
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrolytic degradation of PEG-based thiol–norbornene hydrogels enables multi-modal controlled release
Nathan H.
Dimmitt
and
Chien-Chi
Lin
 *
*
Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN, USA. E-mail: lin711@purdue.edu; Tel: +1(965) 495-7791
First published on 22nd September 2025
Abstract
Poly(ethylene glycol) (PEG) hydrogels crosslinked by orthogonal thiol–norbornene click chemistry have emerged as an ideal platform for tissue engineering and drug delivery applications due to their rapid crosslinking kinetics and excellent biocompatibility. Norbornene-functionalized PEG (PEGNB) is routinely synthesized through the Steglich esterification of 5-norbornene-2-carboxylic acid with hydroxyl-terminated PEG. When crosslinked with thiol-bearing macromers, PEGNB hydrogels undergo slow hydrolytic degradation due to hydrolysis of ester bonds connecting a PEG backbone and a NB moiety. In prior work, we replaced the pungent and nauseous 5-norbornene-2-carboxylic acid with odorless carbic anhydride (CA) for synthesizing PEG–norbornene-carboxylate (PEGNBCA), a new macromer that could be readily photo-crosslinked into thiol–norbornene hydrogels with faster hydrolytic degradation than the PEGNB counterparts. In this contribution, we employed a modular approach to tune the hydrolytic degradation of PEGNBCA hydrogels over days to months. We first demonstrated the diverse crosslinking of PEGNBCA hydrogels using either photopolymerization or enzymatic crosslinking. We characterized the hydrolytic degradation of these hydrogels under different solution pH values and temperatures. Via adjusting crosslinker functionality and the ratio of fast-degrading PEGNBCA to slow-degrading PEGNB, tunable hydrolytic degradation of PEGNBCA hydrogels was achieved from under 2 days to over 3 months. Finally, we designed the highly tunable PEGNBCA hydrogels with varying mesh sizes, degradation rates, and covalent tethering of degradable linkers to afford long-term controlled release of model drugs.
Introduction
Poly(ethylene glycol) (PEG)-based hydrogels are widely used in biomedical applications owing to their excellent biocompatibility and tunable tissue-like elasticity.1 One of the PEG-based hydrogels widely adapted for biomedical applications is chain-polymerized PEG-diacrylate (PEGDA).1,2 Despite its lengthy synthesis steps that require meticulous control of all reaction conditions, especially the slow addition of acryloyl chloride, PEGDA has found numerous biomedical applications as it is adaptable for various chemical modifications. However, chain-growth polymerizations yield heterogeneous polymer networks and prolonged radical propagation that can damage the encapsulated biologics.1 Furthermore, while the ester bond connecting PEG and acrylate is hydrolytically labile, the high-molecular-weight polyacrylate kinetic chains in the chain-polymerized networks are hydrophobic and non-degradable, rendering the PEGDA hydrogels with extremely slow to no in vivo clearance.1 Structurally, chain-polymerized PEGDA hydrogels are mesoporous, with a ‘mesh size’ of the order of a few tens of nanometers.3 As such, the mobility of encapsulated macromolecules (e.g., large proteins and cells) is restricted unless the hydrogels are designed to undergo local or global degradation. In this regard, Hubbell et al. pioneered hydrolytically degradable PEG-based hydrogels via copolymerizing poly(lactic acid) (PLA) with PEG, followed by acrylation of the linear PLA–PEG–PLA copolymers.4 The resulting acryl–PLA–PEG–PLA–acryl macromers were crosslinked into covalent hydrogels with a tunable degradation rate by adjusting the molecular weight of the PLA blocks.5PEG-based hydrogels can also be crosslinked through step-growth polymerizations, such as the Michael addition reactions between multi-arm PEG derivatives (e.g., (meth)acrylate, vinylsulfone, and maleimide) and thiol-bearing crosslinkers.2,6–10 For example, the Hubbell group engineered PEG-based hydrogels by crosslinking multi-arm PEG–acrylate with bis-cysteine-containing peptide linkers labile to matrix metalloproteinases (MMPs).6,8 These hydrogels demonstrated high biocompatibility and enzymatic degradability and were used as carriers to release a recombinant human bone morphogenic protein (rhBMP2) for treating calvarial defects in rats. Metters and Hubbell demonstrated the predictable hydrolytic degradation of thioether–ester bonds in the Michael-type thiol–acrylate hydrogels,5 enabling sustained release of human growth hormone (hGH).7 The crosslinking of thiol–acrylate hydrogels was commonly accelerated by using base catalyst triethanolamine (TEOA)7 or initiated by light and a photoinitiator.11,12 While the network properties of Michael-type hydrogels can be pre-engineered, their crosslinking was not tunable after mixing the mutually reactive macromers. In contrast, radical-mediated step-growth photopolymerization offers unique advantages in creating a well-defined hydrogel network with mild and on-demand tunable gelation.13 For example, the Anseth group synthesized multi-arm PEG-norbornene (PEGNB) for forming thiol–norbornene photopolymerized hydrogels as a versatile synthetic extracellular matrix.14 Our group later characterized and modeled the hydrolytic degradability of PEGNB hydrogels under physiological conditions.15 We also engineered PEGNB hydrogels resistant to hydrolysis by replacing the ester group on 4-arm PEGNB with an amide bond.16 The Kloxin group explored PEGNB hydrogels for protein release, but no attempt was made to control hydrogel degradation.17
While PEGNB has become increasingly used in hydrogel crosslinking, its organic synthesis was laborious and tedious and required stringent containment efforts. Conventionally, PEGNB was synthesized by Steglich esterification between the hydroxyl moiety on the PEG macromer and the acid group on the pungent 5-norbornene-2-carboxylic acid using anhydrous dichloromethane (DCM) as the solvent.14 In this reaction, dicyclohexylcarbodiimide (DCC) is needed for activating 5-norbornene-2-carboxylic acid into norbornene anhydride, whereas dimethylaminopyridine (DMAP) is used as a nucleophilic catalyst for the esterification with norbornene anhydride. Efforts to improve the degree of norbornene substitution on PEG include: (1) azeotropic distillation of bond water by reflux in anhydrous toluene, followed by rotary evaporation to remove toluene; (2) blanking the reaction vessel with inert gas; (3) multiple additions of reactants; and (4) small batch reactions (<5 g).18 Notably, hydrolytic degradation of PEGNB-crosslinked thiol-norbornene hydrogels was achieved without conjugating an additional hydrolytically labile linker, owing to the hydrolytic susceptibility of ester on PEGNB.15
To improve the efficiency of PEGNB synthesis, our lab developed an alternative synthesis route that replaced the pungent 5-norbornene-2-carboxylic acid with odorless carbic anhydride (CA) and used DMAP as the only catalyst.19 Compared with conventional synthesis, the new CA method produced a similar norbornene substitution but with reduced synthesis time (2 days) as no PEG distillation and norbornene acid activation steps were required. This new macromer, PEGNB-carboxylate (PEGNBCA), can be readily crosslinked into hydrogels via a thiol–norbornene photo-click reaction19,20 or a norbornene–tetrazine inverse electron-demand Diels–Alder (iEDDA) click reaction.21,22 Like PEGNB, the new PEGNBCA could also be crosslinkable by an enzyme-initiated thiol–norbornene click reaction.23 The use of enzymes to initiate hydrogel crosslinking may be more advantageous for some biomedical applications due to the specificity and tunability of the catalytic reaction.24,25 However, enzyme-initiated PEGNBCA hydrogel crosslinking has not been demonstrated. Nonetheless, we found that hydrogels crosslinked by PEGNBCA underwent accelerated hydrolytic degradation compared to those crosslinked by PEGNB. Specifically, the hydrolysis rate was approximately 5 times higher in PEGNBCA gels than in PEGNB gels with similar crosslinking density.19
We reason that the accelerated hydrolytic degradation of PEGNBCA hydrogels could be leveraged to produce hydrogels with tunable degradation for controlled release applications. In this contribution, we investigated the gelation kinetics of ester-based PEGNB and PEGNBCA hydrogels with different thiol crosslinkers. From there, we characterized the subsequent hydrogel properties, including degradation over time under different conditions (e.g., pH of the buffer and incubation temperature). The hydrolytic degradation rate was further controlled via modular blending of PEGNB and PEGNBCA crosslinked by different thiol crosslinkers. Finally, we leveraged the higher degradability of the ester bond on PEGNBCA for multi-modal release, including crosslinking gels with varying mesh size and degradation rate, and tethering of thiol-bearing drugs onto PEGNBCA for their degradation-controlled release.
