
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual and sequential drug delivery systems with antimicrobial and bone regenerative therapeutic effects
Miguel A.
Rodrigues
 a,
Carla
Ferreira
b,
João P.
Borges
c,
Beatriz G.
Bernardes
de,
Ana L.
Oliveira
d,
José D.
Santos
a and
Maria A.
Lopes
*a
a,
Carla
Ferreira
b,
João P.
Borges
c,
Beatriz G.
Bernardes
de,
Ana L.
Oliveira
d,
José D.
Santos
a and
Maria A.
Lopes
*a
aLAQV-REQUIMTE, FEUP, Department of Mechanical Engineering, University of Porto, Rua Dr Roberto Frias, s/n, 4200-465, Portugal. E-mail: malopes@fe.up.pt
bDepartment of Chemical and Biological Engineering, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, Porto, 4200-465, Portugal
cCENIMAT|i3N, Department of Materials Science, School of Science and Technology, NOVA University Lisbon, Caparica, Portugal
dUniversidade Católica Portuguesa, CBQF – Centro de Biotecnologia e Química Fina – Laboratório Associado, Escola Superior de Biotecnologia, Rua Diogo Botelho 1327, 4169-005 Porto, Portugal
eAerogelsLab, Department of Pharmacology, Pharmacy and Pharmaceutical Technology, I + D Farma group (GI-1645), iMATUS and Health Research Institute of Santiago de Compostela (IDIS), Universidade de Santiago de Compostela, E-15782 Santiago de Compostela, Spain
First published on 20th June 2025
Abstract
Bone defect healing is often compromised by infections acquired during surgery, hindering regeneration. An effective solution should first prevent infection and then promote bone repair. Localised drug-delivery systems capable of dual and sequential release of antimicrobial and bone-regenerative agents represent a promising solution; however, precisely controlling this sequential release remains an unmet challenge. To address this issue, this study explores a novel approach by developing delivery systems based on either hollow or non-hollow porous bioceramics with an alginate hydrogel matrix, resulting in cutting-edge systems with a controlled, stage-specific release of antimicrobial and bone regenerative agents that meet the clinical needs. Gentamicin served as the antimicrobial agent, while raloxifene and/or alendronate represented hydrophobic and hydrophilic bone-regenerative agents. The systems were evaluated for release profiles, kinetic modelling, and the effects of lyophilisation and sterilisation (using ethylene oxide or supercritical CO2) on drug stability and release kinetics. The release followed a precise dual-sequential pattern: gentamicin was released over 2–3 weeks, followed by another 2–3 weeks of bone-regenerative agents. Kinetic model fitting showed that gentamicin release was driven mainly by diffusion (with or without hydrogel swelling), and raloxifene/alendronate release was dominated by a mixture of diffusion and polymeric matrix swelling/erosion. Lyophilisation and sterilisation preserved release profiles, though timeframes shifted slightly, with supercritical CO2 causing minimal delay. Gentamicin retained strong antimicrobial activity post-processing, confirming the system's potential for infection control and bone repair.
Introduction
The development of delivery systems with dual antimicrobial and bone regenerative action represents an up-and-coming area of research in regenerative medicine and orthopaedics. Such systems aim to address two critical issues in bone healing: preventing infection or avoiding reappearance after infection control, especially in bone injury or implantation cases, while simultaneously promoting bone regeneration.1–4In particular, bioceramic delivery systems have gained prominence in tissue engineering and regenerative medicine, particularly in orthopaedic and dental applications, due to their ability to simultaneously deliver therapeutic agents and serve as scaffolds for tissue regeneration.1,2 The dual release is crucial in bone regenerative surgery since reports show that every year, in Europe and the United States, around 2 million bone regenerative surgeries are performed, with an infection rate during surgery of around 3%.5,6
By delivering antimicrobial or therapeutic agents directly to the site of injury or surgery, bioceramic systems reduce the amount of drug required systemically to achieve effective concentrations.7,8 This localised delivery minimises the risk of systemic side effects to the patient, often associated with the oral or intravenous administration of antibiotics or other drugs, leading to improvement of drug targeting, efficacy, and therefore bioavailability.8–10
In recent years, significant progress has been made in the development of drug delivery systems (DDS) with both antimicrobial and bone regenerative features, with a special focus on localised administration. Several studies have shown that biodegradable hydrogels, 3D printed scaffolds and composites based on polymers such as chitosan or polylactic acid can be functionalised with antibiotics and osteoinductive agents, allowing sustained and localised release, with simultaneous improvement of antimicrobial activity and osteogenesis.11–16 These 3D-printed scaffolds were also incorporated with antibiotics and bioactive nanoparticles (such as mesoporous silica doped with metal oxides) and have shown significant efficacy in eradicating bacterial biofilms and promoting bone regeneration through sequential and synergistic release mechanisms.17 Another approach in this area is the use of materials that are responsive to specific stimuli in the infectious microenvironment, such as local acidity or the presence of reactive oxygen species (ROS), to selectively trigger the release of drugs.18–20 For example, GelMA-based hydrogels (methacrylate gelatine) functionalised with amikacin demonstrated controlled release in response to the acidic pH typical of infected tissues, increasing antimicrobial efficacy and minimising systemic effects.21 However, the outputs of these research works fail to reach the market due to inadequate mechanical properties of the polymeric materials.
This way, research has been made focusing on bioceramic-based solutions for DDSs. One example is scaffolds based on mesoporous bioactive glass doped with zinc oxide (ZnO), capable of releasing Zn2+ ions with antibacterial activity while promoting osteoblastic differentiation. In a recent study, these scaffolds were functionalised with antibiotics such as levofloxacin, vancomycin, rifampicin or gentamicin, demonstrating effective destruction of Staphylococcus aureus and Escherichia coli biofilms and stimulating bone regeneration in vitro.22 In addition, hierarchical scaffolds have been developed with the capacity for sequential antibiotic release. One example is devices composed of bioceramics and polyvinyl alcohol, loaded with rifampicin, levofloxacin and vancomycin. This system has been shown to be effective in destroying resistant bacterial biofilms and inducing osteogenesis due to its high bioactivity and biocompatibility.23 Another relevant approach is the incorporation of antibiotics and angiogenic factors, such as VEGF, into apatite and agarose scaffolds using mesoporous silica nanoparticles. These systems allowed for a dual release of cephalexin and VEGF, resulting in both antibacterial and pro-angiogenic properties.24 S53P4 bioactive glass, on the other hand, stands out for its intrinsic antibacterial properties, which do not require the incorporation of antibiotics. Its composition allows for the release of ions that raise the local pH and osmotic pressure, inhibiting bacterial growth while simultaneously stimulating bone mineralisation. This material has been used successfully in clinical settings to treat chronic bone infections.25 Since these solutions are bioceramic-based, they fulfil the mechanical properties required for the bone healing approach; however, they lack the ability to control the release of agents that are usually accomplished with biopolymer-based carriers. Precise control of the release kinetics of multiple agents and maintaining drug stability during the manufacturing process are areas that require further research. Also, these few systems release antimicrobial and bone regenerative agents in a single burst or over short periods of time, usually hours instead of days, which can be ineffective in addressing the temporal demands of infection control and bone healing.26 An adequate release profile is then critical for the success of such delivery systems. Ideally, antimicrobial agents should be released rapidly yet in a controlled manner, ensuring high local concentrations to eradicate pathogens without promoting systemic toxicity or drug resistance.8,27,28 Following this, bone regenerative agents, such as growth factors or osteoregenerative molecules, should be released over a prolonged period to support the bone healing process. Achieving this delicate balanced sequence requires careful design of the bioceramic matrix and incorporation of delivery technologies that allow for precise control over degradation rates and drug release kinetics.
Lyophilisation and sterilisation are critical steps in developing bioceramic delivery systems, ensuring not only their safety and stability prior to clinical use but also a decrease in transportation costs. Lyophilisation, or freeze-drying, is commonly employed to improve the storage stability of bioceramic scaffolds and bioactive agents encapsulated within them. However, these processes can significantly impact the release profile of the antimicrobial and bone regeneration agents. Lyophilisation can alter the physical properties of the scaffold, potentially affecting porosity, degradation rate, and drug release kinetics. Sterilisation methods, particularly those involving heat or radiation, may degrade sensitive bioactive compounds, reducing efficacy. Therefore, careful optimisation of lyophilisation and sterilisation protocols is necessary to ensure they do not compromise the therapeutic performance of the bioceramic system.29–31
This way, the current work aimed at developing and characterising DDSs for bone regeneration, in which there is a controlled early-stage release of an antimicrobial agent to combat infections in a post-surgery setting and a later-stage release of a bone regenerative agent to accelerate the tissue healing process. Hollow and non-hollow bioceramic microspheres will be used as templates for the DDSs, and the impact of lyophilisation and sterilisation processes on the agents’ release profiles and activity will also be assessed.
Experimental section
Materials
Alginic acid sodium salt, Mw: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–600000 g mol−1 was obtained from PanReac AppliChem; Alendronate sodium trihydrate from TCI Chemicals. Phosphate buffer saline tablets, calcium chloride, sodium chloride, sodium hydroxide, methanol, 2-mercaptoethanol, phthalaldehyde, boric acid, potassium hydroxide, cerium sulphate, sulfuric acid, raloxifene hydrochloride and gentamicin sulphate were obtained from Sigma-Aldrich.
000–600000 g mol−1 was obtained from PanReac AppliChem; Alendronate sodium trihydrate from TCI Chemicals. Phosphate buffer saline tablets, calcium chloride, sodium chloride, sodium hydroxide, methanol, 2-mercaptoethanol, phthalaldehyde, boric acid, potassium hydroxide, cerium sulphate, sulfuric acid, raloxifene hydrochloride and gentamicin sulphate were obtained from Sigma-Aldrich.
Drug delivery systems development
The DDSs produced were based on hollow and non-hollow bioceramic porous microspheres, whose development and characterisation were described previously by the authors (under submission). Briefly, the microspheres were produced by a foaming injection method, where a ceramic foam, composed of a ceramic suspension and a foam solution, was crosslinked into a calcium chloride solution to produce microspheres. The ceramic suspension was made by mixing hydroxyapatite (HA) powder and sodium alginate solution (40% w/w HA in 2% w/v alginate solution), while the foam solution consisted of an aqueous solution (10% w/v) of Pluronic F-127. For hollow microsphere production, a coaxial mould was used, injecting sunflower oil into an inner channel, while the outer channel contained the ceramic foam. After dehydration, both hollow and non-hollow microspheres were sintered, remaining only the inorganic phase (hydroxyapatite), leaving pores where the foaming agent was present, as well as the sunflower oil in the case of hollow microspheres, yielding a core–shell structure in this case. Hollow and non-hollow microspheres were then functionalised to make DDSs. Each DDS is comprised of bioceramic functionalised with an antimicrobial and bone-regenerative agent. A single antimicrobial agent was used as a model (gentamicin), but two therapeutical agents were used as bone-regenerative agents: raloxifene (hydrophobic) and alendronate (hydrophilic). Therefore, the dual and sequential release systems were labelled as follows: (1) hollow delivery system with gentamicin and raloxifene (HDS1); (2) hollow delivery system with gentamicin and alendronate (HDS2); (3) non-hollow delivery system with gentamicin and raloxifene (n-HDS1); and (4) non-hollow delivery system with gentamicin and raloxifene (n-HDS2). The rationale of developing hollow and non-hollow delivery systems was to allow for more possibilities to control the loading and release of the therapeutic agents, meaning that gentamicin should be released first with a delayed release of the raloxifene/alendronate.The functionalisation with the agents was done in reverse order to the expected release, meaning that the bone regenerative agent was loaded first to be released last, and gentamicin was loaded last to be released first. The bone regenerative agent, either raloxifene (10 mg mL−1 in methanol) or alendronate (0.5 mg mL−1), was added to a sodium alginate aqueous solution (2 wt%) in 1:10 volume proportion and mixed at RT overnight. 50 μL of this solution was added per microsphere, and the mixture was submitted to vacuum for 2 hours. The remaining liquid from the vacuum process was collected and saved for later raloxifene/alendronate loading efficiency analysis. The samples were placed in a calcium chloride (CaCl2) solution (2 w/v%, 50 μL per microsphere) and subjected to vacuum for 30 min. To functionalise with gentamicin, the samples were washed with distilled water, and the microspheres were loaded with 50 μL per microsphere of alginate solution followed by 2 hours of vacuum, washing, crosslinking with CaCl2 under vacuum for 30 min, washing, after which was added 50 μL of gentamicin aqueous solution (2 mg mL−1) per microsphere, and put under vacuum for 2 hours, followed by another washing and crosslinking step.
Loading efficiency
The loading efficiency (L.E.) of each therapeutic agent was calculated after each vacuum loading step by determining the ratio of drug mass that remained unloaded to the total drug mass added by the following equation:
Release studies
The DDSs were placed on open flasks (10 systems per flask, 3 flasks per condition) with 1 mL volume of medium (NaCl 0.9% w/v) in an orbital shaker oven (IKA® KS 4000 ic control) with a constant temperature of 37 °C at 60 rpm. The release was recorded after 2, 4, 6, and 24 hours and, thereafter, daily for 43 days. 50% of the release medium was collected and saved for analysis, and the same volume of clean medium was added to the flask to set the final volume at 1 mL again.Gentamicin was quantified by a reaction with o-phthalaldehyde (OPA).32 OPA is a photosensitive reagent that reacts with gentamicin and forms a fluorescent component to detect the drug. Firstly, 65 μL of the sample was diluted with 75 μL of methanol. Then, in the dark, 115 μL of OPA reagent was added to the previous mixture. This reagent was previously diluted in methanol in a volume ratio of 11% OPA reagent to 85% methanol. Finally, the samples were capped, and quantification was carried out by UV spectrophotometry at 330 nm.
Raloxifene was measured by diluting the sample in a water:methanol solution (25:75 v/v) and read at 269 nm.
Alendronate was quantified using an indirect method based on the reduction of cerium IV (Ce(IV)) to cerium III (Ce(III)), as described elsewhere.33 Briefly, Ce(IV) oxidises biphosphonates when added in excess in a medium with sulphuric acid 0.5 M and at room temperature. It is then reduced to Ce(III) and can be measured by UV spectrophotometry at 320 nm. The amount of alendronate present in the solution is obtained based on a stoichiometric ratio of one molecule of alendronate to two molecules of Ce(III).
The release kinetics mechanisms were analysed by fitting the collected data in the zero-order (1), first-order (2), Higuchi (3), and Korsmeyer–Peppas (4) and Weibull (5) models, respectively.
| Qt = Q0 + k0t; | (1) |
| Qt = Q(1 − e−k1t); | (2) |
 | (3) |
| logQt = logKktn; | (4) |
 | (5) |
Lyophilisation and sterilisation
DDSs samples were also lyophilised and sterilised, after which release studies were conducted again to evaluate the impact of these procedures on the release profiles. The samples were frozen at −20 °C for lyophilisation for 3 hours and then freeze-dried for 5 hours at −80 °C (FreeZone 2.5-litre freeze-dryer, Labconco). Two different methods were used for sterilisation on the lyophilised DDSs: ethylene oxide (EtO) and supercritical CO2 (scCO2) sterilisation. EtO sterilisation was conducted on all hollow and non-hollow DDSs at 50 °C and left to aerate for 48 hours. CO2 sterilisation was conducted only on one of the DDSs (HDS2) to validate this methodology as an alternative to EtO.HDS2 DDS was sterilised in a supercritical CO2 reactor. This technology has previously been demonstrated to be effective in sterilising thermolabile materials without altering their physicochemical properties.34 Sterilisation pouches (Tyvek, USA) containing the samples were placed inside a pressure vessel of a 1.2 L-stainless steel autoclave (Parr Instrument Company, Illinois, USA). Premium CO2 Liquid Premier with 99.995% purity was introduced into the pressure vessel via a high-pressure pump at 30 mL min−1, and the pressure was set to 140 bar. H2O2 (35%, Thermoscientific) was used as a co-additive at a concentration of approximately 300 ppm. The temperature was adjusted to 40 °C, and the rotation motor speed was set at 500 rpm. After 4 h of batch operation, the vessel was dried in continuous mode (30 mL min−1) at 100 bar for 30 minutes before depressurising for 30 minutes using a manually operated valve. After treatment, the samples were stored in the desiccator.
Antimicrobial assessment
The systems subjected to lyophilisation and sterilisation, HDS2 and n-HDS2, were tested for antimicrobial activity to assess whether the gentamicin released was still biologically active. The assessment was performed by agar diffusion test on the systems (direct contact), as well as the released media (indirect contact), on days 1, 3, 7, 10, 12, 14 and 16. Petri plate surfaces were inoculated at each time point by spreading 100 μL of Staphylococcus aureus suspension (turbidity set to 0.5 McFarland, ≈1.5 × 108 CFU mL−1) over the entire surface of Mueller–Hinton agar. For each time-point 5 DDSs were tested (in triplicates), as well as three replicates of 10 μL of the released medium. The plates with the samples were incubated for 24 hours at 37 °C.Cytotoxicity preliminary evaluation
To determine whether the delivery systems are biocompatible and non-toxic upon implantation and after, a cytotoxic preliminary evaluation was performed using HDS2 as a model. The cells used were human bone marrow stromal cells (hBMSCs, Lonza, Catalog #: PT-2501). Before the biological evaluation, hBMSCs were characterised by flow cytometry and found to be positive for CD105, CD73, and CD90, and negative for CD45, CD34, and CD31 markers. Cells from the 4th passage were used in this study. The hBMSCs were expanded in Minimal Essential Medium (α-MEM), supplemented with 10% fetal bovine serum (FBS), 100 IU mL−1 penicillin, 100 μg mL−1 streptomycin, and 0.25 μg mL−1 amphotericin B (all from Gibco, Gaithersburg, MD, USA) – referred to as basal culture medium. The cultures were maintained at 37 °C with 5% CO2 in the air. Once the cultures reached approximately 70% confluence, cells were detached and sub-cultured at a density of 5 × 103 cells per cm2. After a 24-hour incubation, ten delivery systems (HDS2) were added to each well of adhered cells. Control cultures were maintained in basal culture medium without any delivery systems. The cultures were kept for up to 27 days, and the culture medium was replaced twice a week.Metabolic activity was quantified by resazurin assay on days 1, 2, 4, 6, 10, 14, 17, 20, 23, and 27. At each time point, the medium in each well was replaced with fresh medium containing 10% resazurin solution (0.1 mg mL−1, Sigma-Aldrich, St. Louis, MO, USA), and the plates were incubated at 37 °C for 4 hours. After incubation, fluorescence intensity was measured in a microplate reader (Synergy HT, Biotek) at excitation and emission wavelengths of 530 and 590 nm, respectively. The assay was performed in quintuplicate across three independent experiments.
Microspheres and DDSs morphology
The microspheres and DDSs (HDS1 and n-HDS1) were visualised through scanning electronic microscopy (SEM). The samples were immobilised on carbon tape coated with a thin gold/palladium film for 120 seconds and with a 15 mA current. All images obtained were made using a FEI Quanta 400 FEG ESEM microscope electron microscope. The SEM/EDS exam was performed using a High-resolution Scanning Electron Microscope with X-Ray Microanalysis: JEOL JSM 6301F/Oxford INCA Energy 350.Statistical analysis
All statistical analyses were performed on SigmaStat 3.5. The number of independent experiments for each condition (N) is described in the corresponding figure label. Data is presented as mean ± standard deviation (SD). Independent two-way analysis of variance (student's t-test: two-sample assuming equal variances) was performed with the significance level set at 0.05. Values between samples were considered statistically different for p < 0.05.Results
The development of hollow DDSs began with the production of porous bioceramic microspheres with hollow conformations, the morphology of which can be seen in Fig. 1(A) and (B). After producing the microspheres, they were functionalised, Fig. 1(C) and (D), by incorporating gentamicin (as an antimicrobial agent) and raloxifene or alendronate (as bone regeneration agents). In this way, and since the difference between the two hollow DDSs, HDS1 and HDS2, was the incorporation of raloxifene in the former and alendronate in the latter, Fig. 1 is representative of both hollow DDSs, whose morphologies are similar. | ||
| Fig. 1 SEM images of: (A) hollow bioceramic microsphere; (B) hollow bioceramic microsphere with core and shell structure exposed; (C) functionalised hollow bioceramic microsphere, drug delivery system HDS2; (D) functionalised hollow bioceramic microsphere, drug delivery system HDS with core and shell structure exposed. Scale bar = 1 mm. | ||
Functionalisation and dual releases for hollow delivery systems
Fig. 1 shows a SEM analysis of hollow microspheres before (Fig. 1(A) and (B)) and after the agents’ incorporation. The core and shell structure of the bioceramic microsphere is clear, and its porous nature is visible. The encapsulation of the microspheres with the alginate hydrogel is also observed (Fig. 1(C) and (D)).The loading efficiency (L.E.) of the antimicrobial agent (gentamicin) and bone regenerative agents, raloxifene for HDS1 and alendronate for HDS2, was determined. For HDS1, gentamicin L.E. was 91.2 ± 1.3% and raloxifene L.E. was 87.4 ± 0.4%. For HDS2, gentamicin L.E. was 96.3 ± 2.1% and alendronate L.E. was 88.7 ± 0.2%.
The dual and sequential release profiles were obtained for both HDS1 and HDS2. Fig. 2 depicts the HDS1 release studies, and it revealed that HDS1 had the desired dual and sequential release of gentamicin and raloxifene.
 | ||
| Fig. 2 Release profiles of therapeutic agents of hollow delivery system with gentamicin and raloxifene (HDS1) along 43 days. (A) Instant release of gentamicin and raloxifene, grey bar is minimum inhibitory concentration interval for gentamicin against Staphylococcus aureus; (B) cumulative release of gentamicin and raloxifene. Release kinetic models fits for gentamicin and raloxifene: zero-order; first-order; Higuchi; Korsmeyer–Peppas and Weibull. * = p < 0.05 statistically difference when compared to previous time point in gentamicin release. + = p < 0.05 statistically difference when compared to previous time point in raolixefene release. t is time in days. N = 3. | ||
Concerning gentamicin release, there was a fast release in the first 2 hours, accounting for around 25% of all the loaded gentamicin (Fig. 2(B)). After this time point, the released concentration decreased and stabilised on day 5, maintaining a steady release until day 18. Up to this point, all the release concentrations of gentamicin are above the minimum inhibitory concentration (MIC) interval for Staphylococcus aureus (0.5–1.5 μg mL−1). Gentamicin continued to be released for an additional 5 days, albeit below the MIC for S. aureus. Raloxifene, on the other hand, had a very low release until day 11 (6% cumulative release, compared to 80% of gentamicin), after which there was a significant increase in release that was kept mostly constant until raloxifene depletion on day 43. This continuous release is evident when analysing the cumulative release (Fig. 2(B)), which presented an almost linear slope between days 11 and 43.
Gentamicin release fits better for the Korsmeyer–Peppas model, with a dissolution constant (Kk) of 50.12 and a liberating exponent (n) of 0.21, suggesting a Fickian diffusion. First-order and Weibull models also fit very well, which suggests concentration-dependent release and a possibly composite mechanism. Concerning raloxifene release, it fits better in the zero-order model despite only ∼10% being released by day 8; the post-lag period is highly linear, which the zero-order model captures very well. Weibull and first-order models underperform, indicating this is not diffusion-controlled in a classic sense and is not concentration-dependent.
The other hollow DDS, HDS2, with gentamicin and alendronate, displayed a similar gentamicin profile to HDS1, with instant release concentrations above the MIC interval until day 20 (Fig. 3). Also, similarly to HDS1, the bone regenerative drug, in this case, alendronate, started the bulk of release (from 25% onwards) only after total depletion of gentamicin, revealing a dual and sequential release of the therapeutic agents. Also, alendronate release happened in a pulsative manner. HDS2 kinetic release was a better fit for zero-order, followed by the Korsmeyer–Peppas model with gentamicin Korsmeyer–Peppas dissolution constant (Kk) being 58.88 with a liberating exponent (n) of 0.16, while for alendronate Kk was 10.23 with a liberating exponent of 0.53. This means gentamicin release followed a pseudo-Fickian diffusion, while alendronate followed a relatively constant release rate (zero-order) after the initial delay with a mixed Fickian transport afterwards.
 | ||
| Fig. 3 Release profiles of therapeutic agents of hollow delivery system with gentamicin and alendronate (HDS2) along 43 days. (A) Instant release of gentamicin and alendronate, grey bar is minimum inhibitory concentration interval for gentamicin against Staphylococcus aureus; (B) cumulative release of gentamicin and alendronate; release kinetic models fits for gentamicin and alendronate: zero-order; first-order; Higuchi; Korsmeyer–Peppas and Weibull. * = p < 0.05 statistically difference when compared to previous time point in gentamicin release. + = p < 0.05 statistically difference when compared to previous time point in alendronate release. t is time in days. N = 3. | ||
Functionalisation and dual releases for non-hollow delivery systems
As with the hollow DDSs, the non-hollow DDSs began with the production of porous bioceramic microspheres but with non-hollow conformations, the morphology of which can be seen in Fig. 4(A) and (B). After producing the microspheres, they were functionalised, Fig. 4(C) and (D), by incorporating gentamicin (as an antimicrobial agent) and raloxifene or alendronate (as bone regeneration agents). In this way, and since the difference between the two non-hollow DDSs, n-HDS1 and n-HDS2, was the incorporation of raloxifene in the former and alendronate in the latter, Fig. 4 is representative of both hollow DDSs, which morphologies are similar. | ||
| Fig. 4 SEM images of: (A) non-hollow bioceramic microsphere; (B) cross section of non-hollow bioceramic microsphere; (C) functionalised non-hollow bioceramic microsphere, drug delivery system n-HDS1; (D) cross-section of functionalised non-hollow bioceramic microsphere, drug delivery system n-HDS. Scale bar = 1 mm. | ||
Similarly to the hollow delivery systems, the loading efficiency (L.E.) of the therapeutic agents for n-HDS1 (gentamicin + raloxifene) and n-HDS2 (gentamicin + alendronate) was determined. For HDS1, gentamicin L.E. was 92.2 ± 1.4% and raloxifene L.E. was 82.2 ± 1.0%. For HDS2, gentamicin L.E. was 97.2 ± 1.7% and alendronate L.E. was 89.9 ± 0.7%.
Fig. 4 shows a SEM analysis of non-hollow microspheres before (Fig. 4(A) and (B)) and after (Fig. 4(C) and (D)) the agents’ incorporation. In the figure, it is possible to observe the highly porous nature of the bioceramic microspheres. In Fig. 3(C), the alginate hydrogel coating can also be seen, revealing a complete encapsulation of the bioceramic microspheres.
Concerning the dual release profiles, for n-HDS1 (Fig. 5), gentamicin presents a release of 25% in the first 2 hours of the experiment, with a slower and steadier release after that. Up to 18 days, the released concentration of gentamicin was above S. aureus MICs and total depletion of gentamicin was realised at day 20, 3 days before the hollow counterpart (HDS1). In this n-HDS1, raloxifene release took off at day 8 (over 10%), at which point gentamicin release was already over 85%, revealing the dual and sequential release nature of n-HDS1.
 | ||
| Fig. 5 Release profiles of therapeutic agents of non-hollow delivery system with gentamicin and raloxifene (n-HDS1) along 43 days. (A) Instant release of gentamicin and raloxifene, grey bar is minimum inhibitory concentration interval for gentamicin against Staphylococcus aureus; (B) cumulative release of gentamicin and raloxifene; release kinetic models fits for gentamicin and raloxifene: zero-order; first-order; Higuchi; Korsmeyer–Peppas and Weibull. * = p < 0.05 statistically difference when compared to previous time point in gentamicin release. + = p < 0.05 statistically difference when compared to previous time point in raloxifene release. t is time in days. N = 3. | ||
The release kinetics of gentamicin fits better in first-order (R2 = 0.9661), with the Korsmeyer–Peppas model as a close second (R2 = 0.9627, n = 0.23), revealing concentration-dependent release with a pseudo-Fickian diffusion. For raloxifene release, zero-order model fits best, showing consistent release post-initial lag. The strong fit on the Korsmeyer–Peppas model (R2 = 0.9076, n = 0.80) implies non-Fickian transport, meaning that both diffusion and polymer relaxation/erosion were involved.
For n-HDS2, the release profiles (Fig. 6) showed a similar release behaviour compared to the hollow DDS counterpart (HDS2). Gentamicin was released close to 30% in the first 2 hours and maintained the concentration levels in the released medium from day 5 to day 16. During the first 16 days, gentamicin concentration was above S. aureus MICs. The release of gentamicin ended on day 21, 4 days earlier than HDS2. The dual and sequential release of n-HDS2 was achieved since the bulk of alendronate release started at day 9, when only 12% of alendronate was released, compared to 80% of gentamicin at the same time point. Similarly to HDS2, alendronate was released in a pulsative manner.
 | ||
| Fig. 6 Release profiles of therapeutic agents of hollow delivery system with gentamicin and alendronate (n-HDS2) along 43 days. (A) Instant release of gentamicin and alendronate, grey bar is minimum inhibitory concentration interval for gentamicin against Staphylococcus aureus; (B) cumulative release of gentamicin and alendronate; release kinetic models fits for gentamicin and alendronate: zero-order; first-order; Higuchi; Korsmeyer–Peppas and Weibull. * = p < 0.05 statistically difference when compared to previous time point in gentamicin release. + = p < 0.05 statistically difference when compared to previous time point in alendronate release. t is time in days. N = 3. | ||
Gentamicin release fitted best the Korsmeyer–Peppas model (R2 = 0.9616, n = 0.23), confirming Fickian diffusion as the primary release mechanism. Alendronate also fitted better for the Korsmeyer–Peppas model (R2 = 0.9508, n = 0.70), indicating non-Fickian anomalous transport again indicative of a mix of diffusion and erosion or swelling of the alginate matrix.
To assess the degradation of the hollow delivery system, HDS2 was analysed by SEM on days 1, 16 and 28 (Fig. 7). On day 1 (Fig. 7(A)), it was possible to observe that the alginate hydrogel covers the shell structure of the microsphere, covering the characteristic porosity that became impossible to see. Also, a clear layer of alginate hydrogel was visible in the sphere's core and within the shell's pores. This confirmed the success in the vacuum impregnation of hydrogel within the different structures of the delivery system. On day 16 (Fig. 7(B)), the porosity of the microsphere was already visible, meaning some degradation of the alginate had occurred; inside the core, it was possible to see some adhered hydrogel, as well as on the shell pores. Finally, on day 28 (Fig. 7(C)), there was evident degradation in the bioceramic structure of the microsphere, and tiny strands of alginate hydrogel were visible inside the core. The same strand forms were also present on the shell pores, revealing heavy degradation of the organic structure.
 | ||
| Fig. 7 SEM images of HDS2 at days 1, 16 and 28. Day 1 – whole system (A1); core and shell structure (A2), shell detail (A3). Day 16 – whole system (B1); core and shell structure (B2), shell detail (B3). Day 28 – whole system (C1); core and shell structure (C2), shell detail (C3). Scale bar for pictures A1, A2, B1, B2, C1 and C2 is 1 mm. Scale bar for pictures A3, B3 and C3 is 100 μm. | ||
Lyophilisation and sterilisation
The impact of lyophilisation and sterilisation by EtO on the release profiles of the delivery systems was also studied. Fig. 8 shows all DDSs (hollow and non-hollow, HDS1, HDS2, n-HDS1 and n-HDS2) with and without lyophilisation and sterilisation treatments. The HDS1 release profile (Fig. 8(A)) for gentamicin and raloxifene was very similar with and without lyophilisation and sterilisation. However, there was a delay in the release for both agents, particularly in the case of raloxifene, which was reflected in the cumulative release reaching 100% 5 days later than the delivery systems not subjected to lyophilisation and sterilisation. | ||
| Fig. 8 Release profiles of hollow and non-hollow delivery systems with and without lyophilisation and sterilisation treatments with ethylene oxide. (A) cumulative releases of hollow HDS1; (B) cumulative releases of hollow HDS2 (C) cumulative releases of non-hollow n-HDS1; (D) cumulative releases of non-hollow n-HDS2. N = 3. | ||
Identical results were obtained for HDS2 (Fig. 8(B)), with the release profiles of both gentamicin and alendronate being similar, albeit delayed. In this case, alendronate release, though still pulsative, was prolonged in time for an additional 12 days, compared to HDS2 not subjected to lyophilisation and sterilisation.
The non-hollow delivery systems release profiles, n-HDS1 (Fig. 8(C)) and n-HDS2 (Fig. 8(D)), were very similar for both gentamicin and raloxifene/alendronate, which was delayed by the lyophilisation and sterilisation processes. In the case of n-HDS1, gentamicin cumulative release reached its peak on day 27, as opposed to day 23 on non-treated n-HDS1. Raloxifene had a longer release for an additional 13 days. On n-HDS2, gentamicin cumulative release had its maximum on the same day for lyophilised/sterilised n-HDS2 as non-lyophilised/sterilised n-HDS2. The alendronate release profile was visible for 43 days, 8 days more than n-HDS2 without lyophilisation and sterilisation.
Parallel to sterilisation with EtO after lyophilisation, n-HDS2 samples were also studied to sterilisation with supercritical CO2 after lyophilisation to assess the validity of supercritical CO2 sterilisation as an alternative to EtO. In Fig. 9, scCO2 treatment slightly accelerates the release of both gentamicin and alendronate to concentration values closer to the non-lyophilised and sterilised n-HDS2. The release profile was preserved after scCO2 treatment, and therefore, it can be affirmed that scCO2 had minimal impact, even smaller than EtO, in the release profile of both gentamicin and alendronate.
 | ||
| Fig. 9 Release profiles of gentamicin and alendronate from HDS2 under no lyophilised and sterilised treatment, under EtO sterilisation and under scCO2 sterilisation. | ||
Cytotoxicity evaluation
HDS2 was used as a model for preliminary cytotoxicity evaluation of all DDSs. In this system, gentamicin is released from the get-go and alendronate is released only later, usually after 2 to 3 weeks (Fig. 8), though the media used for this experiment is different from the release studies, which can alter the release profile. Fig. 10 shows that the metabolic activity of hBMSC is comparable to the control group (hBMSC in α-MEM medium supplemented with 10%FBS), being statistically higher from day 14 onwards. This result indicates that HDS2 presented no cytotoxicity. | ||
| Fig. 10 Metabolic activity of hBMSCs in contact with HDS2 and control group in complete α-MEM medium. *p < 0.05, statistical difference. | ||
To assess whether gentamicin preserves its antimicrobial activity after the sterilisation processes, n-HDS2 (lyophilised and sterilised by EtO) and HDS2 (lyophilised and sterilised by scCO2) were tested in an agar diffusion assay. Fig. 11 represents 4 time points, days 1, 7, 10 and 16 of the delivery systems (direct contact, top row of each plate, visible by the microspheres), as well the released media (indirect contact, bottom row of each plate) at each time point during the release studies. Regardless of which delivery system, all released media maintain gentamicin activity, clearly visible in well-defined inhibition halos at all time points in both systems. This means that the sterilisation processes (EtO or scCO2) did not hinder gentamicin antimicrobial activity.
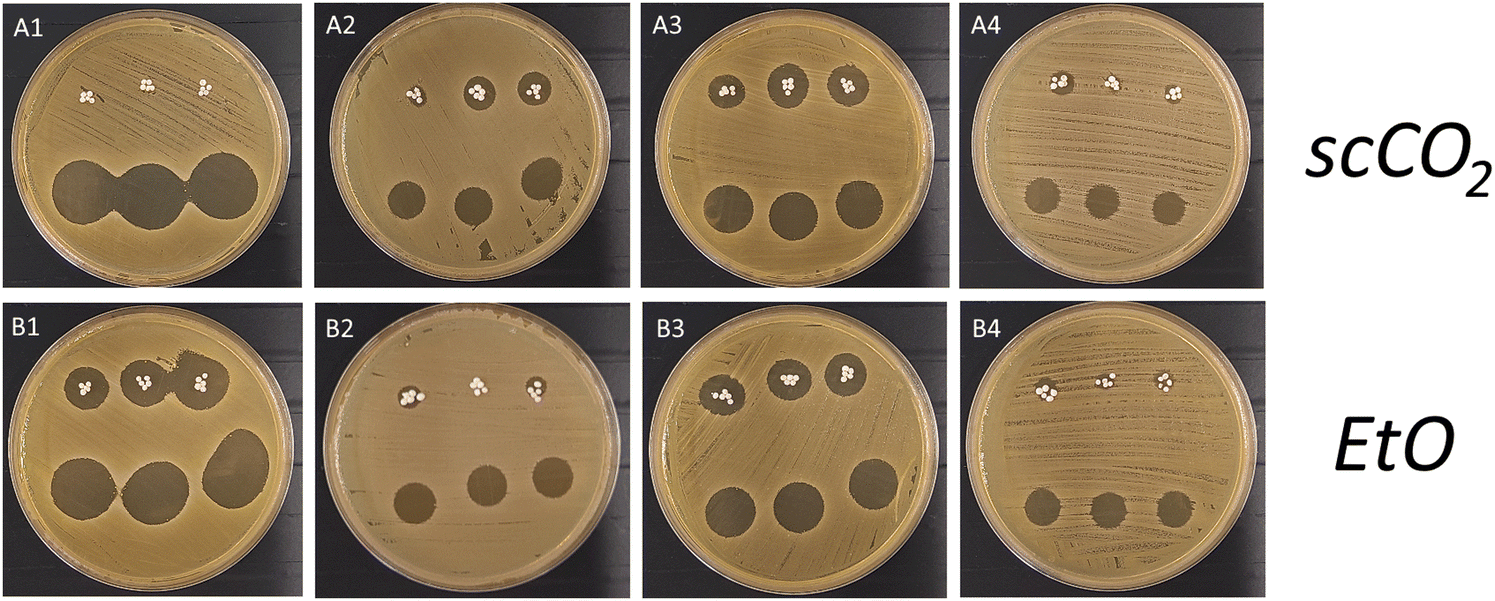 | ||
| Fig. 11 Agar diffusion assay of antimicrobial effectiveness of the delivery systems HDS2 sterilised by scCO2 (A1–A4) and n-HDS2 sterilised by EtO (B1–B4), on days 1, 7, 10 and 16, respectively. | ||
Discussion
The presented work aimed at developing DDSs based on bioceramic porous microspheres with a dual and sequential release of antimicrobial and bone regenerative agents.The DDSs encompassed the incorporation of therapeutic agents in microspheres with two types of morphology: hollow and non-hollow. Given the different morphologies of the two bioceramic porous microspheres, it was expected that the release profiles of the therapeutic agents would be different: in HDS (hollow delivery systems), there is a core and shell structure, where the system was designed to incorporate the maximum possible amount of raloxifene or alendronate into the core, while gentamicin would be incorporated into the pores of the shell and the surface of the microsphere; in n-HDS (non-hollow delivery systems) the pores would be loaded with the bone regenerative agent, and gentamicin would be loaded mainly into the microsphere's surface. To guarantee a sustained release, as opposed to a burst release, the therapeutic agents were encapsulated into an alginate hydrogel.
Taking advantage of the highly porous nature of the microspheres, the therapeutic agents were incorporated into the microspheres through vacuum loading. The delivery systems were designed to release the antimicrobial agent first to prevent possible infections post-surgery, contracted during surgery, and later, the controlled release of a bone regenerative agent to accelerate the bone healing process.
The antimicrobial agent chosen was gentamicin, which is commonly used as a model drug in DDSs for bone infections due to its favourable properties and extensive clinical use in treating severe bacterial infections, particularly those caused by Staphylococcus aureus, a common pathogen in hospital environments, with a probability of resulting in post-surgery infections that can hinder bone regeneration.35–37 Regarding the bone regenerative agents, two molecules were chosen: the hydrophobic raloxifene and the hydrophilic alendronate. The use of molecules with different hydrophilicity is based on the fact that this property may have a major impact on the release mechanism and, therefore, should be considered when choosing model agents in the current DDS setting. The hydrophobic raloxifene has bone-targeting effects and a dual role in osteoporosis treatment and bone healing. It is used as a model therapeutic agent in bone regeneration studies since it is a selective estrogen receptor modulator (SERM), providing oestrogen-like effects on bone tissue without affecting other estrogen-sensitive tissues, making it a promising candidate for enhancing bone formation and reducing bone resorption in drug delivery research.38–40 The hydrophilic alendronate was chosen as a model therapeutic agent due to its high affinity for bone tissue, anti-resorptive action, compatibility with sustained-release carriers, and well-documented safety profile.41–44 Another critical aspect to be considered when developing DDSs with local administration/release of raloxifene or alendronate is the difference in therapeutic concentrations of these agents compared to the oral administration that is currently used in the clinic. Local administration of alendronate and raloxifene can offer several advantages over oral administration, particularly in the context of accelerating bone regeneration on localised bone defects. These advantages, resulting from lower but localised concentrations, are a reduction of systemic side effects, improvement of drug targeting and efficacy, with a more focused and improved bioavailability.45–47
For the dual-release studies, the release medium used was NaCl 0.9% w/v. This formulation was chosen because of the same value in osmotic pressure compared to simulated body fluid (SBF), but without the chemical complexity in composition of SBF that would hinder the quantification of gentamicin and raloxifene/alendronate.48,49 Also, to keep a steady osmotic pressure during the experiment to better approach the physiological conditions, 50% of the medium was replenished at every time point instead of the total volume, which would increase the release of the therapeutic agents from the DDS to the release medium.50,51
The instantaneous (point-to-point) release studies revealed a gentamicin release concentration of interest for all the DDSs throughout at least the first 17 days since gentamicin concentration was higher than the MIC interval for S. aureus.52,53 Sustained gentamicin levels above the MIC ensure continuous suppression of bacterial growth, which is particularly important in preventing the emergence of resistant subpopulations.54 This minimises the selection pressure that can lead to resistance, thereby enhancing treatment efficacy and patient outcomes. Moreover, maintaining concentrations above the MIC for an extended period ensures that the antibiotic remains effective throughout the dosing interval, reducing the likelihood of bacterial regrowth.54
There were small differences when analysing gentamicin release in each delivery system: the hollow systems HDS1 and HDS2 showed a gentamicin therapeutic concentration for 19 and 20 days, respectively, while in non-hollow systems, n-HDS1 and n-HDS2, the therapeutic concentration was maintained for 17 and 18 days, respectively. This 2-day difference between systems is because, in hollow systems, gentamicin is not only on the surface of the microspheres but also within its pores, while in non-hollow systems, gentamicin is more available at the surface of the microspheres since the pores are occupied with the bone regenerative agent, contrarily to the hollow systems where the bone regenerative agent is mainly loaded into the core structure of the microsphere.
Concerning the bone regenerative agents (raloxifene or alendronate), it was clear that regardless of the morphology of the microspheres, the release concentration interval of raloxifene was 10−5–10−6 M and for alendronate was in the range of 10−6 M. These concentrations intervals are, according to the literature, for a therapeutic window. In the case of raloxifene, 10−5–10−6 M was found to enhance bone formation and osteoblast function without adverse effects38,55,56 and in a single DDS from an implantable hydrogel; this localised release concentration interval promoted bone healing and integration without systemic side effects in a 4-week release period.57 For alendronate, the current research team already showed that a localised release concentration of 10−6 M is within range of a therapeutic concentration at which alendronate can differentiate mesenchymal stromal cells into osteoblastic lineage while also inhibiting osteoclastic activity, which can accelerate bone regeneration (scientific paper with findings under submission).
While there are some commercially available products that offer combined antimicrobial and bone regenerative properties,58–60 no product is available that is specifically designed to sequentially release an antimicrobial agent followed by a bone regenerative agent.
The primary aim of the current work regarding the release profiles was obtaining a dual and sequential release. This was accomplished for all developed systems since, in every system, a complete depletion of gentamicin occurred while the bone regenerative agent release was still within 10 to 15% of cumulative release. Also, considering the time window of therapy, gentamicin is reported to be beneficial in a localised antimicrobial effect for 2 to 4 weeks,57,61 and a bone regenerative agent should be released for at least 3 to 6 weeks. These time frames were obtained for all delivery systems. It is also important to restate that the hollow delivery systems had a more prolonged (even delayed) release of raloxifene/alendronate than the non-hollow systems, which was expected since the way the hollow systems were designed made the bone regenerative agents to be concentrated within the core of the microspheres, meaning that the agents had to travel through the shell pores to be released. Also, it is known that in vitro drug release is slower and more controlled, while in vivo drug release is faster due to enzymatic degradation and fluid exchange in physiological conditions; therefore, in vitro models underestimate burst release.62,63 This is particularly important when the release is dependent on alginate hydrogel, which improves sustained release, but in vivo, its enzymatic breakdown can shorten the release duration.64,65 However, the incorporation of the current DDS agents can be tuned to adapt the release kinetics to future in vivo experiment outputs.
Drugs can be released from a hydrogel matrix in several ways, either by diffusion or differences in osmotic pressure, with or without hydrogel swelling. In all systems, gentamicin release kinetics was well characterised by the Korsmeyer–Peppas model, with high correlation coefficients (R2 ≥ 0.96) and release exponent (n) values between 0.16–0.23, indicative of pseudo-Fickian diffusion as the dominant mechanism. This occurs when the release process is primarily diffusion-driven but with a rate that's slower than pure Fickian diffusion (which is defined by n ≈ 0.5). This can happen due to polymer relaxation, meaning that the lower exponent may indicate that the matrix in which the drug is embedded restricts diffusion, possibly due to slow polymer relaxation or tighter structural constraints.66 The slight differences in the fits of the secondary model (e.g. Higuchi in HDS2, first-order in n-HDS1) suggest subtle changes in the diffusional path length or porosity distribution between the hollow and non-hollow microspheres. In particular, hollow microspheres may confer a slightly more controlled release through their macroporous shells, while non-hollow morphologies offer more uniform diffusion pathways.
For all DDSs, gentamicin release presented an initial burst release followed by a stable phase around day 5. The observed rapid release of gentamicin within the initial two hours in all DDSs can be attributed to the burst release phenomenon, commonly associated with drug delivery systems incorporating porous bioceramics and hydrophilic polymeric matrices such as alginate.67,68 This initial surge is primarily due to the desorption of gentamicin located near the outer regions of the alginate matrix.69,70 Additionally, the high porosity of the bioceramic facilitates rapid diffusion of the drug, while the hydrophilic nature of alginate promotes rapid water uptake and swelling upon exposure to an aqueous environment.23 Together, these factors enhance the initial diffusion rate of gentamicin into the surrounding medium, leading to the characteristic burst release. A stable phase follows this initial burst in all DDSs until 100% cumulative release. This sustained release is predominantly governed by diffusion-controlled mechanisms. As the surface-associated drug is depleted, the remaining gentamicin, entrapped within the alginate matrix and the interconnected pores of the porous bioceramic, diffuses outward at a slower, more regulated pace.71 This behavior aligns with the Korsmeyer–Peppas model. The fitting of the release data to these models suggests that the drug release is primarily Fickian in nature, with some contributions from non-Fickian mechanisms related to matrix swelling and degradation. The gelation of alginate in physiological conditions forms a semi-permeable barrier, further moderating drug diffusion.72 Additionally, the reduced concentration gradient over time contributes to the establishment of a steady state, resulting in the observed stabilisation of the release rate.73
There are some similarities but also differences in behaviour when analysing the bone regenerative agents’ (raloxifene and alendronate) kinetic mechanisms. Raloxifene showed a delayed initial release followed by a prolonged, almost zero-order release, particularly in the hollow microsphere system HDS1. For both HDS1 and n-HDS1, the best fits were obtained with zero-order models (R2 = 0.98 and 0.96, respectively), supported by Korsmeyer–Peppas n-values of around 0.80–0.83, suggesting anomalous (non-Fickian) transport mechanisms. These results suggested a release governed by diffusion rather than matrix relaxation or degradation, most likely due to the hydrophobic nature of the raloxifene, contrary to the hydrophilic nature of the polymeric matrix. It is interesting to note that the hollow structure in HDS1 maintained the release of raloxifene for a longer period compared to n-HDS1 since the hollow core–shell structure in HDS1 limits solvent penetration and delays polymer swelling,74 resulting in a significantly extended release profile (42 days to reach 100% cumulative release) compared to the uniformly porous, non-hollow n-HDS1 (27 days to reach 100% cumulative release).
Alendronate presented an interesting and unusual behaviour. In both HDS2 and n-HDS2, alendronate release followed a non-continuous, pulsatile pattern consisting of an initial burst followed by discrete releases at defined intervals. These profiles deviate from classical release kinetics, and none of the standard models fully captures the observed behaviour. However, when the agent was being released from the hollow system (HDS2) the release exponent was 0.53, which is reflective of a Fickian diffusion, while on non-hollow system (n-HDS2) the release exponent was 0.70, meaning a non-Fickian transport. In the case of HDS2, the Fickian diffusion of alendronate indicates that alendronate is released solely by diffusion through the hydrogel matrix since it is “trapped” within the hydrogel matrix inside the core of the microsphere. However, the release of alendronate within the pores of the n-HDS2 is influenced by diffusion with polymer swelling (non-Fickian diffusion). This difference in alendronate behaviour may be due to the closer contact that the release medium has with the hydrogel in the non-hollow system compared to the hydrogel within the core of the hollow system, which directly influences hydrogel swelling. Also, the release exponent of alendronate was always smaller than the one for raloxifene, which may be related to the hydrophilic nature of the alendronate, making it easier to be diffused through a hydrogel when compared to a highly hydrophobic molecule like raloxifene.74–76
Translating the systems developed to clinical practice relies heavily on shelf-life increase, cost-effectiveness and sterilisation processes. Therefore, all delivery systems were also subject to release studies after lyophilisation (for shelf-life and cost-effective purposes) and sterilisation by ethylene oxide. Ethylene oxide was chosen as a sterilisation method due to the lower temperature of sterilisation in the process, which makes it the widely used sterilisation method for medical devices which contain polymeric matrices. In this way, the impact on the hydrogel and the therapeutic agents would be lower. The effect of lyophilisation and sterilisation on the delivery systems occurred through a delay in the release of all agents, although their release profile was maintained. This delay was most likely due to the sterilisation process by EtO. This process was conducted at 50 °C, and it has been shown that Ca+ crosslinked alginate hydrogels undergo further permanent crosslinking at this temperature, altering the diffusion speed of the agents.77 Nevertheless, EtO is still the most used sterilisation method for polymer-containing medical devices; however, there are concerns about its environmental impact and potential health risks, which have prompted regulatory bodies like the U.S. Food and Drug Administration (FDA) and the European Union (EU) to implement specific regulations and explore alternative sterilisation methods.78 The FDA, in particular, has identified and supported alternatives to EtO. In 2019, it launched an innovation challenge to promote such alternatives, selecting technologies including supercritical CO2 (scCO2) sterilisation.79 This technique has shown potential, but it is important to state that it is not yet an FDA-approved sterilisation method for commercial medical devices. In this work, scCO2 was tested on HDS2 as an alternative to EtO sterlisation. ScCO2 is a known sterilising agent and can effectively sterilise alginate hydrogels without introducing high temperatures or harsh chemicals. This is particularly important for biomedical applications.80 It is also significant, when using scCO2 sterilisation, to take note that non-polar agents, such as raloxifene in this work, can be extracted from the polymeric matrix during the sterilisation process, due to the similar intermolecular forces between the agent and the CO2. However, the DDS used for scCO2 contained the polar agent alendronate (HDS2). The results showed that the release profile was maintained, and the release time was much closer to non-sterilised HDS2, most likely due to the protective nature of the polymeric matrix. This finding indicated that scCO2 can be an alternative to EtO sterilisation, particularly in the case of medical devices with a similar composition as the ones presented in this work.
A preliminary cytotoxicity evaluation was also performed using HDS2 to evaluate the safety and compatibility of materials and agents used in these drug-delivery systems. In fact, cytotoxicity testing is a mandatory step in regulatory frameworks (for example, ISO-10993 for medical devices). HDS2 showed compatibility with the cells (hBMSCs), which were metabolically more active than the control group cells, indicating a fast proliferation profile. In addition to the release profiles and the preliminary cytotoxicity tests, assessing whether the therapeutic agents maintained the activity after sterilisation was crucial. The activity assessment of the dual agents (antimicrobial and bone regenerative) from each DDS is already the subject of ongoing research by the current research team. A set-up of dual and sequential in vitro release of the DDSs was developed and will be used to evaluate and show whether these therapeutic agents maintain biological activity after the lyophilisation and sterilisation of each DDS.
To assess whether gentamicin maintained antimicrobial activity after the lyophilisation and sterilisation, an agar diffusion test was carried out on the HDS2 and n-HDS2 samples, using qualitative direct and indirect contact approaches. This assay, based on the Kirby–Bauer diffusion method, evaluates antibacterial activity by comparing the zone of inhibition (ZOI) formed on an agar plate seeded with Staphylococcus aureus. In the case of direct contact, the DDSs were placed directly on the agar surface. This configuration simulates a localised delivery scenario, representative of clinical application, in which the material remains at the implantation site and releases gentamicin locally. Otherwise, the indirect contact method involved adding a medium that was collected during release profile assays to evaluate the antimicrobial potential of the released gentamicin, mimicking a clinical set-up.
The results showed significant zones of inhibition comparable in both conditions, confirming that gentamicin maintains its antimicrobial capacity after lyophilisation and sterilisation either by EtO or supercritical CO2. Furthermore, the comparable activity observed in indirect contact highlights that gentamicin remains bioavailable and functionally active after release from the DDS, which reflects its sustained efficacy throughout the release period.
In the end, and for the first time, different bioceramic delivery systems (hollow and non-hollow) with dual and sequential release of antimicrobials in an early stage and hydrophobic or hydrophilic bone regenerative agents in a later stage were successfully accomplished.
Conclusion
The current work successfully developed and characterised dual and sequential bioceramic porous DDSs in two forms (hollow and non-hollow), with the release of antimicrobial agents in the first 2 weeks, followed by bone regenerative agents up to 4 to 5 weeks. The impact of this work is massive since, in the future, it will allow for a bone regenerative surgery, with localised and sustained release of therapeutic agents to deal with post-surgery infections and accelerate the bone regeneration process, replacing the traditional oral administration of the later agents.Author contributions
Rodrigues: conceptualisation, investigation, methodology, writing original draft; Ferreira: investigation, draft review; Borges: conceptualisation, supervision, draft review; Bernardes: investigation, draft review; Oliveira: draft review; Santos: draft review; Lopes: conceptualisation, supervision, draft review & editing.Conflicts of interest
The authors have no conflicts of interest.Data availability
The raw and processed data underlying this article will be shared on reasonable request to the corresponding author.Acknowledgements
M. A. Rodrigues thanks the Fundação para a Ciência e Tecnologia (FCT), Portugal, for the PhD Grant reference 2021.06440.BD. B.G.B thanks the FCT for PhD Grant 2021.05717.BD. This work also received financial support from FCT/MCTES, UIDB/50006/2020 DOI 10.54499/UIDB/50006/2020, LA/P/0037/2020, and UIDP/50025/2020, UIDB/50025/2020-2023, UIDB/50016/2020 of the Centre for Biotechnology and Fine Chemistry – CBQF, through national funds through national funds. The work was also funded by “Interreg VI A España – Portugal (POCTEP) 2021–2027”, IBEROS+ (0072_IBEROS_MAIS_1_E) – Instituto de Biofabricación en Red para El Envejecimiento Saludable.References
- A. K. VK, S. Ray, U. Arora, S. Mitra, A. Sionkowska and A. K. Jaiswal, Front. Bioeng. Biotechnol., 2022, 10, 969843 CrossRef PubMed.
- D. Lee, M. Wufuer, I. Kim, T. H. Choi, B. J. Kim, H. G. Jung, B. Jeon, G. Lee, O. H. Jeon, H. Chang and D. S. Yoon, Sci. Rep., 2021, 11, 746 CrossRef CAS PubMed.
- M. Farokhi, F. Mottaghitalab, M. A. Shokrgozar, K.-L. Ou, C. Mao and H. Hosseinkhani, J. Controlled Release, 2016, 225, 152–169 CrossRef CAS PubMed.
- T. U. Wani, R. S. Khan, A. H. Rather, M. A. Beigh and F. A. Sheikh, J. Controlled Release, 2021, 339, 143–155 CrossRef CAS PubMed.
- N. Taherpour, Y. Mehrabi, A. Seifi, B. Eshrati and S. S. Hashemi Nazari, BMC Infect. Dis., 2021, 21, 3 CrossRef PubMed.
- M. Motififard, M. Teimouri, K. Shirani, S. Hatami and M. Yadegari, Int. J. Burns Trauma, 2021, 11, 191–196 CAS.
- S. Zhang, M. Xing and B. Li, Acta Biomater., 2019, 93, 135–151 CrossRef CAS PubMed.
- P. P. Kalelkar, M. Riddick and A. J. García, Nat. Rev. Mater., 2022, 7, 39–54 CrossRef CAS PubMed.
- C. B. Ayyanar, T. Bal, Fahaduddin, S. Sharma, B. Gayathri, V. Rinusuba, H. E. Nalini, S. Deepa, M. D. Dharshinii, P. Kharra and A. Sinha, Int. J. Biol. Macromol., 2024, 275, 133691 CrossRef CAS PubMed.
- B. A. Chinnappan, M. Krishnaswamy, T. Bal and A. D. Rajora, Int. J. Biol. Macromol., 2023, 253, 126695 CrossRef CAS PubMed.
- I. Koumentakou, M. J. Noordam, A. Michopoulou, Z. Terzopoulou and D. N. Bikiaris, Biomacromolecules, 2023, 24, 4019–4032 CrossRef CAS PubMed.
- Y. Liu, R. Wang, S. Chen, Z. Xu, Q. Wang, P. Yuan, Y. Zhou, Y. Zhang and J. Chen, Int. J. Biol. Macromol., 2020, 148, 153–162 CrossRef CAS PubMed.
- M. Lazaridou, D. N. Bikiaris and D. A. Lamprou, Pharmaceutics, 2022, 14, 1978 CrossRef CAS PubMed.
- A. Dubey, H. Vahabi and V. Kumaravel, ACS Biomater. Sci. Eng., 2023, 9, 4020–4044 CrossRef CAS PubMed.
- C. Balaji Ayyanar, S. Helaili, S. Mavinkere Rangappa, P. Boonyasopon and S. Siengchin, J. Mech. Behav. Biomed. Mater., 2023, 146, 106086 CrossRef CAS PubMed.
- S. Rajan, K. Marimuthu, C. Balaji Ayyanar, A. Khan, S. Siengchin and S. Mavinkere Rangappa, J. Mater. Res. Technol., 2022, 18, 921–930 CrossRef CAS.
- A. T. Yayehrad, E. A. Siraj, M. Matsabisa and G. Birhanu, Regener. Ther., 2023, 24, 361–376 CrossRef CAS PubMed.
- J. Zhou, C. Fang, C. Rong, T. Luo, J. Liu and K. Zhang, Smart Mater. Med., 2023, 4, 427–446 CrossRef.
- X. Chen, D. Wu and Z. Chen, MedComm, 2024, 5, e643 CrossRef CAS PubMed.
- Y. Zhang, M. Jiang and T. Wang, Front. Bioeng. Biotechnol., 2024, 12, 2024 Search PubMed.
- L. Huang, Z. Guo, X. Yang, Y. Zhang, Y. Liang, X. Chen, X. Qiu and X. Chen, Theranostics, 2025, 15, 460–493 CrossRef CAS PubMed.
- C. Heras, J. Jiménez-Holguín, A. L. Doadrio, M. Vallet-Regí, S. Sánchez-Salcedo and A. J. Salinas, Acta Biomater., 2020, 114, 395–406 CrossRef CAS PubMed.
- R. García-Alvarez, I. Izquierdo-Barba and M. Vallet-Regí, Acta Biomater., 2017, 49, 113–126 CrossRef PubMed.
- J. L. Paris, N. Lafuente-Gómez, M. V. Cabañas, J. Román, J. Peña and M. Vallet-Regí, Acta Biomater., 2019, 86, 441–449 CrossRef CAS PubMed.
- N. Lindfors, J. Geurts, L. Drago, J. J. Arts, V. Juutilainen, P. Hyvönen, A. J. Suda, A. Domenico, S. Artiaco, C. Alizadeh, A. Brychcy, J. Bialecki and C. L. Romanò, Adv. Exp. Med. Biol., 2017, 971, 81–92 CrossRef PubMed.
- M. Seidenstuecker, J. Hess, A. Baghnavi, H. Schmal, D. Voigt and H. O. Mayr, J. Mater. Sci.: Mater. Med., 2024, 35, 40 CrossRef CAS PubMed.
- N. Ahmad, S. N. A. Bukhari, M. A. Hussain, H. Ejaz, M. U. Munir and M. W. Amjad, RSC Adv., 2024, 14, 13535–13564 RSC.
- G. Muteeb, M. T. Rehman, M. Shahwan and M. Aatif, Pharmaceuticals, 2023, 16, 1615 CrossRef CAS PubMed.
- A. Molnar, T. Lakat, A. Hosszu, B. Szebeni, A. Balogh, L. Orfi, A. J. Szabo, A. Fekete and J. Hodrea, Amino Acids, 2021, 53, 917–928 CrossRef CAS PubMed.
- B. Cinici, S. Yaba, M. Kurt, H. C. Yalcin, L. Duta and O. Gunduz, Biomimetics, 2024, 9, 409 CrossRef CAS PubMed.
- Z. Dai, J. Ronholm, Y. Tian, B. Sethi and X. Cao, J. Tissue Eng., 2016, 7, 2041731416648810 CrossRef PubMed.
- J. Gubernator, Z. Drulis-Kawa and A. Kozubek, Int. J. Pharm., 2006, 327, 104–109 CrossRef CAS PubMed.
- M. I. Walash, M. E. Metwally, M. Eid and R. N. El-Shaheny, Chem. Cent. J., 2012, 6, 25 CrossRef CAS PubMed.
- G. C. Soares, D. A. Learmonth, M. C. Vallejo, S. P. Davila, P. González, R. A. Sousa and A. L. Oliveira, Mater. Sci. Eng., C, 2019, 99, 520–540 CrossRef CAS PubMed.
- D. Zapata, J. Higgs, H. Wittholt, K. Chittimalli, A. E. Brooks and P. Mulinti, Pharmaceutics, 2022, 14, 1563 CrossRef CAS PubMed.
- E. J. Sheehy, C. von Diemling, E. Ryan, A. Widaa, P. O’Donnell, A. Ryan, G. Chen, R. T. Brady, A. López-Noriega, S. Zeiter, T. F. Moriarty and F. J. O’Brien, Biomaterials, 2025, 313, 122774 CrossRef CAS PubMed.
- I. D. Athauda, M. G. Shetty, P. Pai, M. Hegde, S. C. Gurumurthy and K. S. Babitha, J. Cluster Sci., 2024, 35, 371–390 CrossRef CAS.
- J. R. Rey, E. V. Cervino, M. L. Rentero, E. C. Crespo, A. O. Alvaro and M. Casillas, Open J. Orthop., 2009, 3, 14–21 CrossRef PubMed.
- P. D’Amelio and G. C. Isaia, Expert Opin. Pharmacother., 2013, 14, 949–956 CrossRef PubMed.
- Z. Guo, R. Afza, M. Moneeb Khan, S. U. Khan, M. W. Khan, Z. Ali, S. Batool and F. U. Din, Heliyon, 2023, 9, e20107 CrossRef CAS PubMed.
- M. Pazianas, C. Cooper, F. H. Ebetino and R. G. Russell, Ther. Clin. Risk Manage., 2010, 6, 325–343 CAS.
- O. Bock and D. Felsenberg, Clin. Interventions Aging, 2008, 3, 279–297 CrossRef CAS PubMed.
- K. Miller, C. Clementi, D. Polyak, A. Eldar-Boock, L. Benayoun, I. Barshack, Y. Shaked, G. Pasut and R. Satchi-Fainaro, Biomaterials, 2013, 34, 3795–3806 CrossRef CAS PubMed.
- S. Shi, H. Duan and X. Ou, Biomed. Pharmacother., 2024, 175, 116699 CrossRef CAS PubMed.
- A. Murthy, P. R. Ravi, H. Kathuria and S. Malekar, Nanomaterials, 2020, 10, 1085 CrossRef CAS PubMed.
- K. M. Hosny, R. H. Bahmdan, N. A. Alhakamy, M. A. Alfaleh, O. A. Ahmed and M. H. Elkomy, J. Pharm. Sci., 2020, 109, 2145–2155 CrossRef CAS PubMed.
- M. R. Newman and D. S. Benoit, Curr. Opin. Biotechnol, 2016, 40, 125–132 CrossRef CAS PubMed.
- K. Touati, F. Tadeo, C. Hänel and T. Schiestel, Desalin. Water Treat., 2016, 57, 10477–10489 CrossRef CAS.
- N. Ozbek and S. Akman, J. Anal. At. Spectrom., 2019, 34, 583–587 RSC.
- M. Vigata, C. Meinert, D. W. Hutmacher and N. Bock, Pharmaceutics, 2020, 12, 1188 CrossRef CAS PubMed.
- Y. Kim, E. J. Park, T. W. Kim and D. H. Na, Pharmaceutics, 2021, 13, 1313 CrossRef CAS PubMed.
- T. S. Sørensen and A. I. Sørensen, Acta Orthop. Scand., 1993, 64, 82–84 CrossRef PubMed.
- L. K. Jensen, T. Bjarnsholt, K. N. Kragh, B. Aalbæk, N. L. Henriksen, S. A. Blirup, K. Pankoke, A. Petersen and H. E. Jensen, Antimicrob. Agents Chemother., 2019, 63, 1–9 CrossRef PubMed.
- G. Bhattacharya, D. Dey, S. Das and A. Banerjee, J. Med. Microbiol., 2017, 66, 762–769 CrossRef CAS PubMed.
- K. M. Beekman, A. G. Veldhuis-Vlug, M. den Heijer, M. Maas, A. M. Oleksik, M. W. Tanck, S. M. Ott, R. J. van’t Hof, P. Lips, P. H. Bisschop and N. Bravenboer, Bone, 2019, 118, 62–68 CrossRef CAS PubMed.
- W. Khovidhunkit and D. M. Shoback, Ann. Intern. Med., 1999, 130, 431–439 CrossRef CAS PubMed.
- S. G. Kala and S. Chinni, Indian J. Pharm. Educ. Res., 2021, 55, s135–s148 CrossRef CAS.
- K. Fraga, M. Maireles, M. Jordan, L. Soldevila and O. Murillo, J. Bone Jt. Infect., 2022, 7, 163–167 CrossRef PubMed.
- C. L. Chia, V. G. Shelat, W. Low, S. George and J. Rao, Int. Surg., 2014, 99, 565–570 Search PubMed.
- J. Syvänen, W. Serlo, J. Jalkanen, I. Kohonen, A. Raitio, Y. Nietosvaara and I. Helenius, J. Bone Jt. Surg., Am. Vol., 2023, 105, 659–666 Search PubMed.
- D. Overstreet, A. McLaren, F. Calara, B. Vernon and R. McLemore, Clin. Orthop. Relat. Res., 2015, 473, 337–347 CrossRef PubMed.
- A. Varela-Moreira, H. van Leur, D. Krijgsman, V. Ecker, M. Braun, M. Buchner, M. H. A. M. Fens, W. E. Hennink and R. M. Schiffelers, Int. J. Pharm., 2022, 618, 121638 CrossRef CAS PubMed.
- S. C. Sundararaj, M. Al-Sabbagh, C. L. Rabek, T. D. Dziubla, M. V. Thomas and D. A. Puleo, J. Biomed. Mater. Res., Part B, 2016, 104, 1302–1310 Search PubMed.
- M. A. S. Abourehab, R. R. Rajendran, A. Singh, S. Pramanik, P. Shrivastav, M. J. Ansari, R. Manne, L. S. Amaral and A. Deepak, Int. J. Mol. Sci., 2022, 23, 9035 CrossRef CAS PubMed.
- D. M. Hariyadi and N. Islam, Adv. Pharmacol. Pharm. Sci, 2020, 2020, 8886095 CAS.
- M. Fosca, J. Rau and V. Uskokovic, Bioact. Mater., 2022, 7, 341–363 CAS.
- P. Diaz-Rodriguez, M. Sánchez and M. Landin, Pharmaceutics, 2018, 10, 272 CrossRef CAS PubMed.
- R. S. Byrne and P. B. Deasy, Int. J. Pharm., 2002, 246, 61–73 CrossRef CAS PubMed.
- A. Nokhodchi, S. Raja, P. Patel and K. Asare-Addo, Bioimpacts, 2012, 2, 175–187 CAS.
- V. Privman, S. Domanskyi, R. A. S. Luz, N. Guz, M. L. Glasser and E. Katz, ChemPhysChem, 2016, 17, 976–984 CrossRef CAS PubMed.
- M. Di Francesco, R. Primavera, D. Romanelli, R. Palomba, R. C. Pereira, T. Catelani, C. Celia, L. Di Marzio, M. Fresta, D. Di Mascolo and P. Decuzzi, ACS Appl. Mater. Interfaces, 2018, 10, 9280–9289 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS PubMed.
- E. Larrañeta, S. Stewart, M. Ervine, R. Al-Kasasbeh and R. F. Donnelly, J. Funct. Biomater., 2018, 9, 13 CrossRef PubMed.
- O. Shafiee, S. G. Jenkins, T. Ito and D. A. Higgins, Phys. Chem. Chem. Phys., 2023, 25, 2853–2861 RSC.
- F. V. Lavrentev, V. V. Shilovskikh, V. S. Alabusheva, V. Y. Yurova, A. A. Nikitina, S. A. Ulasevich and E. V. Skorb, Molecules, 2023, 28, 5931 CrossRef CAS PubMed.
- S. T. Moe, K. I. Draget, G. Skjåk-Bræk and O. Simdsrød, Carbohydr. Polym., 1992, 19, 279–284 CrossRef CAS.
- U. S. F. a. D. Administration, Medical Device Sterilization Town Hall: The Value and Use of Recognised Consensus Standards in Premarket Submissions, https://www.fda.gov/medical-devices/medical-devices-news-and-events/medical-device-sterilization-town-hall-value-and-use-recognized-consensus-standards-premarket, (accessed 2025-01-13, 2025).
- U. S. F. a. D. Administration, FDA Innovation Challenge 1: Identify New Sterilisation Methods and Technologies, https://www.fda.gov/medical-devices/general-hospital-devices-and-supplies/fda-innovation-challenge-1-identify-new-sterilization-methods-and-technologies, (accessed 2025-01-13, 2025).
- C. S. A. Bento, M. C. Gaspar, P. Coimbra, H. C. de Sousa and M. E. M. Braga, Int. J. Pharm., 2023, 634, 122671 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |