
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable biodegradable polysaccharide-based hydrogels for stem cell delivery and cartilage regeneration†
Xiaojie
Lin
 a,
Ruofan
Liu
a,
Yang
Zhou
a,
Jacob
Beitzel
a,
Aya
Noguchi
b,
Masayuki
Kyomoto
b and
Miqin
Zhang
*a
a,
Ruofan
Liu
a,
Yang
Zhou
a,
Jacob
Beitzel
a,
Aya
Noguchi
b,
Masayuki
Kyomoto
b and
Miqin
Zhang
*a
aDepartment of Materials Science and Engineering, University of Washington, Seattle, WA 98195, USA. E-mail: mzhang@uw.edu
bMedical R&D Center, Corporate R&D Group, Kyocera Corporation, 800 Ichimiyake, Yasu, Shiga 520-2362, Japan
First published on 2nd June 2025
Abstract
Current knee osteoarthritis (KOA) treatments mainly provide symptom relief rather than cartilage repair. While regenerative medicine using stem cell therapy holds promise for tissue regeneration and joint function restoration, a significant challenge lies in the efficient and minimally invasive delivery of stem cells to target sites and ensuring high regenerative efficacy. This challenge stems from issues such as cell leakage and reduced cellular activity post-transplantation. In this study, we report the development of an injectable polysaccharide hydrogel (termed Ald-HA/Suc-CS), which is compatible with cells and tissues, and will be suitable to support the proliferation of human adipose-derived stem cells (hADSCs) for cartilage regeneration. The hydrogel is formed on-site at the defect site of articular cartilage by mixing two injectable polymer solutions at physiological temperature post-injection. During the gelation process, hADSCs contained in one of the polymer solutions are encapsulated in the hydrogel. The hydrogel is tailored to create a desired microenvironment with mechanical properties, pore size, and degradation rate suitable for supporting hADSC viability and function. We demonstrated that nearly all of the encapsulated hADSCs remained viable 14 days post-injection and exhibited increased expression of chondrogenic differentiation genes compared to those cultured on 2D surfaces. This hydrogel holds great promise to improve the efficacy of KOA treatment and is potentially applicable to other cell-based therapies.
1. Introduction
Current methods for treating mild and moderate knee osteoarthritis (KOA), a chronic degenerative condition, rely on alleviating symptoms rather than promoting cartilage tissue regeneration.1–4 This is primarily because (a) cartilage has limited self-healing capabilities,5,6 due to its avascular nature and the limited proliferative potential of cartilage cells,7,8 and (b) the lack of effective regenerative medical solutions and established treatment guidelines.9 Stem cell therapy, a form of regenerative medicine that uses immune modulation to repair damaged tissues within the body, holds great potential for tissue regeneration.10–13 In particular, human adipose-derived stem cells (hADSCs) stand out as a favorable choice for cartilage tissue engineering. This is because (a) a large number of hADSCs can be readily isolated and harvested and (b) they possess stem cell characteristics such as self-renewal and multipotency.14,15 In the clinical setting, there is a pressing need for innovative therapeutic strategies using stem cells for both symptomatic alleviation and cartilage regeneration.Typically, therapeutic cells are injected directly into the site of the disease using a syringe or catheter. However, less than 5% of the injected cells would be viable in the injection site a few days after the procedure.16 This diminished viability is largely attributed to the diffusion of the cell suspension and a decrease in cell health caused by the injective procedure and the unfavorable extracellular environment.17 The effectiveness of stem cell therapy in repairing damaged articular cartilage heavily relies on sustaining the presence of injected cells at the disease site over an extended period. One way to address this issue is to use a polymer hydrogel, which serves as both a cell delivery vehicle and an encapsulating matrix where the cells are confined and nurtured within the hydrogel's highly porous structure. Synthetic polymers are commonly used for making hydrogels, because of their ease of production and property control. However, they often lack the necessary compatibility with biological systems and biodegradability when compared to natural polymer hydrogels.18–20 On the other hand, natural polysaccharides are regarded as more suitable materials for the preparation of hydrogels for biomedical applications. Polysaccharide-based hydrogels provide a conducive environment for cell encapsulation and support the growth of various cell types. Their large pore sizes enhance nutrient diffusion and improve cell survival. Additionally, their biodegradability reduces the need for surgical removal, and their widespread availability and cost-effectiveness further contribute to their overall advantages.21,22 Despite their advantages, current natural hydrogel systems often face challenges such as inconsistent gelation, low mechanical strength, and limited tissue retention under physiological conditions. While some have shown promise in cartilage regeneration, the mechanisms by which their material properties influence cell behavior and tissue outcomes are not well understood.23,24 This limits their clinical translation and optimization for specific regenerative applications. Additionally, injectable gels face challenges associated with crosslinking density, gelation time control, and precise cell delivery. Low-density crosslinks can result in excessive softness, whereas high-density crosslinks can lead to rigidity, risking cell death. While slow gelation causes cell dispersion and leakage, fast gelation hinders injection. The effectiveness of cell-based therapies relies on the consistent and accurate cell delivery within the hydrogel to the intended target site.
To address these limitations, we developed a hydrogel system based on chitosan, a polysaccharide with tunable chemical properties, excellent biocompatibility, and proven potential in tissue engineering. Chitosan's amino and hydroxyl functional groups enable chemical modifications like amidation, esterification, and etherification.3,25,26 However, its application as an injectable hydrogel is limited by its poor solubility in physiological solvents, due to its strong intermolecular hydrogen bonding. To overcome this, researchers have explored water-soluble chitosan derivatives, including the N-acetylated chitosan,27O-carboxymethyl chitosan,28 hydroxybutyl chitosan,29N-succinyl chitosan (Suc-CS),30 and poly(ethylene glycol)-g-chitosan.31–35 Suc-CS, a water-soluble chitosan derivative, is produced by introducing succinyl groups at the N-position of chitosan glucosamine units. This modification enhanced its compatibility with biological systems and prolonged retention in vivo, making it a promising candidate as a carrier for cell-base therapy.36 Moreover, Suc-CS can be combined with other polysaccharides, such as hyaluronic acid (HA), to produce a more biomimetic environment for tissue regeneration. HA, another linear polysaccharide, consists of repeating disaccharide units of N-acetyl-D glucosamine and D-glucuronic acid. As a major component of the extracellular matrix (ECM), HA has excellent biodegradability and compatibility with biological systems.3,37 However, its fast degradation rate and high viscosity limit its use in biomedical applications.38,39
Here, we report the development of an injectable, biodegradable, self-healing, and in situ forming polysaccharide hydrogel, as a matrix that encapsulates and maintains the viability of hADSCs for cartilage regeneration (Scheme 1). The hydrogel is composed of two natural polysaccharide polymers, Suc-CS and aldehyde HA (Ald-HA). The carbon–carbon bonds of HA were cleaved through an oxidation reaction, yielding new aldehyde functional groups transforming it into Ald-HA. The aldehyde groups of Ald-HA then reacted with the amino groups of Suc-CS via Schiff base reaction, leading to the formation of a covalent crosslinked hydrogel network. The injectable solutions of these two natural polysaccharide polymers were prepared separately and mixed during injection using a double-barrel syringe to create the hydrogel. The hADSCs were incorporated in the Ald-HA solution and became encapsulated in the hydrogel during the gelation process. Our study investigated the effect of polymer concentrations on the gelation capacity, the physical properties of the hydrogel (such as mechanical strength and pore size), as well as the morphology and viability of the encapsulated hADSCs over time. Furthermore, we examined the temporal changes in gene and protein expression levels associated with chondrogenic differentiation within the encapsulated hADSCs.
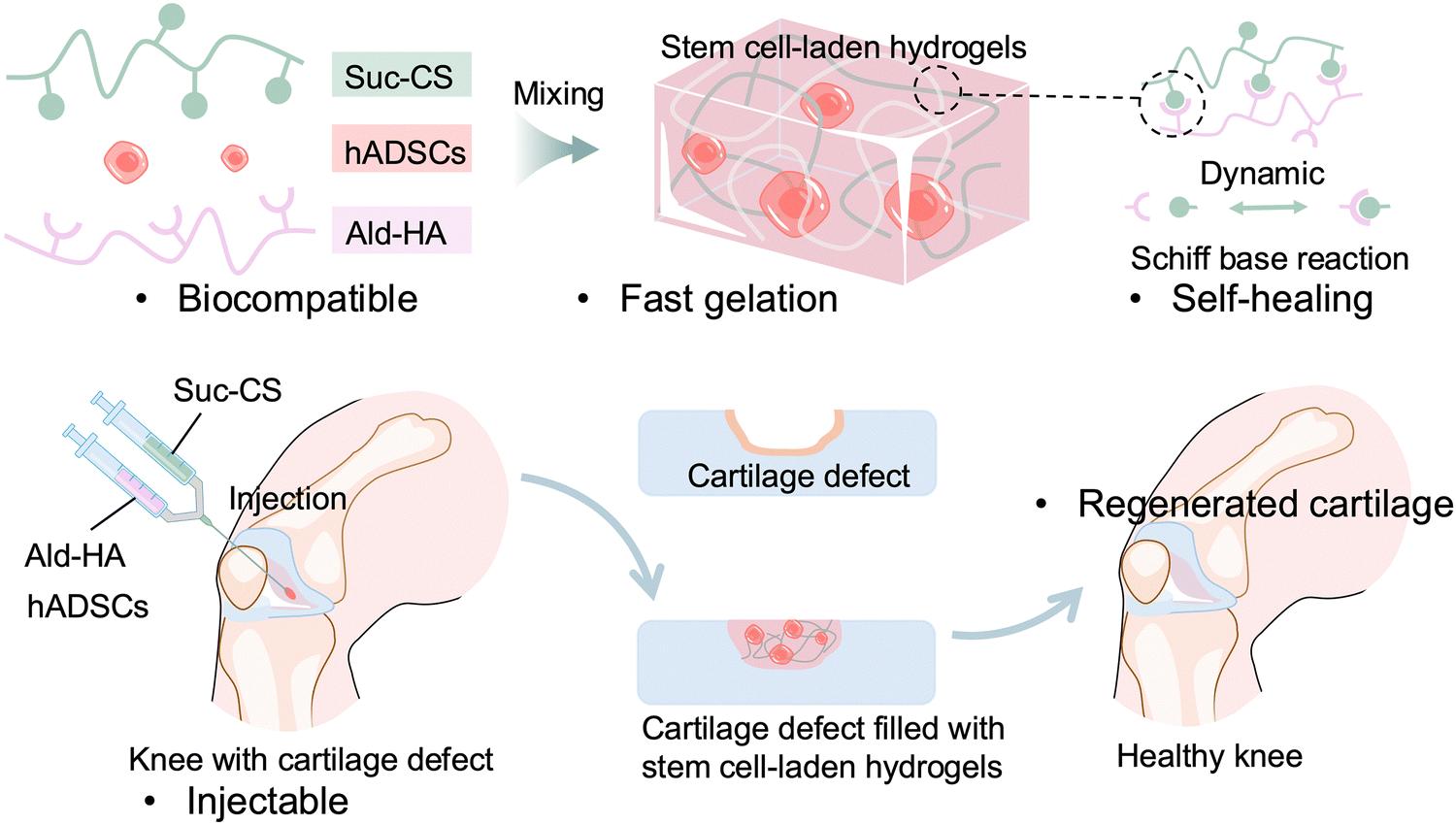 | ||
| Scheme 1 Schematic illustration of an injectable, stem cells-laden hydrogel at cartilage defect site for knee cartilage repair. | ||
2. Materials and methods
2.1. Materials
Hyaluronic acid sodium salt from Streptococcus equi (∼1.5–1.8 × 106 Da), succinic anhydride, sodium periodate, ethylene glycol, and tert-butyl carbazate were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA). Chitosan (medical grade, deacetylation degree: 95%) from Alaska snow crab was purchased from Matexcel (Bohemia, NY, USA). Calcein AM, propidium iodide, aggrecan monoclonal antibody (BC-3), SOX9 recombinant rabbit monoclonal antibody (7H13L8), goat anti-mouse IgG, IgM (H + L) secondary antibody with Alexa Fluor™ 488, and donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody with Alexa Fluor™ 555 were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Human adipose-derived mesenchymal stem cells (hADSCs), Mesenchymal stem cell medium, fetal bovine serum (FBS), penicillin/streptomycin solution (P/S solution), and mesenchymal stem cell growth supplement were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). All chemicals and reagents were used as received.2.2. Synthesis of N-succinyl-chitosan (Suc-CS)
The water-soluble Suc-CS was synthesized through the chemical reaction between chitosan and succinic anhydride. Briefly, chitosan (0.25 g) was dissolved in 5% (v/v) acetic acid solution (20 mL), followed by the addition of methanol (80 mL). Succinic anhydride (0.75 g) was dissolved in acetone (10 mL) and added to the above solution. The reaction was performed at room temperature for 24 h under stirring. After the reaction, the pH of the solution was adjusted to 6 using 1 M NaOH, and the precipitates were collected through centrifugation (4000 rpm, 5 min) and re-dissolved in DI water. The solution was purified by 3-day dialysis against deionized water at room temperature using a Spectra/Por 3 RC dialysis membrane (3.5 K; Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA). Finally, the purified solution was frozen using liquid nitrogen and dried using a lyophilizer (Labconco Co., Ltd., Kansas City, MO, USA). The purified polymer powder was stored at –20 °C. The chemical structure and substitution degree were analyzed and calculated using 1H nuclear magnetic resonance spectroscopy (1H NMR, Bruker AV-500, Bruker BioSpin GmbH, Rheinstetten, Germany).2.3. Preparation of aldehyde hyaluronic acid (Ald-HA)
The hyaluronic acid was oxidized by sodium periodate to form Ald-HA. In brief, hyaluronic acid (2 g) was dissolved in DI water (200 mL) and the mixture was stirred overnight in a 500 mL flask. Sodium periodate solution in DI water (0.5 M, 10 mL) was then added to the solution, and the reaction was performed at room temperature for 2 h, protected from light. Ethylene glycol (2 mL) was then added to the reactant and stirred at room temperature for 1 h to stop the reaction. The solution was purified by 3-day dialysis against deionized water at room temperature using a Spectra/Por 3 RC dialysis membrane. Finally, the purified solution was frozen using liquid nitrogen and dried using a lyophilizer. The purified polymer powder was stored at −20 °C. The chemical structure was analyzed using 1H NMR. The aldehydes content in Ald-HA was quantified using tert-butyl carbazate.40 The peak corresponding to the tert-butyl substituent ((CH3)3COCONHNH–, δ = 1.38 ppm) was compared with the peak of HA acetamide protons at 1.9 ppm.2.4. Fabrication of hydrogels
The Suc-CS and Ald-HA were dissolved separately into DPBS at different concentrations (10, 20, 30, 40, and 50 mg mL−1). The Suc-CS/Ald-HA hydrogels were prepared by mixing the two polymer solutions of the same concentration (1/1, v/v) at room temperature using a double barrel syringe (Duploject syringe, Baxter Healthcare, Deerfield, IL, USA) with a 21 G needle (Becton & Dickinson, Franklin Lakes, NJ). For morphological observation, DPBS was replaced by distilled water to prepare the hydrogel.2.5. Self-healing capability of hydrogels
700 μL of freshly prepared hydrogels was cut into two pieces and one of them (350 μL) was soaked in 500 μL Trypan Blue for 5 min. Then, two pieces of hydrogel were brought together at room temperature for 3 min. The resulting hydrogel piece was picked up using a tweezer.2.6. Rheological analysis of hydrogels
The rheological properties of crosslinked Suc-CS/Ald-HA hydrogels of different concentrations (10, 20, 30, 40, and 50 mg mL−1) were evaluated using a rheometer (Anton Paar MCR 92, Anton Paar GmbH, Graz, Austria). For stress sweeps, the storage (G′) and loss modulus (G′′) were recorded under increasing shear stress from 0.1–10000 Pa at temperature of 37 °C and frequency of 1 Hz. For time sweep, the storage (G′) and loss (G′′) modulus were recorded over time for Suc-CS/Ald-HA hydrogels of different concentrations (10, 20, 30, 40, and 50 mg mL−1) by rheological analysis at temperature of 37 °C, shear strain of 1%, and frequency of 1 Hz.2.7. Gelation times of hydrogels
The inverting tube method was used to determine the gelation times of hydrogels. The polymer solutions of Suc-CS and Ald-HA were incubated at 37 °C before mixing. The Suc-CS and Ald-HA mixture was incubated at 37 °C post mixing, and its flowability was assessed. The gelation time was defined as the point at which the hydrogel solution ceased to flow.41 The evaluation was conducted in triplicate.2.8. Hydrogel morphology
Suc-CS and Ald-HA polymers were separately dissolved in DI water at different polymer concentrations (10, 20, 30, 40, and 50 mg mL−1). Suc-CS/Ald-HA hydrogels were prepared upon mixing two polymer solutions of the same concentration (1/1, v/v) at room temperature. 200 μL of the hydrogel was soaked into 300 μL DI water and incubated at 37 °C for 1, 3, and 7 days. After the incubation, the solution was removed using a pipette and the remaining hydrogel were completely soaked into liquid nitrogen for 30 min and then loaded on the lyophilizer for 2 days. The freeze-dried samples were observed using a SEM with an acceleration voltage of 15 kV (Hitachi TM3000 Tabletop Microscope, Hitachi High-Technologies Corporation, Tokyo, Japan). The pore size was determined by analyzing SEM images using ImageJ. First, the SEM image was imported into ImageJ, and a scale was established based on known dimensions. Second, appropriate tools such as the line tool were utilized to identify individual pores and quantify their diameters. Third, the average pore size was derived from multiple measurements.2.9. In vitro cell culture
The complete cell culture medium was made by mixing 500 mL base medium, 25 mL FBS, 5 mL growth supplement, and 5 mL P/S solution. hADSCs were cultured in the complete medium in an incubator with 5% CO2 at 37 °C. The seeding density of hADSCs was 5000 cells per cm2 in T-75 flask. The hADSCs were passaged every four days, and the cell culture medium was changed every two days. All the cells used in this work were within eight passages.2.10. Live/dead staining
Calcein-AM and propidium iodide were used to stain live and dead cells, respectively. Calcein-AM was dissolved in dimethyl sulfoxide (DMSO) to obtain a stock concentration of 1 mM, and propidium iodide was dissolved in DI water to obtain a stock concentration of 1.5 mM. 96-well plates were used for live/dead staining test. First, hADSCs were suspended in the Ald-HA solution. Then, 75 μL of the Suc-CS solution was mixed with 75 μL of hADSCs-Ald-HA solution in each well of a 96-well plate using a pipette. The total volume of hydrogel in each well was 150 μL, and the total number of hADSCs in each well was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. The concentrations of Suc-CS and Ald-HA were: 10 mg mL−1, 20 mg mL−1, 30 mg mL−1, 40 mg mL−1, and 50 mg mL−1 in a complete cell culture medium. The experiment for each concentration was repeated three times. The hydrogel was incubated at 37 °C for 30 min, then 100 μL complete medium was added on top of the hydrogel. hADSCs in the hydrogel were cultured for 1, 3, 5, 7, and 14 days. The cell culture medium was changed every day, and DPBS was added to the edge of plate to reduce the evaporation. The live/dead staining solution was made by mixing 4 μL of Calcein-AM (2 mM, DMSO), 4 μL PI (2 mM, DI), and 1 mL DPBS. Before adding the staining solution to the hydrogel, the cell culture medium was removed and the hydrogel was gently rinsed with DPBS twice. After removing the DPBS, 100 μL staining solution was added to each well and the solution was incubated for 15 min at room temperature. The dyeing solution was then replaced with an equal volume of DPBS. A fluorescence microscope (Olympus IX81, Olympus Life Science) was used to observe and take images of cells.
000 cells. The concentrations of Suc-CS and Ald-HA were: 10 mg mL−1, 20 mg mL−1, 30 mg mL−1, 40 mg mL−1, and 50 mg mL−1 in a complete cell culture medium. The experiment for each concentration was repeated three times. The hydrogel was incubated at 37 °C for 30 min, then 100 μL complete medium was added on top of the hydrogel. hADSCs in the hydrogel were cultured for 1, 3, 5, 7, and 14 days. The cell culture medium was changed every day, and DPBS was added to the edge of plate to reduce the evaporation. The live/dead staining solution was made by mixing 4 μL of Calcein-AM (2 mM, DMSO), 4 μL PI (2 mM, DI), and 1 mL DPBS. Before adding the staining solution to the hydrogel, the cell culture medium was removed and the hydrogel was gently rinsed with DPBS twice. After removing the DPBS, 100 μL staining solution was added to each well and the solution was incubated for 15 min at room temperature. The dyeing solution was then replaced with an equal volume of DPBS. A fluorescence microscope (Olympus IX81, Olympus Life Science) was used to observe and take images of cells.
2.11. cDNA synthesis and quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
To quantify relative gene expression, qRT-PCR was performed on complementary deoxyribonucleic acid (cDNA) collected from cells cultured in both hydrogels and 2D surfaces after 7 days and 14 days of growth. 24-well plates were used for PCR test. First, hADSCs were suspended in the Ald-HA solution. Then, 350 μL of hADSCs-Ald-HA solution was mixed with 350 μL of Suc-CS solution in each well of a 24-well plate using a pipette. The total volume of hydrogel in each well was 700 μL, and the total number of hADSCs in each well was 140000 cells. The concentrations of Suc-CS and Ald-HA were: 20 mg mL−1 and 30 mg mL−1, and the experiment for each concentration was repeated three times. Cells were cultured in the chondrogenic differentiation medium for 7 days and 14 days. The medium was changed every day. The Qiagen RNeasy Kit (Qiagen Inc., Valencia, CA, USA) was used to extract ribonucleic acid (RNA) from cells and the iScript cDNA synthesis kit was used to make cDNA according to the manufacturer's instruction. In a Bio-Rad CFX96 real-time PCR detection system (Bio-Rad Laboratories, Inc. Hercules, CA, USA), the amplification with a primer for each of the transcripts was evaluated using SYBR Green PCR Mastermix. The reference gene was glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The cycle number at threshold fluorescence intensity was used for quantification. All samples were thermocycled in a 10 μL solution comprising 5 μL SYBR Mastermix, 300 nm primers (Table S1, ESI†), Integrated DNA Technologies, Coralville, IA, and cDNA at 0.04–1 ng μL−1 concentration adjusted for each condition. The thermocycle was 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s. The CFX Manager program was used to examine all RT-qPCR results (Bio-Rad Laboratories, Inc.).
2.12. Immunofluorescence and immunocytochemistry
The samples were prepared using the identical procedure as for the Live/Dead staining method. Specifically, 35 μL of Suc-CS solution was mixed with 35 μL of hADSCs-Ald-HA solution in each well of a 96-well plate using a pipette. The total volume of hydrogel in each well was 70 μL, and the total number of hADSCs in each well was 14000 cells. The concentrations of Suc-CS and Ald-HA were 20 mg mL−1 and 30 mg mL−1, and the experiment for each concentration was repeated three times. Subsequently, the cells were cultivated in a medium designed for chondrogenic differentiation for 14 days, with daily medium replacement. The cells within the hydrogel were fixed with a 4% paraformaldehyde solution for 15 min at room temperature. Following fixation, the cells were permeabilized with 1% (v/v) Triton X-100 in DPBS and blocked with 15% donkey serum albumin and 0.3% Triton X-100 in DPBS for 1 h at room temperature. The cells were then incubated with primary antibodies ACAN and SOX9 overnight at 4 °C, followed by incubation with the corresponding secondary antibodies for 1 h at room temperature in the dark. The nuclei were counterstained with 1 μM DAPI for 15 min. Between each of these steps, thorough washing with cold PBS was carried out at least three times to remove any unbound antibodies or other reagents. All the immunostained samples were imaged using a Nikon TE300 inverted microscope (Nikon Corporation, Tokyo, Japan).
2.13. Statistical analysis
All graphs and bar charts were expressed as the mean ± standard deviation (SD) of more than five repeated experiments as described above. The mean values of the pore sizes of different hydrogels were compared by two-way analysis of variance (ANOVA), followed by Tukey's multiple comparisons test. The mean values of the size of the cell spheroid and the relative expression of RNA were compared by one-factor ANOVA and the significance of the difference was determined by Tukey's multiple comparisons test. Statistical analysis was conducted using Prism 9 software.3. Results and discussion
3.1. Polymer synthesis and characterization
The synthesis of Suc-CS involved the introduction of succinyl groups to the chitosan's N-terminal glucosamine units. The medical grade chitosan, characterized by a 95% deacetylation degree, was dissolved in a 5% (v/v) acetic acid solution, followed by the addition of methanol. The succinic anhydride was dissolved in acetone and added to the above solution. The reaction was allowed to proceed under stirring at room temperature for 24 h. The polysaccharide amine group and the electrophilic carbonyl group of the anhydride underwent a condensation process during the succinylation reaction, which resulted in the formation of an amidic bond and the opening of the anhydride ring.36 The synthesis route and chemical structure of Suc-CS are shown in Fig. 1a. The synthesized Suc-CS exhibited excellent solubility in Dulbecco's phosphate-buffered saline (DPBS) at room temperature at concentrations not exceeding 50 mg mL−1, confirming the successful conjugation of the succinyl group. The substitution degree of Suc-CS was 34%, which was calculated by comparing the integral of the peaks of the CH2–CH2 section of the succinyl group on Suc-CS near 2.5 ppm and the integral of the peaks of the methyl protons around 2.0 ppm (Fig. 1d). | ||
| Fig. 1 Polymer chemical structure and characterizations. (a) Synthesis route and chemical structure of N-succinyl chitosan (Suc-CS). (b) Synthesis route and chemical structure of aldehyde hyaluronic acid (Ald-HA). (c) Chemical structure of crosslinked Suc-CS/Ald-HA hydrogel. (d) 1H NMR spectrum of hyaluronic acid (HA), aldehyde hyaluronic acid (Ald-HA), chitosan (CS), N-succinyl-chitosan (Suc-CS), and Suc-CS/Ald-HA hydrogel. | ||
To synthesize Ald-HA, HA was dissolved in deionized (DI) water, and sodium periodate was then introduced to initiate the reaction at room temperature. By reacting with sodium periodate, which oxidizes the vicinal hydroxyl groups to dialdehydes and opens the sugar ring to create dialdehyde derivatives, aldehyde groups were incorporated into the hyaluronic acid.36 The synthetic route and chemical structure of aldehyde hyaluronic acid are shown in Fig. 1b. The presence of a peak around 5 ppm in the spectrum indicates the presence of aldehyde groups in HA (Fig. 1d). The oxidation percentage was quantified at 14% by measuring the number of aldehydes in the polymer using tert-butyl carbazate. This was determined by comparing the peak corresponding to the tert-butyl substituent ((CH3)3COCONHNH–, δ = 1.38 ppm) with the peak of HA acetamide protons at 1.9 ppm. It is worth noting that the preparation process of hydrogel is facilitated by a substantial reduction in the viscosity of Ald-HA, which results from the disruption of intermolecular hydrogen bonds during the oxidation of HA.42
3.2. Hydrogel characterization
The hydrogels were prepared by gently mixing the solutions of the two modified polysaccharides, Suc-CS and Ald-HA. Both Suc-CS and Ald-HA were dissolved in DPBS at concentrations of 10, 20, 30, 40, and 50 mg mL−1, respectively. The gelation mechanism is thought to be based on the Schiff base reaction between the amino of Suc-CS and aldehyde groups of Ald-HA (Fig. 1c).36 To confirm gelation, rheological analysis was performed using the time-sweep mode immediately after mixing. To establish the appropriate mechanical strain and frequency settings for time-sweep experiments, the gelled samples were initially subjected to incremental strain measurements at a frequency of 1 Hz and a temperature of 37 °C. The result in Fig. S1 (ESI†) showed that strains ranging from 0.1% to 5% fall within the linear viscoelastic region (LVR), where the storage modulus remains unaffected by the applied strain. Consequently, a strain of 1% at 1 Hz was selected as the optimal testing condition to minimize both gel damage and noise during the subsequent time-sweep experiments. Fig. 2a shows the changing of the storage (G′) and loss (G′′) modulus of Ald-HA (20 mg mL−1, DPBS), Suc-CS (20 mg mL−1, DPBS), and Suc-CS/Ald-HA mixture (20 mg mL−1, DPBS) over time at 37 °C. The storage modulus (G′) represents the solid-like behavior of the sample, indicating its elastic characteristics, while the loss modulus (G′′) represents the liquid-like behavior, indicating its viscous characteristics. A viscoelastic solid hydrogel exhibits a higher storage modulus than loss modulus (G′ > G′′) due to the crosslink inside the hydrogel, while a viscoelastic liquid solution exhibits a lower storage modulus than loss modulus (G′ < G′′) because of the lack of strong interactions between polymer chains.43Fig. 2a shows that the storage modulus was larger than loss modulus (G′ > G′′) for the Suc-CS/Ald-HA mixture, confirming the hydrogel formation. On the other hand, both Ald-HA and Suc-CS solutions had a storage modulus smaller than the loss modulus (G′ < G′′), indicating that these individual polymer solutions could not form hydrogels on their own. To demonstrate the occurrence of the Schiff base reaction during crosslinking of Ald-HA and Suc-CS solution, Suc-CS was mixed with HA that lacked aldehyde groups (Suc-CS/HA). The mixture was then left at room temperature overnight after mixing using a vortex to ensure complete interaction between polymer chains. Subsequently, the storage (G′) and loss (G′′) modulus of the fully mixed Suc-CS/HA were measured using rheological analysis in stress sweep mode (unlike the time sweep mode, as the gelation time was not known). The results show that the storage modulus was lower than the loss modulus (G′ < G′′) (Fig. S2, ESI†), indicating no Schiff base reaction between Suc-CS and HA. Thus, the Suc-CS/Ald-HA hydrogel could be readily prepared by mixing two polymer solutions at the same concentrations (1/1, v/v) at 37 °C. | ||
| Fig. 2 Gelation capability. (a) Rheological analysis, with time-sweep mode, of various hydrogel samples including Ald-HA, Suc-CS, and Suc-CS/Ald-HA, post mixing from 0 to 1200 s (temperature: 37 °C, shear strain: 1%, and frequency: 1 Hz). (b) Rheological analysis, with time-sweep mode, of Suc-CS/Ald-HA hydrogels post mixing from 0 to 50 s (temperature: 37 °C, shear strain: 1%, and frequency: 1 Hz). Each polymer (Suc-CS or Ald-HA) was dissolved in DPBS at a concentration of 20 mg mL−1. Suc-CS/Ald-HA hydrogels were prepared by mixing same volume of Suc-CS and Ald-HA solutions. | ||
The preparation of an injectable hydrogel can be achieved through two primary strategies. The first strategy involves in situ gelation of liquid polymers post-injection, wherein precursor solutions are combined, and gelation occurs upon injection into the target site. This method enables a controlled and localized formation of the hydrogel within the body. Alternatively, the second approach employs shear-thinning polymers. In this case, the hydrogel precursor experiences a temporary reduction in viscosity under shear stress during injection, facilitating smooth administration. Subsequently, the polymer system regains its original viscosity after injection, resulting in the formation of a stable hydrogel at the desired location.3 In this study, the hydrogel was created by gently mixing solutions of modified polysaccharides, specifically Ald-HA and Suc-CS utilizing the first strategy. The gelation was confirmed by the rheological analysis using the time-sweep mode immediately following injection. The intersection points of the storage (G′) and loss (G′′) modulus was defined as the gelation time. The results of rheological analysis for Suc-CS/Ald-HA hydrogel in Fig. 2b showed that the Suc-CS/Ald-HA mixture exhibited solid hydrogel behavior as early as 10 s after mixing, guaranteeing immediate encapsulation of hADSCs during the injection process.
Building on the foundational property of rapid gelation, the injectability of a hydrogel also plays a key role in delivering therapeutic agents to the target site with minimal invasiveness.44Fig. 3a schematically illustrates the injection of Suc-CS/Ald-HA hydrogel through a medical grade double-barrel syringe (Duploject syringe, Baxter) fitted with a 21 G needle (Becton Dickinson), allowing for varying concentration from 10 to 50 mg mL−1. This feature ensures that the injection of our hydrogel can be optimized according to the specific requirements of the treatment sites.
 | ||
| Fig. 3 Injectable and self-healing hydrogels. (a) Schematic image of the process of injecting Suc-CS and Ald-HA solutions through a double-barrel syringe with a 21 G needle to form a Suc-CS/Ald-HA hydrogel. Each polymer (Suc-CS or Ald-HA) was dissolved in DPBS at a concentration of 20 mg mL−1. (b) Schematic images of the self-healing capability of a Suc-CS/Ald-HA hydrogel: (1) a 700 μL aliquot of freshly prepared hydrogel was divided into two pieces, (2) one of which was stained with Trypan Blue for identification purposes. (3) Following this, the two pieces of hydrogel were brought together at room temperature for 3 minutes, (4) resulting in the formation of a single hydrogel through spontaneous fusion, which was then picked up using tweezers. | ||
Self-healing hydrogels have found wide applications in the delivery of cells, drugs, biomolecules, and polynucleotides.45 These hydrogels exhibit the remarkable ability to restore their structural integrity and mechanical properties (e.g., elasticity, viscosity, stiffness, and toughness) under stress. Fig. 3b demonstrated the rapid self-healing capability of our Suc-CS/Ald-HA hydrogel, mending within 3 min after two separated pieces of hydrogel were brought together. The self-healing mechanism of Suc-CS/Ald-HA hydrogels relies on the presence of dynamic covalent bonds, particularly imide bonds formed via the Schiff base reaction.46 Moreover, the self-healing capability of this hydrogel would allow it to transform into the shape of and fill the defect site after injection. This property can significantly enhance its therapeutic efficacy, as it ensures complete coverage of the affected area, promoting better tissue repair and regeneration.
To further investigate the effect of the mechanical properties of the hydrogels on stem cell proliferation and phenotype, it is essential to perform rheological analyses on samples after the crosslinking of Suc-CS and Ald-HA. The modulus of the Suc-CS/Ald-HA hydrogel was assessed through stress sweep testing at 37 °C at different polymer concentrations (10, 20, 30, 40, and 50 mg mL−1). For all the Suc-CS/Ald-HA hydrogels prepared at different concentrations and tested at 37 °C, it was observed that the storage modulus (G′) was higher than the corresponding loss modulus (G′′) (Fig. 4a–e). This observation indicated the successful gelation of the Suc-CS and Ald-HA solutions across all concentrations. As the polymer concentrations increased, the storage modulus (G′) within the linear viscoelastic regime (LVR) also increased, ranging from 75 Pa to 5000 Pa (Fig. 4f), due to the increased crosslinking density. The increased crosslinking enhanced the elasticity and allowed the hydrogel to maintain structural integrity even under higher stresses. Notably, the hydrogel matrix stiffness could be tuned by varying the polymer concentration. It is worth mentioning that the Suc-CS and Ald-HA solutions, prepared at different concentrations, formed a hydrogel within just 1 min after merely mixing two polymer solutions at the same concentrations (1/1, v/v) at 37 °C (Fig. S3, ESI†). This rapid gelation capability would enable the in situ gelation immediately after mixing, and allow for the encapsulation of hADSCs contained in the Ald-HA solution in the hydrogel upon injection.
 | ||
| Fig. 4 Mechanical strength of the hydrogels measured by rheology. (a)–(e) The storage (G′) and loss modulus (G′′) were recorded under increasing shear stress from 0.1–10000 Pa for Suc-CS/Ald-HA hydrogels with different concentrations (10, 20, 30, 40, and 50 mg mL−1) (temperature: 37 °C, frequency: 1 Hz). (f) The storage modulus (G′) within linear viscoelastic regime of Suc-CS/Ald-HA hydrogels with different concentrations (10, 20, 30, 40, and 50 mg mL−1). | ||
The hydrogel pore size holds paramount significance as it directly influences stem cell migration, proliferation, and differentiation for tissue regeneration. Scanning electron microscope (SEM) images were acquired to characterize the microstructure of freeze-dried Suc-CS/Ald-HA hydrogels prepared at different polymer concentrations (10, 20, 30, 40, and 50 mg mL−1) after being incubated at 37 °C for 7 days (Fig. 5). The freeze-dried samples exhibited a continuous and porous structure (Fig. 5), with pore diameters ranging from 8–35 μm depending on the polysaccharide content or concentration of the hydrogels (Fig. 6). Notably, the 8 μm pore size is sufficiently large to facilitate the diffusion of nutrients and metabolites for stem cell culture without hindrance. On the other hand, the 0.2 μm pore size acts as a barrier to the diffusion of large nutrients such as 70 kDa dextran.47 Consequently, all the Suc-CS/Ald-HA hydrogels here could ensure the unimpeded diffusion of nutrition and metabolites for stem cell proliferation.
 | ||
| Fig. 5 Porous structures of hydrogels. SEM images of freeze-dried Suc-CS/Ald-HA hydrogels prepared at polymer concentrations of 10, 20, 30, 40, and 50 mg mL−1 and incubated in DI water at 37 °C for 1, 3, and 7 days. The short scale bar represents 20 μm, and the long scale bar represents 100 μm. | ||
 | ||
| Fig. 6 Pore diameters of hydrogels. The pore diameters of the freeze-dried Suc-CS/Ald-HA hydrogels prepared at polymer concentrations of 10, 20, 30, 40, and 50 mg mL−1 and incubated in DI water at 37 °C for 1, 3, and 7 days. The pore diameter was obtained by evaluating SEM images using ImageJ. Two-way ANOVA was used, followed by Tukey's multiple comparisons test (mean ± SD, n > 10; *p < 0.05, **p < 0.01, ****p < 0.0001; ns: nonsignificant). | ||
The pore size and stability of Suc-CS/Ald-HA hydrogel were influenced by the polymer concentration and evolved over the soaking period. On day 0, the pore size inversely correlated with the polymer concentration, as it diminished when the concentration increased from 10 mg mL−1 to 30 mg mL−1. Beyond this point, raising the concentration to 50 mg mL−1 did not further decrease the pore size significantly, suggesting a threshold in the density of the network that governs pore structure. In terms of homogeneity, the hydrogel at higher concentrations, specifically at 40 and 50 mg mL−1, exhibited inhomogeneous porous structure. This inhomogeneity is likely resulting from the high viscosity of the polymer solution, which hindered thorough mixing during gelation. In contrast, at the lower concentration of 10 mg mL−1, the hydrogel presented a loosely formed network with irregularly ordered pores, likely due to insufficient crosslinking between –CHO and –NH2 groups. This hydrogel transformed into a fibric structure after one day of incubation, and completely dissolved by day 3.
At a concentration of 20 mg mL−1, the pore size decreased from day 0 to day 1, followed by an increase from day 1 to day 7, indicative of a phenomenon characterized by hydrogel contraction followed by swelling. This sequence can be attributed to the initial release of loosely bound water molecules from the hydrogel network upon immersion, resulting in contraction or shrinkage along with the structural rearrangements within the polymer network. Subsequently, this initial dehydration phase is typically followed by a subsequent swelling phase as the hydrogel absorbs water from its surroundings, possible driven by osmotic pressure between the polymer chains and water molecules. Consequently, the combined effects of water expulsion and subsequent rehydration contribute to the observed sequence of shrinkage followed by swelling in hydrogel materials. For the concentrations of 30 and 40 mg mL−1, the pore size showed a slight enlargement after soaking in DI, likely due to hydrogel swelling. Conversely, at the high concentration of 50 mg mL−1, the pore size remained relatively consistent throughout incubation, likely attributable to the higher crosslinking density within the hydrogel structure. These results further demonstrate that the degradation rate of hydrogels of different concentrations varies. The hydrogel with the lowest concentration, 10 mg mL−1, degraded the fastest and disappeared completely within 3 days. The 20 mg mL−1 hydrogel, on the other hand, degraded slowly between day 1 and day 7, while there was no significant degradation of hydrogels with higher concentrations (30, 40, and 50 mg mL−1) from day 1 to day 7.
In this study, conventional SEM was used to monitor microscale pore size as an indirect indicator of degradation. However, as it reflects the dehydrated state, this SEM cannot capture hydrated nanostructure or nanoscale changes during degradation. To address this, future studies will employ techniques such as cryo-SEM or small-angle X-ray scattering (SAXS), which better preserve or quantify native nanostructure. These methods will also provide more accurate insight into hydrogel remodeling relevant to regenerative applications. In addition, long-term in vivo degradation will be evaluated to support translational relevance.
3.3. Biocompatibility of hydrogels
The biocompatibility of injectable polymer hydrogels is important for stem cell-based cell therapy, as it directly influences the success of the therapeutic intervention by ensuring a harmonious interaction between the hydrogel matrix and the stem cells within the complex biological milieu. Also, the healthy hADSCs can secret exosomes to regulate the surrounding tissue microenvironment therapeutically,48,49 and they can differentiate into chondrocytes providing an additional cell source to repair cartilage, potentially contributing to the KOA regeneration.50 Here, hADSCs were encapsulated in the hydrogels with different polymer concentrations. To assess the morphology and viability of encapsulated hADSCs, they were stained with live/dead dyes, Calcein-AM and propidium iodide (PI), and visualized using a fluorescence microscope (Olympus IX81, Olympus Life Science, Waltham, MA, USA). Fig. S4 (ESI†) shows the morphological changes of hADSCs encapsulated in the hydrogels of different polymer concentrations on day 0, 1, 3, 5, and 7. On day 0, at a concentration of 10 mg mL−1, some of the hADSCs adhered to the bottom of the cell culture dish, while the remaining cells exhibited a spherical morphology. At concentrations of 20, 30, 40, and 50 mg mL−1, the hADSCs maintained their spherical shape, indicating that the hydrogels could potentially serve as a 3D scaffold for supporting cell growth. On day 1, even at a concentration of 10 mg mL−1, the cells were able to completely attach and grow on the wells of microplates (2D culture). This was likely due to the weak and fibrous structure of the hydrogel at this concentration, as seen in Fig. 4a and 5. Thereafter, all cells started 2D proliferation on the well bottom on day 3, as the hydrogel had dissolved completely on day 3. At concentrations of 20, 30, 40, and 50 mg mL−1, the hADSCs remained spherical for the entire 7-days period.Fig. 7 shows the results of LIVE/DEAD staining of hADSCs encapsulated in hydrogels of different polymer concentrations on days 1, 3, 5, 7, and 14 to evaluate cell viability. On day 1, nearly all hADSCs in all hydrogels remained alive, as indicated by their Calcein-AM staining. hADSCs were shown to be encapsulated homogeneously in the hydrogels with concentrations of 20 and 30 mg mL−1. However, cells were not evenly distributed in hydrogels with concentrations of 40 and 50 mg mL−1 due to the rapid gelation and high viscosity of the hydrogel. At the lower concentration of 10 mg mL−1, the hydrogel degraded completely within 3 days, and the cells started to attach and grow on the 2D well plates. These cells adhered and proliferated well on 2D surfaces, maintaining high viability for 14 days. In the hydrogels with concentration of 20 mg mL−1 and 30 mg mL−1, encapsulated cells started to proliferate with high viability and formed increasingly larger cell spheroids over time (Fig. 8a and b). Notably, through this timeframe, nearly all cells encapsulated in 20 mg mL−1 and 30 mg mL−1 hydrogel were viable. In the hydrogels with concentrations of 40 and 50 mg mL−1, cell viability is low and dead cells (red) increased gradually over time. There is no significant change in the size of cell spheroids over time (Fig. 8c and d), indicating the proliferation of cells was inhibited by the hydrogels at these concentrations. The high crosslinking density and high modulus of these high-polymer concentration scaffolds may hinder the diffusion of nutrients and metabolites causing cell apoptosis.
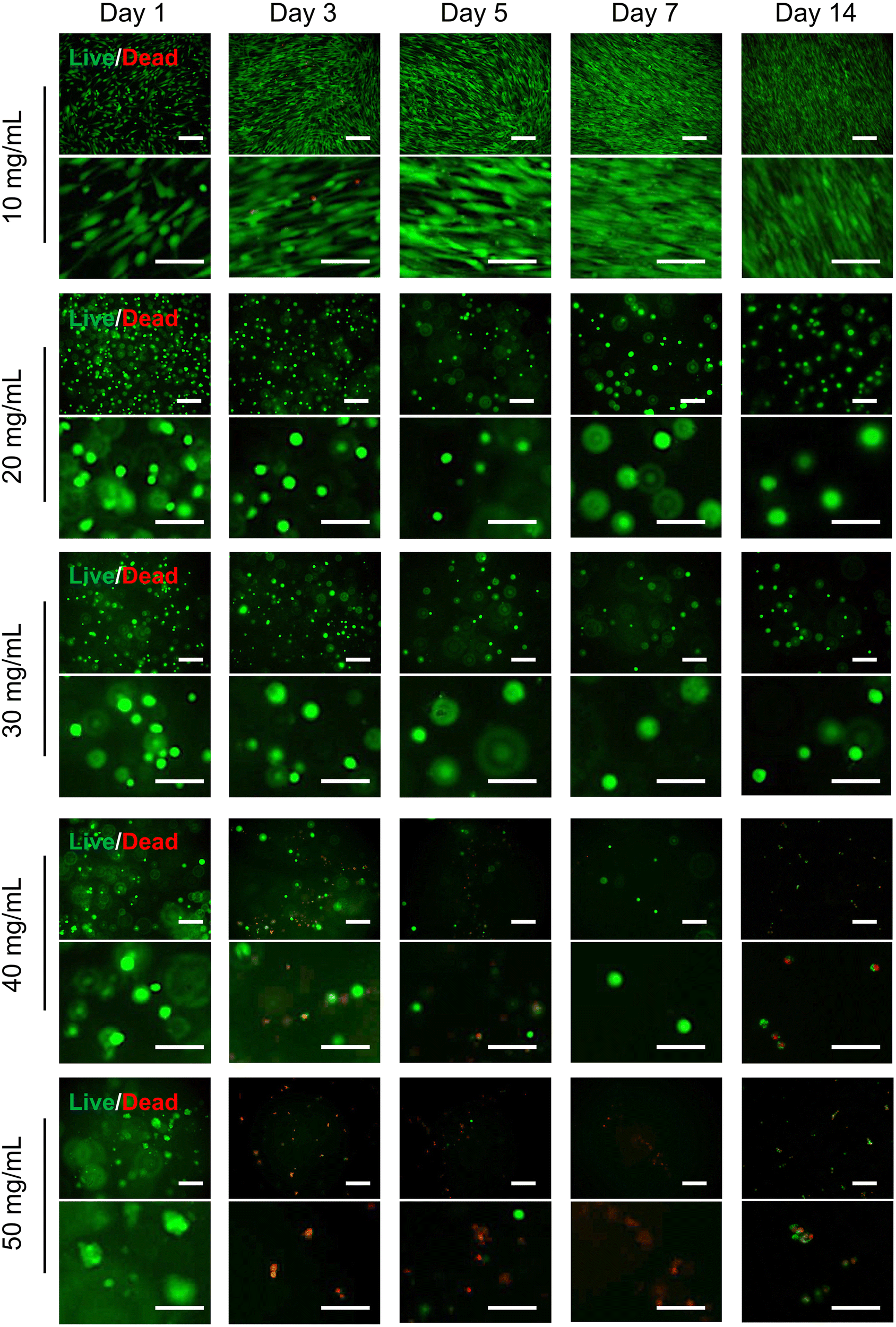 | ||
| Fig. 7 Cell viability in hydrogels. Live/dead staining images of hADSCs cultured in hydrogels prepared at concentrations of 10, 20, 30, 40, and 50 mg mL−1, assessed on day 1, 3, 5, 7, and 14. hADSCs were stained with Calcein-AM (green fluorescence for live cells) and propidium iodide (PI) (red fluorescence for dead cells). The stained cells were visualized using a fluorescence microscope. The short scale bar represents 200 μm, and the long scale bar represents 100 μm. | ||
 | ||
| Fig. 8 The diameters of the cell spheroids assessed from day 1 to day 14. The hADSCs were encapsulated in hydrogels prepared at concentrations of (a) 20 mg mL−1, (b) 30 mg mL−1, (c) 40 mg mL−1, and (d) 50 mg mL−1. One-way ANOVA was used, followed by Tukey's multiple comparisons test (mean ± SD, n ≥ 5; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, nonsignificant). | ||
Cells encapsulated in hydrogels prepared at 20 and 30 mg mL−1, which have a low modulus close to 1000 Pa, exhibited higher viability than those prepared at higher concentrations. This observation aligns with previous studies demonstrating that hydrogels with a softer stiffness of ∼1000 Pa tend to preserve stem cell viability, proliferation, and stemness.51–53 Furthermore, softer hydrogels can help reduce cell death after transplantation, thus enhancing the therapeutic effects of stem cell injections at the intended location.54 Our study suggested that Suc-CS/Ald-HA hydrogels prepared at 20 mg mL−1 and 30 mg mL−1 are particularly suited for the delivery, long-term proliferation, and maintenance of stem cells.
3.4. Expression of genes relevant to cartilage regeneration
Facilitating the chondrogenesis differentiation of hADSCs encapsulated in a hydrogel is pivotal for cartilage regeneration. To comprehensively understand this process, we conducted an examination of hADSC differentiation into chondrocytes at the gene expression level. The expression of specific genes, including ACAN (Aggrecan), SOX9 (SRY-box transcription factor 9), and COL2A1 (Collagen Type II Alpha 1 chain), assumes immense significance due to their pivotal roles. ACAN is crucial for maintaining cartilage integrity, as it encodes the aggrecan protein that forms the foundation of the cartilage extracellular matrix, providing hydration and resistance. SOX9, a transcription factor, regulates chondrocyte differentiation and cartilage formation by controlling the expression of genes like ACAN and COL2A1. Type II collagen (Col-II), as a major component of cartilage matrix, provides structural stability and strength.In this study, we evaluated the relative expression of these genes in various culture conditions. Cells were cultured on 2D tissue culture polystyrene (TCPS), a conventional platform, and in 3D hydrogels at two concentrations: 20 mg mL−1 and 30 mg mL−1 in chondrogenic differentiation medium. Notably, the 3D hydrogel environment closely mimics the native cartilage microenvironment. Fig. 9 shows that cells cultured in differentiation medium exhibited significantly elevated expression levels of ACAN, SOX9, and COL2A1 on both day 7 and day 14 when compared to the cell cultured in the complete medium. Also, the results demonstrated the superior capability of the 3D hydrogel platform in enhancing gene expression associated with chondrogenic differentiation compared to the 2D TCPS surface on both day 7 and day 14. Interestingly, no significant difference in gene expression (ACAN and SOX9) was observed between cells encapsulated in the two different hydrogel concentrations on day 7. However, on day 14, the 3D hydrogel formulated at a concentration of 30 mg mL−1 displayed a remarkable ability to enhance gene expression and chondrogenic differentiation compared to the 20 mg mL−1 hydrogel. This distinctive response on day 14 between the two hydrogel concentrations could be attributed to the mechanical properties and scaffold architecture of the hydrogels. The 30 mg mL−1 hydrogel might offer improved mechanical support and structural cues, facilitating cellular organization and interaction, and ultimately enhancing chondrogenic differentiation and gene expression. Additionally, the higher concentration might provide a more conducive microenvironment for chondrocyte maturation, influencing cellular signaling pathways and extracellular matrix deposition. These findings demonstrated the potential of 3D hydrogel systems, especially at a 30 mg mL−1 concentration, as a promising approach for effective cartilage regeneration, highlighting the significance of microenvironmental factors in guiding chondrogenic differentiation and tissue development. Also, results in Fig. S5 (ESI†) showed that hADSCs encapsulated within the 3D hydrogel exhibited elevated expression levels of ACAN, SOX9, and COL2A1 in comparison to cells cultured on the two-dimensional (2D) surface, even when the cells are maintained in a normal stem cell culture medium rather than a chondrogenic differentiation medium. This further indicates that our hydrogel can stimulate chondrogenic differentiation.
 | ||
| Fig. 9 Gene expression (fold changes) associated with chondrogenic differentiation. Relative expression of RNA: (a) ACAN, (b) SOX9, and (c) COL2A1 contents in hADSCs cultured on 2D TCPS in 3D hydrogels prepared at 20 mg mL−1, and in 3D hydrogels prepared at 30 mg mL−1 in the chondrogenic differentiation medium for a 14-days culture period. Gene expression was first normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (reference gene) and then normalized to the expression associated with 2D TCPS culture on day 0 (set as 1-time). One-way ANOVA followed by Tukey's multiple comparison test was used to determine whether the differences were statistically significant (mean ± SD, n = 3; **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, nonsignificant). | ||
The gene expression fold-changes observed in our 3D hydrogel system, particularly at the 30 mg mL−1 concentration, are comparable to or greater than those reported in other hADSC-based scaffolds for cartilage regeneration. For instance, Kim et al. reported ∼24-fold, ∼177-fold, and ∼533-fold increases in SOX9, ACAN, and COL2A1 expression, respectively, after 21 days in an atelocollagen hydrogel.55 Similarly, Rosadi et al. observed COL2A1 upregulation in the range of 102–103-fold in hADSCs cultured on a silk fibroin scaffold combined with platelet-rich plasma.56 In our study, ACAN, SOX9, and COL2A1 expression levels exceeded 1000-fold on day 14 within the 30 mg mL−1 hydrogel group, indicating a strong chondrogenic differentiation response. These results not only align with but in some cases surpass previously reported outcomes, highlighting the effectiveness of our hydrogel system in supporting cartilage-specific gene expression.
3.5. Expression of proteins relevant to cartilage regeneration
Here, we used immunofluorescence staining to assess the expression of chondrogenic differentiation-associated proteins in hADSCs encapsulated in hydrogels. Specifically, we focused on the evaluation of ACAN and SOX9 expression, which was visualized using green and red fluorescence, respectively. Concurrently, cell nuclei were counterstained with DAPI in blue. ACAN is a key extracellular matrix protein that plays a pivotal role in cartilage formation and maintenance. Its presence is indicative of a mature and functional chondrocyte phenotype, underscoring its importance as a marker for chondrogenic differentiation. SOX9, on the other hand, is a critical transcription factor responsible for the regulation of genes involved in cartilage development and maintenance. The expression of SOX9 serves a fundamental indicator of chondrogenic commitment, as it orchestrates the differentiation process and ensures the proper formation of cartilaginous tissues.1,51Our results in Fig. 10 clearly demonstrated that hADSCs cultured within 3D hydrogels (at concentrations of 20 and 30 mg mL−1) (3D-d) and on a 2D surface in a chondrogenic differentiation medium (2D-d) consistently exhibited robust expression of ACAN (green) and SOX9 (red). In contrast, hADSCs cultured on the 2D surface in a regular cell culture medium (2D) displayed negligible expression of ACAN and SOX9. While hADSCs cultured in both 3D-d and 2D-d environments exhibited expression of chondrogenic differentiation-associated proteins (ACAN and SOX9), it is important to highlight the distinct advantages offered by 3D hydrogels over 2D surfaces. These advantages stem from the hydrogels’ ability to closely mimic the native tissue microenvironment, providing essential spatial cues and mechanical support crucial for cellular organization and function. Unlike 2D surfaces, which limit cell–cell and cell–matrix interactions, 3D hydrogels facilitate multidirectional interactions between cells and the surrounding matrix, thereby promoting physiological responses and offering a more accurate representation of in vivo conditions. Additionally, the porous structure of hydrogels enables enhanced diffusion of nutrients and signaling molecules, thereby further enhancing cellular behavior and promoting the development of complex tissues. Consequently, these findings strongly suggest that the 3D hydrogel environment can effectively support the chondrogenic differentiation of hADSCs, as evidenced by the notable expression of ACAN and SOX9, critical markers associated with commitment to the chondrogenic lineage.
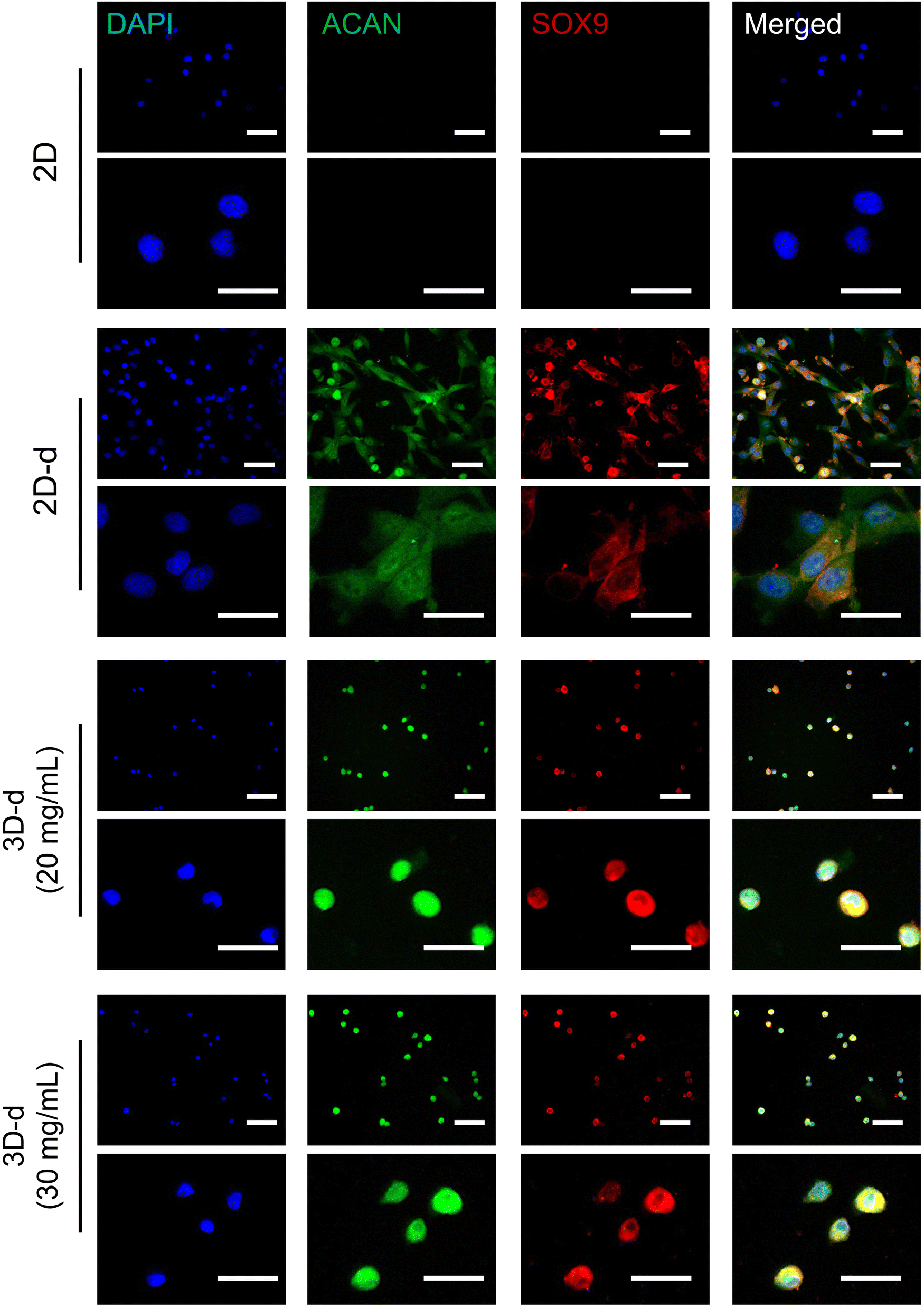 | ||
| Fig. 10 Protein expression associated with chondrogenic differentiation of cells encapsulated in hydrogels. Immunofluorescence staining was applied for Aggrecan (ACAN, green), SRY-box transcription factor 9 (SOX9, red), and cell nuclei (4′,6-diamidino-2-phenylindole (DAPI), blue). The hADSCs were cultured in hydrogels prepared at concentrations of 20 and 30 mg mL−1 in the chondrogenic differentiation medium (3D-d) for a 14-day culture period. The hADSCs cultured on 2D TCPS surface in both chondrogenic differentiation medium (2D-d) and normal cell culture medium (2D) were used as a control. The short scale bar represents 200 μm, and the long scale bar represents 100 μm. | ||
In parallel with the observed gene and protein expression patterns, we evaluated the degradation behavior of the hydrogels to better understand their suitability for supporting dynamic tissue remodelling. The 20 mg mL−1 and 30 mg mL−1 hydrogels both exhibited an initial increase in weight due to swelling, followed by a gradual decrease in mass over a 14-day period (Fig. S6, ESI†). This controlled and sustained degradation profile is critical for injectable hydrogels, as it provides temporary structural support during the early phase of chondrogenic differentiation, while allowing sufficient space for extracellular matrix deposition as new tissue forms. Notably, this degradation timeline coincides with the period of robust ACAN, SOX9, and COL2A1 gene upregulation and strong protein expression, particularly in the 30 mg mL−1 group, indicating that the hydrogel degrades at a rate conducive to cell-mediated matrix production and cartilage regeneration. These findings suggest that the degradation behavior of our hydrogel not only supports but may actively facilitate the transition from a synthetic scaffold to a tissue-integrated construct. To further assess cartilage regeneration beyond gene and protein expression, we plan to conduct in vivo studies comparing our hydrogel system with existing platforms in future work.
4. Conclusions
In this study, we introduced an approach to creating a cell-compatible, biodegradable, injectable, and self-healing polysaccharides-based hydrogel (Suc-CS/Ald-HA) in situ through simultaneous injection of two polymer solutions. The resulting hydrogel not only provided a favorable environment for both delivering and maintaining hADSCs with high viability, demonstrating great promise for cartilage regeneration. The gelation occurred upon mixing the two solutions to form a homogeneous, highly porous structure. By changing the concentrations of the polymer solutions, we were able to tailor the hydrogel to achieve the desired mechanical properties, pore size, and degradation rate, ensuring the sustained viability of encapsulated hADSCs. The encapsulated hADSCs remained highly viable even after 14-days incubation, demonstrating their potential for supporting cartilage regeneration over an extended duration. Moreover, the hADSCs encapsulated within the 3D hydrogels exhibited elevated gene expression levels associated with chondrogenic differentiation. Notably, the 3D hydrogels did not hinder the expression of proteins related to chondrogenic differentiation in the encapsulated hADSCs. This injectable stem cell-laden hydrogel holds promise not only for cartilage regeneration but also for potential applications in other cell-based therapies.Author contributions
Xiaojie Lin: writing – original draft, conceptualization, formal analysis, investigation, methodology, validation, visualization. Ruofan Liu: formal analysis, investigation, methodology, validation, visualization. Yang Zhou: writing – review & editing, formal analysis, investigation, methodology, validation, visualization. Jacob Beitzel: formal analysis, investigation, methodology, validation, visualization. Aya Noguchi: writing – review & editing, conceptualization, formal analysis, investigation, methodology, validation, visualization. Masayuki Kyomoto: writing – review & editing, supervision, conceptualization, formal analysis, investigation, methodology, validation, visualization, funding acquisition, project administration, resources. Miqin Zhang: writing – review & editing, supervision, conceptualization, formal analysis, investigation, methodology, validation, visualization, funding acquisition, project administration, resources.Data availability
We confirm that all the relevant research data is contained with the manuscript and ESI.† No databases have been used and no references to such databases are contained in the manuscript or ESI.†Conflicts of interest
This work was supported by Kyocera Corporation. One or more of the authors (A. N. and M. K.) are employed by Kyocera Corporation.Acknowledgements
The work was supported by Kyocera Corporation. The authors express thanks to Dr Nobuaki Moriguchi and Mr Masanori Enrin of Kyocera Corporation for valuable discussions and suggestions. The authors express special thanks to Ms Shihori Yamane, Dr Narumi Kumamoto, Ms Misato Murata, and Mr Kenichi Saiga of Kyocera Corporation for their technical assistance.References
- N. Gerwin, C. Scotti, C. Halleux, M. Fornaro, J. Elliott, Y. Zhang, K. Johnson, J. Shi, S. Walter, Y. Li, C. Jacobi, N. Laplanche, M. Belaud, J. Paul, G. Glowacki, T. Peters, K. A. Wharton, I. Vostiar, F. Polus, I. Kramer, S. Guth, A. Seroutou, S. Choudhury, D. Laurent, J. Gimbel, J. Goldhahn, M. Schieker, S. Brachat, R. Roubenoff and M. Kneissel, Nat. Med., 2022, 28, 2633–2645 CrossRef.
- C.-Y. Lin, Y.-L. Wang, Y.-J. Chen, C.-T. Ho, Y.-H. Chi, L. Y. Chan, G.-W. Chen, H.-C. Hsu, D. W. Hwang, H.-C. Wu and S.-C. Hung, Nat. Biomed. Eng., 2022, 6, 1105–1117 CrossRef.
- X. Lin, C. T. Tsao, M. Kyomoto and M. Zhang, Adv. Healthcare Mater., 2022, 11, 2101479 CrossRef.
- I. A. Jones, R. Togashi, M. L. Wilson, N. Heckmann and C. T. Vangsness, Nat. Rev. Rheumatol., 2019, 15, 77–90 CrossRef PubMed.
- A. Latourte, M. Kloppenburg and P. Richette, Nat. Rev. Rheumatol., 2020, 16, 673–688 CrossRef.
- R. A. Muzzarelli, F. Greco, A. Busilacchi, V. Sollazzo and A. Gigante, Carbohydr. Polym., 2012, 89, 723–739 CrossRef.
- M. Kim, D. R. Steinberg, J. A. Burdick and R. L. Mauck, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1569–1578 CrossRef PubMed.
- H. B. Sun, Ann. N. Y. Acad. Sci., 2010, 1211, 37–50 CrossRef.
- J. McKay, K. Frantzen, N. Vercruyssen, K. Hafsi, T. Opitz, A. Davis and W. Murrell, J. Clin. Orthop. Trauma, 2019, 10, 59–66 CrossRef PubMed.
- J. Burdick, R. Mauck and S. Gerecht, Cell Stem Cell, 2016, 18, 13–15 CrossRef.
- D. Jevotovsky, A. Alfonso, T. Einhorn and E. Chiu, Osteoarthritis Cartilage, 2018, 26, 711–729 CrossRef.
- J. Gorecka, V. Kostiuk, A. Fereydooni, L. Gonzalez, J. Luo, B. Dash, T. Isaji, S. Ono, S. Liu, S. R. Lee, J. Xu, J. Liu, R. Taniguchi, B. Yastula, H. C. Hsia, Y. Qyang and A. Dardik, Stem Cell Res. Ther., 2019, 10, 87 CrossRef PubMed.
- S. Muthu, J. V. Korpershoek, E. J. Novais, G. F. Tawy, A. P. Hollander and I. Martin, Nat. Rev. Rheumatol., 2023, 19, 403–416 CrossRef.
- R. M. Amsar, A. Barlian, H. Judawisastra, U. A. Wibowo and K. Karina, Fut. Sci. OA, 2021, 7, FSO734 CrossRef.
- N. Buzaboon and S. Alshammary, Stem Cells Cloning, 2020, 13, 117–136 Search PubMed.
- M. Amer, F. Rose, K. Shakesheff, M. Modo and L. White, Npj Regener. Med., 2017, 2, 23 CrossRef PubMed.
- A. Foster, L. Marquardt and S. Heilshorn, Curr. Opin. Chem. Eng., 2017, 15, 15–23 CrossRef PubMed.
- U. S. K. Madduma-Bandarage and S. V. Madihally, J. Appl. Polym. Sci., 2021, 138, 50376 CrossRef.
- A. K. Gaharwar, I. Singh and A. Khademhosseini, Nat. Rev. Mater., 2020, 5, 686–705 CrossRef CAS.
- L. García-Fernández, M. Olmeda-Lozano, L. Benito-Garzón, A. Pérez-Caballer, J. San Román and B. Vázquez-Lasa, Mater. Sci. Eng., C, 2020, 110, 110702 CrossRef PubMed.
- K. Madhusudana Rao, A. Kumar and S. S. Han, Int. J. Biol. Macromol., 2017, 101, 165–171 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 Search PubMed.
- J. Lou and D. J. Mooney, Nat. Rev. Chem., 2022, 6, 726–744 Search PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS.
- M. M. Islam, M. Shahruzzaman, S. Biswas, M. N. Sakib and T. U. Rashid, Bioact. Mater., 2020, 5, 164–183 Search PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- F. Furlani, A. Marfoglia, E. Marsich, I. Donati and P. Sacco, Biomacromolecules, 2021, 22, 2902–2909 CrossRef CAS PubMed.
- Z. Jiang, L. Li, H. Li, L. Xia, H. Hu, S. Wang, C. Liu, J. Chi, Y. Yang, F. Song, W. Liu and B. Han, Carbohydr. Polym., 2022, 280, 119032 CrossRef CAS.
- M. Sun, T. Wang, J. Pang, X. Chen and Y. Liu, Biomacromolecules, 2020, 21, 1351–1367 CrossRef CAS.
- Y. Kato, H. Onishi and Y. Machida, Biomaterials, 2004, 25, 907–915 CrossRef CAS.
- F.-C. Chang, S. L. Levengood, N. Cho, L. Chen, E. Wang, J. S. Yu and M. Zhang, Adv. Ther., 2018, 1, 1800058 CrossRef.
- C. T. Tsao, M. H. Hsiao, M. Y. Zhang, S. L. Levengood and M. Zhang, Macromol. Rapid Commun., 2015, 36, 332–338 CrossRef CAS.
- C.-T. Tsao, F. M. Kievit, K. Wang, A. E. Erickson, R. G. Ellenbogen and M. Zhang, Mol. Pharm., 2014, 11, 2134–2142 CrossRef CAS.
- C.-T. Tsao, F. M. Kievit, A. Ravanpay, A. E. Erickson, M. C. Jensen, R. G. Ellenbogen and M. Zhang, Biomacromolecules, 2014, 15, 2656–2662 Search PubMed.
- X. Lin, R. Liu, J. Beitzel, Y. Zhou, C. Lagadon and M. Zhang, Gels, 2024, 10, 508 CrossRef CAS PubMed.
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Biomaterials, 2009, 30, 2499–2506 CrossRef CAS.
- L. Li, N. Wang, X. Jin, R. Deng, S. Nie, L. Sun, Q. Wu, Y. Wei and C. Gong, Biomaterials, 2014, 35, 3903–3917 CrossRef CAS.
- L. Song, L. Li, T. He, N. Wang, S. Yang, X. Yang, Y. Zeng, W. Zhang, L. Yang and Q. Wu, Sci. Rep., 2016, 6, 1–13 CrossRef.
- A. K. HPS, C. K. Saurabh, A. Adnan, M. N. Fazita, M. Syakir, Y. Davoudpour, M. Rafatullah, C. Abdullah, M. Haafiz and R. Dungani, Carbohydr. Polym., 2016, 150, 216–226 CrossRef CAS PubMed.
- M. Kheirabadi, L. Shi, R. Bagheri, K. Kabiri, J. Hilborn and D. A. Ossipov, Biomater. Sci., 2015, 3, 1466–1474 RSC.
- D. Dehghan Baniani, R. Bagheri and A. Solouk, Carbohydr. Polym., 2017, 174, 633–645 Search PubMed.
- V. Hintze, M. Schnabelrauch and S. Rother, Front. Chem., 2022, 10, 830671 CrossRef PubMed.
- G. Stojkov, Z. Niyazov, F. Picchioni and R. K. Bose, Gels, 2021, 7, 255 CrossRef.
- M. Bhattacharjee, J. L. Escobar Ivirico, H.-M. Kan, S. Shah, T. Otsuka, R. Bordett, M. Barajaa, N. Nagiah, R. Pandey, L. S. Nair and C. T. Laurencin, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120968119 CrossRef.
- H. Rammal, A. GhavamiNejad, A. Erdem, R. Mbeleck, M. Nematollahi, S. Emir Diltemiz, H. Alem, M. A. Darabi, Y. N. Ertas, E. J. Caterson and N. Ashammakhi, Mater. Chem. Front., 2021, 5, 4368–4400 RSC.
- J. Xu, Y. Liu and S. H. Hsu, Molecules, 2019, 24, 3005 CrossRef PubMed.
- F. A. A. Ruiter, J. King, S. Swapnasrita, S. Giselbrecht, R. Truckenmüller, V. L. S. LaPointe, M. B. Baker and A. Carlier, Biomacromolecules, 2023, 24, 604–612 CrossRef.
- K. Zha, X. Li, Z. Yang, G. Tian, Z. Sun, X. Sui, Y. Dai, S. Liu and Q. Guo, npj Regener. Med., 2021, 6, 14 CrossRef PubMed.
- M. Satué, C. Schüler, N. Ginner and R. G. Erben, Sci. Rep., 2019, 9, 10153 CrossRef PubMed.
- X.-N. Xiang, S.-Y. Zhu, H.-C. He, X. Yu, Y. Xu and C.-Q. He, Stem Cell Res. Ther., 2022, 13, 14 CrossRef PubMed.
- M. Bhattacharjee, J. L. E. Ivirico, H.-M. Kan, S. Shah, T. Otsuka, R. Bordett, M. Barajaa, N. Nagiah, R. Pandey, L. S. Nair and C. T. Laurencin, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120968119 CrossRef PubMed.
- Y. Ren, H. Zhang, Y. Wang, B. Du, J. Yang, L. Liu and Q. Zhang, ACS Appl. Bio Mater., 2021, 4, 2601–2613 CrossRef PubMed.
- K. Lee, Y. Chen, X. Li, Y. Wang, N. Kawazoe, Y. Yang and G. Chen, J. Mater. Chem. B, 2019, 7, 7713–7722 RSC.
- L. M. Marquardt and S. C. Heilshorn, Curr. Stem Cell Rep., 2016, 2, 207–220 CrossRef.
- S. A. Kim, Y. J. Sur, M.-L. Cho, E. J. Go, Y. H. Kim, A. A. Shetty and S. J. Kim, Sci. Rep., 2020, 10, 10678 CrossRef CAS PubMed.
- I. Rosadi, K. Karina, I. Rosliana, S. Sobariah, I. Afini, T. Widyastuti and A. Barlian, Stem Cell Res. Ther., 2019, 10, 369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00287g |
| This journal is © The Royal Society of Chemistry 2025 |