Materials and methods
Materials
8-Arm hydroxyl-terminal PEG (PEG-OH, 10 kDa and 20 kDa) and 8-arm thiol-terminal PEG (PEG8SH, 10 kDa) were purchased from JenKem Technology. 4-Arm thiol-terminal PEG (PEG4SH, 10 kDa) was purchased from Laysan Bio Inc. Dithiothreitol (DTT) was purchased from Fisher Scientific. Pyridine, anhydrous dichloromethane (DCM), and 1-(3-dimethylaminopropyl)3-ethylcarbodiimide hydrochloride (EDC) were obtained from Thermo Scientific. 5-Norbornene-2-carboxylic acid, tetrahydrofuran (THF), 4-dimethylaminopyridine (DMAP), N,N′-dicyclohexylcarbodiimide (DCC), and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) were purchased from Sigma-Aldrich. Carbic anhydride was purchased from Acros Organics. Phosphate buffered saline tablets were purchased from Fisher Scientific and prepared according to the manufacturer's recommendation. Fluorescein PEG thiol (MW 3400) was purchased from NANOCS. Bovine serum albumin was purchased from Fisher (Cytiva HyClone™ BSA). The Bradford reagent was purchased from Bio-Rad (Quick Start™ Bradford Protein Assay).Macromer synthesis and purification
Photocrosslinking of PEGNB or PEGNBCAvia thiol–norbornene chemistry
PEGNB or PEGNBCA was crosslinked with DTT, PEG4SH, or PEG8SH at a stoichiometric ratio (i.e., R([SH]/[NB] = 1)) using 2 mM LAP and 365 nm light at 20 mW cm−2 intensity unless specified otherwise. A stoichiometric ratio was chosen to ensure the highest degree of step-growth crosslinking, since previous studies have shown that the off-stoichiometric ratio of thiol to norbornene led to dangling polymer chains that did not contribute to network crosslinking.15,16,18 To prepare hydrogels, 45 μL of hydrogel precursor solution was deposited between two glass slides treated with hydrophobic windshield coating (i.e., Rainaway) separated by 1 mm Teflon spacers. The hydrogel precursor solution contained within the slide was placed under a 365 nm light bar lamp for 2 minutes. Prior to measuring elastic and loss moduli (G′ and G′′), the hydrogels were incubated in pH 7.4 PBS at 37 °C for at least one hour to ensure equilibrium swelling. Strain sweep (0.1 to 5.0% strain) tests were conducted using a Bohlin CVO digital rheometer fitted with an 8 mm diameter parallel geometry plate. The oscillating frequency was fixed at 1 Hz, which falls within the linear viscoelastic region (LVR). The gelation point was defined as the time required for the G′ to surpass the G′′ during in situ photorheometry testing, which was conducted using an Anton-Paar MCR102 rheometer equipped with a UV-curing system fitted with a 25 mm parallel geometry plate. During the test, 200 μL of hydrogel precursor solution was deposited on the rheometer stage, and the plate was lowered to 0.300 mm in height. Time sweep test was then started with a fixed strain at 1% and frequency at 1 Hz over a five-minute period. After 30 seconds, the light (Omnicure S2000, fitted with a 365 nm light filter at 5 mW cm−2) was turned on for two minutes.Enzymatic crosslinking of PEGNBCA using HRP with thiol-based crosslinkers
Enzymatic crosslinking of PEGNBCA was conducted using horse radish peroxidase (HRP) with or without hydrogen peroxide (H2O2), which mediates thiyl radical formation and then subsequently crosslinks to the vinyl bond on the norbornene functional group.23,26 Unless specified otherwise, enzymatically crosslinked PEGNBCA hydrogels were fabricated using 20 U mL−1 HRP and 100 μM of H2O2. For enzymatic crosslinking without H2O2, the concentration of HRP was increased to 1 kU mL−1. For in situ rheology, the Anton Paar MCR102 rheometer was fitted with a peltier platform and a 25 mm parallel plate head with temperature set at 37 °C. The hydrogel precursor solution was deposited on the stage followed by lowering the plate head to 0.300 mm in height. The time sweep test was then started with a fixed strain at 1% and frequency at 1 Hz over a 20-minute period with measurements being taken every 2 seconds. For hydrogel fabrication, the hydrogel precursor solution was placed between two glass slides treated with hydrophobic coating and separated by 1 mm Teflon spacers. The slides containing the hydrogel precursor solution were placed in a sealed container along with a Petri dish of water to increase the humidity to prevent drying. The sealed container was placed in an incubator set at 37 °C and allowed to react for 1 hour. Afterwards, hydrogels were swelled in pH 7.4 PBS and incubated at 37 °C unless specified otherwise.Effect of temperature and pH on hydrolytic degradation of PEGNB or PEGNBCA hydrogels
PBS was prepared using tablets according to the manufacturer's recommendation. Without any addition of a base or an acid, the pH value after preparation was 7.4, which was measured using a pH meter (Mettler Toledo). Using 1 N NaOH, the pH of the PBS was adjusted to 12 and by using 1 N HCl, the pH of the PBS was adjusted to 3. 5 wt% PEGNBCA or PEGNB hydrogels crosslinked with PEG4SH at R = 1 with 2 mM LAP were fabricated through 365 nm exposure at 20 mW cm−2 for 2 minutes. Subsequently, the hydrogels were incubated in PBS with pH values of 3, 7.4, or 12 at 37 °C. Strain sweep tests were conducted at corresponding time points to track the changes in G′ occurring over time. For temperature study, 5 wt% PEGNBCA crosslinked with PEG4SH at R = 1 was utilized and swelled in pH 7.4 PBS at either 37 °C using an incubator or kept at room temperature (i.e., 25 °C). The G′ of these hydrogels was measured at corresponding time points.Effect of crosslinker functionality and polymer content on hydrolytic degradation
To assess the effect of macromer functionality and polymer content, PEGNBCA hydrogels were crosslinked at either 5 wt%, 10 wt%, or 20 wt% with either DTT, PEG4SH, or PEG8SH at R = 1. The hydrogels were crosslinked using 365 nm light at 20 mW cm−2 using 2 mM LAP. Post-crosslinking, the hydrogels were swelled in pH 7.4 PBS at 37 °C. The G′ was measured at corresponding time points of the different hydrogel formulations using the strain sweep test. The G′ values obtained over time were normalized to the G′ value obtained after 1 hour incubation in pH 7.4 PBS at 37 °C. The normalized G′ values were then fitted to exponential decay kinetics using Prism 10 software with an initial value set at 1 and the final plateau value fixed at 0 to indicate complete degradation. From here, the khydrolysis value was derived from the fitting as well as the degradation half-times (t1/2).In addition to G′, we assessed the hydrolytic degradation by measuring changes in the mass swelling ratio (Q) overtime. For this study, the volume of the initial hydrogel was increased to 100 μL and was prepared using water instead of PBS except for hydrogels crosslinked with DTT since gelation was not possible using water. Post-gelation, the hydrogels were dried in vacuo for at least 1 day. Once dried, the hydrogels were weighed using an analytical balance (Mettler Toledo) to obtain dried weight 1 (W1). The hydrogels were then swelled in distilled water overnight. The following day the hydrogels were dried in vacuo for at least one day and were subsequently weighed to obtain dried weight 2 (W2). The hydrogels were then swelled in pH 7.4 PBS at 37 °C and were weighed at corresponding timepoints to obtain the swollen mass (Ws). The equation below was utilized to obtain the mass swelling ratio.
 | (1.1) |
The mass swelling ratios obtained over time were normalized to the initial mass swelling ratio, which was obtained after 1 hour incubation at 37 °C in pH 7.4 PBS. The values were then fitted using exponential growth using Prism 10 software with a fixed initial value of 1.
Tuning the hydrolytic degradation through PEGNBCA/PEGNB blended hydrogels
To tune the hydrolytic degradation of PEG thiol–norbornene crosslinked hydrogels, we blended PEGNBCA with PEGNB at different ratios. The total macromer concentration was fixed at 10 wt%. The different ratios (wt% PEGNBCA/wt% PEGNB) tested were 4/6, 6/4, 8/2, and 9/1. The different ratios of macromers were crosslinked with either DTT, PEG4SH, or PEG8SH. Hydrogels were stored under physiological conditions (i.e., pH 7.4 PBS at 37 °C) for up to 150 days. During this time, changes in G′ were tracked. The G′ values were normalized to the initial values, which were obtained after 1 hour of swelling after gel fabrication. The normalized G′ values were fitted using exponential decay fitting, where the khydrolysis and degradation half-time (t1/2) were obtained.19PEGNBCA based hydrogels for multi-modal release
For release studies, 5 wt% PEGNBCA (8-arm, 10 kDa) was crosslinked with PEG8SH at a stoichiometric ratio unless otherwise noted. To evaluate pure diffusion-controlled release, three proteins of different sizes were chosen as model cargos: lysozyme (∼14 kDa), bovine serum albumin (BSA, ∼65 kDa), and bovine gamma globulin (BGG, ∼150 kDa). All proteins were encapsulated in the hydrogels at a fixed concentration of 20 mg mL−1. Protein-encapsulated hydrogels (50 μL) were incubated in 2 mL PBS at 37 °C. At predetermined time points, 50 μL of solutions were collected and replaced with an equal volume of protein-free PBS. Protein quantification was performed using the protein assay kit (Bio-Rad Quick Start™) according to the manufacturer's recommendation without modification. The accumulated release amount was calculated by quantifying the concentration and amount of protein at the time of collection plus the amounts that were sampled in previous time points.Large size fluorescein isothiocyanate-dextran (FITC-Dextran, 70 kDa or 150 kDa) was utilized for degradation-controlled release studies as these large molecules would be trapped in the hydrogels unless significant hydrogel degradation occurred. Unless specified otherwise, FITC-dextran was encapsulated within the PEGNBCA or PEGNB crosslinked thiol–norbornene hydrogel at a concentration of 0.5 mg mL−1. The hydrogel precursor solution containing FITC-dextran was pipetted in 100 μL increments into 4 separate 2 mL centrifuge tubes. The tubes were then exposed to 365 nm light for 2 minutes to induce gelation. Subsequently, the hydrogels were incubated with pH 7.4 PBS at 37 °C. 200 μL aliquots were taken from the 2 mL tubes at different time points and placed in a 96-well black polystyrene plate, where the fluorescent intensity was measured (485 nm excitation and 528 nm emission) using a microplate reader (Biotek Synergy HT). After measuring, the aliquots were pipetted back into the 2 mL tubes. To recover the fluorescence probe for calculating cumulative release, equivalent hydrogel formulations were degraded at 80 °C (pH 12 for PEGNB hydrogels). If the hydrogel underwent complete degradation, the fluorescence intensities were normalized to this value. For fluorescently labeled dextran, values were normalized to PEGNBCA control post-degradation, since the accelerated degradation reduced the fluorescence intensity of FITC-dextran.
PEG conjugated with fluorescein containing thiol (FITC-PEG-SH, 3.4 kDa) was tethered onto norbornene pendants on PEGNB or PEGNBCA. The macromer content of PEGNB or PEGNBCA was fixed at 10 wt% and was crosslinked with PEG8SH at R = 0.8. The hydrogel precursor solution also contained 2.5 mg mL−1 of PEG-fluorescein-SH and 2 mM LAP. The hydrogel precursor solution was pipetted in 50 μL increments into 2 mL centrifuge tubes and was crosslinked through 365 nm light exposure at 20 mW cm−2 for 2 minutes. The hydrogels were then incubated with 1 mL of PBS for 1 hour to remove any unconjugated FITC-PEG-SH. To measure conjugation efficiency, the PBS was removed and fluorescence intensity was measured and normalized to the unconjugated fluorescent dye at the same concentration. The hydrogels were incubated with pH 7.4 PBS at 37 °C. Fluorescence was measured by pipetting 200 μL aliquots into a 96-well black polystyrene plate. The fluorescence intensity was measured using a microplate reader. The aliquots were pipetted back into the 2 mL centrifuge tubes. The fluorescence intensity was normalized to controls that underwent accelerated degradation.
Statistics/software
All data are presented as the mean with error bars being the standard error of the mean (SEM). For two conditions, the unpaired t-test was performed with statistical significance having p < 0.05. For two or more conditions, one-way or two-way ANOVA was performed using the Tukey post-hoc test. Statistical significance between conditions is represented with p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001). All statistical analysis and data fitting were conducted with GraphPad Prism 10. Initial constraints were placed on the model including Y0 was fixed to 1 and the plateau value was set to equal 0. The equation utilized on Prism software is shown below.Y = (Y0 − Plateau) × e−khydrolysis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×x + Plateau ×x + Plateau |
Results and discussion
Photopolymerization of PEGNB and PEGNBCA hydrogels
Norbornene-functionalized PEG can be synthesized by conjugating norbornene acid (NB) or carbic anhydride (CA) onto hydroxyl-terminal PEG, yielding PEGNB or PEGNBCA, respectively (Fig. 1(A)). Previous work has shown that both PEGNB and PEGNBCA could be crosslinked into thiol–norbornene hydrogels using DTT as a crosslinker under 365 nm light with LAP as the photoinitiator.19 In this study, we achieved high norbornene substitutions (>90%) for 8-arm PEGNBCA with two molecular weights, 10 kDa and 20 kDa (Fig. S1). We varied hydrogel network connectivity using thiol-based crosslinkers with different functionalities (f), including DTT (f = 2), PEG-tetra-thiol (PEG4SH, f = 4), and PEG-octa-thiol (PEG8SH, f = 8) (Fig. 1(B)). Thiol–norbornene photo-click gelation (Fig. 1(C)) was characterized using in situ photorheometry (Fig. 1(D) and (E)), with gel point defined as the time when the storage modulus (G′) surpasses the loss modulus (G′′) (Fig. S2) on the photorheometry plots. At a low LAP concentration of 2 mM and a low 365 nm light intensity of 2 mW cm−2, gel points were reached in under ∼11.3 s, 10.5 s, and 7 s for 20 kDa PEGNB gels were crosslinked with f = 2, 4, and 8, respectively (Fig. S2). For PEGNBCA, the three gel points identified were faster for all three thiol-crosslinkers, at ∼6.3 s, 9 s, and 3.5 s (Fig. S2). Decreasing LAP concentrations from 2 mM to 0.125 mM led to a slower gel point of ∼35 s (Fig. 1(F)). The correlation between the LAP concentration and the gel point could be modeled through one-phase decay fitting, with R2 greater than 0.95 for both PEGNB and PEGNBCA. Higher crosslinker functionality led to substantially stiffer hydrogels, with PEG8SH crosslinked gels reaching ∼120 kPa for PEGNB and ∼70 kPa for PEGNBCA (Fig. 1(G)). Gelation of 10 kDa PEGNBCA was equally efficient as that in the 20 kDa macromer, with gel points falling under 6 seconds for all three thiol crosslinkers (Fig. S3). However, hydrogels crosslinked by 10 kDa PEGNBCA were significantly stiffer than those crosslinked by the 20 kDa counterpart (1.5-fold to 2-fold higher in G′. Fig. 1(H)), which was attributed to the higher norbornene molarity at the same macromer weight content (i.e., 5 wt%).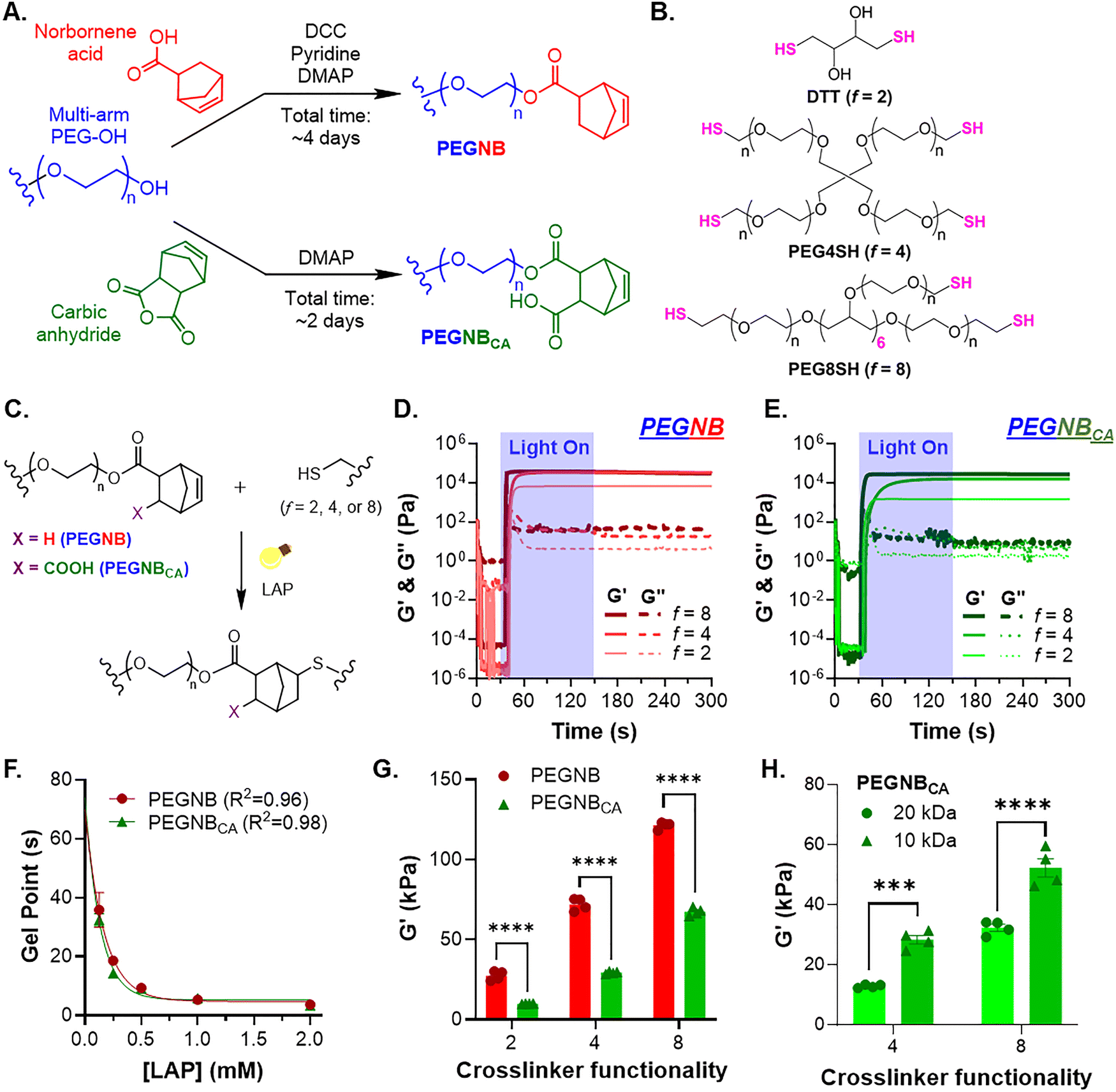 | ||
| Fig. 1 Photocrosslinking of PEGNB and PEGNBCA with different thiol-bearing macromers. (A) Schematics of PEGNB and PEGNBCA synthesis. (B) Chemical structures of thiol-bearing crosslinkers with different functionalities (2, 4, and 8 for DTT, PEG4SH, and PEG8SH, respectively). (C) Schematic of light and radical-mediated thiol-norbornene photopolymerization. (D) and (E) In situ photorheometry of gelation using (D) PEGNB or (E) PEGNBCA (both at 5 wt%) with different thiol crosslinkers at a stoichiometric ratio of thiol to norbornene under 365 nm light at 2 mW cm−2. (F) Effect of LAP concentration on gel points, which were identified from in situ photorheometry using 5 wt% PEGNB or PEGNBCA with PEG4SH at a stoichiometric ratio (R) of thiol and norbornene under 365 nm light at 3 mW cm−2. Symbols represent experimental data; curves represent one-phase exponential decay fitting. (G) Initial elastic shear moduli of 5 wt% PEGNB or PEGNBCA crosslinked with different thiol crosslinkers at R = 1. All PEGNB variants in (D)–(G) were 8-arm and 20 kDa. (H) Initial elastic shear moduli of 5 wt% PEGNBCA (8-arm, 10 kDa or 20 kDa) crosslinked with either PEG4SH or PEG8SH at R = 1. | ||
The high efficiency of step-growth thiol–norbornene gelation was further affirmed by comparing the gel point of chain-growth gelation of PEGDA, which was 4-fold slower (i.e., 20 seconds) using 2.2 mM LAP and a higher light intensity (365 nm light at 10 mW cm−2).27 Higher LAP concentrations generated higher radical concentrations at the onset of photocrosslinking, resulting in faster gelation.27,28 The effects of polymer functionality and molecular weights on hydrogel crosslinking density and stiffness have been described by the rubber elasticity theory, where the elastic modulus is proportional to polymer functionality and inversely proportional to the distance between crosslinks.29,30 However, we noticed that PEGNBCA hydrogels consistently have significantly lower G′ than the equivalent PEGNB hydrogels (Fig. 1(G)). Interestingly, PEGNBCA hydrogels had similar or higher gel fractions and significantly higher mass swelling ratios compared with the equivalent PEGNB hydrogels (Fig. S4), indicating that the lower stiffness of PEGNBCA hydrogels was not due to inefficient crosslinking but could be attributed to higher water absorbability due to the charged NBCA moieties.31 Additionally, we found that photoinitiator LAP only impacted the crosslinking density and elastic moduli of PEGNBCA hydrogels (crosslinked with PEG4SH) at concentrations lower than 0.5 mM (Fig. S5). LAP concentration beyond 0.5 mM (ca. 0.0147 wt%) resulted in no significant changes in G′, highlighting the efficient thiol–norbornene photocrosslinking of PEGNBCA hydrogels.28
Enzymatic crosslinking of PEGNBCA hydrogels
While light and radical-mediated thiol–norbornene hydrogel crosslinking provides many benefits, this gelation mechanism cannot be readily adopted in the clinics for minimally invasive hydrogel delivery where the desired in vivo location is deeper than subcutaneous sites. Thus, developing light-independent gelation is essential for injectable and minimally invasive hydrogel delivery in vivo. To this end, we have shown that the thiol–norbornene click-reaction can be initiated by HRP in the presence or absence of exogenous H2O2.23,26 Similar to the HRP-mediated thiol-crosslinking of PEGNB hydrogels, we found that PEGNBCA could also undergo enzymatic crosslinking with the thiol-crosslinker (Fig. 2(A)).26 Interestingly, gelation with PEGNBCA was one order of magnitude faster than that with PEGNB at 05 kU mL−1 HRP and in the absence of H2O2 (gel points of 2.2 min and 18.5 min for PEGNBCA and PEGNB, respectively. Fig. 2(B)). Unlike photocrosslinking, HRP-mediated gelation in the absence of exogenous H2O2 was slower to reach plateau moduli, with G′ reaching ∼1.2 kPa, ∼3 kPa, and ∼5 kPa after 5, 10, and 20 minutes, respectively (Fig. S6). This could be explained by the multi-step process of HRP-initiated gelation, where HRP self-oxidation produced endogenous H2O2 to catalyze the generation of thiyl radicals from thiol-crosslinkers that eventually reacted with PEGNBCA.26 In the absence of exogenous H2O2, gelation could be significantly accelerated by increasing the HRP concentration to 1 kU mL−1 (Fig. 2(C) and (D)).26 | ||
| Fig. 2 Enzymatic crosslinking of PEGNBCA hydrogels. (A) Picture of an inverted vial containing a gel formed by 5 wt% 8-arm PEGNBCA (10 kDa), PEG8SH (at R = 1), and 0.5 kU mL−1 HRP (no exogenous H2O2). (B) Gel point derived from in situ rheometry data shown in Fig. S6A. (C) In situ rheometry of enzymatic crosslinking of 5 wt% PEGNBCA (8-arm, 20 kDa) under different concentrations of HRP with no exogenous H2O2. (D) Gel points identified from Fig. 2(C). (E) Effect of exogenous H2O2 concentration on gel points of PEGNBCA gels crosslinked by 10 U mL−1 HRP. (F) Effect of the PEGNBCA (8-arm, 20 kDa) concentration of hydrogel moduli. Hydrogels were crosslinked by PEG8SH at R = 1 with 20 U mL−1 HRP and 0.1 mM H2O2. (G) Effect of the crosslinking mechanism on moduli 10 wt% PEGNBCA hydrogels (8-arm, 20 kDa). Hydrogels were crosslinked by PEG8SH at R = 1 through either light-mediated (2 mM LAP, 20 mW cm−2 at 365 nm) or the enzymatic reaction (20 U mL−1 HRP and 0.1 mM H2O2). | ||
While HRP alone at high concentration (e.g., 1 kU mL−1) could induce rapid thiol–norbornene gelation of PEGNBCA (gel point ∼0.5 minute. Fig. 2(D)), the high enzyme concentration is not desirable due to cost and cytotoxicity concerns. As addition of H2O2 has been shown to drastically accelerate HRP-mediated crosslinking of phenol-rich polymers32,33 and thiol-norbornene hydrogels,23,26 we reduce the HRP concentration to 10 U mL−1 while adding a small amount of H2O2 (25 to 75 μM). At a low HRP concentration of 10 U mL−1 and 25 μM H2O2, PEGNBCA hydrogel crosslinking occurred in 12.5 minutes (Fig. 2(E) and Fig. S7). However, increasing H2O2 concentration led to significant acceleration of gelation, with a gel point of under 0.8 and 0.1 minute for 50 μM and 75 μM H2O2, respectively. Additionally, enzymatically crosslinked PEGNBCA hydrogels could be fabricated to exhibit high elastic moduli, from 20 kPa to nearly 100 kPa (Fig. 2(F)). Interestingly, enzymatically crosslinked PEGNBCA hydrogels were softer than their light-crosslinked equivalents (Fig. 2(G)). This was likely due to lower gel fraction (∼0.8) of the enzymatically crosslinked hydrogels compared with the photopolymerized counterparts (∼0.98. Fig. S8). Of note, a gel fraction of 0.8 (i.e., 80% of polymer chains participated in the crosslinking) was still high but nonetheless creates room for improvement in the future. It is also worth noting that both HRP and H2O2 were added at concentrations well below what were used previously for thiol–norbornene hydrogel crosslinking.23,26 When comparing hydrogels formed by the same HRP-mediated crosslinking, PEGNBCA gelation was more effective than PEGNB (Fig. 2(B)). It is likely that the additional carboxylic acid on PEGNBCA decreased the local pH value around the norbornene groups, creating a slightly acidic local environment that enhanced HRP catalytic activity, which has been shown to be optimum at around pH 6.5.34
Hydrolytic degradation of PEGNBCA hydrogels – effects of pH, temperature, and crosslinking methods
Prior work has demonstrated accelerated hydrolytic degradation of PEGNBCA thiol–norbornene hydrogels compared with their PEGNB counterparts (Fig. 3(A)).19 However, no detailed characterization was conducted to evaluate the factors affecting hydrolytic degradation of PEGNBCA hydrogels. In this work, we compared the hydrolytic degradation of PEGNB and PEGNBCA hydrogels in PBS of various pH values – 3 (acidic), 7.4 (physiological), or 12 (basic). Prior to degradation, values of all hydrogels were measured and the ratio of G′ at any given time over
values of all hydrogels were measured and the ratio of G′ at any given time over  was used as a measure of hydrogel degradation. We found that, at pH 12, PEGNB hydrogels degraded rapidly, with complete degradation occurring within 1.5 hours with a hydrolysis reaction constant of 195.3 d−1 (Fig. 3(B) and Table S1).35 In contrast, much slower hydrolysis was observed in PEGNB hydrogels incubated in pH 7.4 or pH 3 buffer solutions, with approximately 40% reduction of their initial moduli over 28-day and a hydrolysis reaction constant of 0.01636 d−1 at pH 7.4 and 0.01755 at pH 3 (Fig. 3(B) and Table S1). These results aligned with previous finding that thiol-norbornene hydrogels crosslinked by PEGNB were highly susceptible to enhanced hydrolytic degradation in basic solution (e.g., pH 12).19,35 Hydrogel degradation was caused by hydrolysis of ester bonds connecting the PEG backbone and the norbornene group, which is known to be accelerated by base-catalyzed nucleophilic acyl substitution of a hydroxide ion (–OH) to the ester's carbonyl carbon. This reaction creates a tetrahedral intermediate that breaks down immediately, releasing an alcohol group and a carboxylic acid group. Interestingly, the equivalent PEGNBCA hydrogels all underwent complete degradation around 21 days, independent of the pH level in which the hydrogels were incubated (Fig. 3(C) and Table S2). PEGNBCA contains an extra carboxylic acid group, which may provide additional buffering effects to reduce the impact of base-catalyzed hydrolysis, leading to pH-independent degradation kinetics.
was used as a measure of hydrogel degradation. We found that, at pH 12, PEGNB hydrogels degraded rapidly, with complete degradation occurring within 1.5 hours with a hydrolysis reaction constant of 195.3 d−1 (Fig. 3(B) and Table S1).35 In contrast, much slower hydrolysis was observed in PEGNB hydrogels incubated in pH 7.4 or pH 3 buffer solutions, with approximately 40% reduction of their initial moduli over 28-day and a hydrolysis reaction constant of 0.01636 d−1 at pH 7.4 and 0.01755 at pH 3 (Fig. 3(B) and Table S1). These results aligned with previous finding that thiol-norbornene hydrogels crosslinked by PEGNB were highly susceptible to enhanced hydrolytic degradation in basic solution (e.g., pH 12).19,35 Hydrogel degradation was caused by hydrolysis of ester bonds connecting the PEG backbone and the norbornene group, which is known to be accelerated by base-catalyzed nucleophilic acyl substitution of a hydroxide ion (–OH) to the ester's carbonyl carbon. This reaction creates a tetrahedral intermediate that breaks down immediately, releasing an alcohol group and a carboxylic acid group. Interestingly, the equivalent PEGNBCA hydrogels all underwent complete degradation around 21 days, independent of the pH level in which the hydrogels were incubated (Fig. 3(C) and Table S2). PEGNBCA contains an extra carboxylic acid group, which may provide additional buffering effects to reduce the impact of base-catalyzed hydrolysis, leading to pH-independent degradation kinetics.
 | ||
| Fig. 3 Hydrolytic degradation of PEGNBCA hydrogels. (A) Reaction schematic of PEGNB or PEGNBCA crosslinked with the thiol macromer undergoing hydrolysis. (B) and (C) Changes in normalized G′ of 5 wt% (B) PEGNB or (C) PEGNBCA (8-arm, 20 kDa) crosslinked with PEG4SH at R = 1 incubated with PBS at different pH levels. (D) Changes in normalized G′ of 5 wt% PEGNBCA (8-arm, 20 kDa) crosslinked with PEG4SH at R = 1 incubated with pH 7.4 PBS at 25 °C or 37 °C over time. (E) Changes in normalized G′ of 10 wt% PEGNBCA (8-arm, 20 kDa) crosslinked with PEG8SH at R = 1. Light based crosslinking occurred using 2 mM LAP with exposure time set at 2 minutes at 20 mW cm−2. Enzymatic crosslinking occurred in the presence of 20 U mL−1 of HRP and 0.1 mM H2O2. Symbols represent experimental data; curves represent one-phase exponential decay fitting. | ||
Next, the effect of solution temperature (25 °C vs. 37 °C) on the rate of degradation was evaluated. At 37 °C, PEGNBCA hydrogels underwent complete degradation after 21 days (Fig. 3(D)). Degradation was slower at 25 °C, with only about 20% drop in initial elastic moduli over the same period, and complete gel degradation did not occur before ending the study at day-80 (Fig. 3(D)). When fitted with one phase decay fitting, we observed that the derived khydrolysis constant was 10 times slower at 25 °C than at 37 °C (Table S3). Previous studies have shown that free radical polymerized PEGDA hydrogels underwent enhanced hydrolytic degradation at higher temperatures, with the reaction/degradation rate described by the classical Arrhenius equation.36 Our results showed that PEGNBCA hydrogels were much more sensitive to temperature change with regard to hydrolysis kinetics than what was previously observed in PEGDA hydrogels.36 The higher sensitivity could result from lower network connectivity at crosslinking sites for step-growth polymerized hydrogels compared to hydrogels crosslinked via chain growth polymerization.37
Finally, the effect of the crosslinking method (i.e., photopolymerization or enzymatic crosslinking) on PEGNBCA hydrogel degradation was compared. PEGNBCA hydrogels were crosslinked with PEG8SH either through LAP-mediated photopolymerization (2 mM LAP with 2-minute light exposure of 365 nm light at 20 mW cm−2) or HRP-mediated enzymatic crosslinking (20 U mL−1 HRP and 0.1 mM H2O2 reacted at 37 °C). The ratio of G′ at any given time over  was used as a measure of hydrogel degradation so the difference in initial hydrogel crosslinking was not factored into the degradation analysis. Regardless of the crosslinking method, PEGNBCA hydrogels from both groups had nearly identical hydrolysis kinetics and completely degraded after approximately 42 days (Fig. 3(E) and Table S4). This was most likely because the hydrogels were still crosslinked via the same thioether bond regardless of the mechanism of initiation.
was used as a measure of hydrogel degradation so the difference in initial hydrogel crosslinking was not factored into the degradation analysis. Regardless of the crosslinking method, PEGNBCA hydrogels from both groups had nearly identical hydrolysis kinetics and completely degraded after approximately 42 days (Fig. 3(E) and Table S4). This was most likely because the hydrogels were still crosslinked via the same thioether bond regardless of the mechanism of initiation.
Hydrolytic degradation of PEGNBCA hydrogels – effects of macromer parameters
As temperature, but not pH nor the crosslinking method, plays a prominent role in dictating the hydrolysis kinetics of PEGNBCA thiol–norbornene hydrogels, all subsequent degradation studies were performed using photopolymerized hydrogels kept at 37 °C and pH 7.4 PBS. Prior work on thiol–norbornene and tetrazine–norbornene PEG-based hydrogels showed that gels crosslinked by a higher macromer content degraded slower.21,22 In order to evaluate the effect of the macromer content on hydrolytic degradation, PEGNBCA hydrogels were formulated into three macromer weight percentages (5 wt%, 10 wt%, and 20 wt%) and crosslinked by thiol-bearing macromers with different functionalities (f = 2, 4, and 8).15 Hydrogels were substantially stiffer when crosslinked with higher macromer contents and with higher crosslinker functionality (Fig. S9). The moduli of these PEGNBCA hydrogels spanned across a wide range of physiologically relevant range (G′ ∼2 kPa to ∼120 kPa).38 When bi-functional DTT was used as the crosslinker, all hydrogels had similarly high gel fraction (>0.95, Fig. S10A), as well as comparable mass swelling ratio (Fig. S10B) and mesh size (Fig. S10C). For PEG-based thiol crosslinkers (PEG4SH and PEG8SH), all hydrogels still exhibited high gel fraction (>0.95, Fig. S11A), but there was an inverse trend between PEGNBCA weight content and mass swelling ratios (Fig. S11B) and calculated mesh size (Fig. S11C). The differences in swelling and mesh size were likely caused by increased water infiltration in the hydrogels at higher content of PEG-based thiol-crosslinkers. Interestingly, even with these differences, PEGNBCA hydrogels degraded at similar rates independent of the macromer content for all three thiol crosslinkers (Fig. 4(A)). Furthermore, the degradation of PEGNBCA hydrogels followed pseudo-first order kinetics and displayed high degree fitting to exponential decay (Fig. 4(A)).19,39 Higher crosslinker functionality drastically delayed hydrolysis kinetics (Fig. 4(A) and Table S5). For example, at 5 wt% PEGNBCA crosslinked with DTT, PEG4SH, or PEG8SH at R = 1, the khydrolysis values were 0.3874 d−1, 0.1812 d−1, and 0.07567 d−1, respectively (Table S5), indicating that adjusting crosslinker functionality, rather than the macromer content, would be a more effective way of tuning degradation of these hydrogels. As a comparison, we evaluated the degradation of conventional PEGNB hydrogels crosslinked with DTT, PEG4SH, or PEG8SH via tracking their moduli (Fig. S12A). As anticipated, PEGNB hydrogels degraded substantially slower than their PEGNBCA counterpart but generally displayed the same pseudo-first-order degradation kinetics, although all gels remained intact after 200 days. Similar to PEGNBCA gels, the degradation of PEGNB gels was faster when crosslinked by DTT or PEG4SH (Fig. S12B). | ||
| Fig. 4 Effect of the macromer content and thiol-crosslinker functionality on hydrolytic degradation of PEGNBCA hydrogels. (A) Normalized changes in G′ or (B) mass swelling ratio over time of PEGNBCA at different macromer concentrations, specifically 5 wt%, 10 wt%, or 20 wt%, crosslinked with either DTT, PEG4SH, or PEG8SH at R = 1 incubated in pH 7.4 PBS at 37 °C. Symbols represent experimental data; curves represent one-phase exponential decay fitting. | ||
We further explored other design parameters that may impact hydrolytic degradation time, such as the stoichiometric ratio of thiol-to-norbornene (i.e., R = 0.6 to 1). In general, hydrogels crosslinked with lower R values were softer (Fig. S13A) and degraded slightly faster (Fig. S13B and C). Another parameter that may impact hydrolysis kinetics of PEGNBCA hydrogels was the molecular weights of PEGNBCA (e.g., 10 kDa vs. 20 kDa). For 10 kDa PEGNBCA crosslinked by PEG4SH, the hydrolysis kinetics were nearly twice as slow as that of the 20 kDa PEGNBCA counterparts (Fig. S14A), with a complete degradation time of 15 days to 35 days for 20 kDa and 10 kDa PEGNBCA. This could be explained by the difference in crosslinking density produced by the 10 kDa macromer, which had twice the NB molarity to that of the 20 kDa macromer. When crosslinked with PEG8SH, which led to stiffer hydrogels, the difference in the degradation rate was less pronounced (Fig. S14B). Nonetheless, these gels degraded slower than those crosslinked by PEG4SH, with complete degradation time falling between 42 to 50 days.
Hydrolytic degradation of PEGNBCA hydrogels – combinatorial effect of crosslinker functionality and macromer degradability
The combination/blending of fast-degrading PEGNBCA and slow-degrading PEGNB offered an opportunity to engineer PEG-based hydrogels with programmable hydrolytic degradation. This approach differs from previous chemical strategies that sought to develop norbornene moieties with different hydrolytic susceptibility, which often require advanced chemical synthetic skills.40 The crosslinking of blended macromers with different thiol crosslinkers also afforded a simple yet robust method to tune the hydrolytic degradation. To this end, we modularly blended PEGNBCA with PEGNB at different weight ratios (i.e., 4/6, 6/4, 8/2, and 9/1, where the numbers indicate the wt% of PEGNBCA and PEGNB, respectively). For all three thiol crosslinkers (DTT, PEG4SH, and PEG8SH), the higher PEGNB content led to stiffer gels (Fig. 5(A)), agreeing with previous observation that pure PEGNBCA gels were softer than pure PEGNB gels (Fig. 1(G)). However, through modularly blending fast-degrading PEGNBCA with slow-degrading PEGNB, the hydrolytic degradation time was extended from ∼49 days (in pure PEGNBCA gels, Fig. 4) to over 150 days and beyond (Fig. 5(B)). Additionally, a wide range of hydrolytic degradation times was achieved through a combinatorial approach of adjusting thiol crosslinker functionality (i.e., f = 2, 4, or 8) and polymer weight ratio. The degradation half-time, defined as the time required to reach half the initial modulus, varied from less than 2 days to over 86 days (Fig. 5(C) and (D)). Overall, this combinatorial approach of thiol crosslinker functionality and the blended PEGNBCA/PEGNB weight ratio was successful in tuning the hydrolytic degradation over a wide period of time. Future studies could explore the development of generalized mathematical models to predict and validate the effect of various parameters on PEGNBCA hydrogel degradation.41 | ||
| Fig. 5 Blending PEGNBCA and PEGNB together to control the rate of degradation using different thiol crosslinkers. (A) Initial G′ and (B) changes in the normalized G′ of blended hydrogels crosslinked with DTT, PEG4SH, or PEG8SH. Hydrogels had a fixed total PEGNB/PEGNBCA (8-arm, 20 kDa) concentration of 10 wt% and was crosslinked at R = 1. Hydrogels were incubated at 37 °C and swelled in pH 7.4 PBS. Symbols represent experimental data; curves represent one-phase exponential decay fitting. (C) Degradation half-times and (D) khydrolysis constants derived from one-phase decay fittings of the changes in G′ of the PEGNBCA/PEGNB blended hydrogels over time. | ||
Multi-modal controlled release
The high tunability of PEGNBCA hydrogel crosslinking and degradation warrants its use in controlled release applications. The release of hydrogel-encapsulated cargos can be adjusted through many mechanisms, including (1) diffusion-controlled, with release governed by the cargo size; (2) degradation-controlled, with release determined by network connectivity; and (3) diffusion-reaction-controlled, with release controlled by degradation and subsequent diffusion of the molecular tether (Fig. 6(A)). The adaptability of PEGNBCA hydrogels in multi-modal controlled release was first evaluated by diffusion-control release using proteins of three sizes: lysozyme (∼14 kDa), BSA (∼65 kDa), and BGG (∼150 kDa) (Fig. 6(B)). The smallest protein lysozyme diffused out of the 5 wt% PEGNBCA hydrogels rapidly, depleting all releasable protein within hours after the onset of the release study. In contrast, the release of medium-size protein BSA exhibited a bi-phasic profile, with ∼30% of burst release in the first 2 hours, followed by a more linear release profile until depleting the releasable protein after 18 days. Finally, the release of the largest protein BGG also exhibited an initial burst of ∼20% but no substantial release was detected following the initial burst. The differences in the three release profiles can be explained by the relative sizes of proteins to the mesh size of the hydrogels used (5 wt% 8-arm PEGNBCA + PEG4SH). As shown in Fig. S14B, the calculated mesh sizes of the hydrogels used in the diffusion-controlled release study were between ∼15 and ∼22 nm. The diameter (2× hydrodynamic radius) of lysozyme, BSA, and BGG was approximately ∼4 nm,42 ∼8 nm,43 and ∼10.5 nm,44 respectively. It can be reasonably expected that the release would be reduced as the size of diffusants approaches the mesh size of the hydrogel. One interesting phenomenon was that only about 22% BGG was released from the hydrogel at the end of the 18-day release study when the hydrogels were nearly completely degraded (Fig. 4(A)). We reason that some non-specific protein–polymer conjugation might occur between PEGNBCA and BGG during the radical mediated thiol-norbornene gel crosslinking. As BGG is an immunoglobulin containing multiple inter-chain and intra-domain disulfide bonds, radicals generated during hydrogel crosslinking may attack some of these disulfide bonds,45 causing its conjugation/immobilization in the thiol–norbornene hydrogel network, hence reducing the amount of total releasable proteins. | ||
| Fig. 6 Controlled release using PEGNB or PEGNBCA thiol–norbornene crosslinked hydrogels. (A) Graphic representing different release strategies using this hydrogel system. (B) Release of lysozyme, BSA, or BGG at 20 mg mL−1 from 5 wt% PEGNBCA (8-arm, 10 kDa) hydrogels. (C) Release of 70 kDa or 150 kDa FITC-dextran from 10 wt% PEGNBCA (8-arm, 10 kDa) hydrogels crosslinked by PEG4SH. (D) Release of PEG-fluorescein-SH (3.4 kDa) conjugated onto 10 wt% PEGNB or PEGNBCA (8-arm, 10 kDa) R = 0.8 with PEG8SH. | ||
To demonstrate degradation-controlled release, we fabricated hydrogels with 10 wt% 8-arm PEGNBCA and PEG8SH, which yielded mesh sizes of ∼10 nm to 13 nm (data not shown). We chose FITC-dextran of 70 kDa (dia. ∼9 nm) and 150 kDa (dia. ∼12 nm) as the model drugs for their larger size and easy fluorescence detection.46 As the relative sizes of FITC-dextran and mesh sizes approached one, no significant burst release was found and the cumulative release profiles approached zeroth-order (i.e., release was linear and the release rate was time-independent) (Fig. 6(C)), suggesting a degradation-controlled release.3 We further tested the release of 70 kDa FITC-dextran from hydrogels crosslinked at 2.5 wt%, 5 wt%, and 10 wt% (Fig. S15).39 Since hydrogels crosslinked by lower weight contents had a larger mesh size,39,47 higher burst release was observed but these formulations also support higher cumulative release than the highly crosslinked hydrogels.3,48
After demonstrating that PEGNBCA hydrogels could be utilized for controlled release of high molecular weight cargos, we tested if this hydrogel system could be engineered to release smaller molecular weight model drugs. However, since simple encapsulation was ineffective in retaining and controlling the release of small size cargos (lysozyme in Fig. 6(B)), we used 3.4 kDa FITC-PEG-SH as a small size model drug to test whether it could be tethered to the hydrogel network for degradation controlled release.9,49 We used the slow-degrading PEGNB hydrogels as a control to evaluate the difference in hydrolysis kinetics on the liberation of the covalently tethered model drug. 3.4 kDa FITC-PEG-SH (dia. <3 nm) was conjugated onto either PEGNBCA or PEGNB during thiol–norbornene hydrogel crosslinking with similar conjugation efficiency (Fig. S16). An off-stoichiometric thiol/norbornene ratio of 0.8 was used to accommodate the conjugation of pendant FITC-PEG-SH. The immobilization of FITC-PEG-SH was visualized and verified via fluorescence imaging (Fig. S17A) and intensity quantification (Fig. S17B). FITC-PEG-SH tethered to slow-degrading PEGNB showed low level (<10%) of release after 35 days. In contrast, near-linear release was found when conjugating to the fast-degrading PEGNBCA (Fig. 6(D)). This was a degradation-controlled release as 3.4 kDa FITC-PEG-SH has a hydrodynamic diameter (less than 3 nm per Sigma-Aldrich) much smaller than the mesh size of the hydrogels used here. Its release was essentially determined by the hydrolytic degradation of ester bond linking the drug and the PEG backbone (Fig. 6(A)). The ability of conventional PEGNB to release tethered FITC-PEG-SH was limited due to the slow hydrolysis kinetics of conventional PEGNB. Through these proof-of-concept release studies, we established PEGNBCA hydrogels as a versatile platform for multi-modal controlled release applications. Potential future applications of PEGNBCA hydrogels in drug delivery space include the formulation of highly crosslinked hydrogels with a tunable hydrolysis rate (via altering the PEG molecular weight or architecture) for degradation-controlled release of large proteins/antibodies. On the other hand, small molecules can be released from highly swollen PEGNBCA hydrogels via covalent tethering as long as the drug contains the free thiol group. Finally, as shown in our previous work, hydrogels crosslinked by PEGNBCA were highly cytocompatible for even highly sensitive induced pluripotent stem cells,19,21,22,50 enabling the design of cell-laden hydrogels with controlled release capability to guide stem cell morphogenesis.
Conclusions
In summary, we demonstrated that PEGNBCA supported rapid gelation with thiol-bearing crosslinkers through the light- or enzyme-mediated thiol-norbornene click reaction. We discovered that the hydrolytic degradation of PEGNBCA hydrogels was pH-independent but temperature-dependent, with higher temperatures accelerating hydrolysis. We systematically characterized the hydrolytic degradation of PEGNBCA hydrogels and developed a simple PEGNBCA/PEGNB blending method to tune the degradation without going through additional chemical synthesis. Finally, this hydrogel system was utilized for multi-modal release of model cargos with different sizes and loading methods (i.e., diffusion-controlled, degradation-controlled release of large molecules, and degradation-controlled release of small tethered molecules). Due to the highly tunable hydrolysis kinetics and the ability to release drug cargo in a controlled release manner, we believe that this hydrogel system will be valuable in the fields of tissue engineering and drug delivery.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information includes additional methods, supporting Fig. S1–S17, supporting Tables S1–S5, and additional references. See DOI: https://doi.org/10.1039/d5tb01524c.Acknowledgements
This work was supported in part by the National Institutes of Health (R01DK127436 to CCL) and the National Science Foundation (NSF GRFP award number: 2141416 to NHD).References
- C. C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- R. Gharios, R. M. Francis and C. A. DeForest, Matter, 2023, 6, 4195–4244 CrossRef CAS PubMed.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed.
- A. S. Sawhney and J. A. Hubbell, J. Biomed. Mater. Res., 1990, 24, 1397–1411 CrossRef CAS PubMed.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- S. C. Rizzi and J. A. Hubbell, Biomacromolecules, 2005, 6, 1226–1238 CrossRef CAS PubMed.
- P. van de Wetering, A. T. Metters, R. G. Schoenmakers and J. A. Hubbell, J. Controlled Release, 2005, 102, 619–627 CrossRef CAS PubMed.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS PubMed.
- L. E. Jansen, L. J. Negron-Pineiro, S. Galarza and S. R. Peyton, Acta Biomater., 2018, 70, 120–128 CrossRef CAS PubMed.
- A. Mora-Boza, Z. Ahmedin and A. J. Garcia, J. Biomed. Mater. Res., Part A, 2024, 112, 1265–1275 CrossRef CAS PubMed.
- N. M. Shah, M. D. Pool and A. T. Metters, Biomacromolecules, 2006, 7, 3171–3177 CrossRef CAS PubMed.
- Y. Hao, H. Shih, Z. Munoz, A. Kemp and C. C. Lin, Acta Biomater., 2014, 10, 104–114 CrossRef CAS PubMed.
- M. R. Arkenberg, H. D. Nguyen and C. C. Lin, J. Mater. Chem. B, 2020, 8, 7835–7855 RSC.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- H. Shih and C. C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- A. Raza and C. C. Lin, Macromol. Biosci., 2013, 13, 1048–1058 CrossRef CAS PubMed.
- M. S. Rehmann, A. C. Garibian and A. M. Kloxin, Macromol. Symp., 2013, 329, 58–65 CrossRef CAS PubMed.
- C. C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- F. Y. Lin and C. C. Lin, ACS Macro Lett., 2021, 10, 341–345 CrossRef CAS PubMed.
- Z. Jiang, F. Y. Lin, K. Jiang, H. Nguyen, C. Y. Chang and C. C. Lin, Biomater. Adv., 2022, 134, 112712 CrossRef CAS PubMed.
- N. H. Dimmitt and C. C. Lin, Macromolecules, 2024, 57, 1556–1568 CrossRef CAS PubMed.
- N. H. Dimmitt, M. R. Arkenberg, M. M. de Lima Perini, J. Li and C. C. Lin, ACS Biomater. Sci. Eng., 2022, 8, 4262–4273 CrossRef CAS PubMed.
- H. D. Nguyen, H. Y. Liu, B. N. Hudson and C. C. Lin, ACS Biomater. Sci. Eng., 2019, 5, 1247–1256 CrossRef CAS PubMed.
- Y. Qi, F. Wang, J. Liu, C. Wang and Y. Liu, Int. J. Biol. Macromol., 2025, 311, 143379 CrossRef CAS PubMed.
- F. Lee, K. H. Bae and M. Kurisawa, Biomed. Mater., 2015, 11, 014101 CrossRef PubMed.
- M. H. Kim and C. C. Lin, Biomed. Mater., 2021, 16, 045027 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- C. Y. Chang, H. C. Johnson, O. Babb, M. L. Fishel and C. C. Lin, Acta Biomater., 2021, 130, 161–171 CrossRef CAS PubMed.
- M. Shibayama, Soft Matter, 2012, 8, 8030–8038 RSC.
- T. Sakai, Y. Akagi, T. Matsunaga, M. Kurakazu, U. I. Chung and M. Shibayama, Macromol. Rapid Commun., 2010, 31, 1954–1959 CrossRef CAS PubMed.
- J. R. Clegg, K. Adebowale, Z. Zhao and S. Mitragotri, Bioeng. Transl. Med., 2024, 9, e10680 CrossRef PubMed.
- D. L. Lefkowitz, K. Mills, A. Castro and S. S. Lefkowitz, J. Leukocyte Biol., 1991, 50, 615–623 CrossRef CAS PubMed.
- J. J. Roberts, P. Naudiyal, K. S. Lim, L. A. Poole-Warren and P. J. Martens, Biomater. Res., 2016, 20, 30 CrossRef PubMed.
- A. J. AL-Sa’ady, M. H. A. Al-Bahrani and G. M. Aziz, Int. J. Curr. Microbiol. Appl. Sci., 2018, 7, 328–339 CrossRef.
- S. E. Holt, A. Rakoski, F. Jivan, L. M. Perez and D. L. Alge, Macromol. Rapid Commun., 2020, 41, e2000287 CrossRef PubMed.
- G. J. Rodriguez-Rivera, M. Green, V. Shah, K. Leyendecker and E. Cosgriff-Hernandez, J. Biomed. Mater. Res., Part A, 2024, 112, 1200–1212 CrossRef CAS PubMed.
- M. W. Tibbitt, A. M. Kloxin, L. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- T. Su, M. Xu, F. Lu and Q. Chang, RSC Adv., 2022, 12, 24501–24510 RSC.
- C. E. Ziegler, M. Graf, M. Nagaoka, H. Lehr and A. M. Goepferich, Biomacromolecules, 2021, 22, 3223–3236 CrossRef CAS PubMed.
- G. R. Sama, M. N. Arguien, T. E. Hoffman, B. D. Fairbanks, M. Trujilo-Lemon, S. Keyser, K. S. Anseth, S. L. Spencer and C. N. Bowman, Biomacromolecules, 2025, 26, 1850–1859 CrossRef CAS PubMed.
- R. Das and D. Kundu, Comput. Mater. Sci., 2025, 247, 113516 CrossRef CAS.
- Y. Zhang; E. Farrell; D. Mankiewic and Z. Weiner, Brookhaven Instruments Application Library, 2019 Search PubMed.
- A. Hawe, W. L. Hulse, W. Jiskoot and R. T. Forbes, Pharm. Res., 2011, 28, 2302–2310 CrossRef CAS PubMed.
- M. Nag, D. Das, D. Bandyopadhyay and S. Basak, Phys. Chem. Chem. Phys., 2015, 17, 19139–19148 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- B. Henry, M. Cheema and S. Davis, Int. J. Pharm., 1991, 73, 81–88 CrossRef CAS.
- E. Axpe, D. Chan, G. S. Offeddu, Y. Chang, D. Merida, H. L. Hernandez and E. A. Appel, Macromolecules, 2019, 52, 6889–6897 CrossRef CAS PubMed.
- A. Metters, K. Anseth and C. Bowman, Polymer, 2000, 41, 3993–4004 CrossRef CAS.
- N. H. Dimmitt and C. C. Lin, Adv. Mater. Interfaces, 2025, 2400952 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |