
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocompatible NaLn(WO4)2 core–shell nanoplatelets for multimodal MRI contrast, NIR imaging, and high sensitivity infrared luminescent ratiometric thermometry†
Carlos
Alarcón-Fernández
a,
Carlos
Zaldo
 a,
Manuel
Bañobre-López
b,
Juan
Gallo
b,
Pedro
Ramos-Cabrer
cd,
Sandra
Plaza-García
c,
Gonzalo
Villaverde
e,
Alejandro
Ruperti
e and
Concepción
Cascales
*a
a,
Manuel
Bañobre-López
b,
Juan
Gallo
b,
Pedro
Ramos-Cabrer
cd,
Sandra
Plaza-García
c,
Gonzalo
Villaverde
e,
Alejandro
Ruperti
e and
Concepción
Cascales
*a
aInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, c/Sor Juana Inés de la Cruz 3, 28049 Madrid, Spain. E-mail: ccascales@icmm.csic.es
bAdvanced (Magnetic) Theranostic Nanostructures Group INL—International Iberian Nanotechnology Laboratory, Av. Mestre José Veiga s/n, 4715-330 Braga, Portugal
cCIC biomaGUNE, Basque Research and Technology Alliance, Paseo Miramón 194, 20014 Donostia-San Sebastián, Spain
dIkerbasque, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
eMatNaBio Group, Departamento de Química en Ciencias Farmacéuticas, Universidad Complutense de Madrid, Plaza Ramón y Cajal 2, 28040 Madrid, Spain
First published on 21st July 2025
Abstract
Multifunctional nanoprobes combining magnetic resonance imaging (MRI) contrast as well as near infrared (NIR) imaging and thermometry are demonstrated by using quasi-bidimensional core–multishell nanostructures based on the scheelite-like NaLn(WO4)2 host (Ln = trivalent lanthanide). These nanostructures are composed of a NaHo(WO4)2 core, plus a first shell of Tm,Yb:NaGd(WO4)2, and a second shell of Nd,Yb:NaGd(WO4)2. Proton nuclear magnetic relaxation dispersion studies and MRI of water dispersions of nanoprobes, whose quasi-bidimensional geometries promote the interaction of Gd3+ with water protons, reveal behaviors evolving from a T1-weighted MR contrast agent (CA) at 1.5 T to a highly effective T2-weighted MR CA at ultrahigh magnetic fields of 7 T and above, and even a dual T1/T2-weighted CA at a clinical 3 T magnetic field. By NIR excitation (λEXC ∼ 803 nm) of Nd3+, luminescence-based thermometry was accomplished at wavelengths within the second biological transparency window (II-BW) through ratiometric analysis of 4F3/2 → 4I11/2 Nd3+ (λ = 1058 nm) and 2F5/2 → 2F7/2 Yb3+ (λ = 996 nm) emissions. Under a biologically safe excitation of 0.68 W cm−2, a chemically stable 2 mg mL−1 nanoprobe water dispersion presents absolute, SA, and relative, SR, thermal sensitivities as remarkable as SA = 480 × 10−4 K−1, and SR = 0.89% K−1 at 40 °C (313 K), and temperature resolution δ ≈ 0.1 K. Moreover, through efficient Nd3+ → Yb3+ → Tm3+ and Nd3+ → Yb3+ → Ho3+ energy transfers, NIR photoluminescence from Tm3+ at ∼1800 nm and Ho3+ at ∼2000 nm facilitates in depth imaging. The low nanoprobe cytotoxicity allows NIR biolabeling during cellular temperature measurement.
1. Introduction
A current direction in bioimaging utilizes hierarchically built nanostructures combining two or even more imaging modalities as exogenous nanoprobes. Among the imaging techniques now in use, photoluminescence (PL) imaging offers real-time precise subcellular and molecular information with high spatial resolution at the wavelength level, but accessible imaging is shallow, and the resolution degrades with the depth, being whole body imaging not possible. Therefore, clinical use of PL imaging is limited to a few applications such as image-guided surgery on tumors and retinal angiography.1,2 On the other hand, MRI is currently one of the most powerful imaging technologies in both preclinical research and clinical diagnosis. MRI is able to report physiological and anatomical information with unlimited penetration depth and high soft tissue contrast. However, MRI has long been affected by low sensitivity owing to the mechanism of measuring the relaxation rates of water protons,3,4 which leads to poor resolution,5 and thus the requirement for long scan times. Despite technical advances in recent years,5,6 the low sensitivity of MRI remains a significant limitation on current clinical examinations. Although MRI has become a popular choice for monitoring magnetically labeled cells,7,8 its low resolution at the cellular level9 has led to the combination with other imaging modalities. By exploiting the complementarity of both MR and PL imaging merged into a single nanoprobe, it will be possible to obtain more complete information from anatomical to molecular levels, required to understand disease mechanisms and drug action.Conventional exogenous probes employed in PL imaging, such as organic dyes, fluorescent proteins, metal complexes and semiconductor quantum dots, among others, require excitation in the ultraviolet (UV) or blue-green visible spectral ranges.10 At these wavelengths, the low penetration of light in biological tissues due to either direct absorption by tissue constituents (water, blood, fat etc.) or light scattering by the non-continuous media (typically described by the Rayleigh law as I ∼ λ−4), seriously limit the potential of these probes for biosensing. Thus, schemes of both excitation and sensing shifted to the near infrared (NIR) are preferred to improve the depth for subcutaneous and in vitro PL-based sensing, simultaneously with reduction of photodamage and improved signal-to-noise ratio through prevention of tissue autofluorescence associated to UV/blue optical excitation.11 These NIR–NIR operating schemes may also profit from the so-called ‘optical biological transparency windows’ (BW) present in biological tissues at defined NIR wavelength ranges, the first, I-BW λ = 700–950 nm, second, II-BW λ = 1000–1400 nm, third, III-BW λ = 1500–1850 nm, and fourth, IV-BW λ = 2100–2300 nm, where the light has lower attenuation.12 I-BW and II-BW are particularly appropriate for PL-based subcutaneous sensing.13
Lanthanide (Ln)-based photoluminescent inorganic crystalline nanoparticles (NPs) are non-photoblinking and non-photobleachable, thus very suitable for long term repetitive PL-based bioimaging.10,14 Other advantages of Ln-NPs in this field are their narrow emission bandwidths, which ease multiplexed imaging, and their high biocompatibility.15,16 Among Lns, Nd3+ owns a favourable energy level distribution to operate within BWs. It can be pumped efficiently at λEXC ∼ 800 nm (I-BW) through the 4I9/2 → 4F5/2,2H9/2 electronic transition, and monitored at different wavelengths, at λEMI ∼ 860–920 nm, ∼1050–1100 nm, ∼1300–1400 nm, and even at ∼2100–2200 nm, Fig. 1a. Under pumping also at λEXC ∼ 800 nm, Tm3+ allows access to the III-BW, at λEMI ∼ 1800 nm, through the 3F4 → 3H6 transition, Fig. 1b.
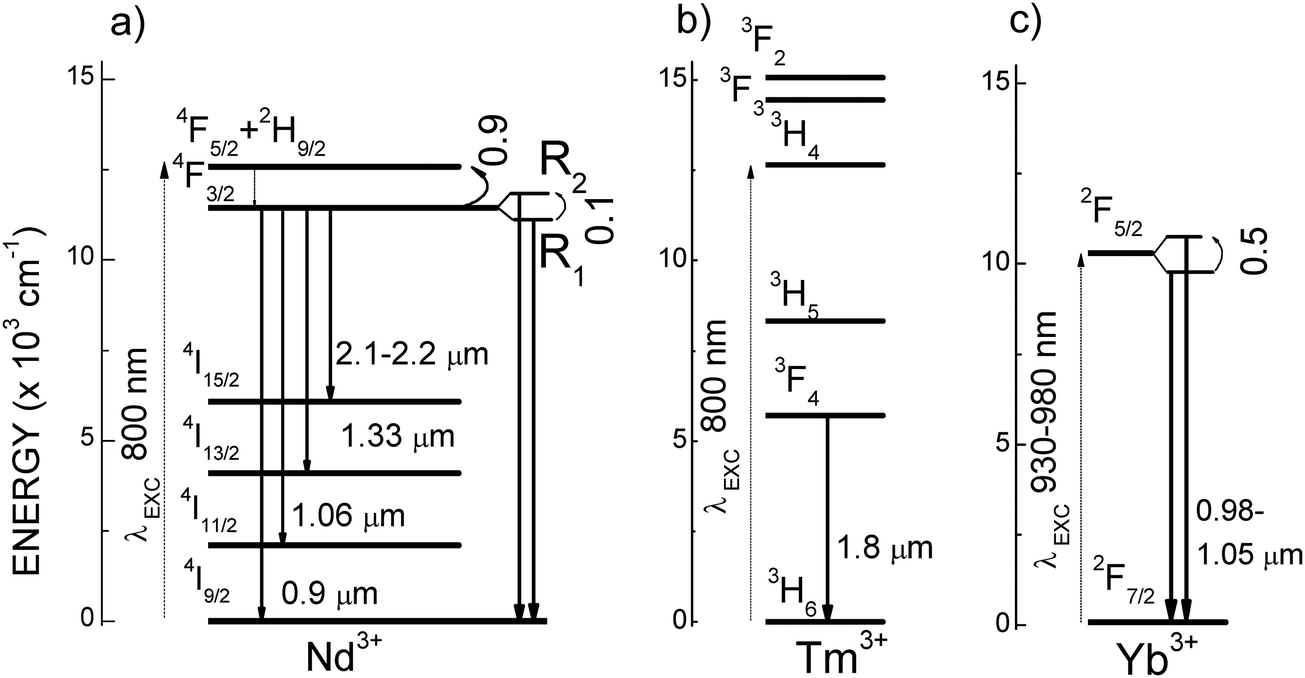 | ||
| Fig. 1 Schemes of lanthanide energy levels with NIR excited photoluminescence emissions within the optical transparency windows for biological tissues, (a) Nd3+, (b) Tm3+, (c) Yb3+. Curved arrows indicate the energy gaps (×103 cm−1) between thermally coupled energy levels used for ratiometric thermal sensing. | ||
Interestingly, thermal properties can be contactless tested at the nanometric scale by using also PL Ln-doped NPs,17–22 being the ratiometric method based on the luminescence intensity ratio (LIR) change of two emissions the most commonly used for temperature sensing. In fact, the intensity changes of the different sub-bands observed for the Nd3+ 4F3/2 → 4I9/2 transitions under λEXC ∼ 800 nm have been studied for thermometric purposes in several hosts, for instance, LaF3,23 YAG,24 YVO4,25 or LiNdP4O12,26,27 but the absolute and relative thermometric sensitivities obtained are low due to the small (≈100 cm−1) energy gap between the two Stark sublevels (R1 and R2), of the Nd3+ 4F3/2 multiplet, see Fig. 1a. Larger thermometric sensitivities have been obtained by using the 4F5/2/4F3/2 Nd3+ PL intensity ratios, but in this case a more energetic excitation in the visible spectra (4I9/2 → 4G5/2 at λ ∼ 577 nm, i.e. outside the BWs), is required,28,29 otherwise the overlap between excitation (resonant to 4F5/2) and emission wavelengths increases the LIR noise to non-acceptable levels.
