
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Addressing the polycation dilemma in drug delivery: charge-converting liposomes
Martyna
Truszkowska
a,
Ahmad
Saleh
ab,
Melanie Lena
Ebert
 a,
Gergely
Kali
a and
Andreas
Bernkop-Schnürch
*a
a,
Gergely
Kali
a and
Andreas
Bernkop-Schnürch
*a
aCenter for Chemistry and Biomedicine, Department of Pharmaceutical Technology, Institute of Pharmacy, Leopold-Franzens-University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria. E-mail: Andreas.Bernkop@uibk.ac.at; Tel: +43 512 507 58 600
bDepartment of Pharmacy, Universitas Mandala Waluya, A.H.Nasution, Kendari 93231, Southeast Sulawesi, Republic of Indonesia
First published on 24th June 2025
Abstract
The aim of this study was to tackle the polycation dilemma in drug delivery by developing charge-converting liposomes capable of permeating the mucus gel layer and enhancing cellular uptake. Positively charged liposomes containing dioleoylphosphatidylethanolamine (DOPE), cholesterol, and oleyl-oligolysine were formulated via the thin-film method. These liposomes were coated with polyphosphate to create negatively charged phosphorylated liposomes (pp-liposomes). Liposomes were characterized regarding droplet size, zeta potential, stability, cytotoxicity, and hemolytic activity. The cleavage of polyphosphates from the surface of liposomes triggered by intestinal alkaline phosphatase (AP) was monitored via malachite green assay and shift in zeta potential. Mucus permeation was assessed using porcine intestinal mucus in Transwell inserts. Cellular uptake was quantified in Caco-2 cells by flow cytometry and confocal microscopy. Liposomes exhibited an average size of 138.7 ± 2.9 nm and a zeta potential of +35.4 ± 1.5 mV, while the size of polyphosphate-coated liposomes increased to 168.4 ± 1.2 nm with a zeta potential of −24.2 ± 2.5 mV; both remained stable over 24 hours. Liposomes were non-toxic and hemolytic in a concentration of 0.1%. pp-Liposomes were less toxic than uncoated liposomes. Significant phosphate release occurred within first 6 hours of incubation with AP, and the zeta potential converted to +12.9 ± 5.19 mV within 24 hours. Mucus permeation studies showed that pp-liposomes exhibited 12-fold increase in permeability in the absence of AP compared to its presence. Cellular uptake of liposomes and pp-liposomes in Caco-2 cells demonstrated comparable levels of internalization. Accordingly, charge-converting liposomes effectively traversed the mucus barrier and improved cellular uptake, indicating a promising approach to resolving the polycation dilemma.
1. Introduction
Efficient drug delivery across mucosal surfaces remains one of the most significant challenges in pharmaceutical technology. Mucosal barriers, comprising the mucus gel layer and the epithelial membrane, serve as the body's first line of defense against pathogens. While this protective function is essential for health, it poses an obstacle to the delivery of therapeutics administered via mucosal routes such as the oral, nasal, and pulmonary pathways.1,2 Overcoming these barriers is crucial for enhancing the bioavailability and efficacy of a wide range of drugs. A critical factor impeding drug delivery is the polycation dilemma arising from the interaction between charged nanocarriers and biological barriers.Cationic nanocarriers generally show improved cellular internalization owing to strong electrostatic attraction to negatively charged cell membranes. This positive charge causes them to become trapped within the negatively charged mucus gel, preventing them from reaching their target cells.3 In contrast, nanocarriers bearing a neutral or negative surface charge permeate the mucus layer more readily, but their cellular uptake is limited.4 This dichotomy presents a significant hurdle in designing nanocarriers that can traverse the mucus barrier and achieve effective cellular internalization.
To address this challenge, the concept of charge-converting nanocarriers has emerged as a promising strategy. These nanocarriers are engineered to possess an initial negative or neutral surface charge, facilitating their diffusion in the mucus layer. Upon reaching the target site, they undergo a charge conversion to positive zeta potential, enhancing cellular uptake through increased electrostatic interactions with cell membranes.5 Several studies have explored this concept using various nanocarrier systems. Polymeric nanocarriers, micelles, and nanoemulsions capable of charge conversion have been developed and have shown promise in improving drug delivery efficacy.6–12 Despite these advancements, however, the concept could so far not been transferred to liposomes. A notable attempt was undertaken by Perttu et al.,13 who developed liposomes using inverse phosphocholine lipids aiming to achieve a charge conversion by the cleavage of the terminal phosphate group by phosphatases. Despite this innovative and elegant concept, a shift in zeta potential from −17 mV to just −7.5 mV could be achieved.
It is therefore the aim of this study to develop liposomes that are able to convert their surface charge from negative to positive. Key to success is likely the surface decoration of liposomes with polycationic peptides that are coated with polyphosphates. Oleyl-oligolysine, a cationic peptide with multiple amino groups, offers a promising approach to achieve this conversion. When incorporated into the liposomal membrane, it is possible to achieve a higher positive zeta potential after the enzymatic cleavage of phosphate groups. This approach can potentially facilitate a more significant charge reversal, enhancing electrostatic interactions with the negatively charged cell membrane and improving cellular uptake. Intestinal alkaline phosphatase (AP), is an enzyme expressed on the epithelial cell surfaces of various tissues, including the intestine, lung, and eye.14,15 AP can cleave phosphate groups from the surface of nanocarriers, triggering a change in zeta potential.
We hypothesize that coating cationic liposomes with sodium polyphosphate will result in liposomes that initially possess a negative surface charge, facilitating their permeation through the mucus layer. Upon contact with IAP expressed on the epithelial cell surface, the phosphate groups will be cleaved, resulting in a shift to a positive zeta potential. This charge reversal will enhance electrostatic interactions with the negatively charged cell membrane, promoting cellular uptake and improving the bioavailability of the encapsulated therapeutic agents.
2. Experimental
2.1. Materials
Sodium polyphosphate (PolyP, Graham's salt), calcein, cholesterol, alkaline phosphatase from bovine intestinal mucosa (7165 units per mg protein, 19.4%), phosphatase inhibitor cocktail 2, Triton X-100, ammonium molybdate tetrahydrate (81–83%), and malachite green (MLG) oxalate salt ≥90% were purchased from Sigma-Aldrich (Vienna, Austria). Phosphate buffered saline (PBS), penicillin, streptomycin, and fetal bovine serum (FBS) were purchased from Merk (Tutzing, Germany). Minimal Essential Medium (MEM), and OptiMEM were obtained from ThermoFisher Scientific, Austria. DiD (DiIC18(5); 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt) and DAPI (4′,6-diamidino-2-phenylindole) were purchased from Biotum (Fremont, CA, USA). All chemicals used were of analytical grade.2.2. Synthesis and characterization of oleyl-oligolysine
Oleyl-oligolysine was synthesized using the following procedure.16 In brief, N∈-(tert-butoxycarbonyl)-L-lysine N-carboxyanhydride (Boc-Lys-NCA) monomer was prepared by reacting Nα,N∈-di-(tert-butoxycarbonyl)-L-lysine (5 g, 14.5 mmol) with triphosgene (2.85 g, 0.009 mol) in 350 mL of ethyl acetate in the presence of triethylamine (1.33 mL, 0.0095 mol) under a nitrogen atmosphere, carried out overnight. Afterward, the reaction mixture was kept at −20 °C for 1 hour, filtered, and washed twice with water and twice with 0.5% aqueous NaHCO3, then precipitated into n-hexane. The white solid product was collected after recrystallization from a mixture of ethyl acetate/n-hexane (20/80), and freeze drying for 48 h, with a yield of 2.2 g (59%).The recovered Boc-Lys-NCA (1.5 g, 5.5 mmol) was polymerized, initiated by oleyl amine (0.15 g, 0.55 mmol) in anhydrous DMF at room temperature under N2 for 5 days. After polymerization, the mixture was precipitated in water, filtered, and lyophilized for 48 hours. Finally, the Boc protecting groups were cleaved with trifluoroacetic acid in methylene chloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio), and the product was precipitated in diethyl ether and dried under vacuum, resulting in a white solid (0.85 g).
:5 ratio), and the product was precipitated in diethyl ether and dried under vacuum, resulting in a white solid (0.85 g).
NMR measurements were performed on a “Mars” 400 MHz Avance 4 Neo spectrometer from Bruker Corporation (Billerica, MA, USA, 400 MHz) in methyl sulfoxide-d6 (DMSO-d6) solution.
2.3. Preparation and characterization of liposomes
Liposomes were prepared by a thin-film hydration technique as described by Coban et al.17 Specifically, DOPE (15 mg) and cholesterol (10 mg) were first dissolved in 5 mL chloroform, and oleyl-oligolysine (2 mg) was dissolved in 0.5 mL methanol, together in a 100 mL round-bottom flask. The solvents were then evaporated at 40 °C under reduced pressure using a rotary evaporator, yielding a dry, homogeneous lipid film after 30 minutes. This film was hydrated with 25 mM HEPES buffer (pH 7.4), and the mixture was stirred at 60 °C (oil bath) for 6 hours. The resulting liposomes were subsequently ultrasonicated to reduce size for 15 minutes in pulses (10 s on, 5 s off; 100 W, 100% amplitude) using a Hielscher UP200Ht sonicator.To obtain polyphosphate-coated liposomes (pp-liposomes), 200 μL of Graham's salt solution (0.25% w/v in water) was added to 1 mL of the liposomes. The mixture was gently agitated on a thermomixer at 300 rpm for 30 minutes at room temperature. Unbound polyphosphate was removed by centrifugation (13400 rpm, 10 min, using Amicon Ultra filters), and the liposomes were resuspended in fresh HEPES buffer for further use.18
Liposomes were labeled with calcein as a fluorescent marker to facilitate further analysis. In detail, a 50 mM calcein solution in phosphate-buffered saline (PBS, pH 7.4) was added to the thin-lipid film during liposome preparation. After 6 h of stirring in an oil bath at 60 °C, liposomes were homogenized for 15 minutes using an ultrasonic homogenizer. To separate unencapsulated calcein from the liposomes, suspension was centrifuged 2 times at 50000 rpm, 10 °C, and under vacuum for 1 hour using an Optima MAX-XP Ultracentrifuge (Beckman Coulter). The supernatant was carefully removed, and the pellet was resuspended in PBS. To ensure complete resuspension, liposomes were gently vortexed.19 Size, polydispersity index (PDI), and zeta potential of liposomes and pp-liposomes were determined by photon correlation spectroscopy and laser Doppler anemometry using a Zetasizer Nano ZS (Malvern Instruments, UK). In brief, 0.05% (m/v) of liposomes were prepared in demineralized water before analysis. Each measurement was performed in triplicate at 37 °C with a detection angle of 173°.
2.4. Phosphate release studies
000 cells per well in 24-well plates and cultured for 10 days, with the medium replaced every other day. Cells were pre-incubated with phosphatase inhibitor cocktail II or buffer for 1 h. Before the addition of test solutions, cells were rinsed twice with 500 μL of physiological HEPES buffer (25 mM, pH 7.4). Subsequently, 500 μL of test solutions prepared by diluting pp-liposomes at a ratio of 1:1 in HEPES buffer were added to each well. At designated time points (0, 1, 3, 6, 12, and 24 h), 50 μL aliquots were collected, and the reaction was halted by adding 5 μL of 3.6 M H2SO4. Phosphate released during the reaction was quantified using the MLG assay, as described above.
2.5. Zeta potential change
To evaluate changes in surface charge, zeta potential of pp-liposomes was determined before and after incubation with AP. Liposomes were prepared as described earlier, and the initial zeta potential was measured. To initiate the reaction, 100 μL of AP solution (10 U mL−1) was added to 1500 μL of pp-liposomes. The mixture was incubated at 37 °C with continuous shaking at 300 rpm, and zeta potential measurements were conducted at predefined time intervals (0, 1, 3, 6, 12, and 24 h) to monitor dynamic changes in surface charge over time. To facilitate the removal of released phosphate during the reaction, the suspension was maintained in a dialysis membrane (Float-A-Lyzer G2, Type Float-A-Lyzer, 8–10 kDa) throughout the incubation process. Control samples, consisting of liposome suspensions without IAP, were incubated under identical conditions. Zeta potential measurements were performed in triplicate at each time point to ensure precision and reproducibility, providing insights into the dynamic changes in liposomal surface charge over the 24-hour period.2.6. Hemolysis
In the hemolysis assay, red blood cell (RBC) membranes serve as a model for the endosomal membrane encountered during cellular uptake.23 The extent of hemolysis (release of hemoglobin) was measured spectrophotometrically to quantify membrane-disruptive activity, following the protocol of Friedl et al.24 A human blood sample, provided by Tirol Kliniken GmbH (Innsbruck, Austria), was diluted in sterile HEPES buffer (20 mM, pH 7.4) at a 1:200 volume ratio. Written informed consent was obtained from all participating blood donors by the Central Institute for Blood Transfusion and Immunological Department, Innsbruck, Austria. The use of anonymized leftover specimens for scientific purposes was approved by the Ethics Committee of the Medical University of Innsbruck. Test solutions, including liposomes and pp-liposomes at varying concentrations (0.5%, 0.25%, and 0.1% in HEPES buffer), were mixed with the RBC suspension at a 1:1 volume ratio. The samples were incubated in a shaked incubator (300 rpm) at 37 °C for 4 and 24 h. Triton X-100 (0.1% v/v) served as the positive control, representing 100% hemolysis. Following incubation, the samples were centrifuged at 2700 rpm for 10 minutes, and the absorbance of the supernatant was measured at 415 nm to quantify hemoglobin release. Hemolysis was calculated using the following equation:24
2.7. Assessmentof cell viability
Caco-2 cell viability after exposure to liposomes was assessed using a resazurin assay, adapted from Perera et al. with minor modifications.12 Cells were incubated at a density of 25000 cells per well in a 24-well plate with serum-supplemented MEM at 37 °C in an atmosphere with 5% CO2 and 95% humidity. The MEM was replaced with fresh medium on alternate days until a cell monolayer was obtained. One day prior to the experiment, liposomes were diluted in HBS to prepare test concentrations of 30 mM, 15 mM, 7.5 mM, 3.25 mM, and 1.8 mM. Afterward, cells were washed twice with HBS, and the samples were added to the wells. Buffer and 0.1% (m/v) Triton X-100 solution were utilized as the positive and negative control, respectively. At predetermined time points (4 and 24 h), cells were washed twice with HBS, and 2.2 mM resazurin solution was added to each well and incubated at 37 °C in dark conditions for 2 h. Subsequently, 100 μL of aliquots from each well was transferred into a 96-well black microtiter plate and fluorescence intensities were measured at excitation wavelength 540 nm and emission wavelength 590 nm (Tecan Infinite M200; Grödig, Austria). Cell viability was assessed according to the following equation:
2.8. Mucus preparation and permeation study of pp-liposomes
Freshly porcine intestines were obtained from a local slaughterhouse, and kept on ice during transport. The intestines were opened longitudinally, rinsed with ice-cold 0.1 M sodium chloride solution to remove debris. The mucus was gently scraped from the intestinal surface. Approximately 1 g of the collected mucus was suspended in 5 mL of 0.1 M sodium chloride and stirred at 100 rpm for 1 hour at 4 °C. The mixture was centrifuged at 10400g for 2 hours at 10 °C, and the clean mucus pellet was resuspended in half the original volume of sodium chloride solution. This process was repeated to obtain purified mucus, which was stored at −20 °C until use.
Mucus permeation experiments were conducted using Transwell® chambers with a pore size of 3 μm were utilized in a 24-well plate format. Approximately 50–60 mg of purified mucus was evenly spread on the Transwell® insert membranes. Calcein labeled liposomes (250 μL, 0.05% w/v in HBS) were added to the donor chambers, while 500 μL of HBS buffer filled the acceptor chambers. For controls, inserts without mucus served as a 100% permeation reference, while buffer alone acted as the 0% ref. 25. The plate was incubated at 37 °C under continuous shaking at 300 rpm for up to 6 hours. At set time points (0, 1, 2, 3, and 6 hours), 100 μL samples were collected from the acceptor chamber, and an equivalent volume of fresh HBS was added to maintain a constant volume. Fluorescence intensity (λex = 485 nm, λem = 535 nm) was measured in a 96-well microplate reader (Tecan Infinite M200; Grödig, Austria). Liposome permeation across the mucus layer was quantified as a percentage relative to the fluorescence intensity of the 100% reference control.
2.9. Cellular uptake studies
000 cells per well in 24-well plates and cultured for 10 days (to reach a fully confluent monolayer). Before exposure, cells were washed twice with warm HBS to remove serum. Fluorescently labeled liposomes (either uncoated or pp-liposomes, 0.05% w/v in HBS) were added to the cells and incubated for 4 hours or 24 hours at 37 °C. After incubation, the cells were washed twice with PBS to remove any liposomes that were not internalized. The cells were then detached by adding 150 μL of trypsin–EDTA solution (0.05%/0.02%) and incubating at 37 °C for ∼7 minutes. The trypsinization was stopped by adding 500 μL of MEM containing FBS. The cell suspension was collected and transferred to 15 mL Falcon tubes, then centrifuged at 800 rpm for 6 minutes to pellet the cells. The supernatant was discarded, and the cells were washed by resuspending in 3 mL of cold PBS followed by a second centrifugation (800 rpm, 6 minutes). The final cell pellet was resuspended in 500 μL of PBS. To ensure single-cell suspension, the cell suspension was passed through a 70 μm cell strainer, followed by a PBS rinse to collect all cells. The samples were analyzed on an Attune NxT flow cytometer (Thermo Fisher Scientific, MA, USA). For each sample, 10000 events were recorded. The relative mean fluorescence intensity (RMFI) of the cell populations was analyzed to quantify uptake on a per-cell basis. Data analysis was performed using FlowJo™ v10.8.1 software.
The live cells were then imaged using a Leica TCS SP8 confocal microscope (Leica Microsystems, Vienna, Austria) with appropriate laser and filter settings for DAPI (blue), DiD (red), and calcein (green). All images were captured using identical gain and exposure settings for consistency. Image analysis was performed using ImageJ software. For each field, z-stacks were collected (optical sections through the cell). Orthogonal yz- and xz-projections were reconstructed from five consecutive XY slices (z-step = 0.2 μm) to verify that green fluorescent signals (calcein-loaded liposomes) were inside the cell. A spectral unmixing algorithm was applied to minimize any bleed-through between channels. Additionally, a mild Gaussian blur filter was applied to improve image clarity and signal-to-noise ratio.29
2.10. Statistical data analysis
In this study, statistical analysis was performed using GraphPad Prism 5.01 software. Differences between two independent groups were assessed using an unpaired Student's t-test. Statistical significance levels were set at *p ≤ 0.05 for significant differences, **p < 0.01 for very significant differences and ***p < 0.001 for highly significant differences. Data were expressed as mean ± standard deviation (SD), calculated from at least three independent experiments.3. Results and discussion
3.1. Synthesis of oleyl-oligolysine
Oleyl-oligolysine was synthesized according to a multistep method16via ring opening polymerization of the beforehand synthesized N∈-(tert-butoxycarbonyl)-L-lysine N-carboxyanhydride (BOC-Lys-NCA) monomer with oleyl amine initiator. The synthetic procedure is summarized in Scheme 1. The chemical structure of the product was confirmed by 1H NMR and NOESY NMR, and the spectra are depicted in Fig. 1(A) and (B), respectively. Based on the integral values of the oleyl double bond (5.35 ppm) and the methine (4.25 ppm) protons of the lysine repeating units, the degree of polymerization of lysine is ∼7.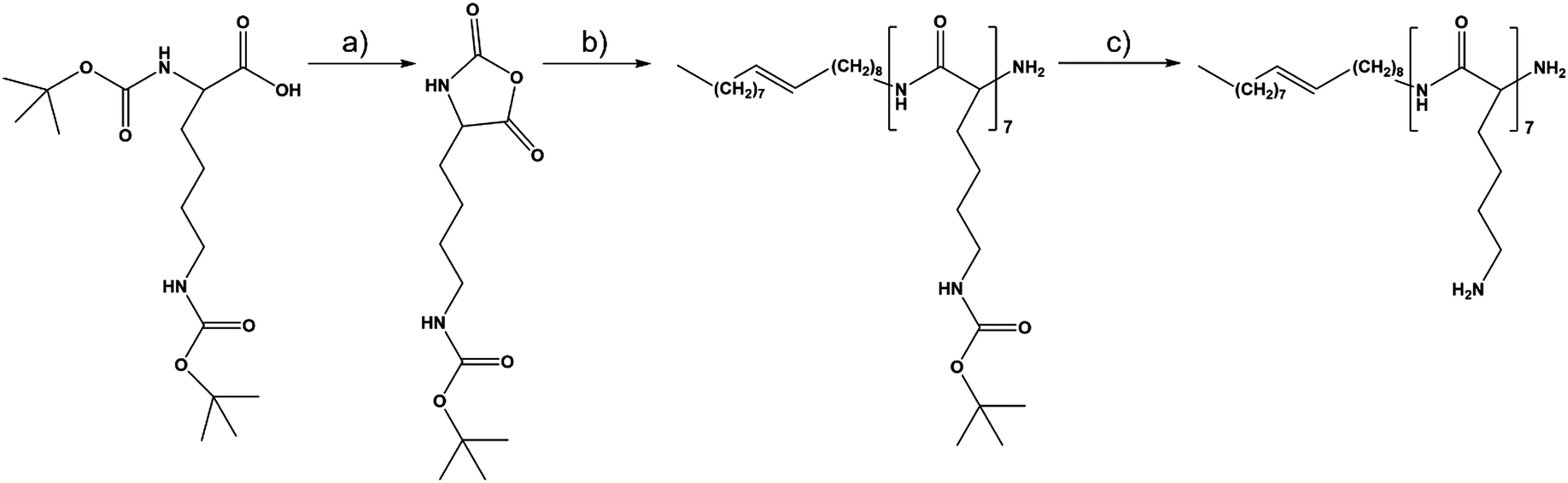 | ||
| Scheme 1 Synthetic pathways of oleyl-oligolysine via triphosgene-induced carboxyanhydride formation of lysine (a), followed by oleylamine-initiated polymerization (b) and subsequent deprotection using trifluoracetic acid in methylene chloride (c). | ||
 | ||
| Fig. 1 (A) 1H NMR and (B) NOESY spectra of the synthesized oleyl-oligolysine in DMSO-d6. | ||
3.2. Development of liposomes
Liposomes were successfully prepared by thin-film hydration, followed by ultrasonication to reduce particle size. The formulation contained DOPE, cholesterol, and oleyl-oligolysine, (1.5:1:0.2), each contributing specific functionalities for optimal stability. This ratio was chosen based on preliminary optimization studies aimed at achieving stability, sufficient membrane fluidity, and cationic surface potential while avoiding cytotoxicity, supported by previous literature.30–32 DOPE provides biocompatibility, low toxicity, and fusogenic properties suitable for effective drug delivery and cellular uptake. Cholesterol enhances membrane fluidity and structural integrity. The concentrations of DOPE and cholesterol were optimized to achieve a balance between membrane stability and flexibility.33,34 Oleyl-oligolysine introduces a cationic surface charge, promoting interactions with negatively charged cellular membranes and mucus layers, which is critical for improved adhesion and uptake. In addition to the size and surface charge differences detailed in Table 1, the stability of liposomes and pp-liposomes was evaluated in 25 mM HEPES buffer at 37 °C over 24 hours (Fig. 2) by measuring particle size and PDI. As shown in Fig. 2, a clear distinction was observed between liposomes and pp-liposomes. The initial size of liposomes was approximately 140 nm, while pp-liposomes displayed a larger size of around 170 nm, attributed to the additional phosphate coating. Throughout the 24-hour incubation period, both liposomes demonstrated remarkable stability, with particle sizes consistently in the ranges of 140–170 nm for liposomes and 170–210 nm for pp-liposomes, showing slight size variation. The PDI values for both formulations remained below 0.4 during the entire period, confirming a uniform size distribution and homogeneity. Both liposomes exhibited no significant aggregation or drastic changes in size over the 24-hour period, indicating good stability under the experimental conditions.
| Formulations | Size (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| Before coating with PP | 138.7 ± 2.9 | 0.106 ± 0.05 | 35.4 ± 1.5 |
| After coating with PP | 168.4 ± 1.2 | 0.121 ± 0.02 | −24.2 ± 2.5 |
 | ||
| Fig. 2 Size (filled bars) and PDI (stripped) of liposomes (pink) and pp-liposomes (green) in 25 mM HEPES buffer at indicated time points at 37 °C. Data are shown as mean ± SD (n = 3). | ||
3.3. Phosphate release studies
The time-dependent release of phosphate groups from pp-liposomes was analyzed using MLG assay in the absence and presence of AP (10 U mL−1), as illustrated in Fig. 3A. In the presence of AP, a rapid release of phosphate was observed during the first 6 hours, reaching 512.6 ± 18.4 μmol g−1. After this initial phase, the rate of phosphate release slowed, culminating in a total release of 537.8 ± 12.7 μmol g−1 at 24 hours. In contrast, negligible phosphate release was detected in the absence of AP, underscoring the pivotal role of enzymatic activity in the cleavage of phosphate groups. The initial rapid release phase within the first 6 hours can be attributed to efficient enzymatic hydrolysis of surface-bound phosphate groups. The subsequent plateau phase suggests either the depletion of readily accessible phosphate groups or reduced enzymatic efficiency due to substrate limitations or structural changes in the liposomal surface. These findings align with previous studies investigating the enzymatic cleavage of polyphosphate chains, which demonstrated a time-dependent release of free phosphate associated with the catalytic activity of AP. Similar trends have been reported for polyphosphates with chain lengths ranging from 12–23 residues, where the majority of phosphate release occurred within the first few hours of enzymatic treatment. Caco-2 cell monolayers have been shown to express AP. The presence of AP in these cells was verified by Prüfert et al. using immunocytochemistry.35 To evaluate phosphate release under physiological conditions, pp-liposomes were incubated on Caco-2 cell monolayers, as depicted in Fig. 3B. In the absence of a phosphatase inhibitor cocktail, phosphate release followed a kinetic profile similar to that observed with isolated AP, reaching 122.8 ± 6.3 μmol g−1 at 24 hours. However, when the cells were treated with a phosphatase inhibitor cocktail, phosphate release was significantly reduced, achieving only 74.6 ± 4.1 μmol g−1 at 24 hours. This reduction highlights the inhibitory effect of the cocktail on phosphatase activity, as well as the role of cellular alkaline phosphatases in mediating phosphate release. Caco-2 cells cleaved the phosphate group to a significantly lower extent compared to isolated AP. This observation might be explained by several washing steps prior to the experiment, which can lead to a partial loss of AP by Caco-2.18 | ||
| Fig. 3 Phosphate release from pp-liposomes by enzymatic cleavage with (blue) and without (red) isolated AP (10 U mL−1) at 37 °C over time (A), phosphate release from pp-liposomes on Caco-2 cell monolayer in the absence (blue) and presence (red) of phosphate inhibitor cocktail II as a function of time. (B) Data are shown as means ± SD (n = 3), ***p ≤ 0.001, **p ≤ 0.01. | ||
3.4. Charge conversion
Phosphate release studies were supported by time-dependent zeta potential measurements of pp-liposomes. As illustrated in Fig. 4, upon incubation with AP, a notable zeta potential shift of Δ 37.1 mV was observed over 24 hours. In contrast, negligible changes were detected without AP, indicating that charge conversion is directly related to enzymatic activity. The most pronounced change occurred during the first 6 hours, during which the zeta potential rapidly increased from −24.2 ± 2.6 mV to −1.4 ± 4.45 mV. This initial phase corresponds to efficient enzymatic hydrolysis of surface-bound phosphate groups, leading to a substantial reduction in the anionic charge of the liposomal surface. Beyond the 6-hour mark, the rate of zeta potential change slowed down, suggesting the onset of a plateau phase where fewer accessible phosphate groups remained for enzymatic cleavage. By 24 hours, liposomes exhibited a stabilized positive zeta potential of approximately +13 mV. This charge conversion over time was in good agreement with the phosphate release from pp-liposomes. The observed shift in zeta potential highlights the dynamic nature of the liposomal surface charge in response to phosphatase activity. The removal of negatively charged phosphate groups exposes underlying cationic structures causing charge conversion. This behavior aligns with previous findings demonstrating similar zeta potential shifts in phosphorylated nanoparticles after phosphatase treatment. Nguyen Le et al.7 confirmed that PP-coated nanoemulsions exhibit charge conversion from a negative value up to +8.5 mV due to cleavage of phosphates by AP. | ||
| Fig. 4 Time-dependent charge conversion of pp-liposomes with (blue) and without (red) isolated AP (10 U mL−1). Data are shown as means ± SD (n = 3), ***p ≤ 0.001. | ||
3.5. Cytotoxicity and hemolysis
The cytotoxicity of liposomes was evaluated by resazurin assay on Caco-2 cells after 4 and 24 h of exposure at various concentrations. As depicted in Fig. 5, for both incubation periods, pp-liposomes demonstrated significantly lower cytotoxicity compared to uncoated liposomes. At the highest tested concentration (30 μmol mL−1), cell viability was markedly reduced for both liposomes, with uncoated liposomes showing a more pronounced effect. At lower concentrations (1.86–7.5 μmol mL−1), pp-liposomes consistently maintained higher cell viability, highlighting their improved biocompatibility. Over 24 h a similar trend was observed, with pp-liposomes showing lower cytotoxic effects compared to uncoated liposomes across all concentrations. The hemolytic activity of the liposomes was assessed to evaluate their effect on red blood cells. After 4 hours of incubation, liposomes exhibited significantly higher hemolytic activity compared to pp-liposomes, especially at higher concentrations (0.5% and 0.25%), with hemolysis exceeding 80% for liposomes at 0.5%. In contrast, pp-liposomes caused considerably lower hemolysis, demonstrating improved membrane compatibility. At lower concentrations (0.1%), both formulations showed lower hemolysis, with pp-liposomes maintaining a more favorable profile. After 24 hours of incubation, pp-liposomes showed lower hemolytic activity compared to uncoated liposomes at all tested concentrations. A dose-dependent decrease in hemolysis was observed for both liposomes, pp-liposomes exhibited a markedly safer profile. | ||
| Fig. 5 Cell viability of Caco-2 cells exposed to uncoated liposomes (pink) and pp-liposomes (green) for 4 h (A) and 24 h (B) and hemolysis effect on red blood cells after 4 h (C) and 24 h (D) of incubation at 37 °C. All results are shown as mean ± SD, n = 3, ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05. | ||
DOPE enhances liposome biocompatibility by reducing toxicity, promoting membrane stability, and mitigating the harmful effects of cationic surfactants.36 Cholesterol further supports membrane integrity and fluidity without causing significant hemolytic activity, ensuring the liposomes remain structurally stable and non-toxic.36,37 In contrast, oleyl-oligolysine, while beneficial for imparting a positive surface charge and enhancing interactions with negatively charged biological membranes, is known to exhibit cytotoxic properties. Its strong cationic nature can destabilize cell membranes, resulting in increased hemolysis and limited biocompatibility.32 The observed hemolytic and cytotoxic effects in this study are primarily attributed to the presence of oleyl-oligolysine.
3.6. Mucus permeation
The mucus permeation ability of calcein-loaded pp-liposomes was evaluated using an in vitro model incorporating purified porcine intestinal mucus and Transwell® inserts. This model mimics the complex physiological properties of intestinal mucus, characterized by negatively charged and hydrophobic domains that pose significant challenges to nanocarrier transport. Fig. 6 showed that after 6 h, pp-liposomes displayed an approximate 12-fold increase in mucus permeability, compared to de-pp-liposomes, which were generated by treating pp liposomes with AP to cleave the phosphate groups. This enhanced permeation of pp-liposomes can be attributed to the electrostatic repulsion between negatively charged phosphate groups on liposomes and sialic and sulfonic acid moieties of the mucus gel layer. These repulsive forces facilitate liposome movement through the mucus, overcoming the immobilization observed with positively charged nanocarriers. The hydrophilic nature of the phosphate groups on pp-liposomes further enhances mucus permeability by preventing hydrophobic interactions that can hinder effective transport through the mucus layer.38 | ||
| Fig. 6 Permeation studies of calcein-labeled pp-liposomes through native porcine intestinal mucus using transwell chambers at 37 °C for 6 h with (yellow) and without AP (blue). Data is presented as mean ± SD (n = 3), ***p ≤ 0.001, **p ≤ 0.01. | ||
This observation aligns with previous findings by Akkus et al.,18 who reported superior mucus diffusion of pp-nanocarriers compared to de-pp-nanocarriers. Similarly, studies by Mahlert et al.39 demonstrated that nanoparticles with hydrophilic and muco-inert coatings, such as poly(ethylene)glycol (PEG), exhibited greater mucus permeation than hydrophobic or positively charged coatings, such as chitosan. The results of this study provide evidence for the ability of the coated particles to permeate the mucus layer.
3.7. Cellular uptake
By combining flow cytometry and confocal microscopy, both quantitative and qualitative analyses of liposome uptake and intracellular localization were conducted, providing a robust and comprehensive understanding of cellular internalization dynamics. Liposomes interact with cells through multiple pathways: adsorption onto the cell surface with subsequent extracellular release of their contents; endocytosis via clathrin-mediated or clathrin-independent mechanisms; lipid exchange, where lipophilic molecules transfer from the liposomal bilayer to the cellular membrane; and fusion with intracellular membranes, resulting in direct cytosolic delivery of encapsulated agents.40In confocal laser scanning microscopy experiments, the nuclei of the cells were stained with DAPI, a nucleic acid-binding dye that emits blue fluorescence, allowing for clear visualization of nuclear regions. The cellular membranes were labeled with DID, a red fluorescent membrane stain. The internalization of calcein-loaded liposomes was indicated by green fluorescence signals observed within the cytoplasmic regions, confirming successful uptake. As shown in Fig. 7A, liposomes exhibited higher fluorescence intensity after 4 h of incubation when compared to pp-liposomes. This suggests that cationic nanoparticles, such as liposomes, demonstrate enhanced cellular uptake. The observed dotted green signals within the cytoplasm and near the nuclei further indicate endocytic uptake, aligning with the size range of the liposomes.41
 | ||
| Fig. 7 Confocal laser scanning microscopy images of calcein-loaded liposomes. Nucleus was stained with DAPI, while cellular membranes were labeled with DID. The white bar on the bottom of the image represents a length of 20 μm (A). Relative mean fluorescent intensity (RMFI) of pp-liposomes (green) and liposomes (red) in Caco-2 cell line at 37 °C after 4 h of incubation and 24 h (striped green and striped red) (B). Shifts in fluorescence intensity after 4 h (C) and 24 h (D). Indicated values are means ± SD, n = 3, ***p ≤ 0.001. | ||
Flow cytometry analysis confirmed substantial cellular uptake of both liposomes. Quantitative analysis of RMFI further validated these trends (Fig. 7B), showing that liposomes exhibited 2-times greater uptake after 4 h of incubation. However, after 24 h, both liposomes demonstrated comparable levels of internalization. The shift in fluorescence intensity through higher values indicated improved cellular uptake (Fig. 7C and D). The initially lower fluorescence intensity of pp-liposomes after 4 h may be attributed to their anionic surface charge, which hinders interaction with the negatively charged cellular membranes. These findings suggest that the charge conversion of pp-liposomes improved electrostatic interactions with cellular membranes, ultimately facilitating more efficient cellular internalization.
4. Conclusion
This study confirms the successful development of charge-converting liposomes, providing a novel and effective solution to the polycationic dilemma in drug delivery. By leveraging polyphosphate coatings, we achieved stable, negatively charged liposomal carriers that efficiently traversed the mucus barrier. The hypothesis that enzymatic dephosphorylation of the polyphosphate layer by AP would induce a charge conversion was validated, as liposomes transitioned from a negatively charged state to a significantly positive zeta potential upon enzymatic activation. The use of oleyl-oligolysine as a polycationic peptide incorporated into the liposomal membrane played a pivotal role in this charge transition. The enzymatic cleavage of surface polyphosphates triggered a controlled and substantial shift in zeta potential, markedly enhancing electrostatic interactions with epithelial cells while maintaining biocompatibility. This mechanism facilitated improved cellular uptake without excessive cytotoxicity or hemolytic effects, addressing a critical challenge in the design of polycationic drug carriers.These findings underscore the potential of charge-converting liposomes as a highly promising platform for mucosal drug delivery, capable of enhancing intestinal absorption and therapeutic bioavailability.
Author contributions
Martyna Truszkowska: conceptualization, methodology, investigation, writing – original draft, visualization, writing – rewiev & editing. Ahmad Saleh: methodology, investigation. Melanie Lena Ebert: methodology, investigation. Gergely Kali: methodology, investigation, writing – review & editing. Andreas Bernkop-Schnürch: conceptualization, writing – review & editing, funding acquisition, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the experimental part.Acknowledgements
Martyna Truszkowska and Ahmad Saleh received a doctoral scholarship for the promotion of young researchers from “Doktoratsstipendium aus der Nachwuchsförderung programme” of Leopold-Franzens-University of Innsbruck, Austria.References
- S. K. Lai, Y.-Y. Wang and J. Hanes, Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues, Adv. Drug Delivery Rev., 2009, 61(2), 158–171, DOI:10.1016/j.addr.2008.11.002.
- H. H. Sigurdsson, J. Kirch and C.-M. Lehr, Mucus as a barrier to lipophilic drugs, Int. J. Pharm., 2013, 453(1), 56–64, DOI:10.1016/j.ijpharm.2013.05.040.
- F. Veider, E. Sanchez Armengol and A. Bernkop-Schnürch, Charge-Reversible Nanoparticles: Advanced Delivery Systems for Therapy and Diagnosis, Small, 2024, 20(3), 1–19, DOI:10.1002/smll.202304713.
- J. Griesser, G. Hetényi and A. Bernkop-Schnürch, Thiolated hyaluronic acid as versatile mucoadhesive polymer: From the chemistry behind to product developments-What are the capabilities?, Polymers, 2018, 10, 243, DOI:10.3390/polym10030243.
- S. Bonengel and A. Bernkop-Schnürch, Thiomers—From bench to market, J. Controlled Release, 2014, 195, 120–129, DOI:10.1016/j.jconrel.2014.06.047.
- A. Arshad, et al., Zeta potential changing self-nanoemulsifying drug delivery systems: A newfangled approach for enhancing oral bioavailability of poorly soluble drugs, Int. J. Pharm., 2024, 655, 123998, DOI:10.1016/j.ijpharm.2024.123998.
- N. M. Nguyen Le, et al., Charge-Converting Nanoemulsions as Promising Retinal Drug and Gene Delivery Systems, ACS Appl. Mater. Interfaces, 2022, 14(39), 44981–44991, DOI:10.1021/acsami.2c11649.
- E. Salimi, B. Le-Vinh, F. Zahir-Jouzdani, B. Matuszczak, A. Ghaee and A. Bernkop-Schnürch, Self-emulsifying drug delivery systems changing their zeta potential via a flip-flop mechanism, Int. J. Pharm., 2018, 550(1), 200–206, DOI:10.1016/j.ijpharm.2018.08.046.
- S. Bonengel, F. Prüfert, M. Jelkmann and A. Bernkop-Schnürch, Zeta potential changing phosphorylated nanocomplexes for pDNA delivery, Int. J. Pharm., 2016, 504(1), 117–124, DOI:10.1016/j.ijpharm.2015.10.021.
- I. Nazir, C. Leichner, B. Le-Vinh and A. Bernkop-Schnürch, Surface phosphorylation of nanoparticles by hexokinase: A powerful tool for cellular uptake improvement, J. Colloid Interface Sci., 2018, 516, 384–391, DOI:10.1016/j.jcis.2018.01.082.
- S. Bonengel, F. Prüfert, G. Perera, J. Schauer and A. Bernkop-Schnürch, Polyethylene imine-6-phosphogluconic acid nanoparticles – a novel zeta potential changing system, Int. J. Pharm., 2015, 483(1), 19–25, DOI:10.1016/j.ijpharm.2015.01.041.
- G. Perera, M. Zipser, S. Bonengel, W. Salvenmoser and A. Bernkop-Schnürch, Development of phosphorylated nanoparticles as zeta potential inverting systems, Eur. J. Pharm. Biopharm., 2015, 97, 250–256, DOI:10.1016/j.ejpb.2015.01.017.
- E. K. Perttu, A. G. Kohli and F. C. Szoka, Inverse-phosphocholine lipids: A remix of a common phospholipid, J. Am. Chem. Soc., 2012, 134(10), 4485–4488, DOI:10.1021/ja210989h.
- B. Le-Vinh, Z. B. Akkuş-Dağdeviren, N. M. N. Le, I. Nazir and A. Bernkop-Schnürch, Alkaline Phosphatase: A Reliable Endogenous Partner for Drug Delivery and Diagnostics, Adv. Ther., 2022, 5, 202100219, DOI:10.1002/adtp.202100219.
- J. L. Millán, Alkaline Phosphatases, Dyn. Bone Cartilage Metab., 2006, 153–164, DOI:10.1007/s11302-005-5435-6.
- A. Saleh, D. Stengel, M. Truszkowska, M. Blanco Massani, G. Kali and A. Bernkop-Schnürch, Nanostructured lipid carriers decorated with polyphosphate coated linear and loop cell-penetrating peptides, Int. J. Pharm., 2024, 667, 124844, DOI:10.1016/j.ijpharm.2024.124844.
- Ö. Çoban and Z. Deǧim, Liposomes containing imatinib mesylate and dexketoprofen trometamol: Development and characterization, FABAD J. Pharm. Sci., 2013, 38(3), 121–126 Search PubMed.
- Z. B. Akkus, I. Nazir, A. Jalil, M. Tribus and A. Bernkop-Schnürch, Zeta Potential Changing Polyphosphate Nanoparticles: A Promising Approach to Overcome the Mucus and Epithelial Barrier, Mol. Pharm., 2019, 16(6), 2817–2825, DOI:10.1021/acs.molpharmaceut.9b00355.
- B. Maherani, E. Arab-Tehrany, A. Kheirolomoom, D. Geny and M. Linder, Calcein release behavior from liposomal bilayer; influence of physicochemical/mechanical/structural properties of lipids, Biochimie, 2013, 95(11), 2018–2033, DOI:10.1016/j.biochi.2013.07.006.
- J. Feng, et al., An improved malachite green assay of phosphate: Mechanism and application, Anal. Biochem., 2011, 409(1), 144–149, DOI:10.1016/j.ab.2010.10.025.
- A. Saleh, Z. B. Akkuş-Dağdeviren, J. D. Friedl, P. Knoll and A. Bernkop-Schnürch, Chitosan – Polyphosphate nanoparticles for a targeted drug release at the absorption membrane, Heliyon, 2022, 8, e10577, DOI:10.1016/j.heliyon.2022.e10577.
- C. Jumarie and C. Malo, Alkaline phosphatase and peptidase activities in Caco-2 cells: Differential response to triiodothyronine, In Vitro Cell. Dev. Biol.: Anim., 1994, 30(11), 753–760, DOI:10.1007/BF02631298.
- B. C. Evans, et al., Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs, J. Vis. Exp., 2013, 73, 6–10, DOI:10.3791/50166.
- J. D. Friedl, C. Steinbring, S. Zaichik, N. M. N. Le and A. Bernkop-Schnürch, Cellular uptake of self-emulsifying drug-delivery systems: Polyethylene glycol versus polyglycerol surface, Nanomedicine, 2020, 15(19), 1829–1841, DOI:10.2217/nnm-2020-0127.
- I. Pepić, J. Lovrić and J. Filipović-Grčić, How do polymeric micelles cross epithelial barriers?, Eur. J. Pharm. Sci., 2013, 50(1), 42–55, DOI:10.1016/j.ejps.2013.04.012.
- P. Knoll, N. Hörmann, N. M. Nguyen Le, R. Wibel, R. Gust and A. Bernkop-Schnürch, Charge converting nanostructured lipid carriers containing a cell-penetrating peptide for enhanced cellular uptake, J. Colloid Interface Sci., 2022, 628, 463–475, DOI:10.1016/J.JCIS.2022.07.160.
- K. Zöller, A. Karlegger, M. Truszkowska, D. Stengel and A. Bernkop-Schnürch, Fluorescent hydrophobic ion pairs: A powerful tool to investigate cellular uptake of hydrophobic drug complexes via lipid-based nanocarriers, J. Colloid Interface Sci., 2024, 654, 174–188, DOI:10.1016/j.jcis.2023.10.001.
- J. D. Friedl, C. Steinbring, S. Zaichik, N. M. N. Le and A. Bernkop-Schnürch, Cellular uptake of self-emulsifying drug-delivery systems: Polyethylene glycol versus polyglycerol surface, Nanomedicine, 2020, 15(19), 1829–1841, DOI:10.2217/nnm-2020-0127.
- Ö. Kaplan, et al., Thiolated α-cyclodextrin: The likely smallest drug carrier providing enhanced cellular uptake and endosomal escape, Carbohydr. Polym., 2023, 316, 121070, DOI:10.1016/j.carbpol.2023.121070.
- A. Rodríguez-Pulido, F. Ortega, O. Llorca, E. Aicart and E. Junquera, A physicochemical characterization of the interaction between DC-Chol/DOPE cationic liposomes and DNA, J. Phys. Chem. B, 2008, 112(39), 12555–12565, DOI:10.1021/jp804066t.
- M. R. Krause and S. L. Regen, The structural role of cholesterol in cell membranes: From condensed bilayers to lipid rafts, Acc. Chem. Res., 2014, 47(12), 3512–3521, DOI:10.1021/ar500260t.
- E. Fröhlich, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int. J. Nanomed., 2012, 7, 5577–5591, DOI:10.2147/IJN.S36111.
- V. M. B. Grace, D. D. Wilson, C. Guruvayoorappan, J. P. Danisha and L. Bonati, Liposome nano-formulation with cationic polar lipid DOTAP and cholesterol as a suitable pH-responsive carrier for molecular therapeutic drug (all-trans retinoic acid) delivery to lung cancer cells, IET Nanobiotechnol., 2021, 15(4), 380–390, DOI:10.1049/nbt2.12028.
- M. L. Briuglia, C. Rotella, A. McFarlane and D. A. Lamprou, Influence of cholesterol on liposome stability and on in vitro drug release, Drug Delivery Transl. Res., 2015, 5(3), 231–242, DOI:10.1007/s13346-015-0220-8.
- F. Prüfert, et al., ζ Potential Changing Nanoparticles As Cystic Fibrosis Transmembrane Conductance Regulator Gene Delivery System: an In Vitro Evaluation, Nanomedicine, 2017, 12(22), 2713–2724, DOI:10.2217/nnm-2017-0115.
- A. Rodríguez-Pulido, F. Ortega, O. Llorca, E. Aicart and E. Junquera, A physicochemical characterization of the interaction between DC-Chol/DOPE cationic liposomes and DNA, J. Phys. Chem. B, 2008, 112(39), 12555–12565, DOI:10.1021/jp804066t.
- M. R. Krause and S. L. Regen, The structural role of cholesterol in cell membranes: From condensed bilayers to lipid rafts, Acc. Chem. Res., 2014, 47(12), 3512–3521, DOI:10.1021/ar500260t.
- J. Leal, H. D. C. Smyth and D. Ghosh, Physicochemical properties of mucus and their impact on transmucosal drug delivery, Int. J. Pharm., 2017, 532(1), 555–572, DOI:10.1016/j.ijpharm.2017.09.018.
- L. Mahlert, J. Anderski, D. Mulac and K. Langer, The impact of gastrointestinal mucus on nanoparticle penetration – in vitro evaluation of mucus-penetrating nanoparticles for photodynamic therapy, Eur. J. Pharm. Sci., 2019, 133, 28–39, DOI:10.1016/j.ejps.2019.03.010.
- E. Ducat, B. Evrard, O. Peulen and G. Piel, Cellular uptake of liposomes monitored by confocal microscopy and flow cytometry, J. Drug Delivery Sci. Technol., 2011, 21(6), 469–477, DOI:10.1016/S1773-2247(11)50076-0.
- K. Cleal, L. He, P. D. Watson and A. T. Jones, Endocytosis, Intracellular Traffic and Fate of Cell Penetrating Peptide Based Conjugates and Nanoparticles, Curr. Pharm. Des., 2013, 19(16), 2878–2894, DOI:10.2174/13816128113199990297.
| This journal is © The Royal Society of Chemistry 2025 |