperature.We also prepared an additional set of BCP colloidal solutions to investigate the effect of prior centrifugation of the t-LHP solutions on the complexation and crystallization of perovskite NCs. The procedures for preparing the t-LHP solutions were identical to those described above, except that these solutions were stirred continuously for 7 days to produce t-LHP7 solutions, where the subscript denotes the stirring time in days if specified (Scheme 1B′). After prolonged stirring, the t-LHP7 solutions were centrifuged at 7000 rpm for 10 minutes to remove undissolved PbBr2 crystals, producing clear supernatant precursor solutions (denoted as c-LHP7, Scheme 1C′).
The centrifuged c-LHP7 solutions were used for the synthesis of perovskite NCs. Specifically, 20 μL of either the MABr/methanol or MABr/DMF precursor solution was added dropwise to the stirred c-LHP7 solutions. After stirring for 30 minutes at 800 rpm, the resulting solutions were labelled as t-PERc7 (Scheme 1D′). Here, the subscript “c” indicates prior centrifugation of the t-LHP7 solution. These solutions were then centrifuged to separate the encapsulated perovskite NCs from unreacted precursors and by-products. The supernatants (c-PERc7) and precipitates were collected separately for further characterization. A list of abbreviations in this study is shown in Table 1.
| Abbreviation | Description |
|---|---|
| t-LHP | Turbid lead halide precursor solution (consists of BCP colloidal micelles and PbBr2) after 1 day of stirring |
| t-PER | Turbid perovskite solution after the addition of MABr aliquots |
| c-PERMx | Clear perovskite solution after subsequent centrifugation of t-PER solution synthesized by the addition of MABr/methanol aliquots to the t-LHP solution (x = 5, 10, 20) |
| c-PERDx | Clear perovskite solution after subsequent centrifugation of t-PER solution synthesized by the addition of MABr/DMF aliquots to the t-LHP solution (x = 5, 10, 20) |
| t-LHP7 | Turbid lead halide precursor solution after 7 days of stirring |
| c-LHP7 | Clear lead halide precursor solution after 7 days of stirring and subsequent centrifugation |
| t-PERc7 | Turbid perovskite solution after the addition of MABr aliquots to the c-LHP7 solution |
| c-PERc7,Mx | Clear perovskite solution after subsequent centrifugation of t-PERc7 solution synthesized by the addition of MABr/methanol aliquot to the c-LHP7 solution (x = 20) |
| c-PERc7,Dx | Clear perovskite solution after subsequent centrifugation of t-PERc7 solution synthesized by the addition of MABr/DMF aliquot to the c-LHP7 solution (x = 20) |
Material characterization and instruments
UV-vis absorption spectra were measured with a JASCO V-770 UV-vis/near-IR spectrophotometer (JASCO Analytical Instruments), equipped with both a halogen and deuterium lamp. The fluorescence of the perovskite solutions was probed using a UV lamp (UVGL-25 4W compact UV lamp, λ = 365 nm). Photoluminescence (PL) spectra at room temperature were measured using an F-2500 FL Spectrophotometer (Hitachi), equipped with an Xe lamp (λexc: 375 nm). Prior to UV-vis and PL measurements, all perovskite solutions were diluted to a ratio of 1:10 in TMB to accommodate detector resolution. PLQY were determined using an absolute PLQY measurement system (FluoroMax® spectrometer, HORIBA Scientific). Prior to the PLQY measurements, all samples were diluted to a ratio of 1:30 in TMB.
Ten microliters of solutions were deposited on a carbon film-coated Cu grid. The grids were dried in an oven at 50 °C overnight before imaging. Morphologies of encapsulated perovskite nanocubes were characterized by low-resolution transmission electron microscopy (TEM) (H-7500) at National Cheng Kung University (NCKU), Taiwan. The low-resolution TEM was operated at 80 kV. High-resolution TEM (HR-TEM) was performed with a JEOL JEM-1400 (120 kV) at the NCKU Instrument Center, Taiwan. The analysis of elemental composition in perovskite nanocubes was performed using energy dispersive X-ray spectroscopy (EDS) attached to the HR-TEM. The elemental composition of N, Pb, and Br was quantified in each point scan. X-ray diffraction of precipitates was performed using CuKα1 radiation (λ = 0.154 nm) on a Rigaku Ultima IV-9407F701 X-ray diffractometer at 40 kV. Precipitates were carefully transferred from centrifuge tubes to glass substrates, which were then dried in an oven at 70 °C overnight. The 2θ scanning range was from 10° to 60° with a step size of 0.02° and a scan rate of 5° min−1.
Simultaneous small-angle X-ray scattering (SAXS) and wide-angle X-ray diffraction (WAXD) measurements of the BCP/perovskite solutions were conducted at the TPS 13A end station of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan).44 Data were collected at an energy of 15 keV (λ = 0.827 Å) using two detectors: Eiger X 9 M for SAXS and Eiger X 1 M for WAXD. Silver behenate and polyethylene standards were used to calibrate the sample-to-detector distance and absolute SAXS intensity, respectively. Solutions were sealed in capillary tubes (Φ = 2 mm) and mounted on a modified Linkam force stage for measurements. Background subtraction was performed using scattering data from empty capillary tubes and tubes containing neat solvents or solvent/methanol mixtures. An exposure time of 10 s was used for all measurements. One-dimensional SAXS profiles were obtained from data reduction. Absolute intensity (I(q)), as a function of the scattering vector (q), was used to describe scattering features. q is defined as  , where λ is the X-ray wavelength and 2θ is the scattering angle. Structural parameters were determined by fitting the SAXS data using SASview software.45 Two models were used: the two-level Beaucage model and the polydisperse core–shell sphere model.46–48 Based on the Beaucage model,46 SAXS fitting quantifies hierarchical structures, with each level having structural parameters defined by the radius of gyration (Rg,i) and power-law exponent (df,i), where the subscript “i” represents the i-th level structure. Rg,i indicates the size of the irregular structure, while df,i reflects its dimensionality. For the polydisperse core–shell sphere model, SAXS fitting provides structural parameters, including core radius (rc), shell thickness (rs), size dispersity, and the scattering length density (ρc, ρs, and ρsol) of the core, shell, and solvent, respectively. Of the structural parameters, size dispersity is based on the Schultz size distribution function.45
, where λ is the X-ray wavelength and 2θ is the scattering angle. Structural parameters were determined by fitting the SAXS data using SASview software.45 Two models were used: the two-level Beaucage model and the polydisperse core–shell sphere model.46–48 Based on the Beaucage model,46 SAXS fitting quantifies hierarchical structures, with each level having structural parameters defined by the radius of gyration (Rg,i) and power-law exponent (df,i), where the subscript “i” represents the i-th level structure. Rg,i indicates the size of the irregular structure, while df,i reflects its dimensionality. For the polydisperse core–shell sphere model, SAXS fitting provides structural parameters, including core radius (rc), shell thickness (rs), size dispersity, and the scattering length density (ρc, ρs, and ρsol) of the core, shell, and solvent, respectively. Of the structural parameters, size dispersity is based on the Schultz size distribution function.45
Results and discussion
Characterization of the BCP, t-LHP and c-LHP solutions
The synthesis of encapsulated perovskite nanocrystals (NCs) involves three stages in sequence: (1) PS-b-P2VP micellization, (2) precursor complexation in multiple emulsion, and (3) confined crystallization. The micellization of block copolymers (BCP) is governed by the interplay between the solubility parameters of the solvent and the constituent blocks. PS-b-P2VP exhibits strong micellization in TMB due to the selective solvation of the PS blocks. This selectivity arises because the solubility parameter of TMB (δTMB = 18 MPa1/2)49 is lower than those of both PS (δPS = 18.6 MPa1/2)50 and P2VP (δP2VP = 19.8 MPa1/2).51To quantitatively analyze the micellar structure, TEM and SAXS measurements were performed on dry films and original BCP solutions in TMB, respectively. The neat BCP solution appeared bluish due to the Rayleigh scattering of micellar nanostructures52 (inset of Fig. 1A). TEM image and size distribution analysis in Fig. 1A reveals well-defined dark spheres with an average diameter of 47 nm. The SAXS profile (the black curve in Fig. 1D) of the neat BCP solution exhibits a Guinier regime in the low-q region (q < 0.008 Å−1), followed by form factor oscillations in the intermediate-q region (0.008–0.08 Å−1). The oscillations locate at 0.0287, 0.0456, 0.0614 and 0.0794 Å−1, respectively. In the high-q region (q > 0.08 Å−1), a power-law intensity decay of I ∼ q−2 is observed. This intensity decay indicates polymer brushes forming within the micelle corona.48 The black SAXS curve was fitted using the polydisperse core–shell spherical model to extract structural parameters for micelles in the BCP solution. The structural parameters are summarized in Table 2. A core radius of 19.59 nm and a shell thickness of 10.74 ± 0.09 nm were obtained from the fitting. The WAXD profile of the neat BCP solution shows a broad amorphous halo due to inter-chain or intra-chain correlations (the black curve in Fig. 1E).
| Model fitting | Solutions | ||
|---|---|---|---|
| Neat BCP | t-LHP | c-LHP | |
| Beaucage model | |||
| R g,1/nm | — | 152.07 | — |
| d f,1 | — | 3.89 | — |
| R g,2/nm | — | 258.07 | — |
| d f,2 | — | 3.99 | — |
| Core–shell sphere | |||
| r c/nm | 19.59 ± 0.00 | 23.80 ± 0.05 | 20.59 ± 0.00 |
| PDIc | 0.065 | 0.110 | 0.085 |
| r s/nm | 10.74 ± 0.09 | 10.47 ± 0.01 | 11.40 ± 0.13 |
| ρ core (×10−6 Å−2) | 8.720 | 8.705 | 8.720 |
| ρ shell (×10−6 Å−2) | 8.111 | 8.072 | 8.090 |
| ρ solvent (×10−6 Å−2) | 8.057 | 8.057 | 8.057 |
Next, we characterized a t-LHP solution with visual examination and TEM. The t-LHP solution appeared turbid (the insert in Fig. 1B). The turbidity is attributed to Mie scattering from microstructures with dimensions comparable to the wavelength of visible light.53Fig. 1B shows a representative morphology of a microparticle surrounded by a collection of BCP micelles with an average diameter of ∼45 nm. This morphology is similar to that observed in Fig. S1A and E (ESI†), which was measured on a t-LHP7 solution in its dried state. EDS 2D maps (Fig. S1B–D, ESI†) and 1D profile (Fig. S1G, ESI†) demonstrates that the microparticle is enriched with N, Pb and Br elements, suggesting that the PbBr2 microparticle is adsorbed with PS-b-P2VP micelles. We believe that strong repulsions imparted by the PS shells effectively disperse the microparticles as a microemulsion within the TMB, causing the solution turbidity. Note that most micelles tend to cluster in the t-LHP solution (Fig. 1B). In comparison, the 1D profile (Fig. S1F, ESI†) of a selected PS-b-P2VP micelle reveals that each micelle contains Pb and Br elements with an atomic ratio of 1:3. This ratio suggests that PbBr2 exists as complexes inside the PS-b-P2VP micelles.
The SAXS profile (the blue curve in Fig. 1D) of the t-LHP solution exhibits a low-q upturn with I ∼ q−3.9 in intensity at q < 0.008 Å−1. This strong low-q upturn is likely due to PbBr2 microparticles and micellar clustering in the t-LHP solution. Damped oscillations in the intermediate-q range (0.008–0.08 Å−1) correspond to an increase in size dispersity of micelles. This fringe dampening is likely due to adsorption of P2VP cores onto the surface of microparticles for microscale emulsion, or micellar clustering. The surface adsorption or micellar clustering may cause micellar deformation or distortion, thus leading to the dampening of the fringes. The WAXD pattern of the t-LHP in Fig. 1E (blue curve) shows several diffractions, which can be assigned to orthorhombic PbBr2 crystals (PDF #031-0679). This result indicates that microparticles are comprised of orthorhombic PbBr2 crystals.
We further performed SAXS fitting on the blue curve shown in Fig. 1D for quantitative analysis. The fitting is based on a combination of the two-level Beaucage model and core–shell sphere model. The two-level Beaucage model fits the scattering of inorganic particles and micellar clustering, while core–shell sphere model fit the scattering of core–shell micelles. The fitted curve, based on structural parameters shown in Table 2, reveals that the inorganic particles are quantified with Rg,1 ∼ 258.07 nm and df,1 ∼ 3.99 and the micellar clustering is quantified with Rg,2 ∼ 152.07 nm and df,2 ∼ 3.89. The presence of these large particles indeed leads to an increase in the size distribution from 0.065 to 0.11 for the micelles in the t-LHP solution.
Yoon et al.54 demonstrated the complexation of PbBr2 when dissolved in DMF. In our study, PbBr2 partially dissolves in TMB added with PS-b-P2VP, resulting in the formation of a microemulsion with BCP micelles. This microemulsion, however, is transient due to the large size of the PbBr2 microcrystals. To further investigate how complexation occurred in the t-LHP solution, a c-LHP solution was prepared by centrifuging the t-LHP solution. The c-LHP solution (inset of Fig. 1C) exhibits a bluish color similar to the neat BCP solution, suggesting the removal of PbBr2 microparticles. This is supported by the WAXD profile (the green curve in Fig. 1E) of the c-LHP solution, which closely resembles that of the neat BCP solution. Fig. 1C reveals a morphology of dark spheres with an average diameter of ∼46 nm. The dark spheres correspond to P2VP cores. Fitting the SAXS profile (the green curve in Fig. 2D) of the c-LHP solution shows that the radius (∼20.6 nm) of the P2VP core and thickness (∼11.4 nm) of the PS shell slightly increased for micelles in the c-LHP solution. Without PbBr2 microparticles, the core–shell spherical micelles have a small size distribution at 0.085.
We further characterized the three solutions with UV-vis absorbance spectrometer. Interestingly, the UV-vis absorption spectra of the t-LHP and c-LHP solutions display an additional peak centered at ∼315 nm (Fig. 1F). This broad band is assigned to the absorbance of [PbBr3]− complexes bound with ligands,38 suggesting coordination between PbBr2 and 2-vinylpyridine segments within the micelle core. This coordination suggests nanoscale emulsions, by which [PbBr3]− complexes are encapsulated within the PS-b-P2VP micelles.
Synthesis of perovskite nanocrystals
To investigate the influence of the precursor medium on the synthesis of encapsulated perovskite nanoparticles, varying volumes (5, 10, and 20 μL) of MABr aliquots (300 mg mL−1) in either methanol or DMF were added to t-LHP solutions. The resulting solutions were labelled as t-PERMx and t-PERDx solutions. As shown in Fig. 2Ai–iii and Aiv–vi, the t-PERMx and t-PERDx solutions appeared milky yellow after 30 minutes of stirring, indicating perovskite formation. Under UV exposure (Fig. 2Bi–vi), a significant difference in luminescence was observed between the t-PERMx and t-PERDx solutions prepared with two types of MABr solutions. The t-PERMx solutions exhibited uniform green luminescence, while t-PERDx solutions displayed inhomogeneous luminescence, with only the top layer emitting green light (Fig. 2Biv–vi). This inhomogeneity suggests a higher concentration of MAPbBr3 microparticles with low fluorescence in the t-PERDx solutions.Centrifugation at 7000 rpm for 10 minutes was performed to remove large particles. The resulting c-PER supernatants (c-PERMx and c-PERDx) exhibited uniform green luminescence (Fig. 2C and D). Notably, the c-PERDx solutions displayed a paler yellow coloration compared to c-PERMx solutions. A centrifugation speed of 7000 rpm was found to be optimal for producing uniform, high-quality MAPbBr3 nanocrystals with superior optical properties. Deviations from this speed, whether too low or too high, failed to yield perovskite colloidal solutions with comparable optical quality (Fig. S2, ESI†). Insufficient centrifugation left residual microcrystals in the solution, while excessive centrifugation likely disrupted the micelle encapsulation of the nanocrystals. The c-PERMx and c-PERDx solutions were further analyzed by UV-vis and PLQY.
Fig. 3A and B shows the optical properties of the c-PERMx and c-PERDx solutions, as measured by UV-vis absorbance and PL spectroscopy. The c-PER solutions are separated supernatants after centrifugation was performed on t-PER solutions to remove precipitates. By comparing the intensities of emitted and absorbed light during PL measurements, PLQY can be quantified (Fig. 3C). Several prominent features are evident from Fig. 3. With the exception of the c-PERM5 solution, the absorbance spectra of the c-PERM10 and c-PERM20 solutions show an Urbach tail extending from 350 to 500 nm followed by an absorbance edge at ∼527 nm. These features indicate the formation of perovskite crystals in the c-PERM10 and c-PERM20 solutions (Fig. 3A). Due to the formation of perovskite crystals, the c-PERM10 and c-PERM20 solutions emit intense PL signals with superior PLQY of 74.5 and 77.3% (Fig. 3C). In comparison, the UV-vis absorbance of the c-PERM5 solution still shows an intense absorbance at 325 nm but a weak absorbance edge at ∼527 nm. The absorbance at 325 nm can be assigned to the absorbance of [PbBr3]− complexes.38 This result indicates that the c-PERM5 solution contained abundant [PbBr3]− complexes coexisting with a small amount of perovskite crystals. Because of the low content of perovskite crystals, the c-PERM5 solution emits a weak PL signal with a PLQY of approximately 59%. The weak PL and low PLQY are due to the insufficient MABr content. In contrast, the UV-vis absorbance spectra of the c-PERDx solutions show intense absorbance band at 325 nm and their PL spectra show extremely low signals with PLQY values of less than 3% (Fig. 3B and C). The result indicates that using DMF to solvate MA+ cations cannot effectively form perovskite crystals in the c-PERDx solutions. The comparison between the c-PERMx and c-PERDx solutions highlights the crucial role of solvent choice in the efficiency of perovskite crystallization and the resulting photoluminescence performance.
PLQY is a crucial parameter for quantitatively assessing the optical performance of perovskite nanocrystals. Table 3 summarizes the PLQY values of various perovskite materials encapsulated in BCP micelles reported in this work, alongside data from other studies.29–32 As shown in Table 3, our study demonstrates a higher PLQY compared to other perovskite materials synthesized via BCP-templating approaches.
In addition to PLQY, the long-term stability of perovskite solutions is crucial. After 90 days of ambient storage, no observable precipitate was found in the c-PERM5, c-PERM10, and c-PERM20 solutions (Fig. S3A, ESI†). Furthermore, the c-PERMx solutions continued to exhibit strong absorbance at 520 nm and photoluminescence (PL) at approximately 529.3 nm (Fig. S3B, ESI†), features attributed to the encapsulated MAPbBr3 nanocubes. These findings demonstrate that the encapsulated MAPbBr3 nanocubes in the c-PERMx solutions maintain good stability in air. Particularly, the c-PERMx solutions showed increased PLQY values of 61.2%, 87.9%, and 85.6%, respectively, after 90 days of ambient storage (Fig. S3C, ESI†).
Note that the freshly prepared c-PERM10 solution exhibited a PLQY of 77%, which is already the highest among the c-PERMx solutions. However, after three months of ambient storage, the PLQY increased to 88%, suggesting that the encapsulated MAPbBr3 nanocubes underwent further passivation during storage. Prolonged storage likely allows residual ions or block copolymer chains in the solution to interact with the nanocube surfaces, passivating additional defects and further reducing non-radiative recombination pathways.
Structural characterization of perovskite nanocrystals
Next, we characterized the c-PERMx and c-PERDx supernatants using WAXD and SAXS. Fig. 4A shows the WAXD profiles of the c-PERMx and c-PERDx supernatants. All of the c-PERMx and c-PERDx supernatants reveal a broad halo in Fig. 4A. This broad halo corresponds to the inter-chain correlation of polymer chains within micelles.55 Furthermore, the WAXD profiles of the c-PERMx supernatants display MAPbBr3 diffractions, while those of the c-PERDx supernatants show no MAPbBr3 diffractions. This discrepancy indicates that the c-PERMx supernatants contain a mixture of micelles and MAPbBr3, whereas the c-PERDx supernatants contain only micelles with little to no MAPbBr3.Fig. 4B shows the SAXS profiles of the c-PERMx and c-PERDx supernatants. Several scattering features are prominent. First, they show a Guinier regime at q < 0.008 Å−1 and a series of oscillations in the intermediate q regime (0.02 < q < 0.08 Å−1), corresponding to form factor scattering of core–shell spheres.47,48,55 The series of oscillations indicates a monodisperse distribution of nanostructures. Second, the oscillations in the c-PERMx solutions damped more quickly than those in the c-PERDx solutions (Fig. 4B). This difference in oscillation prominence is attributed to the coexistence of PS-b-P2VP micelles and inorganic nanoparticles within the c-PERMx solutions. The dampened fringes observed in the c-PERMx solutions, compared to those in the c-PERDx solutions, reflect an increase in size dispersity.
We further fitted the SAXS data using a core–shell spherical model47,48 to extract the structural parameters of the micelles in both types of c-PER solutions, with the results summarized in Table 4. The fits based on the structural parameters in Table 4 are consistent with the SAXS curves (Fig. 4B). Table 4 demonstrates that the c-PERMx solutions contain PS-b-P2VP micelles with a higher dispersity compared to the c-PERDx solutions. This comparison suggests that the coexistence of micelles and nanoparticles contributes to the dampening of the scattering fringes.
| Model fitting | c-PER solutions | |||
|---|---|---|---|---|
| c-PERM10 | c-PERM20 | c-PERD10 | c-PERD20 | |
| Core–shell sphere | ||||
| r c/nm | 20.55 ± 0.05 | 20.41 ± 0.05 | 20.61 ± 0.03 | 20.63 ± 0.03 |
| PDIs | 0.090 | 0.100 | 0.045 | 0.052 |
| r s/nm | 10.25 ± 0.11 | 10.19 ± 0.11 | 11.35 ± 0.06 | 11.44 ± 0.06 |
| ρ core (×10−6 Å−2) | 10.951 | 10.953 | 10.851 | 10.851 |
| ρ shell (×10−6 Å−2) | 8.208 | 8.208 | 8.112 | 8.114 |
| ρ solvent (×10−6 Å−2) | 8.057 | 8.058 | 8.057 | 8.057 |
The c-PER solutions were characterized by TEM in a dried state. Fig. 5 shows TEM images of the dried c-PER solutions. While the addition of both types of MABr solutions to the t-LHP solutions produced MAPbBr3 cubic crystals, the morphology and encapsulation behavior differed significantly. Using methanol as the solvent produced numerous monodispersed encapsulated MAPbBr3 nanocubes in the c-PERM10 and c-PERM20 solutions (Fig. 5B and C). Adding a 5 μL MABr/methanol aliquot mainly produced distorted nanocubes (Fig. 5A). In contrast, using DMF as the solvent resulted in little to no formation of MAPbBr3 nanocubes (Fig. 5E–G). This difference in crystal growth and encapsulation likely accounts for the observed variation in fluorescence intensity between the c-PER solutions (Fig. 3).
Note that not all PS-b-P2VP micelles encapsulated MAPbBr3 nanocubes (Fig. 5A–D). A large portion of PS-b-P2VP micelles contained no nanocubes, indicating that the nanocubes grew inside the P2VP cores of only a portion of the micelles. Moreover, the growth of encapsulated MAPbBr3 nanocubes was almost prohibited for the c-PERDx solutions. Little to no MAPbBr3 nanocrystals can be found neighboring with empty micelles (Fig. 5E–H).
Note that there is a significant contrast in the appearance of empty micelles between c-PERMx and c-PERDx in Fig. 5. This discrepancy should be due to different contents of Pb and Br elements trapped within P2VP cores. To address this issue, we further performed HR-TEM and EDS characterization on the nanostructures formed in the c-PERM10 and c-PERD20 solutions. Fig. 6B, C and E, F show representative EDS elemental analysis imposed on the c-PERM10 and c-PERD20 solutions in a dried state. The atomic compositions of N, Pb, and Br were detected at two different positions, labeled 1 and 2 in Fig. 6A and D.
EDS elemental analysis reveals three key findings (Fig. 6B, C and E and F). First, the MAPbBr3 nanocubes that formed in the c-PERM10 solution contained significant amounts of Pb and Br, although the Pb ratio did not precisely match the stoichiometric ratio of cubic MAPbBr3 crystals. This stoichiometric imbalance suggests the presence of defects in the cubic MAPbBr3 crystals. Second, the spherical micelles in the c-PERM10 solution appeared to contain a low amount of Pb. One selected spherical micelle displayed a high Br ratio (Fig. 6C), indicating that each MAPbBr3 nanocube in c-PERM10 likely grew at the expense of [PbBr3]− complexes released from multiple micelles. Third, the micelles in the dried c-PERD20 solution contained abundant Pb elements with an atomic ratio of approximately 1:3 (Fig. 6E and F). Analysis of the UV-vis absorbance spectrum (blue curve in Fig. 3B) and the WAXD profile (blue curve in Fig. 4A) indicates that the micelles in the c-PERD20 solution primarily retained [PbBr3]− complexes, with only a negligible amount of encapsulated MAPbBr3 nanocubes. The Pb:Br ratio determined by EDS elemental analysis aligns closely with the stoichiometry of [PbBr3]− complexes. Since the formation of MAPbBr3 nanocubes using DMF was significantly inhibited (Fig. 5G and 6D), it is likely that most of the [PbBr3]− complexes remained trapped within the spherical micelles (Fig. S4, ESI†).
Furthermore, a comparison of the EDS profiles indicates that the significant contrast in the appearance of empty micelles between c-PERMx and c-PERDx in Fig. 5 and 6A, D is attributed to the retention of [PbBr3]− complexes in the P2VP cores of the micelles in the c-PERDx solutions. In contrast, the perovskite-free micelles in the c-PERM10 solution contained negligible amounts of Pb and Br elements in their P2VP cores. As a result, the P2VP cores with negligible Pb and Br content appear as gray nanodomains (Fig. 5D and 6A), while those containing [PbBr3]− complexes appear as dark nanodomains (Fig. 5H and 6D) under high-magnification TEM imaging.
These findings raise two key questions: how do the different solvents influence the nucleation and growth of encapsulated MAPbBr3 nanocubes? What is the underlying mechanism driving the selective encapsulation of MAPbBr3 nanocubes within PS-b-P2VP micelles? Further investigation is required to elucidate the impact of solvent quality on MAPbBr3 crystallization and its relationship to micelle encapsulation efficiency.
Fig. 4–6 demonstrate that MAPbBr3 nucleation and growth differ in methanol and DMF. In methanol, MA+ cations effectively bind with P2VP-bound [PbBr3]− complexes, resulting in the formation of encapsulated MAPbBr3 nanocubes. In contrast, in DMF, MA+ cations do not effectively bind with [PbBr3]− complexes, likely due to preferential interactions with more accessible PbBr2 microparticles. To explore this hypothesis further, we characterized the precipitates separated from the t-PERMx and t-PERDx solutions. Fig. 7A and B show that the dried precipitates appear orange and exhibit green fluorescence. However, their XRD patterns differ significantly. The XRD patterns of the precipitates separated from the t-PERMx solutions show diffractions corresponding to a mixture of cubic MAPbBr3 perovskite (ICSD#268785) and orthorhombic PbBr2 (PDF#031-0679) (Fig. 7C). In contrast, the precipitates from the t-PERDx solutions exhibit WAXD patterns with diffractions corresponding solely to cubic MAPbBr3 crystals (Fig. 7D). Note that the additional peak at q = 1.86 Å−1 is attributed to the diffraction of the glass substrate.56 These results suggest that in methanol, MA+ cations easily migrate to the P2VP core to bind with encapsulated [PbBr3]− complexes through the PS shell, producing encapsulated MAPbBr3 nanocubes via crystallization. Excess MA+ cations are further captured by PbBr2 microcrystals, leading to the formation of MAPbBr3 microparticles. However, the formation of MAPbBr3 microparticles competes with the formation of MAPbBr3 nanocubes encapsulated within P2VP cores. This competition explains why the precipitates from the t-PERMx solutions show diffractions corresponding to a mixture of MAPbBr3 and PbBr2. In contrast, in DMF, most MA+ cations preferentially bind with free PbBr2 microparticles, leading to the formation of MAPbBr3 microparticles rather than MAPbBr3 nanocubes. The precipitates primarily consisted of these MAPbBr3 microparticles. As a result, the precipitates obtained from the t-PERDx solutions exhibited visible green photoluminescence under UV light (Fig. 7Biii–iv), while the supernatants separated from the t-PERDx solutions emitted PL with low quantum yields (Fig. 3B and C). The low quantum yields displayed by the c-PERDx solutions are attributed to the low density of encapsulated MAPbBr3 nanocubes (Fig. 5E–H).
Effect of centrifugation to remove PbBr2 microparticles
The first section demonstrates that the hierarchical emulsion of PbBr2 in TMB with PS-b-P2VP forms dispersed PbBr2 microparticles and [PbBr3]− complexes. During the micro-scale emulsion process, the PbBr2 microparticles are coated with PS-b-P2VP micelles, where the P2VP cores adhere to the PbBr2 microparticles, and the PS shells swell in TMB. The [PbBr3]− complexes are encapsulated within the P2VP cores through nanoscale emulsion. These encapsulated [PbBr3]− complexes are well-dispersed in TMB due to the strong repulsive forces provided by the swollen PS shells. As a result, the hierarchical emulsion of PbBr2 in TMB with PS-b-P2VP offers two key resources—dispersed PbBr2 microparticles and [PbBr3]− complexes—for the formation of MAPbBr3. To eliminate the influence of dispersed PbBr2 microparticles on the formation of encapsulated MAPbBr3 nanocubes, centrifugation was performed on the t-LHP solutions before adding MABr aliquots.To saturate the loading of [PbBr3]− complexes within the P2VP cores, lead halide precursor solutions (denoted as t-LHP7 solutions) were prepared by mixing PbBr2 with PS-b-P2VP in TMB, followed by prolonged stirring for 7 days. After 7 days, excess PbBr2 microparticles were removed by centrifugation at 7000 rpm for 10 minutes, resulting in clear supernatants (denoted as c-LHP7 solutions). This centrifugation step ensured that MA+ and Br− ions interacted exclusively with [PbBr3]− complexes, eliminating the influence of excess PbBr2 microparticles while preventing the disruption of micellar encapsulation efficiency by harsh centrifugation.
The c-LHP7 solutions were then added with 20 μL of MABr aliquots to produce clear perovskite solutions (denoted as c-PERc7,M20 or c-PERc7,D20). The c-PERc7,M20 solution appeared fluorescent green and emitted strong PL (Fig. S5A and B, ESI†), whereas the c-PERc7,D20 solution appeared light yellow and emitted weak PL (Fig. S5C and D, ESI†). The absorbance spectrum of the c-PERc7,D20 solution exhibited a strong absorbance band at ∼320 nm, indicating the presence of abundant [PbBr3]− complexes (red curve in Fig. S5E, ESI†). The c-PERc7,M20 solution showed a prominent band at ∼310 nm, followed by a Urbach tail in the range of 340–500 nm and an absorbance edge at 528 nm. A review article has demonstrated that the Urbach tail is associated with defects in semiconductors.57 Therefore, the Urbach tail and the band at 528 nm indicate the formation of perovskite crystals with defects in the c-PERc7,M20 solution. The 310-nm band is more pronounced in the c-PERc7,M20 solution (black line in Fig. S5E, ESI†) compared to the c-PERM20 solution (blue line in Fig. 3A). Previous studies have shown that the 310-nm band arises from [PbBr6]4− octahedral clusters.58–64 We believe that adding MABr to the c-LHP7 solution after the removal of PbBr2 microparticles may also lead to the formation of excess [PbBr6]4− octahedral clusters coexisting with MAPbBr3 nanocubes.
Fig. S6A and B (ESI†) shows that the c-PERc7,M20 solution forms encapsulated MAPbBr3 nanocubes with a slightly increased content. However, most of the PS-b-P2VP micelles remain a spherical shape without MAPbBr3 nanocubes. EDS analysis of three different regions enriched with spherical micelles demonstrates that the spherical micelles contain high Br residues but low Pb residues (Fig. S6C–E, ESI†), indicative of [PbBr6]4− complexes. While the optical properties of c-PERc7,M20 and c-PERM20 are comparable, the former exhibits reduced long-term stability. After three months of storage, the c-PERc7,M20 solution became transparent due to perovskite degradation (Fig. S7, ESI†).
Mechanistic insights on micellization, hierarchical emulsion, PbBr2 complexation, and formation of encapsulated MAPbBr3 nanocubes
It is generally understood that the formation of perovskite colloidal NCs occurs via ion exchange through a crystalline state.65 The mechanism of ion exchange allows us to explain how dispersed PbBr2 microparticles bind with MA+ cations to form MAPbBr3 microparticles. However, our findings indicate that amorphous [PbBr3]− complexes within the P2VP cores are the primary species to grow encapsulated perovskite nanocubes. This mechanism of the nucleation and growth of perovskite nanocubes in our system differs from the classical model of ion exchange.Based on the results in Fig. 1–7, we propose an alternative mechanism to explain how PS-b-P2VP colloidal micelles mediate PbBr2 complexation and MAPbBr3 crystallization in TMB when MABr aliquots, either in methanol or DMF, are added. In TMB, a good solvent for PS, the micellization of PS-b-P2VP produces core–shell micelles (Fig. 8Ai). Two types of emulsions occur at different scales: microscale and mesoscale (Fig. 8Aii). At the microscale, P2VP chains preferentially adsorb onto PbBr2 microparticles while PS chains extend into TMB. At the mesoscale, PbBr2 undergoes dissociation and complexation, followed by coordination within the P2VP cores. This process results in a hierarchical emulsion comprising a microemulsion of dispersed PbBr2 microparticles and a nanoemulsion of [PbBr3]− complexes within P2VP cores. It is important to note that the surface adsorption of PS-b-P2VP micelles is a dynamic and transient process. Frequent exchanges between complex-captured micelles and complex-free micelles occur at the interface of PbBr2 microcrystals and PS-b-P2VP micelles due to Brownian motion in the solution (see Fig. 8Aii where desorption and adsorption are depicted by red and blue arrows, respectively). This dynamic adsorption behavior contributes to a more uniform distribution of [PbBr3]− complexes throughout all P2VP cores (Fig. 8Aiii). Without BCP micelles, hierarchical emulsion could not form. Instead, adding a MABr/methanol aliquot led to the complete conversion of PbBr2 into MAPbBr3 microcrystals with poor dispersion in TMB (Fig. S8, ESI†).
 | ||
| Fig. 8 Schematic illustration of structural evolutions: (A) micellization, hierarchical emulsion, and inter-micelle fusion and fission in a t-LHP solution, and the nucleation and growth of MAPbBr3 in a t-PER solution added with (B) methanol-based or (C) DMF-based MABr aliquots. | ||
Next, we explain how encapsulated MAPbBr3 nanocubes form at the expense of [PbBr3]− complexes inside P2VP cores. Separately dissolving MABr in methanol is crucial for the formation of encapsulated MAPbBr3 nanocubes in non-polar solvents. This dissolution step generates solvated MA+ and Br− ions, which are necessary for the subsequent nucleation and growth of perovskite nanocubes confined within P2VP cores. Without this solvent-mediated ion formation, MABr would predominately bind with dispersed PbBr2 microparticles in TMB to form MAPbBr3 microparticles, thus preventing the formation of encapsulated MAPbBr3 nanocubes inside P2VP cores (Fig. S9, ESI†). As a result, the P2VP cores still retain a high content of [PbBr3]− complexes. When using methanol to dissolve MABr, solvated MA+ cations can diffuse from the non-polar phase into the P2VP cores filled with [PbBr3]− complexes. The coordination between solvated MA+ cations and [PbBr3]− complexes initiates the crystallization of MAPbBr3 (Fig. 8Bi). This mechanism facilitates the formation of encapsulated MAPbBr3 nanocubes.
The confinement of MAPbBr3 crystallization within P2VP cores imparts a space for homogeneous nucleation. This nucleation and growth promote the formation of stable cubic crystals (Fig. 8Bii). The stability of perovskite structures can be predicted using the Goldschmidt tolerance factor (t) and the octahedral factor (μ), which are determined by the ionic radii of the constituent atoms.66 The values of t and μ are calculated using eqn (1) and (2), respectively:
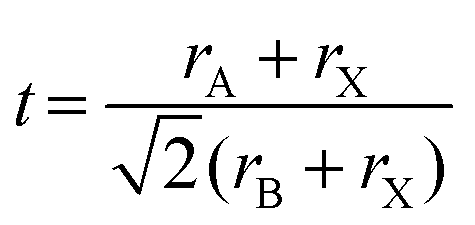 | (1) |
 | (2) |
Interestingly, the observation that nanocubes are encapsulated within some individual micelles, while others remain empty, suggests that inter-micelle fusion and fission events occur during MAPbBr3 crystallization (Fig. 8Biii). This dynamic process could explain the observed distribution of nanocubes, where some micelles encapsulate multiple nanocubes, while others do not contain any [PbBr3]− complexes. The uniform capture of [PbBr3]− complexes by micelles in the t-LHP solution further supports this mechanism.
The choice of solvent significantly influences the crystallization kinetics of MAPbBr3. Using methanol yields MAPbBr3 nanocrystals with narrow size dispersity, well dispersed in the supernatants. In contrast, using DMF quickly results in MAPbBr3 microcrystals (Fig. 8Ci), which are found only in the precipitates. Only a few MAPbBr3 nanocubes can form within P2VP cores when DMF is used to solvate MA+ and Br− ions (Fig. 8Cii). This difference arises from the solvents' impact on MA+ ion diffusion through the PS shells. Methanol facilitates MA+ diffusion through the PS shells, promoting coordination with [PbBr3]− complexes within the P2VP cores, allowing the encapsulated nanocubes to be well dispersed in the supernatants. Conversely, DMF-solvated MA+ cations have difficulty penetrating and diffusing through the PS shells, thus limiting their access to the [PbBr3]− complexes. Instead, DMF-solvated MA+ cations preferentially bind to PbBr2 microcrystals, leading to the rapid growth of MAPbBr3 microcrystals, which then exist as precipitates after centrifugation (Fig. 7).
Several factors can explain the observed differences in crystallization kinetics. The first factor is the distinct precursor-solvent interactions, quantified by Guttman's donor number (DN).69 A significant difference in DN values of methanol (DN = 19)70 and DMF (DN = 27)70 reflects their electron-donating abilities. Previous work by Hamill et al.71 demonstrated that DN serves as a strong predictor of a solvent's ability to solvate perovskite precursors. Solvents with higher DN values can more effectively compete with halide ions for coordination sites around the Pb2+ center. This competition might suppress the formation of ion complexes, potentially retarding perovskite nucleation and growth due to a reduced concentration of precursor ion complexes. In our case, MA+ cations strongly coordinate with DMF to form solvated species. While DMF's higher DN could explain its stronger coordination with MA+ ions compared to methanol, the high DN value does not fully account for the preferential formation of MAPbBr3 microcrystals in t-PERDx solutions. The interaction between MA+ cations and PbBr2 microcrystals, which governs microcrystal formation, should not be directly affected by DN. Therefore, we can exclude the difference in Guttman's donor number as the principal cause for the distinctive crystallization pathways observed in the two solvent systems.
Here, it is important to consider that the swollen PS shells act as barriers to the diffusion of solvated MA+ cations from the liquid matrix into the P2VP cores. As a result, the diffusion rate of solvated MA+ cations is significantly faster in the liquid matrix than within the PS shells. The diffusion rate of solvated MA+ cations dispersed in a solvent can be quantified by:
 | (3) |
In solution, methanol-solvated MA+ cations can more quickly promote the formation of MAPbBr3 microcrystals at the expense of PbBr2 microcrystals adsorbed by PS-b-P2VP micelles, compared to DMF-solvated MA+ cations. Microscale emulsion disperses the PbBr2 microcrystals, which are partially adsorbed by PS-b-P2VP micelles. However, there should be sufficient voids that are free from PS-b-P2VP micelle adsorption. These BCP-free voids allow the solvated MA+ cations to bind with PbBr2 microcrystals, facilitating the rapid formation of MAPbBr3 microcrystals. This observation explains why the precipitates are predominantly composed of cubic MAPbBr3 microcrystals.
For the formation of encapsulated MAPbBr3 nanocubes within P2VP cores through nanoscale emulsion, the additional barriers imparted by PS shells must be carefully considered. The penetration and diffusion of solvated MA+ cations are analogous to the permeation of gas through a polymer.73 Two factors affect permeability: the solubility and diffusivity of the permeating species through the polymer along the concentration gradient. In our system, solubility should not be a key factor as the contrast in solubility parameters between methanol (29.6 MPa1/2)49 and TMB (18 MPa1/2)49 is greater than the contrast in solubility parameters between DMF (24.8 MPa1/2)49 and TMB. Therefore, we believe that diffusivity plays a more critical role in the permeation of solvated MA+ cations through PS shells (18.6 MPa1/2).50 Diffusivity is influenced by polymer packing and the kinetic diameter of the diffusing molecule itself.
Quantitative analysis of the SAXS data demonstrates that the addition of DMF or methanol has no significant effect on the radii of the P2VP core and PS shell, indicating that the spatial packing of the shell-forming corona remains unchanged. In other words, the polymer packing of PS shells swollen in TMB-rich solutions is consistent, regardless of whether DMF or methanol is added.
In this study, we hypothesize that the inhibition of encapsulated MAPbBr3 nanocube formation is linked to the possibility that DMF-solvated MA+ cations have a larger kinetic diameter. As a result, the penetration of PS shells with a brush conformation by DMF-solvated MA+ cations may cause a significant entropic penalty (Fig. 8Ci), which is unfavorable for the diffusion of DMF-solvated MA+ cations through the PS shell. This could explain why [PbBr3]− complexes remained within the P2VP cores without forming MAPbBr3 nanocubes when DMF was used to produce solvated MA+ and Br− ions.
Next, we explain why the c-PERc7,M20 solution exhibits lower PL stability compared to the c-PERM20 solution. PbBr2 microcrystals likely serve as a reservoir, continuously replenishing [PbBr3]− complexes. Without PbBr2 microcrystals, Br− ions are left unbound, leading to a significant reduction in [PbBr3]− complexes. This is supported by the distinct optical properties in freshly prepared c-PERc7,M20 solutions compared to c-PERM20 (see Fig. S5 (ESI†) and Fig. 3). Unlike the broad shoulder displayed by the c-PERM20 solution, a sharp peak at ∼308 nm suggests the presence of abundant [PbBr6]4− octahedral clusters, likely due to excess Br− ions in the c-PERc7,M20 solution. A high residue of [PbBr6]4− octahedral clusters may lead to the degradation of MAPbBr3 nanocubes (see Fig. S7, ESI†). This explains why the PL performance of the c-PERc7,M20 solution degraded after three months of storage at room temperature.
Conclusions
We have demonstrated a straightforward method for synthesizing MAPbBr3 nanocubes encapsulated within polymeric micelles. This approach capitalizes on the interaction between [PbBr3]− complexes and solvated MA+ cations within a confined micellar environment. The choice of solvent to solvate MA+ cations significantly influences the crystallization kinetics and the quality of the nanocubes. Methanol, with its smaller molecular size, facilitates faster diffusion of solvated MA+ ions, promoting the formation of well-defined nanocubes. In contrast, DMF leads to slower diffusion of solvated MA+ ions and the formation of less desirable microcrystals. The MAPbBr3 colloidal solutions synthesized using methanol-based aliquots exhibit remarkable stability. After 90 days of storage under ambient conditions, no decrease in PLQY was observed. In fact, the PLQY measurements indicated a 14% increase compared to the freshly prepared solutions.Furthermore, our findings highlight the crucial role of excess PbBr2 microcrystals in synthesizing encapsulated nanocubes and maintaining their long-term stability. Although removing these microcrystals through centrifugation initially produces solutions with comparable optical properties, it disrupts the stoichiometric balance of MA, Pb, and Br elements necessary for forming MAPbBr3 crystals. This stoichiometric imbalance ultimately undermines the stability of the nanocubes, leading to degradation over time. These insights underscore the importance of carefully controlling crystallization conditions and understanding the underlying stoichiometric equilibria to achieve stable, high-quality perovskite nanomaterials for optoelectronic applications.
Author contributions
Belda Amelia Junisu: conceptualization, writing – original draft, data curation, methodology, investigation, visualization; Ya-Sen, Sun: conceptualization, writing – review and editing, funding acquisition, resources, supervision; Bo Cheng Zhao: investigationData availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science and Technology Council (grant numbers: NSTC 111-2221-E-006-232-MY3 and 112-2811-E-006-042-MY2) is acknowledged. We further thanks Dr Orion Shih, Dr Yi-Qi Yeh, Mr Je-Wei Chang, and Ms Kuei-Fen Liao for SAXS and WAXD experiments at TPS 13A. We also express our gratitude for the use of EM025800 (NSTC 113-2740-M-006-002) at the Core Facility Center of National Cheng Kung University (NCKU). Additionally, we thank Mr Yu-Hsuan Cheng for performing the TEM and EDS measurements at NCKU.References
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- S. D. Adhikari, A. F. G. Reyes, S. Paul, J. Torres, B. Escuder, I. Mora-Seró and S. Masi, Chem. Sci., 2023, 14, 8984–8999 RSC.
- D. Park, W. Kim, C. Park, J. Choi, A. Ghorai, G. Lee, S. Choi, W. Moon and U. Jeong, Small, 2024, 20, 2402281 CrossRef CAS.
- F. Wang, X. Zou, M. Xu, H. Wang, H. Wang, H. Guo, J. Guo, P. Wang, M. Peng, Z. Wang, Y. Wang, J. Miao, F. Chen, J. Wang, X. Chen, A. Pan, C. Shan, L. Liao and Q. Hu, Adv. Sci., 2021, 8, 2100569 CrossRef CAS.
- Y. Liu, Y. Liu and Y. Guo, Mater. Chem. Front., 2023, 7, 5215–5246 RSC.
- S. Luo and W. A. Daoud, J. Mater. Chem. A, 2015, 3, 8992–9010 RSC.
- B. W. Boote, H. P. Andaraarachchi, B. A. Rosales, R. Blome-Fernández, F. Zhu, M. D. Reichert, K. Santra, J. Li, J. W. Petrich, J. Vela and E. A. Smith, Chem. Phys. Chem., 2019, 20, 2647–2656 CrossRef CAS.
- D. Han, K. Yang, C. Bai, F. Chen, Z. Sun, Y. Wang, H. Ji, Z. Yang and X. Tang, J. Chem. Eng., 2023, 475, 146209 CrossRef CAS.
- D. Liu, Z. Shao, C. Li, S. Pang, Y. Yan and G. Cui, Small Struct., 2021, 2, 2000089 CrossRef CAS.
- A. Ghorai, A. Das, Y. Choi, B. Roy and U. Jeong, Small, 2024, 20, 2404384 CrossRef CAS.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- W. K. Chan, J. Chen, D. Zhou, J. Ye, R. J. Vázquez, C. Zhou, G. C. Bazan, A. Rao, Z. Yu and T. T. Y. Tan, Nanomaterials, 2023, 13, 815 CrossRef CAS PubMed.
- J. You, Y. M. Yang, Z. Hong, T.-B. Song, L. Meng, Y. Liu, C. Jiang, H. Zhou, W.-H. Chang, G. Li and Y. Yang, Appl. Phys. Lett., 2014, 105, 183902 CrossRef.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- M. L. Petrus, J. Schlipf, C. Li, T. P. Gujar, N. Giesbrecht, P. Müller-Buschbaum, M. Thelakkat, T. Bein, S. Hüttner and P. Docampo, Adv. Energy Mater., 2017, 7, 1700264 CrossRef.
- W. Zhao, D. Yang and S. F. Liu, Small, 2017, 13, 1604153 CrossRef.
- X. Sheng, G. Chen, C. Wang, W. Wang, J. Hui, Q. Zhang, K. Yu, W. Wei, M. Yi, M. Zhang, Y. Deng, P. Wang, X. Xu, Z. Dai, J. Bao and X. Wang, Adv. Funct. Mater., 2018, 28, 1800283 CrossRef.
- J. A. Laranjeira, S. A. Azevedo, G. S. Fabris, J. R. Sambrano and M. M. Ferrer, Comput. Mater. Sci., 2024, 239, 112977 CrossRef CAS.
- H. Li, L. Xiong, J. Li, Y. Lu, Z. Shen, D. Song, S. Zhao, Z. Xu, Z. Liang and B. Qiao, J. Phys. Chem. Lett., 2023, 14, 5481–5488 CrossRef CAS PubMed.
- S. Wang, A. A. Yousefi Amin, L. Wu, M. Cao, Q. Zhang and T. Ameri, Small Struct., 2021, 2, 2000124 CrossRef CAS.
- M. Meyns, M. Perálvarez, A. Heuer-Jungemann, W. Hertog, M. Ibáñez, R. Nafria, A. Genç, J. Arbiol, M. V. Kovalenko, J. Carreras, A. Cabot and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2016, 8, 19579–19586 CrossRef CAS.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z. H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef PubMed.
- H. Kim, N. Hight-Huf, J. H. Kang, P. Bisnoff, S. Sundararajan, T. Thompson, M. Barnes, R. C. Hayward and T. Emrick, Angew. Chem., Int. Ed., 2020, 59, 10802–10806 ( Angew. Chem. , 2020 , 132 , 10894–10898 ) CrossRef CAS PubMed.
- Y. He, Y. J. Yoon, Y. W. Harn, G. V. Biesold-McGee, S. Liang, C. H. Lin, V. V. Tsukruk, N. Thadhani, Z. Kang and Z. Lin, Sci. Adv., 2019, 5, eaax4424 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- Y. Zou, X. Zhou, J. Ma, X. Yang and Y. Deng, Chem. Soc. Rev., 2020, 49, 1173–1208 RSC.
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- L. Hui, C. Beswick, A. Getachew, H. Heilbrunner, K. Liang, G. Hanta, R. Arbi, M. Munir, H. Dawood, N. Isik Goktas, R. LaPierre, M. C. Scharber, N. S. Sariciftci and A. Turak, ACS Appl. Nano Mater., 2019, 2, 4121–4132 CrossRef CAS.
- S. Hou, Y. Guo, Y. Tang and Q. Quan, ACS. Appl. Mater. Interface, 2017, 9, 18417–18422 CrossRef CAS.
- K. Lee, J. Y. Kim and B.-H. Sohn, RSC Adv., 2022, 12, 6389–6395 RSC.
- Y. Nah, D. Jang and D. H. Kim, Chem. Commun., 2021, 57, 1879–1882 RSC.
- M. G. Greiner, A. Singldinger, N. A. Henke, C. Lampe, U. Leo, M. Gramlich and A. S. Urban, Nano Lett., 2022, 22, 6709–6715 CrossRef CAS.
- V. A. Hintermayr, C. Lampe, M. Löw, J. Roemer, W. Vanderlinden, M. Gramlich, A. X. Böhm, C. Sattler, B. Nickel, T. Lohmüller and A. S. Urban, Nano Lett., 2019, 19, 4928–4933 CrossRef CAS PubMed.
- Q. Jiang, Y. Ren, Z. Cui, Z. Li, L. Hu, R. Guo, S. Duan, F. Xie, G. Zhou and S. Xiong, ACS Appl. Nano Mater., 2023, 6, 8655–8667 CrossRef CAS.
- C. Collantes, W. Teixeira, V. G. Pedro, M.-J. Bañuls and Á. Maquieira, Appl. Mater. Today, 2023, 31, 101775 CrossRef.
- H. Zamani, T.-H. Chiang, K. R. Klotz, A. J. Hsu and M. M. Maye, Langmuir, 2022, 38, 9363–9371 CrossRef CAS PubMed.
- S. K. Balakrishnan and P. V. Kamat, Chem. Mater., 2018, 30, 74–78 CrossRef CAS.
- M. Wang, Q. Yu, T. Yu, S. Zhang, M. Gong and Y. Liu, RSC Adv., 2023, 13, 5158–5167 RSC.
- K. G. Stamplecoskie, J. S. Manser and P. V. Kamat, Energy Environ. Sci., 2015, 8, 208–215 RSC.
- H. Han, J. W. Oh, H. Lee, S. Lee, S. Mun, S. Jeon, D. Kim, J. Jang, W. Jiang, T. Kim, B. Jeong, J. Kim, D. Y. Ryu and C. Park, Adv. Mater., 2024, 36, 2310130 CrossRef CAS.
- N. Pradhan, ACS Phys. Chem. Au., 2022, 2, 268–276 CrossRef CAS PubMed.
- P. C. Sadek, The HPLC solvent guide, Wiley-Interscience, 2002, ISBN: 978-0 Search PubMed.
- O. Shih, K.-F. Liao, Y.-Q. Yeh, C.-J. Su, C.-A. Wang, J.-W. Chang, W.-R. Wu, C.-C. Liang, C.-Y. Lin, T.-H. Lee, C.-H. Chang, L.-C. Chiang, C.-F. Chang, D.-G. Liu, M.-H. Lee, C.-Y. Liu, T.-W. Hsu, B. Mansel, M.-C. Ho, C.-Y. Shu, F. Lee, E. Yen, T.-C. Lind and U. Jenga, J. Appl. Cryst., 2022, 55, 340–352 CrossRef CAS PubMed.
- M. Doucet, J. H. Cho, G. Alina, Z. Attala, J. Bakker, P. Beaucage, W. Bouwman, R. Bourne, P. Butler, K. Campbell, T. Cooper-Benun, C. Durniak, L. Förster, P. Gilbert, M. Gonzalez, R. Heenan, A. Jackson, S. King, P. Kienzle, J. Krzywon, T. Nielsen, L. O’Driscoll, W. Potrzebowski, S. Prescott, R. Ferraz Leal, P. Rozyczko, T. Snow, A. Washington and C. Wolf, SasView (version 5.0.3), 2020 Search PubMed.
- G. Beaucage, J. Appl. Crystall., 1995, 28, 717–728 CrossRef CAS.
- S. Förster, A. Timmann, M. Konrad, C. Schellbach, A. Meyer, S. Funari, P. Mulvaney and R. Knott, J. Phys. Chem. B, 2005, 109, 1347–1360 CrossRef PubMed.
- I. Akiba and K. Sakurai, J. Polym., 2021, 53, 951–973 CAS.
- C. M. Hansen, Hansen solubility parameters: a user's handbook, CRC press, 2007 Search PubMed.
- A. F. M. Barton, Chem. Rev., 1975, 75, 731–753 CrossRef CAS.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- J.-Y. Liou and Y.-S. Sun, Macromolecules, 2012, 45, 1963–1971 CrossRef CAS.
- C. M. Septani, M.-F. Ku, C.-Y. Chen, J.-M. Lin and Y.-S. Sun, Colloids Surf., A, 2022, 652, 129857 CrossRef CAS.
- S. J. Yoon, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2016, 7, 1368–1373 CrossRef CAS PubMed.
- R.-J. Roe, Methods of X-ray and neutron scattering in polymer science, Oxford University Press, 2000, pp. 140–145, pp 160–162 Search PubMed.
- P. Ponce-Peña, M. A. González-Lozano, M. Á. Escobedo-Bretado, D. M. Núñez-Ramírez, A. Rodríguez-Pulido, Z. V. Quiñones Jurado, M. Poisot and B. Sulbarán-Rangel, Crystals, 2022, 12, 574 CrossRef.
- L. Wang, H. Liu, Y. Zhang and O. F. Mohammed, ACS Energy Lett., 2020, 5, 87–99 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS PubMed.
- M. Nikl, E. Mihokova, K. Nitsch, F. Somma, C. Giampaolo, G. Pazzi, P. Fabeni and S. Zazubovich, Chem. Phys. Lett., 1999, 306, 280–284 CrossRef CAS.
- Y. Zhang, M. I. Saidaminov, I. Dursun, H. Yang, B. Murali, E. Alarousu, E. Yengel, B. A. Alshankiti, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 961–965 CrossRef CAS.
- S. K. Sharma, C. Phadnis, T. K. Das, A. Kumar, B. Kavaipatti, A. Chowdhury and A. Yella, Chem. Mater., 2019, 31, 3111–3117 CrossRef CAS.
- S. Seth and A. Samanta, J. Phys. Chem. Lett., 2017, 8, 4461–4467 CrossRef CAS PubMed.
- D. Chen, Z. Wan, X. Chen, Y. Yuan and J. Zhong, J. Mater. Chem. C, 2016, 4, 10646–10653 RSC.
- H. Jiang, S. Cui, Y. Chen and H. Zhong, Nano Select, 2021, 2, 2040–2060 CrossRef CAS.
- E. Akman, T. Ozturk, W. Xiang, F. Sadegh, D. Prochowicz, M. M. Tavakoli, P. Yadav, M. Yilmaz and S. Akin, Energy Environ. Sci., 2023, 16, 372–403 RSC.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- T. J. Jacobsson, J.-P. Correa-Baena, M. Pazoki, M. Saliba, K. Schenk, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1706–1724 RSC.
- V. Gutmann and V. Gutmann, The donor-acceptor approach to molecular interactions, Springer, 1978 Search PubMed.
- F. Cataldo, Eur. Chem. Bull., 2015, 4, 92–97 Search PubMed.
- J. C. Hamill Jr, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2018, 3, 92–97 CrossRef.
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368–7373 CrossRef CAS.
- L. H. Sperling, Introduction to Physical Polymer Science, John Wiley & Sons, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM image, EDS 2D maps, and EDS 1D profiles of the dried state of t-LHP7 solution (Fig. S1); photos, UV-vis, and PL spectra of c-PERM20 solutions at various centrifugation speeds (Fig. S2); photos and PLQY of the c-PERM solutions after being stored in an ambient environment for 3 months (Fig. S3); TEM image and EDS 1D profiles of the c-PERD20 solution in dried state (Fig. S4); UV-vis absorbance and PL spectra of fresh c-PERc7,M20 and c-PERc7,D20 solutions (Fig. S5); HR-TEM image and EDS 1D profiles of the fresh c-PERc7,M20 solution in dried state (Fig. S6); UV-vis absorbance and TEM image of c-PERc7,M20 solution with prolonged storage (Fig. S7); photos of solutions during the synthesis of perovskite in the absence of PS-b-P2VP (Fig. S8); characterization of solutions and its precipitates during the synthesis of perovskite without MABr dissolution (Fig. S9). See DOI: https://doi.org/10.1039/d4tc03885a |
| This journal is © The Royal Society of Chemistry 2025 |