Yb3+ is an alternative for NIR excitation (λEXC = 930–980 nm), and monitoring (λEMI = 980–1050 nm) within the II-BW, based on 2F7/2 ↔ 2F5/2 electronic transitions, Fig. 1c. However, the spectrally broader nature of the Yb3+ luminescence, and again the small energy gap of the Stark sublevels inside of the excited multiplet impose low limits on the sensitivity and resolution of the thermometric determinations. A more promising thermometric approach is to use the electronic transfer between 4F3/2 Nd3+ and 2F5/2 Yb3+ quasi-resonant energy levels.30–32 In the latter case, the results are greatly dependent on the distance between Nd3+ and Yb3+, i.e. the Nd3+ and Yb3+ concentrations.33 The transfer efficiency is assisted by the absorption or emission of lattice phonons, thus the use of fluoride hosts with small cutoff phonon energy is no longer advantageous. In fact, scheelite-type tetragonal double tungstates NaT(WO4)2, T = Y, La, Gd, Lu (or isostructural molybdates), with cutoff phonon energy ħϖ ≈ 920 cm−1, have shown much higher ratiometric sensitivity than the commonly used β-NaYF4 fluorides for the Yb3+-sensitized Er3+ PL-based thermometry, even for sensing in the physiological temperature range.34,35
Regarding MRI, a number of Gd3+-based chelates are currently used as T1-weighted contrast agents (CA),36–38 due to Gd3+ capability to shorten T1 relaxation times of coordinated and/or nearby water protons, resulting in brighter (positive) contrast generation. On the other hand, NPs of superparamagnetic Fe-containing oxides mainly Fe3O4, but also CoFe2O4, MnFe2O4 and ZnFe2O4, are used to shorten T2 relaxation times in the neighbouring regions, producing darker (negative) contrast in T2-weighted MR images.39–42 With the purpose of synergizing the contrast enhancement provided by T1-weighted images, especially for dynamic contrast-enhanced MRI analyses43 with the detailed morphological information obtained with T2-weighted images, T1/T2 dual-mode CAs for MRI have been developed to yield complementary data, with the additional benefit of a single dose for both imaging modes. These CAs are mainly constructed by combining Gd3+-chelates as the bright T1 CA, and superparamagnetic iron oxide NPs as T2 CA, in a single nanoprobe.44,45 However, since Gd3+-chelates have optimal performance below 1 T, and superparamagnetic iron oxide NPs saturate their magnetization at ∼1.5 T,46,47 these dual T1/T2 CAs are no efficient in high magnetic field MRI instruments developed to gain higher signal-to-noise ratio, higher speed and higher resolution imaging.5,6 On the other hand, mild adverse reactions associated to Gd3+ deposition, accumulation and retention on tissues and organs after Gd3+-chelates administration have been evidenced.48 Alternatively, Gd3+-based crystalline NPs have demonstrated advantageous T1-weighted imaging properties compared to small molecule Gd3+-chelates,49 mostly because they contain a very high density of Gd3+ probing centers, thus providing higher T1 relaxivity and good imaging contrast through lower doses, along with longer circulation times in blood. On the other hand, NPs incorporating Dy3+ and Ho3+, with high effective magnetic moments (μeff = 10.6 and 10.4μB, respectively) and short electronic relaxation times (around 0.5 ps), show improved capability to relax water protons when the strength of the external magnetic field increases, thus being suitable T2 CAs even in high magnetic fields. In fact, some Dy3+ and Ho3+-containing fluoride and oxide NPs have been already proposed as high field T2 MRI CAs.50–55
Unfortunately, in Nd3+-sensitized (λEXC ∼ 800 nm) Ln-doped NPs, PL quenching occurs through efficient energy back-transfers from Lns to Nd3+, due to the closeness of these cations within NPs.56–58 On the other hand, problems associated with a reduction of the expected contrasts in T1/T2 dual-mode CAs are unavoidable if the magnetic Lns of interest, Gd3+ for T1, Ho3+ or Dy3+ for T2, are integrated nearby into a single nanoprobe, since magnetic coupling between T1 and T2 effects will appear.59 The proper separation of optically active Lns in rationally designed core–shell nanostructures, in which Yb3+ is also incorporated, is a successful strategy to optimize the Ln PL brightness through the prevention of energy back-transfers to Nd3+.56–58 In these core–shells, Yb3+ acts as an efficient energy mediator to facilitate energy transfers from the sensitizer Nd3+, lying in the outer shell, to the other Lns. Moreover, core–shell nanostructures offer the possibility of achieving magnetically decoupled T1/T2 dual mode contrasts in a single nanoprobe, by separating effects of T2, Ho3+ or Dy3+ at the core, and T1, Gd3+ at the shell in contact with water.60
To address the challenge of developing multifunctional nanoprobes combining MRI contrast, as well as NIR–NIR PL-related imaging and high sensitivity contactless ratiometric thermometry, herein we present the design, preparation and evaluation of a series of materials from single ultra-small Ln-containing NPs to multilayer core–shell nanostructures, built on double tungstates NaLn(WO4)2 with the tetragonal scheelite-like structure.61 The choice of this double tungstate crystalline phase over commonly studied fluorides56–58 is based on multiple factors, some of which have already been highlighted in previous studies,34,35 namely the possibility of preparing individualized NPs with ultra-small sizes, even below 10 nm, without the appearance of competitive phases, its high PL efficiency, which is considerably better retained with respect to the bulk material, and the aforementioned superior thermometric sensitivity. Concerning MRI requirements, the main factor favouring longitudinal relaxivity r1 is the easy access of water molecules to Gd3+ centers at the NPs surface; thus, morphologies with the highest surface-to-volume ratio possible must be sought because they will allow higher r1 values. From this point of view, the planar, quasi-bidimensional diamond-shaped morphology shown by NPs of NaLn(WO4)2 will be better suited to provide high r1 values than 3D NPs of fluorides, either hexagonal β-NaYF4 or tetragonal LiYF4 phases.
The Fig. 2 displays the schematic representation of the nanoprobe system proposed in this study.
 | ||
| Fig. 2 Architecture of the proposed series of planar diamond-shaped nanoparticles and successive core–shell nanostructures based on the scheelite-like double tungstate NaLn(WO4)2 host, showing the distribution of incorporated lanthanides. | ||
By choosing Ho3+ to provide T2-MRI contrast, single NaHo(WO4)2 NPs will constitute the core, C, of the multi-layered system. The first nanostructure, C@1S, is prepared by using NaHo(WO4)2 NPs as seeds, which are coated with a Tm3+,Yb3+-doped NaGd(WO4)2 shell (1S). C@1S is suitable for additional generation of T1-MRI contrast due to surface–lying Gd3+ centers. The confinement of Ho3+ and Gd3+ in the core and the first shell, respectively, should help weaken the magnetic interaction between T1 and T2 CAs, thus avoiding contrast quenching. C@1S were the seeds for the second nanostructure C@1S@2S, resulting from the coating with a Nd3+,Yb3+-doped NaGd(WO4)2 second shell (2S), which in turn will provide the thermometric function. Further, the presence of Yb3+ in both the 2S and 1S shells allows the excitation of Tm3+ and Ho3+ through efficient Nd3+ → Yb3+ → Tm3+ and Nd3+ → Yb3+ → Ho3+ energy transfers, giving rise to NIR PL emissions from Tm3+ at ∼1800 nm (within the III-BW, 3F4 → 3H6 transition)62 and Ho3+ at ∼2000 nm (5I7 → 5I8 transition),63,64 of interest for high penetration NIR optical imaging purposes. The spatial separation between surface-lying Nd3+ and Tm3+ or Ho3+cations minimizes energy back-transfers to Nd3+, and thus avoids the quenching of their emissions, even at the high Nd3+ doping levels in the nanoprobe. Moreover, the 2S layer is protected from its degradation in diluted water dispersions by further growth of an isostructural optically inert NaGd(WO4)2 outermost layer to produce C@1S@2S@i3S, which avoids the quenching of Nd3+ and Yb3+ PL emissions to be used for LIR thermometry.
TWEEN80, a safe food additive for human consumption,65 has been used as surfactant in the procedure to render luminescent double tungstate NPs and core–shell nanostructures dispersible in water. MRI contrast and PL-based thermometric sensing characteristics in proposed nanoprobes were optimized through the evaluation and control of the concentration of incorporated Lns. MRI experiments were performed at 1.5, 3, 7 and 11.7 T by using water-stabilized dispersions of the nanoprobes to determine their behaviours as CAs from clinical up to research magnetic fields. PL spectra of these diluted dispersions were excited under low laser power density, typically 0.68–2.5 W cm−2, and used for LIR-based thermometry at temperatures within the range of physiological interest. Furthermore, to ascertain the safe use of developed multifunctional nanoprobes for biomedical applications, such as diagnostic imaging and NIR biolabeling during cellular temperature measurement, their metabolic interactions with pre-osteoblastic MC3T3 healthy cells and HeLa human cancer cells have been analyzed in vitro through viability, cytotoxicity and nuclear visualization by DAPI (4′,6-diamidino-2-phenylindole) staining assays.
2. Experimental section
2.1 Chemicals
Lanthanide acetate hydrates (LnAc) Ho(CH3CO2)3·4H2O (Alfa Aesar, 99.9%), Yb(CH3CO2)3·xH2O (Sigma-Aldrich 99.95%), Tm(CH3CO2)3·4H2O (Alfa Aesar, 99.9%), Gd(CH3CO2)3·4H2O (Strem Chemicals, 99.9%) and Nd(CH3CO2)3·4H2O, (Strem Chemicals, 99.9%), tungsten hexacarbonyl W(CO)6 (Acros Organics, 99%) and NaOH were used as reagents in a mixture of oleic acid (OA) (Aldrich, technical grade 90%), 1-octadecene (ODE) (Alfa Aesar, technical grade 90%) and oleylamine (OLE) (Sigma Aldrich, 98%) solvents.To extract and purify the prepared NPs, n-hexane (LabKem, 99%+) and absolute ethanol (EssentQ, Scharlau) were used. TWEEN 80 (Sigma-Aldrich) and CHCl3 (Merck, Emprove Essential 99%) were used to transform the oleate-capped hydrophobic prepared nanomaterials into water dispersible ones.
Dulbecco's modified Eagle's medium (DMEM), anti–anti (penicillin–streptomycin), trypsin–EDTA medium, fetal bovine serum (FBS), 4′,6-diamidino-2-phenylindole (DAPI), AlamarBlue®, and CyQUANT™ LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific, Cat. No. C20300) were used in nuclear stain, viability, and cytotoxicity assays of MC3T3 and HeLa cells.
2.2 Prepared nanomaterials
Core NPs, C, with composition NaHo(WO4)2, were prepared by coprecipitation synthesis in organic solvents with high boiling points, according to a protocol based on previously described methods,21,22 which have been adapted to the present case as described below. Shells 1S, 2S and i3S with the following selected compositions:| 1S ≡ 8 at% Tm,15 at% Yb:NaGd(WO4)2 |
| 2S ≡ x at% Nd,20 at% Yb:NaGd(WO4)2, x = 20, 30 |
| i3S ≡ NaGd(WO4)2 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for two times. The result is a yellowish-colored pellet, which can be easily resuspended in n-hexane, yielding a transparent yellow dispersion.
:1 for two times. The result is a yellowish-colored pellet, which can be easily resuspended in n-hexane, yielding a transparent yellow dispersion.
For the preparation of C@1S-b, the described procedure with the 1S-precursor was repeated twice.
NaHo(WO4)2@8 at% Tm:15 at% Yb:NaGd(WO4)2@20 at% Nd (30 at% Nd):20 at% Yb:NaGd(WO4)2 (C@1S@2S), as well as the nanostructure with a further NaGd(WO4)2 shell (C@1S@2S@i3S), were prepared following procedures similar to the described above, and all details are included in the ESI.†
This ligand exchange procedure can be also carry out with TWEEN40, but to prevent aggregation of the current ultrasmall NPs, the bigger TWEEN80 ligands were preferred.
2.3 Characterization of nanomaterials: phase, size, morphology and concentration of lanthanides
The phase purity of all synthesized materials, NPs, core–shell nanostructures and polycrystalline solid-state prepared samples, was tested by room temperature powder X-ray diffraction (XRD) analysis performed in a Bruker AXS D-8 advance diffractometer, using Cu Kα radiation. To identify the crystalline phase, the angular range scanned was 2θ = 10–65 deg, with a step size Δ2θ = 0.05 deg and an integration time of 3 s at each step.The morphology of prepared nanomaterials was studied by transmission electron microscopy using JEOL JEM1010 and JEM3000F microscopes. To obtain evidence of the formation of core–shell designs from described syntheses, concentration profiles of Lns were obtained by combined scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDX) analyses along a line in individual NPs of the largest nanostructure, C@1S@2S@i3S. For this, a JEOL JEM Arm200cF microscope operated at 200 kV, with corrected spherical aberration in the condensed lens and equipped with an X-EDS Oxford X-MaxN 100 TLE detector with active area of at least 50 mm2 and resolution of 129 eV, was used. The thickness of single NPs was measured with a Bruker Multimode AFM Nanoscope III A in contact mode on a mica substrate, and evaluated with the Bruker NanoScope Analysis software.
Hydrodynamic sizes and size distributions of NPs in hexane, as the long-lasting storage medium of prepared oleate-capped NPs and nanostrosctures, and in water dispersions after surface modification with TWEEN80, which were used in proton nuclear magnetic relaxation dispersion studies and MRI analyses as well as in PL NIR nanothermometry, were evaluated overtime by dynamic light scattering (DLS) measurements with a Cordouan Technologies particle size analyzer, model Vasco 2, powered by the NanoQ proprietary software.
Magnetic properties were characterized using a SQUID magnetometer (MPMS-3) from quantum design. 80 to 160 μL of sample solutions were dried inside SQUID capsules with cotton. The magnetization of prepared nanomaterials as a function of the applied magnetic field was measured at 300 K under fields up to ±7 T.
The concentration of Lns in water dispersions was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) with a PerkinElmer Optima 2100 ICP equipment, after digestion of the samples in HNO3 and dilution with doubly distilled water.
2.4 Photoluminescence measurements
Down-shifted near infrared (NIR) PL (λEMI > 900 nm) was excited with a tunable Ti-sapphire Spectra Physics laser model 3900S. The PL emission was dispersed with a SPEX f = 34 cm spectrometer and detected either with a 77 K cooled Applied Detector Corporation Ge photodiode model 403R with spectral response in the λ = 850 to 1650 nm range or with a 77 K cooled Hamamatsu InAs photovoltaic detector model P7163, with spectral response in the λ = 1500 to 3500 nm range. Optical excitation was modulated with a chopper and the detector signal evaluated in phase with a lockin amplifier.In all cases, the optical excitation density was kept at low levels (IEXC = 0.68 W cm−2 for NPs dispersed in water or IEXC ≤ 2.5 W cm−2 for n-hexane dispersions) in order to avoid the heating of samples. Proper optical filters were used to avoid optical harmonics and other undesired light contributions. All spectra were corrected by the instrumental responses. For every measured combination of sample/concentration/temperature at least three acquisitions were performed in order to estimate the measurement error.
Liquid colloidal samples were hold in a 10 × 10 × 50 mm3 glass cuvette, and heated with hot water generated by a Lauda bath model RE-420 with temperature resolution of 0.01 °C. The actual temperature of the dispersion was monitored with a K-type thermocouple immersed in the liquid with a reading accuracy of 0.1 °C. Powdered solid samples were set on the horizontal surface of an Instec hot plate (with nominal resolution of 0.01 °C) and the actual temperature was monitored by a K-type thermocouple buried in the powder with a reading accuracy of 0.1 °C. The emission was collected by a parabolic mirror, and sent to the SPEX monochromator through a two lenses optical system.
2.5 In vitro cellular assays
000 cells and 50000 cells per well, respectively. The cells were incubated at 37 °C (310 K) in a humidified atmosphere of 5% CO2 and 95% humidity, using Dulbecco's modified Eagle medium (DMEM) supplemented with appropriate growth factors, antibiotics and glutamine. The cells were allowed to adhere overnight under these controlled conditions. Following this incubation period, the cells were treated with increasing concentrations of C@1S@2S@i3S in the water dispersion, up to 200 μg mL−1, for 24 h. Before the viability assay, both NPs-exposed and control (untreated) cells were washed twice with phosphate-buffered saline (PBS) to remove non-internalized NPs.
Cell viability was quantified using the AlamarBlue® (resazurin-based reagent) assay, a sensitive and non-toxic method suitable for assessing cell metabolism. The AlamarBlue® reagent was added to a 10% working solution in DMEM, and the cells were incubated for 30 minutes at 37 °C (310 K). The principle behind this assay is based on the reduction of the AlamarBlue® reagent by NADH/H+ produced by metabolically active, viable cells. The reduced form of the reagent (resorufin) regains aromaticity, and emits very bright orange fluorescence, which can be measured as a direct indicator of cell viability. Thus, after the incubation period, the medium containing the AlamarBlue® reagent was carefully removed, and using a plate reader set, the fluorescence at 590 nm was measured by excitation at 560 nm. The fluorescence intensity correlates directly with the number of viable cells in the sample, providing quantitative data on cell viability. This assay allows for the determination of cell viability without interference from the NPs themselves, ensuring accurate results.
The percentage of viable cells was calculated by normalizing the fluorescence values from treated samples to those of controls, with data presented as the mean ± standard deviation from four independent experiments (N = 4).
2.6 Relaxivity measurements and MRI imaging
Relaxivities of the different compounds in dispersion at different concentrations (solvent: water HPLC Gradient grade, Fischer Scienctific) were measured at magnetic fields B of: (a) 1.5 T, at 37 °C (310 K), using a benchtop relaxometer Bruker Minispec MQ60 (Bruker Biospin GmbH, Ettlingen, Germany). T1 relaxation time was measured using an inversion recovery sequence (TR = 0.01–10.000 ms, recycle delay = 2 s, number of scans = 4, TE = 0.05 ms, points for fitting = 20). T2 relaxation time was measured using the Carr–Purcell–Meiboom–Gill sequence (TR = 10.000 ms, recycle delay = 2 s, number of scans = 8, TE = 0.05 ms, points for fitting = 30.000). Relaxivities r1 and r2 were calculated using the least squares curve fitting of R1 = 1/T1 and R2 = 1/T2 relaxation rates (s−1) versus iron concentration (mmol L−1); (b) B = 7 T, at room temperature, using a horizontal bore Bruker Biospec USR 70/30 MRI system (Bruker Biospin GmbH, Ettlingen, Germany), interfaced to an AVANCE III console, and with a BGA12-S imaging gradient insert (maximal gradient strength 400 mT m−1, switchable within 80 μs), and a 40 mm inner diameter volume coil from Bruker, for RF transmission and reception. For T1 maps imaging of the phantoms the following parameters were adopted: spin echo saturation recovery using a variable repetition time Bruker's RAREVTR method. Images were acquired at 9 different TR values of 150, 400, 750, 1100, 1500, 2000, 3000, 5000 and 10000 ms, TE = 8.2 ms, RARE factor 1. For T2 maps imaging of the phantoms the following parameters were adopted: Bruker's MSME (multi slice spin echo) sequence. The TE values were varied in 64 steps of 5 ms, ranging from 5 ms to 320 ms, and TR 4.8 s; (c) B = 11.7 T, at room temperature, using a horizontal bore Bruker Biospec USR 117/16 MRI system (Bruker Biospin GmbH, Ettlingen, Germany), interfaced to an AVANCE III console, and with a BGA12-S imaging gradient insert (maximal gradient strength 600 mT m−1, switchable within 80 μs), and a 40 mm inner diameter volume coil from Bruker, for RF transmission and reception. For T1 maps imaging of the phantoms the following parameters were adopted: spin echo saturation recovery using a variable repetition time Bruker's RAREVTR method. Images were acquired at 9 different TR values of 150, 500, 1000, 1500, 2000, 3000, 5000, 8000 and 14000 ms, TE 5.7 ms, RARE factor 1. For T2 maps imaging of the phantoms the following parameters were adopted: Bruker's MSME sequence was used. The TE values were varied in 64 steps of 5 ms ranging from 5 ms to 320 ms, and TR 4.8 s.
All the above data were acquired with 3 averages, 280 × 280 points and a field of view of 2.8 cm × 2.8 cm, 3 slices with a slice thickness of 1.0 mm and 1 mm gap between slices. The images were fitted into Levenberg–Margardt method to calculate T1 and T2 values using Bruker's Paravision 6.0.1 software.
T 1 and T2 parametric maps at 3.0 T were acquired in a horizontal bore MR Solutions Benchtop scanner equipped with 48 G cm−1 actively shielded gradients with a 56 mm diameter quadrature bird-cage coil in transmit/receive mode, using MPRAGE (magnetization prepared rapid gradient echo imaging) and MEMS (multi-echo multi-slice) sequences, respectively. Images were acquired with an image matrix of 256 × 252 and FOV of 60 × 60 mm. The following parameters were used for the MEMS sequences: NE = 10 (TE = 0.015, 0.03, 0.045, 0.06, 0.075, 0.09, 0.105, 0.12, 0.135, and 0.15 s), TR = 1400 ms. For the MPRAGE sequences, the parameters were as follows: TI = 11 values (0.275, 0.3, 0.35, 0.45, 0.65, 1.05, 1.85, 3.45, 6.65, 12.45, 26.25 s), TE = 5 ms, TR = 10 s. Image analysis was performed using ImageJ software (https://imagej.nih.gov/ij) and the “MRI analysis calculator” plugin by Karl Schmidt.
3. Results and discussion
3.1 Characterization of nanoparticles and core–shell nanostructures
XRD patterns of C, C@1S, C@1S@2S and C@1S@2S@i3S confirm that they correspond to the tetragonal space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) scheelite-like crystal phase described for the isostructural NaGd(WO4)2 single crystal,61 with no foreign peaks, see Fig. S1a, b, c and d, respectively, in ESI.† The values of their mean crystalline domain sizes calculated by the Scherrer's equation, ds, evolved as ∼7 nm, ∼10 nm ∼12 nm and ∼17 nm for C, C@1S, C@1S@2S and C@1S@2S@i3S, respectively, in accordance with the corresponding continuous decrease observed in the width of the Bragg reflections.
scheelite-like crystal phase described for the isostructural NaGd(WO4)2 single crystal,61 with no foreign peaks, see Fig. S1a, b, c and d, respectively, in ESI.† The values of their mean crystalline domain sizes calculated by the Scherrer's equation, ds, evolved as ∼7 nm, ∼10 nm ∼12 nm and ∼17 nm for C, C@1S, C@1S@2S and C@1S@2S@i3S, respectively, in accordance with the corresponding continuous decrease observed in the width of the Bragg reflections.
Fig. 3a–d show a panel of representative transmission electron microscopy (TEM) images of these NPs, with statistical histograms for size distribution, and corresponding high magnification images (HRTEM) in Fig. 3e–h reveal their well-defined crystalline characteristics. Diamond-like shape is observed in all cases, with diagonal sizes of ∼6.2 × 4 nm, ∼8.7 × 5 nm, ∼13.0 × 7, and ∼16.5 × 8 nm for C, C@1S, C@1S@2S and C@1S@2S@i3S, respectively, and this size increase would suggest successful growth of 1S, 2S and i3S coating layers, with ∼1.2 nm, ∼2.1 nm and ∼1.7 nm, respectively. These NPs are mostly individualized, highly monodisperse in size, and very thin, see atomic force microscopy (AFM) images for C@1S@2S NPs presented in Fig. 3i and j, whose evaluation indicates thicknesses ranging between 1.6 and 2.5 nm, see Fig. 3k.
 | ||
| Fig. 3 Morphological characteristics of scheelite-like double tungstate-based nanoprobes. TEM images of (a) C, (b) C@1S, (c) C@1S@2S, and (d) C@1S@2S@i3S NPs, with corresponding histograms for nanodiamond largest diagonal size distribution. High-resolution TEM images of (e) C, (f) C@1S, (g) C@1S@2S, and (h) C@1S@2S@i3S NPs. AFM images, (i) and (j), and height profiles, (k), of C@1S@2S NPs. | ||
Qualitative high-angle annular dark field HAADF-STEM images of prepared nanostructures did not provide a complete evidence of contrast differences between the core and shell(s), Fig. 4a shows a typical HAADF image for C@1S. In fact, this was expected since the contrast depends on the atomic number Z, which is very close for Lns distributed in current nanostructures, and the reduced thickness may also minimize the contrast. Therefore, to ascertain the formation of proposed core–shell structures, concentration profiles of Lns in the largest nanostructure, C@1S@2S@i3S, were obtained by STEM-EDX analyses along a line in individual NPs of such material, see Fig. 4b. As it can be seen, the Ho3+ ions are well-localized at the central part of the profile, indicating their sole presence at the core. On the other hand, the presence of Gd3+ ions has been found to be maximum at the edges of its profile, in accordance with the existence of a stoichiometric Gd3+ (i3S) external shell, although the EDX scan indicated also some extended presence to the interior as due to Gd3+-containing 2S and 1S shells. The Nd3+ profile shows also maxima near the edges, but somewhat displaced to the interior, as expected from Nd3+ presence as the major Ln component in 2S. For simplicity, we chose to exclude in this figure the concentration profiles of Yb3+, present in 1S (15 at%) and 2S (20 at%), and of Tm3+, with very weak concentration (8 at%) in the inner 1S, for better focusing in the characterization of the overall nanostructure.
 | ||
| Fig. 4 (a) HAADF image of NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2 (C@1S); (b) EDX line scan across a single nanoparticle of NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)@30 at% Nd, 20 at% Yb:NaGd(WO4)2@NaGd(WO4)2 (C@1S@2S@i3S). | ||
Results of the measurement of each Ln concentration in water dispersions of TWEEN80-modified C, C@1S and C@1S@2S NPs by using inductively coupled plasma optical emission spectroscopy (ICP-OES) are included in Table S1 (ESI†). Corresponding Ln concentrations for C@1S-b and C@1S@2S appear in Table S2 (ESI†). These data indicate that detected amounts of Tm3+ and Yb3+ are lower than expected according to proposed stoichiometries for the syntheses. Table S3 (ESI†) provides the comparison of relationships of interest between the concentrations of Ho, Gd and Nd in the studied nanoprobes.
Mean hydrodynamic sizes, dDLS, of C, C@1S, C@1S@2S and C@1S@2S@i3S and their size distributions in hexane, the storage medium, and in the water dispersions after surface modification with TWEEN80, used in current MRI and nanothermometry studies, were assessed by overtime DLS measurements, and results can be viewed in Fig. S2–S5 (ESI†). The dDLS values in hexane are well preserved even 18 months after synthesis, and those in water dispersions, ∼20 nm for C, ∼24 nm for C@1S, ∼29 nm for C@1S@2S, and ∼79 nm for C@1S@2S@i3S, are similar for freshly prepared samples and after five weeks of storage, having polydispersity index (PDI) values typically in the 0.12–0.20 range, indicating that they are satisfactorily water dispersible. It must be taken into account that the morphology of current NPs is not spherical but elongated, with higher aspect ratio when the number of shells increase, and dDLS results reflect this fact for the very thin C@1S@2S@i3S, whose diffusion will be further hindered, leading to an overestimation of size by DLS.
3.2 Magnetic field dependent MRI contrast properties
To quantitatively evaluate the feasibility of C, C@1S and C@1S@2S nanocrystalline materials as MRI contrast agents (CAs), their longitudinal (r1) and transverse (r2) relaxivity values were determined through T1- and T2-weighted image acquisitions by using water dispersions of the above nanomaterials. Measurements at current clinical (1.5 T and 3 T) as well as at research (7 T and 11.7 T) magnetic fields have been carried out. Relaxivity values r1 and r2 of proposed nanoprobes are calculated by plotting relaxation rates R1 or R2 (corresponding to the inverse of the T1 or T2 relaxation times) versus the concentration of the Ln responsible for the change in the relaxation. Plots to calculate r1 and r2 values at 1.5 T, 7 T and 11.7 T appear in Fig. S6, S7 and S8, respectively, and in Fig. S9 for data at 3 T (ESI†).The r1 value of NPs containing Lns other than Gd3+ is usually very small when no direct coordination of the Ln to water molecules occurs, and especially at magnetic fields higher than 1 T,66 therefore we calculate r1 values from the concentration of Gd3+ in C@1S and C@1S@2S nanostructures. On the other hand, the effect of Gd3+ on the r2 relaxivity of water protons is negligible, being the high anisotropy of the 5I8 ground state of Ho3+, and hence the very short relaxation times τR of Ho-containing materials, along with its high magnetic moment (10.6μB), the main factors leading to an important Curie-spin contribution to the r2 relaxivity of water protons,67 and this effect will increase considerably for ultra-high magnetic fields. Thus, r2 will be determined from corresponding Ho3+ concentrations in the water dispersions of C, C@1S and C@1S@2S. Table 1 shows the r1, r2 relaxivity values of water protons caused by C, C@1S and C@1S@2S, and corresponding r2/r1 ratios.
| 1.5 T | 7 T | 11.7 T | |||||||
|---|---|---|---|---|---|---|---|---|---|
| r 1 | r 2 | r 2/r1 | r 1 | r 2 | r 2/r1 | r 1 | r 2 | r 2/r1 | |
| [Gd3+] | [Ho3+] | [Gd3+] | [Ho3+] | [Gd3+] | [Ho3+] | ||||
| C | — | 12.1 | — | — | 70.5 | — | — | 84.4 | — |
| C@1S | 7.4 | 13.4 | 1.8 | 1.8 | 155.5 | 86 | 1.1 | 182.7 | 171 |
| C@1S@2S | 6.0 | 35.8 | 6.0 | 0.6 | 112.8 | 179 | 0.6 | 194.0 | 353 |
From these values, it is clear that r1 of C@1S and C@1S@2S are lower than their r2 at any magnetic field, and that for each material r1 tends to decrease when the magnetic field increases. On the contrary, the r2 values show a steady increase with the magnetic field in all cases, and reach maxima of 182.7 and 194.0 mM−1 s−1, for C@1S and C@1S@2S respectively, at 11.7 T. These magnetic field-related evolutions constitute the classical behavior that can be expected for MRI CAs.68,69
The generation of T1 contrast is produced by the coupling of the electron spins of the surface lying Gd3+ and the nuclear spins of protons of the water in contact. In the current system, r1 is greater for C@1S than for C@1S@2S, at any field. This fact is, in principle, surprising since: (i) per NP, C@1S@2S offers higher availability of Gd3+ centers at the water contact surface; (ii) the ratio between Gd3+ and Ho3+ concentrations is considerably higher in C@1S@2S (1:0.53) than in C@1S (1:1.18), see Table S2 (ESI†); (iii) the estimated distances between Ho3+ centers at the core and the Gd3+ ones in contact with water are also higher in C@1S@2S than in C@1S (the coating thickness of 1S + 2S layers being ∼3.3 nm, compared to ∼1.2 nm coating thickness of 1S), thus (ii) and (iii) suggest that the perturbation caused by the magnetic field of Ho3+ would be lessened for C@1S@2S with respect to C@1S. The observed lower r1 value in the water dispersion of C@1S@2S compared to that of C@1S can be attributed to the additional disturbance that the magnetic field generated by Nd3+ centers exerts on the coupling between the spins of neigbouring 2S-Gd3+ and the water protons.70 This effect, therefore, depends on the number of Nd3+ centers, which is related to both the stoichiometry of Nd3+ in the 2S layer and the thickness of the layer. An alternative interpretation of the data is supported by considering the surface mechanism of T1 contrast generation at the macroscopic sample level rather than by individual NP surface area. In this scenario, at equivalent Gd3+ concentration, the total surface would be larger for the smaller NPs, C@1S, because the number of these NPs would be larger than that of C@1S@2S. Thus, the reduction of the total available surface in C@1S@2S, combined with the partial substitution of Gd3+ by Nd3+ ions, may explain the r1 reduction observed for the C@1S@2S water dispersion.
It is worth noting that at 1.5 T magnetic field, the r1 value of the C@1S, r1 = 7.4 mM−1 s−1, exceeds three times the r1 value determined for the fluoride core–shell counterpart, NaHoF4@Yb,Tm:NaGdF4, r1 = 1.93 mM−1 s−1 (at 1.41 T),68 and the comparison between r1 data is likewise favorable for the C@1S@2S nanostructure. These large r1 values for NaLn(WO4)2-based nanostructures are attributable to the high surface-to-volume ratios derived from their quasi-bidimensional geometry, which facilitate the interaction of Gd3+ cations with water protons.
On the other hand, at each measured magnetic field, an increase in r2 is observed when going from single Ho-containing NPs, C, to nanostructures incorporating Gd3+ either in one layer, C@1S, or in two layers, C@1S@2S, see Table 1. According to a classical behavior following the quantum-mechanical outer-sphere theory,71 and from a oversimplified consideration, larger Ho-containing NPs will present better r2 because water diffusion around them is slower and the magnetic field from the particle is higher. However, the studied system is not ideal to make this kind of comparison since it is not only the size that changes, but also the composition, which is not spatially uniform, with Ho3+ located only at the NPs core, and Gd3+ in 1S and 2S outer shells. Instead, it can be considered that the Ho3+ Curie-spin contribution to the r2 relaxivity of water protons should be subjected to a reduction when increasing the number of layers, due to the larger distance between magnetic Ho3+ centers at the core and the surface in contact with water. Also, in large NPs and owing to the presence of Gd3+ in 1S and 2S shells, lower magnetization, see plots in Fig. S10 (ESI†), and thus lower Curie-spin effect would contribute to the worsening of r2 values. In fact, these plots of the magnetization vs. the applied magnetic field indicate that, for a same magnetic field, the magnetization decreases as C > C@1S > C@1S@2S. Otherwise, it has been observed in other systems that the interaction between T1 and T2 CAs depends on the nanoscale, and that instead of inducing dual T1/T2 behaviors, it causes a significant increase in r2.68,72,73 Thus, the r2 increase in C@1S with regards to C can be assigned to Ho3+–Gd3+ dipolar interactions, given the small concentration of the other paramagnetic Lns at 1S, Table S1 (ESI†). The additional increase observed with C@1S@2S would be due to: (i) the existence of an optimal interaction at somewhat longer distances than with 1S, from the balance between the Hocore–Gd1S and Hocore–Gd2S interactions, which would result in an increase in r2, or (ii) the effect of the contribution of additional dipolar interactions between neigbouring Gd3+, as these between Gd1S–Gd2S, to the Curie-spin effect on r2. This second possibility has been described in a hybrid compound of Fe3O4@SiO2 NPs that incorporates a Gd3+-chelate immobilized inside the channels of SiO2 pores, to account for the strong increase in r2,74 and is plausible in the present C@1S@2S system, in which a number of Gd3+ centers can be similarly considered as confined in the 1S.
The behavior of a CA as T1, T2 or dual T1/T2 agent in MRI is a consequence of the r2/r1 value, being a T1 CA characterized by low r2/r1 ratios, while an efficient T2 CA will have a high r2/r1 ratio. According to Table 1, it can be concluded that C@1S would behave as T1 CA at 1.5 T (r1 = 7.4 mM−1 s−1; r2/r1 = 1.8), while at high fields both C@1S and C@1S@2S would be efficient T2 CAs, Fig. 5. Interestingly, C@1S@2S exhibit dual T1/T2 behavior already at 1.5 T (r1 = 6 mM−1 s−1, r2/r1 = 6). Thus, it seems relevant to examine the behavior of these nanoprobes at an intermediate magnetic field between 1.5 T and 7 T.
 | ||
| Fig. 5 Evolution of r2/r1 at 1.5 T, 7 T and 11.7 T magnetic fields of (a) NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2 (C@1S), squares, and (b) NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2@20 at% Nd, 20 at% Yb:NaGd(WO4)2 (C@1S@2S), circles, dispersed in water. Pink, gray and blue symbols are for T1, dual T1/T2 and T2 behaviors, respectively. Hollow square and hollow circle correspond to r2/r1 values of C@1S-b and C@1S-b@2S-b in water dispersions, respectively, measured at 3 T. | ||
To corroborate the T2 character of C, C@1S and C@1S@2S at high magnetic fields, T2-weighted images of their water dispersions at different concentrations were collected at 7 T, Fig. 6a, and at 11.7 T, Fig. 6b. These images show that the three nanoprobes switch from hyper- to hypo-signal intensity with the increase in the concentration.
 | ||
| Fig. 6 T 2-weighted images of C, C@1S and C@1S@2S dispersed in water at (a) 7 T and (b) 11.7 T. T1-maps of C@1S and C@1S@2S nanoprobes dispersed in water at (c) 7 T and (d) 11.7 T. T2-maps of C, C@1S and C@1S@2S nanoprobes dispersed in water at (e) 7 T and (f) 11.7 T. | ||
T 1-maps of C@1S and C@1S@2S nanoprobes dispersed in water are shown in Fig. 6c for 7 T magnetic field, and in Fig. 6d for 11.7 T. T2-maps of C, C@1S, and C@1S@2S are displayed in Fig. 6e for 7 T magnetic field, and in Fig. 6f for 11.7 T. As expected, a linear relation is observed between concentrations of Gd3+ and Ho3+ and inverse of relaxation times T1 and T2, respectively, of protons for all nanoprobes, Fig. S6–S8 (ESI†).
From the above data, the r1 and r2 values and their evolution with the magnetic field appear to be closely related to Gd:Ho and Gd:Nd relative concentrations as well as to distances between these magnetic cations according to their distribution into the nanostructure. These two factors are related to the thicknesses of 1S and 2S shells, thus two optimized core–shell nanostructures, C@1S-b, and C@1S-b@2S-b, have been additionally prepared to be evaluated at the intermediate 3 T magnetic field. 1S-b and 2S-b shells have the same composition as 1S and 2S in previously studied nanostructures, but their thicknesses are different. Taking into account the previous results, a greater thickness of the surface shell in C@1S-b will increase the distance between Ho3+ and the surface in contact with the water, thus decreasing the magnetic perturbation over surface-lying Gd3+, which translates into an increase of r1. On the other hand, since the presence of Nd3+ in the 2S surface shell represents an important disturbance over the coupling of spins of the closest Gd3+ with the water protons, to minimize this effect it is desirable a smaller Nd3+ content in the surface shell, i.e., a smaller thickness for S-2b. Concentrations of Lns in C@1S-b and C@1S-b@2S-b, measured by ICP-OES, are included in Table S2 (ESI†), and Table S3 (ESI†) shows the comparison of relationships of interest between the concentrations of Ho, Gd and Nd in these optimized nanoprobes with regard to previous C@1S and C@1S@2S ones. The inspection of data in Table S2 (ESI†) reveals the achievement of the above-indicated modifications in both first and second shells of prepared nanoprobes. The relationship of Gd:Ho concentrations is about 33% higher in C@1S-b than in C@1S, and being the nominal stoichiometry of the first shell the same in both samples, it is attributable to a greater shell thickness in C@1S-b.
On the other hand, higher Gd:Nd and Gd:Ho + Nd ratios in C@1S-b@2S-b than in C@1S@2S would mean less disturbance from Ho3+ and Nd3+ magnetic fields over the coupling of surface Gd3+ spins and water protons in C@1S-b@2S-b. Results of measured r1 and r2 relaxivities and r2/r1 values at 3 T by using water dispersions of C@1S-b and C@1S-b@2S-b are shown in Table 2. The highest measured r1 has been achieved with C@1S-b@2S-b, r1 = 10.5 mM−1 s−1, due to its optimized distribution of Gd3+ centers. At this magnetic field, the planar geometries of C@1S-b and C@1S-b@2S-b can also explain their high r1 values when compared to the NaHoF4@Yb,Tm:NaGdF4 nanostructure (r1 = 1.50 mM−1 s−1, at 2.82 T).68 Further, r2/r1 values indicate that both C@1S-b and C@1S-b@2S-b would behave as dual T1/T2 CAs at 3 T magnetic field. Fig. 5 includes the r2/r1 values of water dispersed C@1S-b and C@1S-b@2S-b nanoprobes at 3 T.
| 3 T | |||
|---|---|---|---|
| r 1 | r 2 | r 2/r1 | |
| [Gd3+] | [Ho3+] | ||
| C@1S-b | 2.2 | 33.1 | 15.0 |
| C@1S-b@2S-b | 10.5 | 70.1 | 6.7 |
To confirm the dual T1/T2 character of C@1S-b and C@1S-b@2S-b nanoprobes at 3 T magnetic field, T1- and T2-weighted images of their water dispersions with different concentrations were recorded under this magnetic field, see Fig. 7a–d. Both nanoprobes alter the signal intensity of the contrast of the T1-weighted images, from dark to gray (C@1S-b), or to light gray (C@1S-b@2S-b), and of T2-weighted images, from light gray to dark, with Gd3+ or Ho3+ concentrations, respectively. T1 and T2-maps of these C@1S-b and C@1S-b@2S-b water dispersions are presented in Fig. 7e and f. A linear relationship is observed between concentrations of Gd3+ and Ho3+ and inverse of relaxation times T1 and T2, respectively, of protons, for both nanoprobes, Fig. S9 (ESI†).
 | ||
| Fig. 7 T 1-weighted images of water dispersions of (a) C@1S-b, (b) C@1S-b@2S-b. T2-weighted images of water dispersions of (c) C@1S-b, (d) C@1S-b@2S-b. In all cases the images were acquired at 3 T magnetic field. T1 maps (e), and T2 maps (f), of C@1S-b and C@1S-b@2S-b nanoprobes in water dispersions. The images were recorded at 3 T magnetic field. | ||
According to the above results for water dispersions of the series of MRI nanoprobes based on NaLn(WO4)2 nanostructures, the optimization of the separation between the Ln responsible for T1-weighted contrast, Gd3+ in the surface layer, and T2-weighted contrast, Ho3+ in the core, provided by the core–shell design, and the control of the relative concentrations of these cations, are critical parameters in the evolution of the behavior as MRI CAs with the magnetic field. Thus, behaviors evolving from a T1-weighted MR CA at 1.5 T to a highly effective T2-weighted MR CA at ultrahigh magnetic fields of 7 T and above, and even a dual T1/T2-weighted CA at a clinical 3 T magnetic field, have been observed in this studied system. Furthermore, the distinctive quasi-bidimensional geometry of NaLn(WO4)2 nanostructures maximizes the surface-to-volume ratio, thereby optimizing the interaction between Gd3+ cations on the surface of the nanoprobe and the protons of the water, leading to higher r1 values than those observed for other bulkier Ln-doped core–shell nanostructures, such as those based on NaLnF4 fluorides, for magnetic fields up to 3 T.68
3.3 Photoluminescence of C@1S@2S nanoprobe within I-, II-, III-, and IV-BWs wavelength ranges
When excited at λEXC ∼ 800 nm, through the 4I9/2 → 4F5/2, 2H9/2 Nd3+ transition, C@1S@2S emits light across a wide spectral range spanning the UV, visible and NIR optical regions. However, improved subcutaneous PL sensing requires the use of NIR emissions, mainly within BW wavelength ranges, thus the current interest in C@1S@2S is focused on the evaluation of Nd3+, Yb3+, and Ho3+ NIR PL emissions within these BW ranges.Fig. 8a shows schemes of electronic configurations of Nd3+, Yb3+, Tm3+ and Ho3+, as well as known energy transfer (ET) processes and electronic transitions yielding NIR down-shifted PL emissions. Under NIR excitation at λEXC ∼ 800 nm, external 2S-lying Nd3+ are pumped to 4F5/2, 2H9/2 energy levels, and after non radiative relaxation to 4F3/2 level, NIR light is emitted from Nd3+ at λEMI ∼ 870–930 nm (4F3/2 → 4I9/2, within I-BW), ∼1.06 μm (4F3/2 → 4I11/2, II-BW), ∼1.33 μm (4F3/2 → 4I13/2, II-BW), and at 2–2.15 μm (4F3/2 → 4I15/2, IV-BW), Fig. 8b–d. Moreover, from 4F3/2 Nd3+ the energy is transferred to close 2S-Yb3+ and to nearby 1S-Yb3+, populating the excited 2F5/2 Yb3+ state, from which the excited emitting levels of 1S-Tm3+ and C-Ho3+ are populated by ET. PL bands peaking at λEMI ∼ 1 μm (2F7/2 → 2F5/2, Yb3+, II-BW), 1.8 μm (3F4 → 3H6, Tm3+, III-BW), and ∼2.1 μm (5I7 → 5I8, Ho3+ IV-BW) have been observed simultaneously in the prepared nanoprobes, Fig. 8b–d. Profiles of the emission cross-sections (σEMI) of Tm3+ and Ho3+ in isostructural NaLn(WO4)2 single crystals,62,75 have been also included in Fig. 8d for help with maxima assignment. Therefore, C@1S@2S would result very suitable for deep tissue NIR optical imaging. It must be noted that the direct excitation of Tm3+ at λEXC = 795.5 nm, i.e. at the 3H4 state, does not allow the observation of any Tm3+ emission in the PL spectrum of C@1S@2S. This fact points to the decisive role of Nd3+ and Yb3+ as the sensitizer and energy transmitter elements, respectively, in the mechanism for PL in C@1S@2S nanoprobe.
 | ||
| Fig. 8 (a) Schemes of the electronic configurations of involved trivalent lanthanides and energy transfer processes describing proposed mechanisms for NIR excited (λEXC ∼ 800 nm) down-shifted PL emissions (thick black arrows) of Nd3+, Yb3+, Tm3+ and Ho3+ in C@1S@2S. (b), (c) and (d) Room temperature luminescence spectra (λEXC = 803.4 nm, Ti-sa laser) of C@1S@2S NPs dispersed in CCl4 (black lines), showing relevant Nd3+, Yb3+, Tm3+ and Ho3+ emissions within BWs. Red circles and blue diamonds are the emission cross-sections (σEMI) of Tm3+ and Ho3+, respectively, in the isostructural NaY(WO4)2 and NaBi(WO4)2 hosts, respectively. The blue continuous line is the Ho3+ emission spectrum obtained after substraction of the Nd3+ contribution, to be compared with its σEMI shape. | ||
3.4 Photoluminescence-based nanothermometry in the II-BW
In order to determine the Nd3+ and Yb3+ concentrations that optimize the PL-based temperature sensing properties in the NaGd(WO4)2 host, as a preliminary step we have evaluated polycrystalline samples prepared by solid-state synthesis (SSS), as well as nanocores (6 × 4 nm) prepared by the coprecipitation route, of NaGd(WO4)2 doped with different Nd3+ and Yb3+ contents. The description of the SSS procedure, prepared compositions, and room temperature XRD patterns for these SSS-samples, Fig. S11, are included in the ESI.†Upon Nd3+ excitation at λEXC = 803.4 nm, all these materials show simultaneously Nd3+ and Yb3+ PL, see Fig. 9a for the x at% Nd, 20 at% Yb:NaGd(WO4)2 series of nanocores dispersed in n-hexane. Among the three Nd3+ PL emissions from 4F3/2 within I- and II-BWs, the one with the largest emission cross-section is the corresponding to the 4F3/2 → 4I11/2 transition peaking at λEMI = 1058 nm,76 thus it was selected for the ratiometric comparison with the Yb3+ emission peaking at λEMI = 996 nm in the evaluation of the temperature sensing potential of C@1S@2S nanoprobes. In this case, a point of concern is the overlap between both PL contributions at λ = 1058 nm, which is particularly critical when the Nd3+ PL intensity is low in comparison to the Yb3+ one, i.e., when the Nd3+ → Yb3+ energy transfer is very efficient, as observed for high Yb3+ and Nd3+ concentrations. To fix this point, the Yb3+ contribution at λEMI = 1058 nm has been evaluated in each spectrum by extrapolating the shape of the main observed Yb3+ emission with the reference emission band profile measured under identical conditions in polycrystalline single doped x at% Yb:NaGd(WO4)2, and this contribution was discounted to evaluate the true Nd3+ PL intensity.
 | ||
| Fig. 9 (a) Evolution of the PL in the x at% Nd, 20 at% Yb:NaGd(WO4)2 series of nanocores dispersed in n-hexane at 20 °C (293 K) upon excitation of Nd3+ at λEXC = 803.4 nm. (b) Comparison of the spectral distributions at 20 °C (293 K) (black line) and 78 °C (351 K) (red line) of 7.5 at% Nd, 7.5 at%Yb:NaGd(WO4)2 polycrystalline SSS sample, λEXC = 803.4 nm. The 300 K absorption spectrum of single doped Yb:NaGd(WO4)2 is given (hollow circles). (c) Slope of dLIR/dT (being the absolute sensitivity SA the corresponding |dLIR/dT|) value (black squares), and relative sensitivity SR (red spheres) of Nd:Yb:NaGd(WO4)2 polycrystalline SSS materials as a function of the average Yb–Nd distance in the lattice. (d) Evolution with the Nd concentration of the absolute sensitivity, SA (points), of x at% Nd, 20 at% Yb:NaGd(WO4)2 single nanocores dispersed in n-hexane. The line is a visual help. | ||
In the ratiometric method, the thermometric properties of a material derive from the thermal evolution of the PL intensity ratio (LIR = I2/I1) of two emission bands.77,78 The main thermometric figures of merit of the material are described by the absolute, SA, and relative, SR, sensitivities, defined as SA = |dLIR/dT|, and SR = SA/LIR,77–79 where T is the temperature.
In the present case the emissions come from two different ions, Nd3+ and Yb3+, thus their relative electronic populations do not follow the Boltzmann law. Nd3+ was selected as ion 1, I1058nm, and Yb3+ as ion 2, I996nm.
For all studied Nd–Yb concentrations, the PL intensity of Yb3+ decreases with the temperature while the PL of Nd3+ basically remains constant within the temperature range of interest for biomedical applications, Fig. 9b. The as-defined LIR(T) evolution is nearly linear with a negative slope. As an example, Fig. S12 (ESI†) shows LIR(T) for 7.5 at% Nd, 7.5 at% Yb:NaGd(WO4)2 SSS sample.
From LIR analyses, the Fig. 9c shows the evolution of SA and SR as a function of the Nd–Yb average lattice distance,

In a next step, PL spectra (λEXC = 803.4 nm) of colloidal dispersions in n-hexane of NaGd(WO4)2 nanocores doped with 20 at% Yb and Nd concentration ranging from 2 at% to 30 at% were analyzed in the 20 °C–70 °C (293 K–343 K) temperature range. For a given temperature, the intensity distribution of the spectral Nd3+ and Yb3+ bands resembles those of polycrystalline materials with lower Nd3+ and Yb3+ concentrations, suggesting that some loss of one or both optically active Ln occurs during the coprecipitation synthesis. In fact, Ln compositional analyses by ICP-OES confirm a lower than nominal Yb3+ incorporation in prepared nanomaterials, see Tables S1 and S2 (ESI†). LIR(T) analyses indicated that the highest SA = 573 × 10−4 K−1 corresponds to the colloidal dispersion in n-hexane with composition 25 at% Nd, 20 at% Yb:NaGd(WO4)2, Fig. 9d.
Finally, the LIR(T) characterization of the NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2@20 at% Nd, 20 at% Yb:NaGd(WO4)2 (C@1S@2S) nanostructure was carried out in the temperature range of biological interest. Colloidal dispersions in n-hexane, as well as in water with increasing dilution degree, i.e., 5 mg mL−1, 0.5 mg mL−1 and 0.05 mg mL−1, have been used for the PL measurements under Ti-sa excitation at λEXC = 803.4 nm. Optical excitation densities of 2.4 W cm−2 and 0.68 W cm−2 were used for n-hexane and water dispersions, respectively. Fig. 10a shows the PL distribution at room temperature for the three water dispersions. Obviously the PL signal-to-noise ratio decreases when the dilution increases, but even at the lowest tested concentration, 0.05 mg mL−1, the shape of the spectrum allows the unequivocal determination of a confident LIR value for each measured temperature. Fig. 10b shows the thermal evolution of experimental LIR results in both n-hexane and water colloidal dispersions. It is worth noting that when the nanoprobes are transferred to the water medium the SA value decreases, see in Fig. 10b the different slopes of LIR(T) for both kinds of colloidal media.
 | ||
| Fig. 10 Thermometric PL response of colloidal dispersions of NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2@20 at% Nd, 20 at% Yb:NaGd(WO4)2 (C@1S@2S): (a) PL emissions of dispersions in water with different dilutions, λEXC = 803.4 nm, IE = 0.68 W cm−2. (b) Comparison of the thermal evolution of the Yb/Nd based LIR in n-hexane, blue squares, IE = 2.4 W cm−2, and after transfer to water, red circles, IE = 0.68 W cm−2, the symbols are the experimental results and the lines are the linear fits used for calculation of SA sensitivity. Thermometric PL response of colloidal dispersions of NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2@30 at% Nd, 20 at% Yb:NaGd(WO4)2@NaGd(WO4)2 (C@1S@2S@i3S): (c) comparison of the spectral distributions at 15 °C (288 K), black line, and at 70 °C (343 K), red line, of the colloidal dispersion in water (2 mg mL−1). (d) Thermometric characterization in n-hexane, blue squares, and in the water dispersion, red circles, IE = 0.68 W cm−2, λEXC = 803.4 nm, the lines are the linear fits of the experimental results. Values of SR at 40 °C (313 K) are 0.58% K−1 and 0.89% K−1, for colloidal dispersions in n-hexane and water, respectively. (e) Stability of the PL response upon seven heating/cooling cycles between 20 °C (293 K) and 60 °C (333 K), λEXC = 803.4 nm, IE = 0.68 W cm−2. | ||
To prevent this undesired degradation of the LIR sensitivity in aqueous/biological media, an additional isostructural undoped NaGd(WO4)2 outermost shielding layer was grown on the nanoprobe. Fig. 10c depicts the temperature-dependent (15 °C–70 °C/288 K–343 K) intensity distribution of Yb3+ and Nd3+ PL bands of a diluted (2 mg mL−1) colloidal water-stabilized dispersion of TWEEN80-modified NaHo(WO4)2@8 at% Tm, 15 at% Yb:NaGd(WO4)2@30 at% Nd, 20 at% Yb:NaGd(WO4)2@NaGd(WO4)2 (C@1S@2S@i3S), and Fig. 10d displays the corresponding LIR thermometric characterization. The optically inert NaGd(WO4)2 shell induces an efficient enhancement of the thermometric sensitivities in comparison with those of similar nanostructures lacking it, and importantly, the SA value remains unchanged after transfer from the n-hexane to the diluted water dispersion. The small LIR change upon this transfer is due to the selective optical absorption of water at λ = 995 nm. In both dispersive liquid media, remarkable high SA= 400–500 × 10−4 K−1 and SR = 0.6–0.9% K−1 thermometric sensitivities are determined, specifically SA = 480 (± 33) × 10−4 K−1 and SR = 0.89% K−1 at 40 °C (313 K) in the water dispersion. To further assess the performance for temperature sensing, the stability of the spectral response of the above water dispersed C@1S@2S@i3S nanothermometer was analyzed through seven heating/cooling cycles. No degradation of LIR values occurs, see Fig. 10e. Under the present measurement conditions, a LIR uncertainty value σLIR ≈ 0.1% has been established from the observed fluctuations of the mean LIR value for each heating/cooling cycle. From the data collected for water dispersions of C@1S@2S@i3S, the temperature resolution, δT, defined as δT = σLIR/SR,79 is ≈0.1 K.
The comparison of luminescent Ln-based nanothermometers performance is carried out through the evaluation of the above-described thermal parameters, i.e., SA, SR, δT, and reproducibility in luminescence measurements. However, when nanothermometers are intended for temperature biosensing purposes, other important aspects need to be considered in this assessment. Among these factors the main ones are the wavelength ranges in the excitation–emission operation scheme, the luminescence efficiency at biologically interesting temperatures, which must be high enough to allow keeping low irradiation power densities, and the cellular viability upon nanothermometer intake when in vivo applications are envisaged, which additionally implies the use of highly diluted and stable water dispersions of the nanoprobe. Thus, disregarding the plethora of studied Ln-doped nanothermometers based on visible and I-BW PL emission LIR analyses,79,80 the comparison for current C@1S@2S@i3S will focused on a handful of described λ ∼ 800 nm-excited thermal sensors similarly operated within the II-BW, through Nd3+ (∼1060 nm) and Yb3+ (∼1000 nm) LIR measurements,30–32,81 and even so the comparison is not unequivocal. It should be noted that all issues usually mentioned as hindrances for using SA as the figure of merit80 disappear in the current comparison. Unfortunately, direct SA values, that would be illustrative of the potential of compared nanothermometers, are not explicitly provided for these systems, and furthermore the normalization of LIR values in reported figures showing LIR vs. T plots does not allow the corresponding SA estimations.
Regarding SR results, the highest SR = 2.1% K−1 is indicated for 808 nm-excited Er,Yb:NaYF4@Yb, Nd:NaYF4 (LIR = Nd1060/Yb980) nanothermometer at 97 °C (370 K),31 but this thermal probe has considerably lower SR values at biologically interesting temperatures, what required the use of the visible Er520/Er540 LIR channel to achieve SR ∼ 1% K−1 for temperature sensing in the 17 °C–37 °C (290 K–310 K) range. Furthermore, LIR measurements were carried out with dried films of prepared NPs, instead of using their colloidal water dispersions. On the other hand, the thermometric performance of the core@multishell system Yb,Tm:SrF2@Yb:SrF2@Yb,Nd,Er:SrF2@Nd:SrF2 (colloidal water dispersions of ∼27 nm NPs, 1 wt%) is evaluated through Yb980/Nd1060 LIR analysis, yielding very high SR values, up to 1.62% K−1 at 50 °C (323 K), and δT = 1.7 K, but in this case the power density of the 806 nm cw excitation laser diode was 31 W cm−2,32 well beyond the limit for biologically safe exposure. For diluted PBS dispersions of simple codoped Nd, Yb:LaF3 NPs, and two LaF3-based core–shell nanostructures, one having Nd3+ at the core, with Yb3+ at the shell, and the other with Yb3+ at the core and Nd3+ at the shell, the evaluation of SR (from 790 nm-excited LIR = Nd1060/Yb1000, values obtained under 0.7 W cm−2 power density) indicated a better performance of the latter core–shell nanostructure, SR = 0.41% K−1, with regards to simple NPs, SR = 0.1% K−1, in the physiological range of temperatures, with δT = 1.61 K.30 Similar SR values are found for Nd3+ and Yb3+ codoped complexes with pyridine, 3,5 carboxylate (PDC).81 In this case, the Nd1052/Yb1005 LIR analysis of the complex Gd1.45Nd0.40Yb0.15-PDC yielded SR values between 0.48% K−1 and 0.439% K−1 in the 25 °C–46 °C (298 K–318 K) range, with δT = 0.08 K.
The above results from NIR–NIR LIR thermometric analyses show that C@1S@2S@i3S behaves as a well-suited nanothermometer for biologically safe operation within the II-BW, exhibiting advantageous thermal sensitivities, SA = 480 (± 33) × 10−4 K−1 and SR = 0.89% K−1 at 40 °C (313 K), and temperature resolution, δT ≈ 0.1 K, with regards to other Yb, Nd-based LIR thermometric systems.
3.5 In vitro assays with cells exposed to core–shell nanoprobes
Establishing the biocompatible nature of developed double tungstate-based nanoprobes in both healthy and cancerous cell lines is a critical step toward validating their potential for intended biomedical applications, such as diagnostic imaging and NIR biolabeling during cellular temperature measurement. For this purpose, HeLa and MC3T3 cells lines were chosen as representative models of human cancer cells and of healthy, non-tumorigenic cells, respectively. In vitro assays of viability by AlamarBlue tests, and cytotoxicity by measuring the catalytic activity of lactate dehydrogenase (LDH), were carried out for these cellular lines by incubation with water dispersions of nanoprobes, at concentrations up to 200 μg mL−1 for viability, and up to 135 μg mL−1 for cytotoxicity, for 24–48 h.Results of AlamarBlue assays in HeLa cells treated with TWEEN80-modified C@1S@2S@i3S water dispersions are shown in Fig. 11a. After 24 h of the nanoprobe dispersion addition, cell viability is preserved for concentrations of the nanoprobe at least up to 50 μg mL−1. When this concentration reaches 100 μg mL−1, a significant (p < 0.05 Student t-test) reduction in viability to 60% is observed. Interestingly, an enhancement of the AlamarBlue dye reduction appears with increasing nanoprobe doses above 100 μg mL−1, indicating an increase in the metabolism of alive cells. This same effect in metabolic cell activity of living cells has been previously observed in MTT assays with human mesenchymal stem cells (hMSCs) cultured with water dispersions of similar TWEEN80-modified Yb, Er-doped NaGd(WO4)2 NPs, and it has been attributed to the TWEEN80 content,34 through its effects on the cellular membrane fatty acid composition.82–85 Thus, this phenomenon is restricted to cell metabolism, and it does not lead to a cell proliferation increase or overall cell viability, as confirmed by the fluorescence microscopy images shown in Fig. 12a. In any case, the half maximal inhibitory concentration of the nanoprobe, IC50, is even beyond that the most concentrated water dispersion analyzed, 200 μg mL−1.
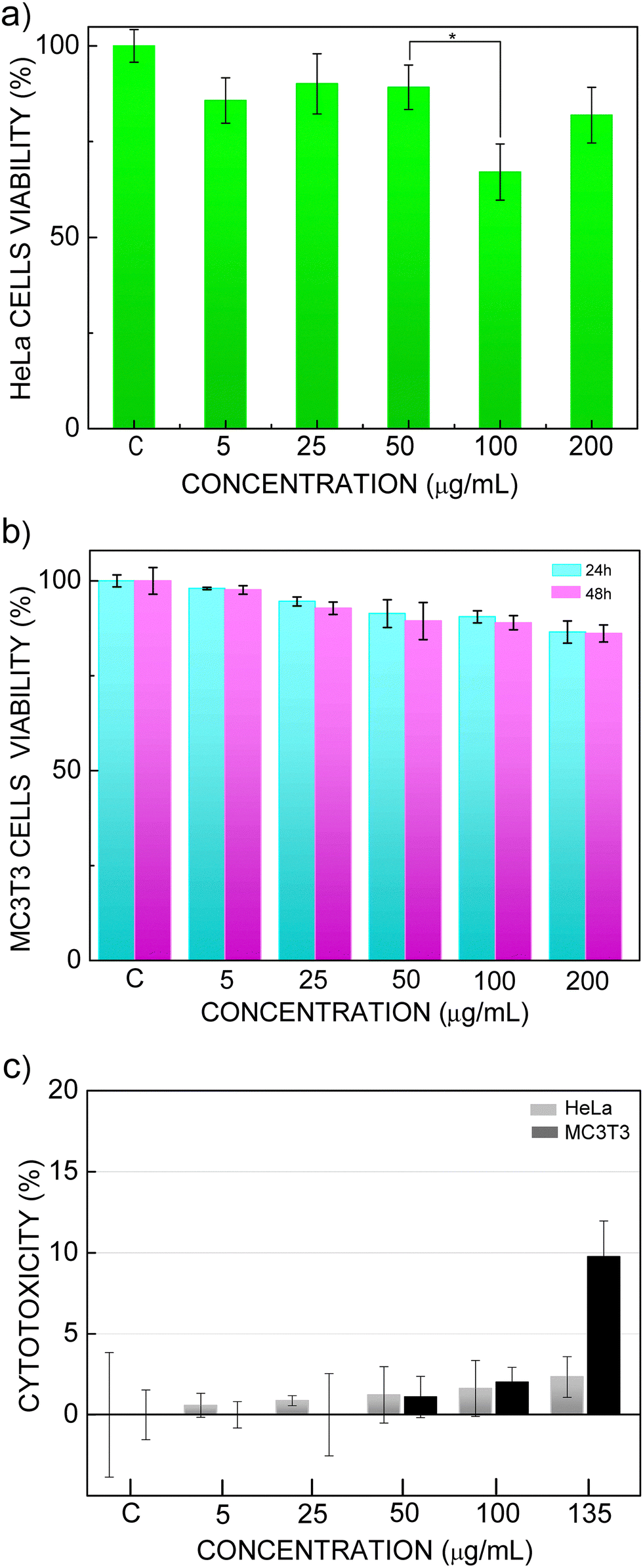 | ||
| Fig. 11 Effect of C@1S@2S@i3S water dispersions on cells viability (AlamarBlue assay): (a) HeLa cells, after 24 h incubation compared with control HeLa cells. (b) MC3T3 cells, after 24 h and 48 h incubation compared with control MC3T3 cells. Error bars represent the standard deviation of four independent samples. *p < 0.05 (Student's t-test). Effect of C@1S@2S@i3S water dispersions on cells cytotoxicity (LDH release): (c) HeLa and MC3T3 cells after 24 h incubation. | ||
 | ||
| Fig. 12 Fluorescence microscopy images of HeLa (left block) and MC3T3 (right block) cells stained with DAPI: (a)–(c) untreated HeLa cells; (d)–(f) 24 h HeLa cultured cells with 200 μg mL−1 of C@1S@2S@i3S water dispersion; (g)–(i) untreated MC3T3 cells: (j)–(l) 24 h MC3T3 cultured cells with 200 μg mL−1 of C@1S@2S@i3S water dispersion. Scale bars correspond to 200 μm. | ||
Viability assays conducted in healthy MC3T3 cells revealed their remarkable robustness in the presence of the nanoprobe, Fig. 11b. Even at extended incubation periods, cellular integrity and metabolic activity remained largely unimpaired. For instance, with a nanoprobe concentration of 200 μg mL−1, a significant 85% cell viability was maintained even after a prolonged 48 h incubation period.
To further investigate and confirm the non-toxic effects of C@1S@2S@i3S nanoprobe on both HeLa and MC3T3 cells, cytotoxicity assays by measuring the catalytic activity of lactate dehydrogenase (LDH) were also carried out. This assay is a well-established and reliable method for quantifying cell death. The underlying principle of the LDH assay is based on the cytosolic presence of this enzyme. When the plasma membrane of a cell is damaged or compromised, LDH is released into the cell culture medium. Therefore, the amount of LDH activity detected in the extracellular medium is directly proportional to the number of dead or dying cells. By measuring the extracellular LDH activity, the extent of cytotoxicity induced by a given treatment or condition on the cells can be evaluated accurately. Fig. 11c shows the results of this assay. For both cell lines, the cytotoxicity was almost negligible even at the highest measured concentration, 135 μg mL−1, at 24 h of incubation, with HeLa cells showing a 2.3%, and MC3T3 a 9.7% cytotoxicity.
Fluorescence microscopy using DAPI staining was employed to qualitatively observe the nuclei of viable cells and chromatin condensation at the 24-hour endpoint. Fig. S13 and S14 (ESI†) show the comparison between fluorescence microscopy images of the control (untreated) HeLa and MC3T3 cells and images corresponding to cultured cells with increasingly concentrated nanoprobe water dispersions, and Fig. 12 is a simplified view displaying images of cultured cells only with the highest concentrate nanoprobe dispersion, 200 μg mL−1. The HeLa control sample exhibits a high number of stained nuclei, which represents 100% viability under the established conditions. The presence of intact nuclei, with evenly distributed chromatin, is an indication of healthy cells, and very similar images have been collected for HeLa cells treated during 24 h with a 25 μg mL−1 nanoprobe water dispersion, Fig. S13 (ESI†). In contrast, images of HeLa cells treated with highly concentrated nanoprobe dispersions, 100 μg mL−1 and 200 μg mL−1, exhibit significantly lower levels of stained nuclei, with some cells displaying blue fluorescence brighter than others, due to chromatin condensation, and nuclei shrinkage and fragmentation are also observed, which in all cases indicate cellular apoptosis occurring to some extent. For MC3T3 cell samples, the images indicate that the number of stained nuclei is comparable between the control group and samples for all tested concentrations of the nanoprobe dispersion, suggesting that membrane integrity is not adversely affected by the treatment up to the highest nanoprobe concentration, 200 μg mL−1.
4. Conclusions
Nanoprobes integrating capabilities for magnetic resonance imaging contrast as well as NIR photoluminescence-based imaging and contactless thermometry have been demonstrated through Ln-doped core–shell nanostructures based on the scheelite-like double tungstate NaLn(WO4)2 crystal host. The synthesis strategy was the confirmed coating of a NaHo(WO4)2 core with shells, the first of Tm,Yb:NaGd(WO4)2, and the second of Nd, Yb:NaGd(WO4)2, to properly separate the various Lns responsible for the different functionalities of the nanoprobe. Interestingly, these nanostructures offer a large surface-to-volume relationship due to their quasi-bidimensional geometry, which favors T1-weighted contrast. Furthermore, the surface modification of nanoprobes by coating them with an undoped isostructural crystalline layer and then by the biocompatible emulsifier TWEEN80 facilitates their transfer to water dispersion while preserving their radiative properties.Results on relaxativity measurements and magnetic resonance imaging of water dispersions of prepared nanoprobes indicate an increase of more than 50 times in r2/r1 values and the associated contrast behavior evolution from positive to negative when the strength of the magnetic field increases from 1.5 to 11.7 T. Furthermore, r2/r1 = 15 was achieved at 3 T by optimizing the thickness of the first shell, and in this case magnetic resonance imaging demonstrates a dual T1/T2-weighted behavior as CA at this clinically relevant magnetic field strength.
By NIR excitation (λEXC ∼ 803 nm) of Nd3+ in the second shell, contactless thermometry was performed at wavelengths within the II-BW (λEMI ∼ 1000–1400 nm) through LIR analysis of the pair of emissions 4F3/2 → 4I11/2 Nd3+ (λEMI = 1058 nm) and 2F5/2 → 2F7/2 Yb3+ (λEMI = 996 nm). The second shell surface degradation in diluted water dispersions, which leads to Nd3+ and Yb3+ emissions quenching, is fully avoided by a further coating with an optically inactive NaGd(WO4)2 external shell. Absolute, SA, and relative, SR, thermal sensitivity values as remarkable as SA = 480 × 10−4 K−1, and SR = 0.89% K−1 at 40 °C (313 K), and temperature resolution δ ≈ 0.1 K, were delivered by chemically stable 2 mg mL−1 water dispersion of the nanoprobe under a biologically safe excitation power density of 0.68 W cm−2. Moreover, through efficient Nd3+ → Yb3+ → Tm3+ and Nd3+ → Yb3+ → Ho3+ energy transfers, NIR PL emissions from Tm3+ at ∼1800 nm (3F4 → 3H6 transition, III-BW) and Ho3+ at ∼2000 nm (5I7 → 5I8 transition) useful for NIR optical imaging are also observed, being energy back-transfers to Nd3+ minimized by the spatial separation between these emitting Lns and Nd3+.
The metabolism of HeLa cells treated with water dispersions of the nanoprobe is preserved for concentrations at least up to 50 μg mL−1 in incubations during 24 h, and healthy MC3T3 cells maintain a significant 85% cell viability even after a prolonged 48 h incubation period with a nanoprobe concentration of 200 μg mL−1. Moreover, cytotoxicity levels induced by the nanoprobe are very reduced, 2.3% for HeLa cells and 9.7% for MC3T3 cells, after 24 h incubation with 135 μg mL−1 of the nanoprobe dispersion. Thus, biomedical applications of the developed nanoprobe, in diagnostic imaging and NIR biolabeling during cellular temperature measurement, for example in hyperthermia processes, would be facilitated by its demonstrated safety profile.
Author contributions
C. Alarcón-Fernández: investigation, methodology, writing – original draft, writing – review & editing; C. Zaldo: conceptualization, investigation, methodology, formal analysis, validation, funding acquisition, writing – original draft, writing – review & editing; M. Bañobre-López: investigation, methodology, formal analysis, writing – original draft, writing – review & editing; J. Gallo: investigation, methodology, formal analysis, writing – original draft, writing – review & editing; P. Ramos-Cabrer: investigation, methodology, formal analysis, writing – original draft, writing – review & editing; S. Plaza-García: investigation, writing – original draft, writing – review & editing; A. Ruperti: investigation, writing – review & editing; G. Villaverde: investigation, formal analysis, writing – review & editing; C. Cascales: conceptualization, investigation, formal analysis, validation, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Data will be made available upon reasonable request. The data supporting this article have been included as part of the ESI.†Acknowledgements
This work has been supported by the Ministerio de Ciencia, Innovación y Universidades (MICIU) of Spain under project PID2021-128090OB-C21 funded by MICIN/AEI/10.13039/501100011033, and by ‘ERDF A way of making Europe’, and C. A. F. acknowledges his PhD MICIU grant PRE2019-090446. The National Center for Electron Microscopy (CNME)-Universidad Complutense de Madrid facility, and E. Urones contribution to TEM and STEM-EDX measurements are gratefully acknowledged.References
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, Advanced Fluorescence Imaging Technology in the Near-Infrared-II Window for Biomedical Applications, J. Am. Chem. Soc., 2020, 142(35), 14789–14804 CrossRef CAS PubMed.
- A. Refaat, M. L. Yap, G. Pietersz, A. P. G. Walsh, J. Zeller, B. del Rosal, X. Wang and K. Peter, In vivo fluorescence imaging: success in preclinical imaging paves the way for clinical applications, J. Nanobiotechnol., 2022, 20, 450, DOI:10.1186/s12951-022-01648-7.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957–1057, DOI:10.1021/acs.chemrev.8b00363.
- H. Li and T. J. Meade, Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances, J. Am. Chem. Soc., 2019, 141, 17025–17041, DOI:10.1021/jacs.9b09149.
- M. E. Ladd, P. Bachert, M. Meyerspeer, E. Moser, A. M. Nagel, D. G. Norris, S. Schmitter, O. Speck, S. Straub and M. Zaiss, Pros and cons of ultra-high-field MRI/MRS for human application, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 1–50, DOI:10.1016/j.pnmrs.2018.06.001.
- D. A. Feinberg, A. J. S. Beckett and A. T. Vu, et al., Next-generation MRI scanner designed for ultra-high-resolution human brain imaging at 7 Tesla, Nat. Methods, 2023, 20, 2048–2057, DOI:10.1038/s41592-023-02068-7.
- I. de Vries, W. Lesterhuis and J. Barentsz, et al., Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy, Nat. Biotechnol., 2005, 23(11), 1407–1413, DOI:10.1038/nbt1154.
- Y. Ning, I. Y. Zhou and P. Caravan, Quantitative in Vivo Molecular MRI, Adv. Mater., 2024, 36, 2407262, DOI:10.1002/adma.202407262.
- J. Yang, M. Yan, Z. Wang, C. Zhang, M. Guan and Z. Sun, Optical and MRI Multimodal Tracing of Stem Cells In Vivo, Mol. Imaging, 2023, 2023, 4223485, DOI:10.1155/2023/4223485.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Upconversion Nanoparticles: Design, Nanochemistry, and Applications in Theranostics, Chem. Rev., 2014, 114(10), 5161–5214, DOI:10.1021/cr400425h.
- N. N. Dong, M. Pedroni, F. Piccinelli, G. Conti, A. Sbarbati, J. E. Ramírez-Hernández, L. Martínez Maestro, M. C. Iglesias-de la Cruz, F. Sanz-Rodriguez, A. Juarranz, F. Chen, F. Vetrone, J. A. Capobianco, J. García Solé, M. Bettinelli, D. Jaque and A. Speghini, NIR-to-NIR Two-Photon Excited CaF2:Tm3+,Yb3+ Nanoparticles: Multifunctional Nanoprobes for Highly Penetrating Fluorescence Bio-Imaging, ACS Nano, 2011, 5(11), 8665–8671, DOI:10.1021/nn202490m.
- S. L. Jacques, Optical properties of biological tissues: a review, Phys. Med. Biol., 2013, 58, 5007, DOI:10.1088/0031-9155/58/14/5007 ; see also K. Calabro, Modeling Biological Tissues in LightTools, Synopsis 2020, https://www.synopsys.com/content/dam/synopsys/optical solutions/documents/modeling-tissues-lighttools-paper.pdf.
- S. Smith, M. Mancini and S. Nie, Second window for in vivo imaging, Nat. Nanotechnol., 2009, 4, 710–711, DOI:10.1038/nnano.2009.326.
- S. Wu, G. Han, D. J. Milliron, S. Aloni, V. Altoe, D. V. Talapin, B. E. Cohen and P. J. Schuck, Non-blinking and photostable upconverted luminescence from single lanthanide-doped nanocrystals, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10917–10921, DOI:10.1073/pnas.0904792106.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Upconverting nanoparticles: assessing the toxicity, Chem. Soc. Rev., 2015, 44, 1561–1584, 10.1039/c4cs00177j.
- Y. Sun, W. Feng, P. Yang, C. Huang and F. Li, The biosafety of lanthanide upconversion nanomaterials, Chem. Soc. Rev., 2015, 44, 1509–1525, 10.1039/c4cs00175c.
- D. Jaque and F. Vetrone, Luminescence nanothermometry, Nanoscale, 2012, 4, 4301–4326, 10.1039/c2nr30764b.
- C. Zaldo, in Advanced Nanomaterials, Lanthanide-based multifunctional materials: From OLEDs to SIMs, ed. P. Martín-Ramos and M. Ramos-Silva, Elsevier, 2018, ch. 10 DOI:10.1016/B978-0-12-813840-3.00010-7.
- T. Bai and N. Gu, Micro/Nanoscale Thermometry for Cellular Thermal Sensing, Small, 2016, 12, 4590–4610, DOI:10.1002/smll.201600665.
- Q. Wang, M. Liao, Q. Lin, M. Xiong, Z. Mu and F. Wu, A review on fluorescence intensity ratio thermometer based on rare earth and transition metal ions doped inorganic luminescent materials, J. Alloys Compd., 2021, 850, 156744, DOI:10.1016/j.jallcom.2020.156744.
- C. Wang, Y. Jin, R. Zhang, Q. Yao and Y. Hu, A review and outlook of ratiometric optical thermometer based on thermally coupled levels and non-thermally coupled levels, J. Alloys Compd., 2022, 894, 162494, DOI:10.1016/j.jallcom.2021.162494.
- H. Suo, X. Zhao, Z. Zhang, Y. Wang, J. Sun, M. Jin and C. Guo, Rational Design of Ratiometric Luminescence Thermometry Based on Thermally Coupled Levels for Bioapplications, Laser Photonics Rev., 2021, 15, 2000319, DOI:10.1002/lpor.202000319.
- U. Rocha, C. Jacinto, K. U. Kumar, F. J. López, D. Bravo, J. G. Solé and D. Jaque, Real-time deep-tissue thermal sensing with sub-degree resolution by thermally improved Nd3+:LaF3 multifunctional nanoparticles, J. Lumin., 2016, 175, 149–157, DOI:10.1016/j.jlumin.2016.02.034.
- A. Benayas, B. Rosal, A. Pérez-Delgado, K. Santacruz-Gómez, D. Jaque, G. A. Hirata and F. Vetrone, Nd:YAG near-infrared luminescent nanothermometers, Adv. Opt. Mater., 2015, 3, 687–694, DOI:10.1002/adom.201400484.
- I. E. Kolesnikov, E. V. Golyeva, M. A. Kurochkin, E. Lähderanta and M. D. Mikhailov, Nd3+-doped YVO4 nanoparticles for luminescence nanothermometry in the first and second biological windows, Sens. Actuators, B, 2016, 235, 287–293, DOI:10.1016/j.snb.2016.05.095.
- L. Marciniak, K. Prorok, A. Bednarkiewicz, A. Kowalczyk, D. Hreniak and W. Strek, Water dispersible LiNdP4O12 nanocrystals: New multifunctional NIR–NIR luminescent materials for bio-applications, J. Lumin., 2016, 176, 144–148, DOI:10.1016/j.jlumin.2016.03.034.
- Ł. Marciniak, A. Bednarkiewicz, D. Hreniak and W. Strek, The influence of Nd3+ concentration and alkali ions on the sensitivity of non-contact temperature measurements in ALaP4O12:Nd3+ (A = Li, K, Na, Rb) nanocrystalline luminescent thermometers, J. Mater. Chem. C, 2016, 4, 11284, 10.1039/c6tc03396b.
- L. A. O. Nunes, A. S. Souza, L. D. Carlos and O. L. Malta, Neodymium doped fluoroindogallate glasses as highly-sensitive luminescent non-contact thermometers, Opt. Mater., 2017, 63, 42–45, DOI:10.1016/j.optmat.2016.08.038.
- X. Tian, X. Wei, Y. Chen, C. Duan and M. Yin, Temperature sensor based on ladder-level assisted thermal coupling and thermal-enhanced luminescence in NaYF4: Nd3+, Opt. Express, 2014, 22, 30333, DOI:10.1364/OE.22.030333.
- E. C. Ximendes, W. Querioz Santos, U. Rocha, U. K. Kagola, F. Sanz-Rodríguez, N. Fernández, A. S. Gouveia-Neto, D. Bravo, A. M. Domingo, B. del Rosal, C. D. S. Brites, L. D. Carlos, D. Jaque and C. Jacinto, Unveiling in vivo subcutaneous thermal dynamics by infrared luminescent nanothermometers, Nano Lett., 2016, 16, 1695–1703, DOI:10.1021/acs.nanolett.5b04611.
- L. Marciniak, K. Prorok, L. Francés-Soriano, J. Pérez-Prieto and A. Bednarkiewicz, A broadening temperature sensitivity range with a core–shell YbEr@YbNd double ratiometric optical nanothermometer, Nanoscale, 2016, 8, 5037–5042, 10.1039/c5nr08223d.
- P. Cortelletti, A. Skripka, C. Facciotti, M. Pedroni, G. Caputo, N. Pinna, M. Quintanilla, A. Benayas, F. Vetrone and A. Speghini, Tuning the sensitivity of lanthanide-activated NIR nanothermometers in the biological windows, Nanoscale, 2018, 10, 2568–2576, 10.1039/c7nr06141b.
- Ł. Marciniak, A. Bednarkiewicz, M. Stefanski, R. Tomala, D. Hreniak and W. Strek, Near infrared absorbing near infrared emitting highly-sensitive luminescent nanothermometer based on Nd3+ to Yb3+ energy transfer, Phys. Chem. Chem. Phys., 2015, 17, 24315, 10.1039/c5cp03861h.
- C. Cascales, C. L. Paíno, E. Bazán and C. Zaldo, Ultrasmall, water dispersible, TWEEN80 modified Yb:Er:NaGd(WO4)2 nanoparticles with record upconversion ratiometric thermal sensitivity and their internalization by mesenchymal stem cells, Nanotechnology, 2017, 28, 185101, DOI:10.1088/1361-6528/aa6834.
- C. Zaldo, M. D. Serrano, X. Han, C. Cascales, M. Cantero, L. Montoliú, E. Arza, V. R. Caiolfa and M. Zamai, Efficient up-conversion in Yb:Er:NaT(XO4)2 thermal nanoprobes. Imaging of their distribution in a perfused mouse, PLoS One, 2017, 12(5), e0177596, DOI:10.1371/journal.pone.0177596.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications, Chem. Rev., 1999, 99, 2293–2352, DOI:10.1021/cr980440x.
- M. Bendszus, A. Laghi, J. Munuera, L. N. Tanenbaum, B. Taouli and H. C. Thoeny, MRI gadolinium-based contrast media: Meeting radiological, clinical, and environmental needs, J. Magn. Reson. Imaging, 2024, 60, 1774–1785, DOI:10.1002/jmri.29181.
- K. Dydak, T. Zalewski, M. Kempka, P. Florczak, G. Nowaczyk, Ł. Przysiecka, J. Jagielski, B. Loppinet, M. Banaszak and D. Flak, Nanoassemblies with Gd-chelating lipids (GMO@DTPA-BSA-Gd) as a potential new type of high molecular weight contrast agents, J. Mater. Chem. B, 2024, 12, 12017, 10.1039/d4tb01684j.
- A. Banerjee, B. Blasiak, E. Pasquier, B. Tomanek and S. Trude, Synthesis, characterization, and evaluation of PEGylated first-row transition metal ferrite nanoparticles as T2 contrast agents for high-field MRI, RSC Adv., 2017, 7, 38125, 10.1039/c7ra05495e.
- R. Lapusan, R. Borlan and M. Focsan, Advancing MRI with magnetic nanoparticles: a comprehensive review of translational research and clinical trials, Nanoscale Adv., 2024, 6, 2234–2259, 10.1039/D3NA01064C.
- A. F. O. Aboushoushah, Iron oxide nanoparticles enhancing magnetic resonance imaging: A review of the latest advancements, J. Sci.:Adv. Mater. Devices, 2025, 10(2), 100875, DOI:10.1016/j.jsamd.2025.100875.
- P. Pawar and A. Prabhu, Smart SPIONs for Multimodal Cancer Theranostics: A Review, Mol. Pharmaceutics, 2025, 22(5), 2372–2391, DOI:10.1021/acs.molpharmaceut.5c00411.
- M. Behar, K. K. Peck, O. Yildirim, J. Tisnado, A. Saha, J. Arevalo-Perez, E. Lis, Y. Yamada, A. I. Holodny and S. Karimi, T1-Weighted, Dynamic Contrast-Enhanced MR Perfusion Imaging Can Differentiate between Treatment Success and Failure in Spine Metastases Undergoing Radiation Therapy, Am. J. Neuroradiol., 2023, 44(12), 1451–1457, DOI:10.3174/ajnr.A8057.
- Y. Zhuang, Y. Sun, A. Dai, X. Shi, D. Wu, F. Li, H. Hu and S. Yang, Targeted dual-contrast T1- and T2 weighted magnetic resonance imaging of tumors using multifunctional gadolinium-labeled superparamagnetic iron oxide nanoparticles, Biomaterials, 2011, 32, 4584–4593, DOI:10.1016/j.biomaterials.2011.03.018.
- K. Wang, L. An, Q. Tian, J. Lin and S. Yang, Gadolinium-labelled iron/iron oxide core/shell nanoparticles as T1–T2 contrast agent for magnetic resonance imaging, Gadolinium-labelled iron/iron oxide core/shell nanoparticles as T1–T2 contrast agent for magnetic resonance imaging, RSC Adv., 2018, 8, 26764, 10.1039/c8ra04530e.
- P. Caravan, Strategies for increasing the sensitivity of gadolinium based MRI contrast agents, Chem. Soc. Rev., 2006, 35, 512–523, 10.1039/b510982p.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Challenges for molecular magnetic resonance imaging, Chem. Rev., 2010, 110, 3019–3042, DOI:10.1021/cr100025t.
- N. Iyad, M. S. Ahmad, S. G. Alkhatib and M. Hjouj, Gadolinium contrast agents-challenges and opportunities of a multidisciplinary approach: Literature review, Eur. J. Radiol. Open, 2023, 11, 100503, DOI:10.1016/j.ejro.2023.100503.
- K. B. Ghaghada, M. Ravoori, D. Sabapathy, J. Bankson, V. Kundra and A. Annapragada, New dual mode gadolinium nanoparticle contrast agent for magnetic resonance imaging, PLoS One, 2009, 4(10), e7628, DOI:10.1371/journal.pone.0007628.
- Y. Zhang, V. Vijayaragavan, G. K. Das, K. K. Bhakoo and T. T. Y. Tan, Single-phase NaDyF4:Tb3+ nanocrystals as multifunctional contrast agents in high-field magnetic resonance and optical Imaging, Eur. J. Inorg. Chem., 2012, 2044–2048, DOI:10.1002/ejic.201101203.
- Y. Feng, Q. Xiao, Y. Zhang, F. Li, Y. Li, C. Li, Q. Wang, L. Shi and H. Lin, Neodymium-doped NaHoF4 nanoparticles as near-infrared luminescent/T2-weighted MR dual-modal imaging agents in vivo, J. Mater. Chem. B, 2017, 5, 504–510, 10.1039/C6TB01961G.
- G. K. Das, N. J. J. Johnson, J. Cramen, B. Blasiak, P. Latta, B. Tomanek and F. C. J. M. van Veggel, NaDyF4 nanoparticles as T2 contrast agents for ultrahigh field magnetic resonance imaging, J. Phys. Chem. Lett., 2012, 3, 524–529, DOI:10.1021/jz201664h.
- X. Zhang, B. Blasiak, A. J. Marenco, S. Trudel, B. Tomanek and F. C. J. M. van Veggel, Design and regulation of NaHoF4 and NaDyF4 nanoparticles for high-field magnetic resonance imaging, Chem. Mater., 2016, 28, 3060–3072, DOI:10.1021/acs.chemmater.6b00264.
- E. Gómez-González, C. Caro, D. Martínez-Gutiérrez, M. L. García-Martín, M. Ocaña and A. I. Becerro, Holmium phosphate nanoparticles as negative contrast agents for high-field magnetic resonance imaging: Synthesis, magnetic relaxivity study and in vivo evaluation, J. Colloid Interface Sci., 2021, 587, 131–140, DOI:10.1016/j.jcis.2020.11.119.
- E. Gómez-González, C. Caro, N. O. Núñez, D. González-Mancebo, J. D. Urbano-Gámez, M. L. García-Martín and M. Ocaña, Sodium lanthanide tungstate-based nanoparticles as bimodal contrast agents for in vivo high-field MRI and CT, J. Mater. Chem. B, 2024, 12, 11123, 10.1039/d4tb01157k.
- X. J. Xie, N. Y. Gao, R. R. Deng, Q. Sun, Q. H. Xu and X. G. Liu, Mechanistic investigation of photon upconversion in Nd3+-sensitized core-shell nanoparticles, J. Am. Chem. Soc., 2013, 135, 12608–12611, DOI:10.1021/ja4075002.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, Nd3+-sensitized upconversion nanophosphors: Efficient in vivo bioimaging probes with minimized heating effect, ACS Nano, 2013, 7, 7200–7206, DOI:10.1021/nn402601d.
- J. Shen, G. Y. Chen, A. M. Vu, W. Fan, O. S. Bilsel, C. C. Chang and G. Han, Engineering the upconversion nanoparticle excitation wavelength: Cascade sensitization of tri-doped upconversion colloidal nanoparticles at 800 nm, Adv. Opt. Mater., 2013, 1, 644–650, DOI:10.1002/adom.201300160.
- J. Estelrich, M. J. Sánchez-Martín and M. A. Busquets, Nanoparticles in magnetic resonance imaging: From simple to dual contrast agents, Int. J. Nanomed., 2015, 10, 1727–17416, DOI:10.2147/IJN.S76501.
- T. H. Shin, Y. Choi, S. Kim and J. Cheon, Recent advances in magnetic nanoparticle-based multi-modal imaging, Chem. Soc. Rev., 2015, 44, 4501–4516, 10.1039/c4cs00345d.
- C. Cascales, M. D. Serrano, F. Esteban-Betegon, C. Zaldo, R. Peters, K. Petermann, G. Huber, L. Ackermann, D. Rytz, C. Dupre, M. Rico, J. Liu, U. Griebner and V. Petrov, Structural, spectroscopic, and tunable laser properties of Yb3+-doped NaGd(WO4)2, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 74, 174114, DOI:10.1103/PhysRevB.74.174114.
- J. M. Cano-Torres, M. Rico, X. Han, M. D. Serrano, C. Cascales, C. Zaldo, V. Petrov, U. Griebner, X. Mateos, P. Koopmann and C. Kränkel, Comparative study of crystallographic, spectroscopic, and laser properties of Tm in NaT(WO4)2 (T = La, Gd, Y, and Lu) disordered single crystals, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 84, 174207, DOI:10.1103/PhysRevB.84.174207.
- A. A. Lagatsky, X. Han, M. D. Serrano, C. Cascales, C. Zaldo, S. Calvez, M. D. Dawson, J. A. Gupta, C. T. A. Brown and W. Sibbett, Femtosecond (191 fs) NaY(WO4)2 Tm, Ho-codoped laser at 2060 nm, Opt. Lett., 2010, 35, 3027–3029, DOI:10.1364/OL.35.003027.
- A. Méndez-Blas, M. Rico, V. Volkov, C. Zaldo and C. Cascales, Crystal field analysis and emission cross sections of Ho3+ in the locally disordered single-crystal laser hosts M+Bi(XO4)2 (M+ = Li, Na; X = W, Mo), Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 75, 174208, DOI:10.1103/PhysRevB.75.174208.
- US Food&Drug Administration, Code of Federal Regulations Title 21, sec. 172.840, https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=172.840.
- M. Norek and J. A. Peters, MRI contrast agents based on dysprosium or holmium, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 64–82, DOI:10.1016/j.pnmrs.2010.08.002.
- S. Liu, T. Tegafaw, H. Yue, S. L. Ho, S. Kim, J. A. Park, A. Baek, M. Y. Ahmad, S. H. Yang, D. W. Hwang, S. Kim, A. Khamis, A. Al Saidi, D. Zhao, Y. Liu, S. W. Nam, K. S. Chae, Y. Chang and G. H. Lee, Paramagnetic ultrasmall Ho2O3 and Tm2O3 nanoparticles: characterization of r2 values and in vivo T2 MR images at a 3.0 T MR field, Mater. Adv., 2022, 3, 5857–5870, 10.1039/d2ma00322h.
- S. Biju, J. Gallo, M. Bañobre-López, B. B. Manshian, S. J. Soenen, U. Himmelreich, L. Vander Elst and T. N. Parac-Vogt, A magnetic chameleon: Biocompatible lanthanide fluoride nanoparticles with magnetic field dependent tunable contrast properties as a versatile contrast agent for low to ultrahigh field MRI and optical imaging in biological window, Chem. – Eur. J., 2018, 24, 7388–7397, DOI:10.1002/chem.201800283.
- P. Caravan, C. T. Farrar, L. Frullano and R. Uppal, Influence of molecular parameters and increasing magnetic field strength on relaxivity of gadolinium- and manganese-based T1 contrast agents, Contrast Media Mol. Imaging, 2009, 4, 89–100, DOI:10.1002/cmmi.267.
- B. Divband, N. Gharehaghaji and M. Takhiri, Effect of Nd doping on MRI relaxitivity of gadolinium oxide nanoparticles, J. Biomed. Phys. Eng., 2020, 10, 580–596, DOI:10.31661/jbpe.v0i0.2008-1165.
- H. Du, Q. Y. Wang, Z. Y. Liang, Q. L. Li, F. Y. Li and D. S. Ling, Fabrication of magnetic nanoprobes for ultra high field magnetic resonance imaging, Nanoscale, 2022, 14, 17483–17499, 10.1039/d2nr04979a.
- N. A. Keasberry, M. Bañobre-López, C. Wood, G. J. Stasiuk, J. Gallo and N. J. Long, Tuning the relaxation rates of dual-mode T1/T2 nanoparticle contrast agents: a study into the ideal system, Nanoscale, 2015, 7, 16119–16128, 10.1039/c5nr04400f.
- J. Gallo, B. I. Harriss, J. Hernández-Gil, M. Bañobre-López and N. J. Long, Probing T1–T2 interactions and their imaging implications through a thermally responsive nanoprobe, Nanoscale, 2017, 9, 11318–11326, 10.1039/c7nr01733b.
- C. C. Huang, C. Y. Tsai, H. S. Sheu, K. Y. Chuang, C. H. Su, U. S. Jeng, F. Y. Cheng, C. H. Su, H. Y. Lei and C. S. Yeh, Enhancing transversal relaxation for magnetite nanoparticles in MR imaging using Gd3+-chelated mesoporous silica shells, ACS Nano, 2011, 5, 3905–3916, DOI:10.1021/nn200306g.
- X. Han, F. Fusari, M. D. Serrano, A. A. Lagatsky, J. M. Cano-Torres, C. T. A. Brown, C. Zaldo and W. Sibbett, Continuous-wave laser operation of Tm and Ho co-doped NaY(WO4)2 and NaLu(WO4)2 crystals, Opt. Express, 2010, 18, 5413–5419, DOI:10.1364/OE.18.005413.
- A. Méndez-Blas, M. Rico, V. Volkov, C. Zaldo and C. Cascales, Optical emission properties of Nd3+ in NaBi(WO4)2 single crystal, Mol. Phys., 2003, 101(7), 941–949, DOI:10.1080/0026897021000046780.
- S. F. Collins, G. W. Baxter, S. A. Wade, T. Sun, K. T. V. Grattan, Z. Y. Zhang and A. W. Palmer, Comparison of fluorescence-based temperature sensor schemes: Theoretical analysis and experimental validation, J. Appl. Phys., 1998, 84, 4649–4654, DOI:10.1063/1.368705.
- S. A. Wade, S. F. Collins and G. W. Baxter, Fluorescence intensity ratio technique for optical fiber point temperature sensing, J. Appl. Phys., 2003, 94, 4743–4756, DOI:10.1063/1.1606526.
- A. Nexha, J. J. Carvajal, M. C. Pujol, F. Díaz and M. Aguiló, Lanthanide doped luminescence nanothermometers in the biological windows: Strategies and applications, Nanoscale, 2021, 13, 7913–7987, 10.1039/d0nr09150b.
- C. D. S. Brites, S. Balabhadra and L. D. Carlos, Lanthanide-Based Thermometers: At the cutting-edge of luminescence thermometry, Adv. Opt. Mater., 2019, 7, 1801239, DOI:10.1002/adom.201801239.
- X. Zhou, Y. Wang, H. Wang, L. Xiang, Y. Yan, L. Li, G. Xiang, Y. Li, S. Jiang, X. Tang and X. Zhou, Nd3+ and Nd3+/Yb3+-incorporated complexes as optical thermometer working in the second biological window, Sens. Biosens. Res., 2020, 29, 100345, DOI:10.1016/j.sbsr.2020.100345.
- B. B. Zhang and P. C. K. Cheung, A mechanistic study of the enhancing effect of Tween 80 on the mycelial growth and exopolysaccharide production by Pleurotus tuber-regium, Bioresour. Technol., 2011, 102, 8323–8326, DOI:10.1016/j.biortech.2011.06.021.
- D. Reitermayer, T. A. Kafka, C. A. Lenz and R. F. Vogel, Interrelation between Tween and the membrane properties and high pressure tolerance of Lactobacillus plantarum, BMC Microbiol., 2018, 18, 72, DOI:10.1186/s12866-018-1203-y.
- R. D. Pietersen, I. du Preez, D. T. Loots, M. van Reenen, D. Beukes, G. Leisching and B. Baker, Tween 80 induces a carbon flux rerouting in Mycobacterium tuberculosis, J. Microbiol. Methods, 2020, 170, 105795, DOI:10.1016/j.mimet.2019.105795.
- D. Zaręba and M. Ziarno, Tween80™-induced changes in fatty acid profile of selected mesophilic lactobacilli, Acta Biochim. Pol., 2024, 71, 13014, DOI:10.3389/abp.2024.13014.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedure for preparing core–shell nanoprobes C@1S@2S and C@1S@2S@i3S. Characterization of nanomaterials prepared by coprecipitation: room temperature X-ray diffraction patterns. Overtime sizes and size distributions by DLS measurements of nanoprobes in hexane and water solutions. Tables with concentrations of Lns in nanomaterials by ICP-OES. Plots of the magnetization vs. the applied magnetic field for prepared nanoprobes. Plots of R1 and R2vs. [Ln] concentration for r1 and r2 calculations. Solid state synthesis of Yb, Nd:NaGd(WO4)2 polycrystalline samples, description and characterization. Thermal evolution of LIR Yb/Nd of the polycrystalline sample with composition 7.5 at% Nd, 7.5 at% Yb:NaGd(WO4)2, λEXC = 803.4 nm. Fluorescence images of HeLa and MC3T3 cells treated with water dispersions of the C@1S@2S@i3S nanoprobe and stained with DAPI. See DOI: https://doi.org/10.1039/d5tb00548e |
| This journal is © The Royal Society of Chemistry 2025 |