
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced antimicrobial protection through surface immobilization of antibiotic-loaded peptide multicompartment micelles†
Shabnam
Tarvirdipour
 ab,
S. Narjes
Abdollahi
a,
Joachim
Köser
c,
Maryame
Bina
a,
Cora-Ann
Schoenenberger
a and
Cornelia G.
Palivan
*ab
ab,
S. Narjes
Abdollahi
a,
Joachim
Köser
c,
Maryame
Bina
a,
Cora-Ann
Schoenenberger
a and
Cornelia G.
Palivan
*ab
aDepartment of Chemistry, University of Basel, Mattenstrasse 22, Basel-4058, Switzerland. E-mail: cornelia.palivan@unibas.ch
bNCCR-Molecular Systems Engineering, Mattenstrasse 24a, Basel-4058, Switzerland
cSchool of Life Sciences, Institute for Chemistry and Bioanalytics, University of Applied Sciences and Arts Northwestern Switzerland, Hofackerstrasse 30, Muttenz-4132, Switzerland
First published on 9th April 2025
Abstract
The escalating global threat of antibiotic-resistant bacterial infections, driven by biofilm formation on medical device surfaces, prompts the need for innovative therapeutic strategies. To address this growing challenge, we develop rifampicin-loaded multicompartment micelles (RIF-MCMs) immobilized on surfaces, offering a dual-functional approach to enhance antimicrobial efficacy for localized therapeutic applications. We first optimize the physicochemical properties of RIF-MCMs, and subsequently coat the optimal formulation onto a glass substrate, as confirmed by quartz crystal microbalance and atomic force microscopy. Surface-immobilized RIF-MCMs facilitate sustained antibiotic release in response to biologically relevant temperatures (37 °C and 42 °C). In addition, their heterogeneous distribution enhances the surface's roughness, contributing to the antibacterial activity through passive mechanisms such as hindering bacterial adhesion and biofilm formation. In vitro antimicrobial testing demonstrates that RIF-MCM-modified surfaces achieve a 98% reduction in Staphylococcus aureus viability and a three-order-of-magnitude decrease in colony formation compared to unmodified surfaces. In contrast, RIF-MCMs exhibit minimal cytotoxicity to mammalian cells, making them suitable candidates for medical device coatings. Our dual-function antimicrobial strategy, combining sustained antibiotic release and enhanced surface roughness, presents a promising approach to locally prevent implant-associated infections and biofilm formation.
1. Introduction
Biomedical implants are indispensable in modern healthcare, offering essential solutions for patients with missing or impaired body parts. However, they are prone to infections, which can lead to severe complications and often result in implant failure, necessitating additional surgeries.1 The most common cause of biomaterial-centered infection is Staphylococcus aureus, which, upon binding to medical devices, forms biofilms enabling bacteria to avoid antimicrobial activity and host immune response. Major challenges in combating bacterial infections in healthcare include preventing bacterial adhesion to surfaces, ensuring their removal, and addressing their ability to form biofilms, especially on medical implants.1The prevention and treatment of bacterial infections on medical implants primarily rely on passive and active strategies.2–4 Passive strategies focus on creating pathogen-repellent surfaces or environments inhospitable to bacteria, aiming to prevent adhesion, colonization, and biofilm formation without directly killing bacteria.5 These methods are particularly effective in the application of medical devices like catheters and implants, where reducing infection risks is crucial.5,6 However, their effectiveness is limited in high-bacterial-load environments or established infections, as they do not directly target or kill bacteria.7 In contrast, active strategies focus on directly targeting bacteria to kill them or inhibit their growth.8 These approaches typically employ antimicrobial agents, biomolecules, or biocidal coatings to disrupt bacterial processes, including contact-killing surfaces, controlled antimicrobial agent release, and photodynamic or photothermal therapies.9,10 Although highly effective in reducing bacterial populations, active strategies may face limitations, such as toxicity to human cells, limited duration of action, and an enhanced risk of inducing bacterial resistance.11,12
To overcome the limitations associated with either approach and enhance antibacterial efficacy, modern antibacterial strategies increasingly employ strategies that combine passive and active mechanisms.5,6 The dual-functional approach is particularly sought after in the design of implants and wound care solutions, where both prevention and eradication of infections are critical.13,14 Dual-functionality has the potential to maximize antibacterial performance, extend protective lifespan of antibiotics, and reduce resistance development by targeting bacteria through multiple mechanisms.8 Recent advances utilize nanomaterials, chemical surface modifications, and micro- and nano-structuring to create effective chemical, physical, or hybrid barriers against microbial colonization.9,15–18
Nanomaterials are of particular interest for developing advanced antibacterial surfaces that can effectively combat bacterial infections owing to their unique physicochemical properties.9,19 A diverse range of nanomaterials and coatings including metal-based nanoparticles, carbon-based nanomaterials, polymeric, lipidic, and hybrid nanostructures, as well as surface-immobilized antibacterial peptides has demonstrated promising antibacterial activities.9,18,20,21 For instance, nanomaterials enable sustained release of antimicrobial agents over extended time periods and facilitate disruption of bacterial membranes through various mechanisms, thereby enhancing the potency of existing antibiotics.22–25 In addition, by increasing the local concentration of antimicrobial agents at the implant-tissue interface and minimizing the systemic side effects associated with conventional antibiotics, nanomaterials hold transformative potential to significantly reduce implant-related infection risks and improve patient outcomes.15,25–27 Other solutions include nanostructured surface topographies, self-cleaning and photocatalytic coatings, and quaternary ammonium compound (QAC)-based coatings, each utilizing distinct mechanisms to combat bacterial adhesion and biofilm formation.21,28 However, the growing prevalence of antibiotic-resistant strains, which pose significant challenges in clinical settings, further highlights the urgent need for high-performance nanomaterials suitable for combined passive and active antibacterial performance.14,29
Here, we present antibacterial surfaces based on the immobilization of antibiotic-loaded nanoassemblies on a solid support, to combine passive and active strategies for enhanced antimicrobial efficacy. These surfaces feature solid supports with non-uniformly immobilized peptidic multicompartment micelles (MCMs), which encapsulate antibiotics within their hydrophobic core (Fig. 1). Peptide-based supramolecular assemblies due to their inherent biocompatibility, biodegradability, and ability to form colloidally stable nanostructures, are expected to serve as effective and versatile platforms for antibiotic entrapment. Despite these advantages, the application of these platforms for antibiotic delivery in implants remains underexplored.30,31 The (HR)3(WL)6W peptide consists of 3 sequential histidine–arginine (HR) units followed by tryptophan–leucine (WL) repeats, imparting amphiphilic properties that facilitate favorable interactions with both hydrophilic and hydrophobic environments. This amino acid composition facilitates the self-assembly of peptides into MCMs, which can disassemble in response to temperature.32 The tryptophan and leucine residues originated from a truncated gramicidin A sequence.33 They contribute to hydrophobic interactions with hydrophobic compounds and facilitate their entrapment into nanoassemblies in the process of their formation. The dual-action antibacterial performance of our surfaces arises the increased surface roughness resulting from MCM immobilization, combined with stimuli-responsive antibiotic release.
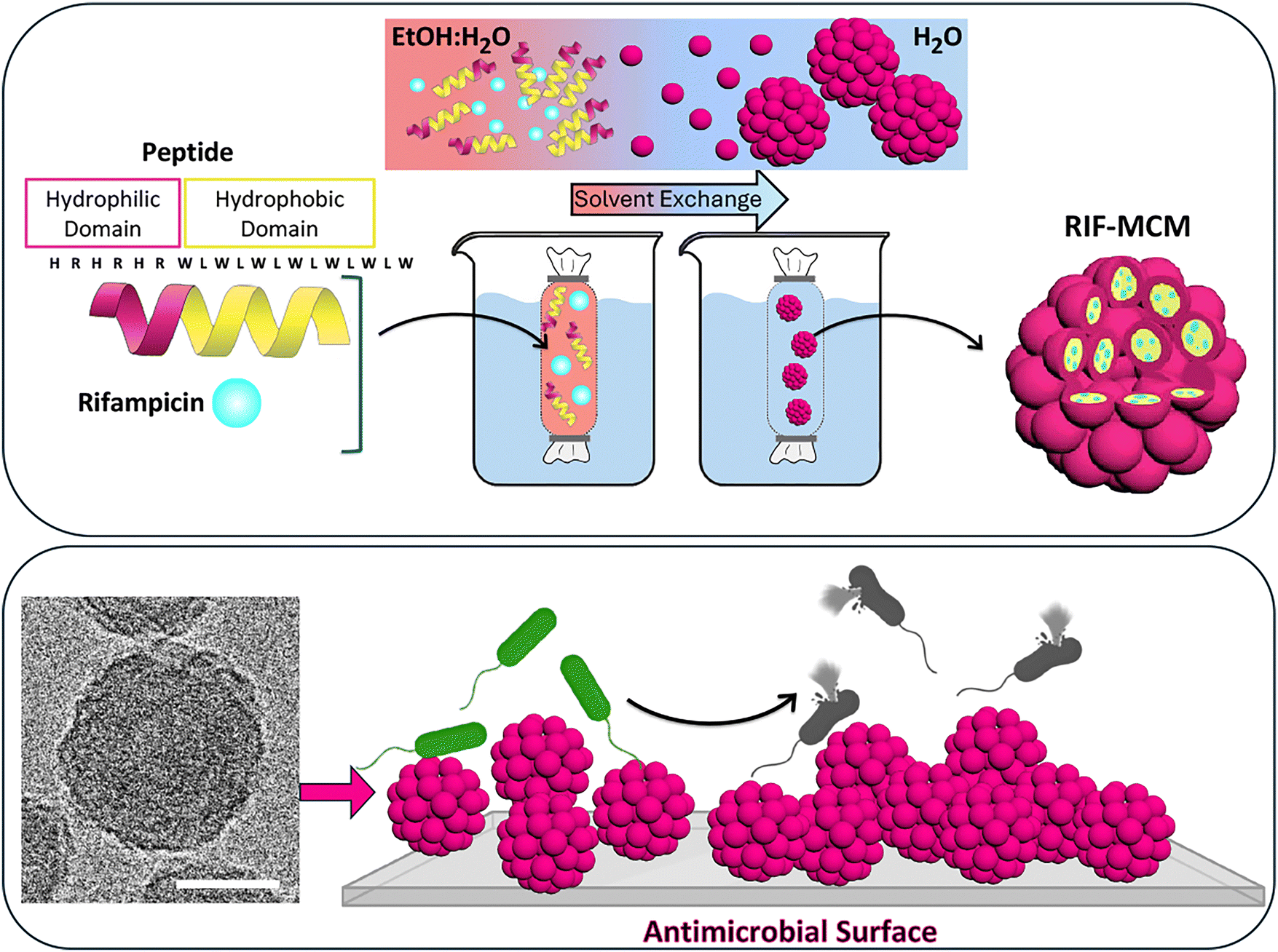 | ||
| Fig. 1 Top panel, schematic representation of RIF-loaded multicompartment micelle (RIF-MCM) assembly from (HR)3(WL)6W peptide and rifampicin (RIF). Bottom panel, cryo-transmission electron micrograph of RIF-MCM (left). Scale bar = 50 nm; (right) Schematic representation of surfaces decorated with these RIF-MCMs to exhibit antimicrobial activity. | ||
As a model antibiotic, we selected a potent bactericidal antibiotic, rifampicin (RIF), known for its strong efficacy against Staphylococcus aureus and Staphylococcus epidermidis, two key pathogens in implant-associated infections.34–36 Rifampicin derived from Amycolatopsis rifamycinica, possesses a complex macrocyclic structure featuring a naphthalene core bridged by an aliphatic chain, a key determinant of its antimicrobial activity. Its broad-spectrum antibacterial efficacy is primarily attributed to its ability to inhibit bacterial DNA-dependent RNA polymerase, thereby suppressing RNA synthesis and leading to bacterial cell death.37 This mechanism is particularly effective against Gram-positive bacteria, which lack an outer membrane, allowing rifampicin to readily diffuse across the cell membrane and reach its intracellular target. In contrast, Gram-negative bacteria feature an outer membrane that acts as a permeability barrier, significantly restricting rifampicin uptake and reducing its effectiveness against them. In addition, rifampicin exhibits potent activity against biofilm-forming bacteria, a critical advantage in mitigating persistent implant-associated infections, where biofilm resistance poses a significant therapeutic challenge.38 However, its direct administration is hindered due to its hydrophobic nature, susceptibility to degradation under environmental conditions (such as exposure to light and pH changes), and potential cytotoxicity at high concentrations.39–41 Therefore, the controlled release of RIF in implant coatings has been explored through encapsulation within polymer- or lipid-based supramolecular assemblies or incorporation into composite materials.42–44 We are advancing this field by entrapping RIF in peptide MCMs and immobilizing them on a solid support, utilizing the unique advantages offered by such assemblies. Our aim was to develop a functional surface decorated with nanoassemblies that facilitate sustained release of RIF, preserving its stability and enhancing its antimicrobial efficacy over time, while minimizing the risk of cytotoxicity.
The self-assembly process of the (HR)3(WL)6W peptide into unique supramolecular architecture of MCMs facilitates the straightforward encapsulation of hydrophobic payloads—here, rifampicin—within the hydrophobic cores of individual micelles. The subsequent formation of MCMs is expected to yield a higher local concentration of the hydrophobic antibiotic by spatially confining it within multiple micelles, resulting in an enhanced entrapment capacity compared to individual micelles. Considering that hydrophobic interactions are the primary driving force behind the entrapment process, we systematically optimized the rifampicin-to-peptide mass ratios to maximize entrapment efficiency while maintaining the structural integrity of the self-assembled MCMs. This process involved a detailed study of physico-chemical properties of the self-assembled MCMs. Once optimized, RIF-MCMs were immobilized on a glass substrate to generate antimicrobial surfaces. Then, we investigated the stability, antimicrobial activity and cell toxicity of surface immobilized RIF-MCMs. These surfaces' ability to combine passive and active antimicrobial effects highlights their potential for developing implant coatings that provide robust infection prevention while minimizing toxicity.
2. Experimental
2.1 Self-assembly of RIF-MCMs
(HR)3(WL)6W peptide synthesis and purification was performed as described elsewhere.32 The self-assembly of rifampicin-loaded and empty MCMs was achieved via a solvent exchange method. Briefly, for the rifampicin (RIF)-loaded nanocarriers, 100 μL of peptide stock solution (1 mg mL−1 in a 50/50 v/v ethanol/water mixture) were mixed at various RIF-to-peptide mass ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 1:2, and 1:1) from a RIF stock solution (2 mg mL−1 in dimethylformamide) for 15 minutes. Each mixture was then adjusted to a final volume of 500 μL with a 35/65 v/v ethanol/water solution and transferred to a prewashed 500–1000 MWCO dialysis tube. For the empty nanocarriers, 100 μL of the peptide stock solution were diluted to 500 μL with the same 35/65 v/v ethanol/water solution and transferred to a dialysis tube. All samples were dialyzed overnight at 4 °C for approximately 20 hours, with two changes of 500 mL Milli-Q water.
:4, 1:2, and 1:1) from a RIF stock solution (2 mg mL−1 in dimethylformamide) for 15 minutes. Each mixture was then adjusted to a final volume of 500 μL with a 35/65 v/v ethanol/water solution and transferred to a prewashed 500–1000 MWCO dialysis tube. For the empty nanocarriers, 100 μL of the peptide stock solution were diluted to 500 μL with the same 35/65 v/v ethanol/water solution and transferred to a dialysis tube. All samples were dialyzed overnight at 4 °C for approximately 20 hours, with two changes of 500 mL Milli-Q water.
2.2 Transmission electron microscopy
Nanocarrier aliquots were placed onto carbon-coated 400-mesh copper grids for 2 minutes to allow adsorption. After blotting the excess liquid, the grids were negatively stained with 2% uranyl acetate for 10 seconds. Following staining, the grids were rinsed with water, dried, and analyzed using a Philips CM100 transmission electron microscope (Philips Healthcare, Netherlands) at an acceleration voltage of 80 kV.2.3 Dynamic light scattering
The hydrodynamic mean diameter (z-average) and polydispersity index (PDI) of the nanocarriers were measured using a Zeta Sizer Nano ZSP (Malvern Instruments Ltd, UK) with a laser wavelength of 633 nm and a detection angle of 173° at 25 °C. All dynamic light scattering (DLS) measurements were conducted in triplicate.2.4 Zeta-potential
The electrophoretic mobility of the nanocarriers in water was analyzed using a Zeta Sizer Nano ZSP (Malvern Instruments Ltd, UK). Measurements were taken at room temperature using a cuvette. The zeta potential values represent the average of three consecutive measurements taken after each polyelectrolyte layer was deposited.2.5 Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was carried out using the NanoSight NS 300 instrument (NanoSight Ltd, UK). Peptide nanocarrier samples were diluted 1:50 in water and introduced into the viewing chamber via a 1 mL syringe. For each measurement, three 60-second videos were recorded at room temperature to track the movement of individual particles frame by frame. The NTA software (version 3.4, NanoSight, UK) was then used to analyze particle motion and calculate the concentration of nanocarriers in the solution.
2.6 Absorption spectroscopy
To determine the entrapment efficiency (EE) for various formulations of RIF within MCM nanocarriers, 200 μL of each sample were mixed with 800 μL of DMSO and then exposed to 4 hours of sonication to release the encapsulated RIF. The absorbance of each sample was then measured in triplicate at 475 nm using a SpectraMax iD3 plate reader (Molecular Devices, US). A calibration curve of free RIF in DMSO was used to determine the RIF concentration in the samples and the EE (%) was calculated using the following formula:
The resulting EE (%) was multiplied by the initial RIF concentration of each sample to calculate the total mass of RIF encapsulated in the nanocarriers. EE values from three samples were averaged to determine the overall amount of RIF entrapped in the MCMs.
2.7 ART-FTIR spectroscopy
Infrared spectra were measured using a Bruker Alpha II IR spectrometer in attenuated total reflection (ATR) mode. ATR-FTIR spectra were recorded between 400 and 4000 cm−1 over 24 scans. Prior to each measurement, the pure diamond surface was recorded as a background over 24 scans. MCMs and RIF-MCMs spectra were measured in solution form, while the RIF spectrum was acquired from Rifampicin in powder form. All spectra were recorded by placing the sample directly on the diamond detector. For MCMs and RIF-MCMs, 3 μL of solution was deposited onto the detector. Specifically, for the RIF-MCMs sample, 40 cycles of solution casting followed by evaporation were performed to concentrate the compound of interest. Data processing was carried out using OPUS software. The water signal was attenuated by subtracting water from the MCMs and RIF-MCMs spectra using the built-in subtraction function of the software. All measurements were conducted at room temperature.2.8 In vitro payload release experiments
To monitor the release behavior of RIF-MCMs, in vitro drug release experiments were conducted at both 37 °C and 42 °C using a dialysis bag setup. In this setup, 5 mL of diluted RIF-MCMs were placed into prewashed dialysis bags (Spectrum Spectra/Por, MWCO 12–14 kDa). Each bag was immersed in 20 mL of HEPES buffer (25 mM HEPES, pH 7.4, containing 150 mM NaCl and 0.5% ascorbic acid) under continuous stirring at the respective temperature. For comparison, free RIF, in amounts equivalent to those encapsulated in 5 mL of RIF-MCMs, was placed into separate dialysis bags and dialyzed under the same conditions. At predetermined intervals, 1 mL of release buffer was collected and replaced with 1 mL of fresh buffer to maintain sink conditions. The absorbance of the released RIF was measured at 475 nm using a SpectraMax iD3 plate reader (Molecular Devices, US).To determine the cumulative release of RIF from RIF-MCMs over time using the dialysis method, first, at each sampling time point ti, the concentration of RIF in the external release buffer (Cti) was measured using spectroscopy technique. Subsequently, amount of RIF at each time point was calculated as:
| RIF amount at ti = Cti × Vs |
Since the removed volume was replaced with fresh buffer, the cumulative amount of RIF released up to time tn was calculated as:

• Qtn is cumulative amount of RIF released up to time tn
• Vext is total volume of the external release buffer (20 mL)
• D is dilution factor
To express the cumulative release as a percentage of the total RIF initially loaded into the RIF-MCMs (Mtotal):

2.9 Quartz-crystal microbalance with dissipation
Quartz-crystal microbalance with dissipation (QCM-D) experiments were performed using a QSense E1 and QSense Analyzer, allowing for up to four simultaneous measurements, with QSoft 401 (version 2.8.5) serving as the control software (Biolin Scientific, Göteborg, Sweden). A four-channel peristaltic pump (Reglo Digital, Ismatec, Glattbrugg, Switzerland) was utilized, connected via Tygon MHSL tubing with two stoppers and PTFE tubing (ID 0.75 mm). During the measurements, resonance frequencies and dissipation of the fundamental harmonic, along with odd-numbered overtones (3rd to 9th), were recorded at room temperature. Prior to experimentation, the QCM-D SiO2 sensors (QSX 303; 5 MHz; Biolin Scientific, Göteborg, Sweden) were sequentially sonicated in baths of ethanol and water for 10 minutes each, followed by drying under a stream of nitrogen. To remove residual impurities and enhance surface hydrophilicity, the sensors were treated with UV-ozone (Jelight Company Inc., Irvine, USA) for 20 minutes. The QCM-D sensors were placed into the QCM-D flow cell and used immediately after activation.The QCM-D technique was employed to study the attachment of RIF-MCMs, unloaded MCMs, and free peptide (HR)3(WL)6W to SiO2-coated sensor surfaces. The free peptide was dissolved in filtered MilliQ water at a concentration of 5 μg mL−1, while the RIF-MCMs and unloaded MCMs were used after self-assembly. The injection sequence for QCM-D measurements was as follows: after calibration, the sensors were equilibrated in filtered Milli-Q water at a flow rate of 100 μL min−1. Then, 501 μL of each sample (RIF-MCMs, unloaded MCMs and free peptide) was injected into separate flow cells at 100 μL min−1. Finally, the sensors were washed with 501 μL of filtered Milli-Q water at the same flow rate to remove any unbound components.
The average frequency shift (ΔF) and dissipation shift (ΔD) values for each step of the protocol were calculated by averaging the data over the final 1 minute of each step. For each sample, replicates measured at the 5th overtone (n = 5) were used, and the relative ΔFsample were calculated as follow:
| ΔFsample = ΔFafterwashing − ΔFbeforedeposition |
The frequency shift, corresponding to the attachment of each sample was then converted into mass per unit area (Δm/A) using the Sauerbrey equation:

Using the calculated Δm/A values, the sensor surface area (diameter = 12 mm), and data from nanoparticle tracking analysis (NTA) for the concentration of nanoparticles per mL, and the initial concentration of peptide used for self-assembly, we calculated the approximate mass of each MCM (mMCM). Finally, the mass per unit area was converted into the number of MCMs per unit area (N/A) using the following equation:
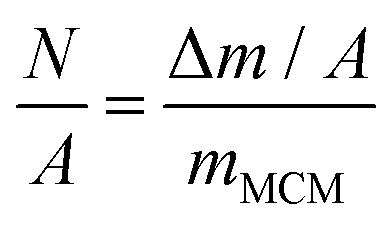
2.10 Atomic force microscopy
Topographical images of the RIF-MCMs immobilized on glass slides were captured using a JPK NanoWizard 3 atomic force microscope (AFM; Bruker, Germany). Prior to imaging, the glass slides underwent a thorough cleaning process using SDS, followed by activation through a 20-minute UV-ozone (UV-O3) treatment to ensure a clean and reactive surface. After treatment, the glass slides were incubated at 4 °C overnight in a solution containing RIF-MCMs. Following incubation, the slides were carefully washed with Milli-Q water to remove any unbound particles and stored at 4 °C in Milli-Q water until imaging. AFM was performed in AC mode using gold-coated silicon nitride cantilevers (DNP-S10 A, with a resonance frequency of 65 kHz and a spring constant of 0.35 N m−1). The measurements were conducted in Milli-Q water at a line rate of 0.5 Hz. Image analysis was carried out using open-source software Gwyddion (version 2.65). In addition, JPKSPM data processing software (version 8.1.29+; JPK Instruments AG) has been used for verification of the roughness values. Both software tools rely on standardized algorithms for roughness calculation, and the consistency between the two supports the reliability of our measurements.2.11 ISO 22196/JIS Z 2801 based antimicrobial activity assays
The antimicrobial activity of the MCM-modified surfaces was evaluated according to with ISO 22196/JIS Z 280145,46 with adjustments for smaller sample sizes. The ISO norm specifies an inoculum of 400 μL on 4 × 4 cm samples, whereas we adapted it to 125 μL for 2 × 2 cm samples to maintain a consistent inoculum-to-surface ratio. In brief, Staphylococcus aureus DSM 2569 was cultured overnight in LB medium at 37 °C, and an inoculum of 106 CFU per ml was prepared in 0.2% LB medium with 145 mM NaCl (OD600 = 1 correspond to 109 CFU per mL). Then, aliquots of 125 μL were pipetted onto MCM-modified and unmodified glass slides (20 × 20 mm) and covered with parafilm for incubation at 37 °C for 24 h. To maintain humidity, Petri dishes were lined with pre-moistened paper towels and sealed with parafilm (approximately 4 × 3 cm). After incubation, the surviving bacteria was quantified using two complementary methods:Resazurin-based metabolic assay: samples with inoculum were incubated in 3 mL of 5% LB medium containing 0.015 mg mL−1 resazurin at 37 °C under 90 rpm agitation for approximately 5 h. The resorufin fluorescence produced by metabolically active bacteria was quantified by fluorimeter (excitation 536 nm, emission 588 nm) with untreated glass serving as the 100% viability control. This method captures signals from both planktonic and surface-adherent bacteria, providing a comprehensive activity assessment.
(ii) Plate counting: bacteria in the inoculum and on the sample surface were manually rubbed off the surface in 5 mL of 5% LB medium with145 mM NaCl, followed by serial dilution, plating on agar plates, incubation at 37 °C for 24 h and counting colony formation. In this method, unlike the resazurin assay, oxidizing or reducing molecules in the sample or inoculum will not interfere with the assay providing a robust and complementary approach for quantifying bacterial viability.
2.12 MTS cell viability assay
The cytotoxicity of RIF-MCMs towards A549 cancer cells was assessed using the tetrazolium compound-based Cell Titer 96® AQueous One Solution Cell Proliferation assay (MTS; Promega). MTS assays were performed according to the manufacturer's protocol, with measurements taken after 24 and 48 h of treatment with peptide MCMs at different concentrations. In brief, A549 cells were seeded in a 96-well plate at 3 × 103 cells per well in 100 μL RPMI medium and cultured for 24 h at 37 °C in a 5% CO2. Subsequently, the cells were treated with RIF-MCMs, unloaded MCMs and free RIF under three treatment dosages (Dose I, Dose II, Dose III). Dose I corresponded to the concentration of RIF-MCMs required to achieve full surface coverage when developing antibacterial surfaces, Dose II was double, and Dose III was triple this concentration. The dosages for unloaded MCMs corresponded to the peptide MCM concentration in corresponding RIF-MCM samples, while for free RIF, doses were equivalent to the entrapped RIF in corresponding RIF-MCM samples. Control cells were incubated correspondingly but without treatment as a reference value for 100% viability. All experiments were performed in triplicate wells for each condition. After 24 and 48 h of incubation at 37 °C, 20 μL of MTS solution were added to each well, and the plate incubated for an additional 2 h at 37 °C. The absorbance of the plate was then read at λ = 490 nm using a Spectramax plate reader (Molecular Devices LLC, USA). Background absorbance from wells without cells was subtracted from all test wells. The viability in treated wells was determined by normalizing the absorbance to that of untreated control cells.3. Results and discussion
3.1 Antibiotic-loaded peptide MCMs
To obtain antibiotic-loaded peptide nanoassemblies, we used (HR)3(WL)6W, an amphiphilic peptide, which forms multi-compartment micellar (MCM) nanostructures upon self-assembly via the solvent-exchange method.32 To optimize the conditions for the self-assembly of MCMs with high RIF loading, we investigated different mass ratios of RIF to (HR)3(WL)6W peptide. Specifically, we selected 1:4, 1:2, and 1:1 RIF to peptide to assess the impact of varying drug loads on the self-assembly process, the encapsulation efficiency and the properties of the resulting supramolecular assemblies. We were interested to identify the optimal balance between maximizing RIF loading and preserving the structural integrity of the supramolecular assemblies.
Transmission electron microscopy (TEM) analysis of negatively stained samples prepared at different RIF to peptide ratios, revealed a spherical morphology in all cases with indications of a multimicellar architecture (Fig. 2, top). This architecture was further supported by cryo-TEM images of unstained RIF-MCMs (Fig. 1 and Fig. S1A, ESI†). Dynamic light scattering (DLS) and zeta potential measurements were used to characterize the size and colloidal stability of the resulting MCMs (Fig. 2, bottom, distribution profiles in Fig. S2, ESI†). Depending on the mass ratio of RIF to peptide (Pep), the hydrodynamic diameter (DH) of loaded MCMs varied slightly. At 1:4 RIF:Pep, the average DH of MCMs was 105 ± 2 nm with a polydispersity index (PDI) of 0.17. At 1:2 RIF:Pep, the DH slightly increased to 113 ± 1 nm, while the PDI remained similar at 0.16. With further relative increase of RIF (1:1 RIF:Pep), the DH value increased to 117 ± 4 nm with a PDI of 0.19. MCMs assembled in the absence of RIF exhibited a DH of 101 ± 5 nm with a PDI of 0.17 (Fig. S1C, ESI†). These results suggest that while the resulting MCMs slightly increase in size as the mass ratio of RIF to peptide increases, the self-assembly into MCMs is not perturbed and did not lead to aggregation under the conditions tested. The zeta potential remained positive and relatively high for all RIF-MCMs, indicating stable colloidal systems. Specifically, the RIF-MCMs exhibited a zeta potential of +27 ± 2 mV at the 1:4, +22 ± 3 mV at the 1:2, and +24 ± 2 mV at the 1:1 RIF:Pep (Fig. 2, bottom). The zeta potential of MCMs assembled in the absence of RIF was +29 ± 3 mV (Fig. S1D, ESI†).
 | ||
| Fig. 2 Physico-chemical characterization of peptide MCMs formed at different mass ratios of rifampicin to peptide. Upper panels, TEM micrographs of supramolecular assemblies containing RIF, scale bars = 200 nm. Lower panels: (left) Hydrodynamic diameter (DH) and polydispersity index (PDI); (right) zeta potential and RIF entrapment efficiency (EE) of RIF-MCMs. | ||
To investigate the RIF entrapment efficiency in peptide MCMs, we established a standard curve of free RIF in dimethyl sulfoxide (DMSO) (Fig. S3, ESI†) and measured nanoparticle concentration using nanoparticle tracking analysis (Table S1, ESI†). RIF was successfully integrated into the MCM architecture in all assemblies, driven by hydrophobic interactions between RIF and the micelle cores. The entrapment efficiency of RIF-MCMs varied depending on the mass ratio of RIF to peptide. At 1:4 RIF:Pep, the entrapment efficiency was 46% (Fig. 2, bottom). Increasing the RIF:Pep ratio to 1:2 resulted in an entrapment efficiency of 84%. However, at 1:1 RIF:Pep, the entrapment efficiency decreased to 67%. This decrease can be attributed to the saturation of entrapment capacity at the micelle cores and/or the destabilization of the MCM structure at higher RIF concentrations. Notably, NTA showed a decrease in RIF-MCM concentration with increasing RIF:Pep ratios, supporting the notion that a higher RIF content reduces the overall MCM assembly yield (Table S1, ESI†). Based on entrapment efficiency results, the optimal balance between RIF and the peptide for obtaining MCMs with maximum RIF load is 1:2. This data highlights the importance of optimizing the drug-to-carrier ratio to maximize drug loading without compromising the formation or integrity of the nanoassemblies. In addition, the interaction between RIF and MCMs was investigated by ATR-FTIR spectroscopy. The spectra of RIF, MCMs, and RIF-MCMs (1:2 RIF:Pep) (Fig. S4, ESI†) demonstrated characteristic peak differences, confirming RIF incorporation into the micellar structure. A slight shift in peak positions around 3000 cm−1 and 1250 cm−1 in the RIF-MCMs spectra compared to MCMs indicates interactions between RIF and MCMs. Additionally, the presence of RIF in RIF-MCMs is supported by the peak at ∼1250 cm−1, which appears with lower intensity compared to pure RIF but is absent in MCMs.
We previously reported that the MCMs, which maintain their multimicellar architecture over months at 4 °C, disassemble into smaller MCMs and/or individual micelles over time at 37 °C.32 Therefore, we exploited this temperature responsiveness to examine the release of RIF from MCMs in solution by a cumulative release study47 at 37 °C and 42 °C using the dialysis tube method (Fig. 3A). For each temperature, RIF-MCMs were placed in a dialysis tube, immersed in 20 mL of HEPES buffer (25 mM HEPES, pH 7.4, containing 150 mM NaCl and 0.5% ascorbic acid) and RIF release was monitored over time by measuring absorbance at 475 nm. A standard curve of free RIF in buffer was used to quantify the release (Fig. S5, ESI†). RIF-MCMs assembled at 1:2 and 1:4 RIF:Pep, showed a sustained release profile, with 100% cumulative release achieved in 9 h (Fig. 3A). At 1:1 RIF:Pep, complete RIF release took 12 h. In contrast, 90% of free RIF was released from the dialysis tube within the first 2 h at 37 °C.
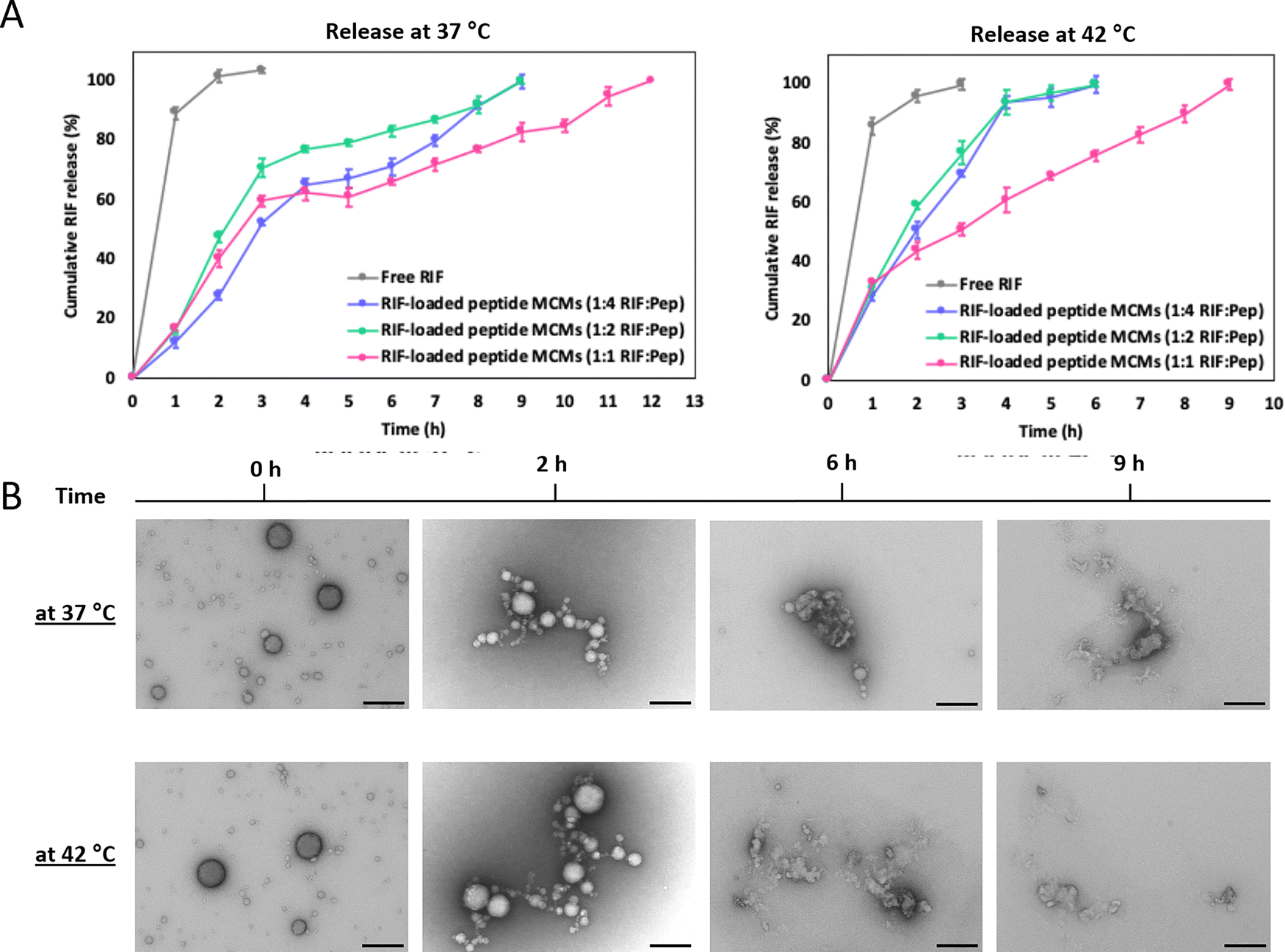 | ||
| Fig. 3 Release of RIF from RIF-MCMs. (A) Cumulative release of RIF at 37 °C (left panel) and 42 °C (right panel) from RIF-MCMs formed at 1:4, 1:2 and 1:1 RIF:Pep. (B) TEM micrographs of RIF-MCMs (1:2 RIF:Pep) after 0, 2, 6 and 9 h incubation at 37 °C and 42 °C. Scale bars = 200 nm. | ||
At 42 °C, RIF release occurred more rapidly due to the accelerated dissociation of MCMs at the elevated temperature (Fig. 3A). The 1:2 and 1:4 formulations reached 100% release in 6 h, while the 1:1 formulation took 9 h. Consistent with diffusion-driven release, free RIF displayed a fast release behavior similar to that at 37 °C. The temperature-dependent release profile of RIF-MCMs, with faster release observed at 42 °C compared to 37 °C, is attributed to the increased kinetic energy and diffusion rates at the higher temperature, which facilitate the dissociation of the MCMs and the subsequent release of RIF. We further monitored the change in the architecture of MCMs during the release of RIF by TEM analysis of RIF-MCMs after 2, 6, and 9 h at both, 37 °C and 42 °C (Fig. 3B). Ultrastructural analysis confirmed that the disintegration of RIF-MCMs occurred more rapidly at the higher temperature. At 42 °C, the assemblies completely disintegrated within 6 hours, whereas at 37 °C, slower disassembly was observed, with complete breakdown occurring by 9 hours (Fig. 3B). These observations are consistent with the cumulative release studies. The temperature-induced release behavior demonstrates that RIF-MCMs are capable of maintaining sustained RIF levels over an extended period of time. The faster release at 42 °C highlights the potential for a temperature-regulated release in certain applications.
3.2 Immobilization of RIF-MCMs on solid support
After the systematic investigation into the impact of various RIF-to-peptide mass ratios on encapsulation efficiency and physicochemical properties, the 1:2 ratio was identified as the optimal formulation for developing an antimicrobial surface. Because the chemistry of silicon oxide (SiO2) solid supports, such as those used in quartz crystal microbalance with dissipation (QCM-D) sensors, closely resembles that of glass substrates, we interchangeably used them depending on the corresponding experimental requirements. We considered glass and silicon oxide (SiO2) surfaces to be suitable models for the initial development of an antimicrobial surface. Specifically, both supports share a silica-based composition and the capacity to form hydroxyl groups upon activation through processes such as UV-O3 treatment. Besides introducing hydroxyl functional groups that enhance the surface reactivity,48,49 UV-O3 treatment efficiently cleans the substrate surfaces by removing organic contaminants.50 The resulting clean, hydrophilic, negatively charged, and highly functionalized surfaces51,52 are particularly suited for the immobilization of our positively charged RIF-MCMs. Moreover, the amine functionalities of histidine groups exposed on the surface of MCMs facilitate strong interactions with the activated silica or glass substrates. Besides, glass is a useful preliminary model for coatings in medical device research due to its uniformity and optical properties.53
To investigate the attachment dynamics of RIF-MCMs and unloaded MCMs on solid supports, we employed quartz crystal microbalance with dissipation monitoring (QCM-D) using silicon oxide-coated sensors. First, the frequency signal of bare silica substrates mounted in parallel flow cells was stabilized in water to establish a baseline. Subsequently, RIF-MCMs and unloaded MCMs in aqueous solution were introduced into separate flow cells, and real-time changes in frequency (ΔF) and dissipation (ΔD) were recorded to evaluate attachment behavior and immobilization efficiency over time (Fig. 4A). Both, RIF-MCMs and unloaded MCMs resulted in frequency decreases relative to the bare substrate, indicating a mass increase on the sensor surface due to successful immobilization. Once the signal had stabilized, the system was flushed with water (for 40 minutes under continuous flow of 100 μL min−1) to remove loosely adhered MCMs and to assess the stability of the immobilized layers. The immobilized RIF-MCMs and unloaded MCMs remained stably attached to the substrate, with only a slight frequency decrease observed during the washing step. This slight decrease suggests further attachment of MCMs to the surface under flow conditions. After the washing step, the frequency shift measured for RIF-MCMs was −105 Hz, while unloaded MCMs exhibited a shift of −67 Hz, consistent with the higher mass of RIF-MCMs due to the encapsulated rifampicin (Fig. 4B). The frequency shifts were converted into mass per unit area (Δm/A) using the Sauerbrey equation (see Method section). The calculated mass per unit area (Δm/A) was 372 ± 18 ng cm−2 for RIF-MCMs and 237 ± 14 ng cm−2 for unloaded MCMs (Fig. 4C). The difference in mass, 134 ± 10 ng cm−2, corresponds to the amount of rifampicin encapsulated within the RIF-MCMs, assuming comparable attachment efficiencies for both MCM types. To further investigate the surface coverage of MCMs, the nanoparticle density (N/A) was calculated using NTA data and the known mass of the peptide used for self-assembly (see Method section). Based on these parameters, we derived a surface density of (2.8 ± 0.2) × 107 nanoparticles per cm2 for RIF-MCMs and (2.6 ± 0.3) × 107 nanoparticles per cm2 for unloaded MCMs, reflecting a similar attachment density for both MCM types (Fig. 4C). Additionally, similar experiments were performed for free peptide adsorption onto the SiO2 substrate (Fig. S6, ESI†). As expected, the free peptide exhibited a smaller frequency decrease compared to MCMs due to its lower mass. Notably, like the MCMs, the free peptide remained stably attached to the substrate after the washing procedure, further demonstrating the robust interaction between the peptide and the SiO2 surface (Fig. S6, ESI†).
 | ||
| Fig. 4 QCM-D monitoring of MCM binding to SiO2 sensors. (A) QCM-D sensograms showing frequency shifts (ΔF) during the deposition of RIF-MCMs (top) and unloaded MCMs (bottom), followed by a water wash at room temperature. Dashed lines with arrows indicate corresponding injections. Frequency shifts were recorded for the 3rd, 5th, 7th, and 9th harmonics. (B) Average ΔF before and after the washing step, reflecting the binding of RIF-MCMs and unloaded MCMs to the solid support. (C) Nanoparticle density (N/A) and mass per area (Δm/A) of MCMs after the washing step, calculated using the Sauerbrey equation. | ||
As we aim to maximize the antibacterial performance of our RIF-MCM-based surface through dual functionality (passive and active), we also examined the spatial distribution and topography of RIF-MCMs and unloaded MCMs incubated overnight on UV-O3-treated glass substrates. After carefully rinsing off unbound MCMs, we analyzed the modified surface topography using atomic force microscopy (AFM) in AC mode. The untreated glass surface was studied as a reference (Fig. 5A and Fig. S7A(i–iii), ESI†). AFM images confirmed the successful surface immobilization of both unloaded MCMs and RIF-MCMs (Fig. 5B, C and Fig. S7B(i–iii)C(i–iii), ESI†). The immobilization results from a combination of electrostatic interactions between the negatively charged glass substrate and the positively charged peptide-modified MCMs, as well as hydrogen bonding between the hydroxyl groups on the glass surface and the amine functionalities present on the surface of the peptide MCMs.
 | ||
| Fig. 5 AFM topography of RIF-MCM modified surfaces. AFM topography image of (A) bare glass substrate, (B) unloaded MCMs and (C) RIF-MCMs immobilized on a glass surface. (D) AFM height image at higher magnification of a patch of immobilized RIF-MCMs. The inset shows the height profile measured along the dashed line. (E) The corresponding height distribution histogram of RIF-MCMs immobilized on a glass surface shown in (C), where ρ represents the normalized density of height values. The inset provides a magnified view of the histogram, highlighting the height data specific to the surface-immobilized MCM population. (F) Roughness data (Ra and Rq) of the glass substrate, and unloaded MCMs and RIF-MCMs immobilized on a glass surface. For each sample, three randomly selected spots were analyzed by AFM, and the corresponding roughness values were obtained with Gwyddion software. The analyzed areas are shown in Fig. S6 (ESI†). | ||
AFM topography micrographs revealed that RIF-MCMs and unloaded MCMs were heterogeneously distributed on the surface (Fig. 5B and C). In some regions, several MCMs formed compact patches by attaching next to or on top of one another, while other regions showed sparsely immobilized, single MCMs. High-resolution AFM images (Fig. 5D) demonstrated that native RIF-MCMs were composed of individual micelles, providing evidence that the multimicellar architecture of the RIF-MCMs observed by cryo-TEM remained intact after attachment to the solid support.
Height distribution analysis of surfaces with immobilized RIF-MCMs revealed two distinct peaks: one at 17 nm corresponding to the base glass substrate, and another at 90 nm corresponding to the RIF-MCMs (Fig. 5E, inset). Such nanoscale topographical features are expected to synergize with the active antimicrobial functionality of RIF-MCMs and enhance the overall antibacterial performance of the surface. Surface roughness analysis further showed that RIF-MCM attachment resulted in a moderately rough surface. The mean roughness (Ra) and root mean square roughness (Rq) values measured using Gwyddion for the glass substrate were 312.8 pm and 397.5 pm, respectively. Upon immobilization of unloaded MCMs, these values increased to 16.3 nm (Ra) and 23.4 nm (Rq), and further increased to 19.9 nm and 27.7 nm, respectively, upon immobilization of RIF-MCMs (Fig. 5F). Ra and Rq values for each condition were obtained by analyzing three different areas per sample (Fig. S7, ESI†), and the mean values were calculated from three independently prepared samples. The slight difference in roughness between unloaded MCMs and RIF-MCMs is likely due to their size difference, as previously demonstrated by the DH distribution profiles (Fig. S1 and S2, ESI†). The enhanced roughness of the substrate resulting from RIF-MCM immobilization is particularly advantageous in passive antibacterial strategies, as it increases the surface area and may hinder bacterial adhesion or create physical barriers.54
To assess the stability of surface-immobilized MCMs over time, we performed the self-assembly of peptides in the presence of hydrophobic fluorescein isothiocyanate (FITC) to produce fluorescent MCMs (FITC-MCMs) for visualization using confocal laser scanning microscopy (CLSM). Untreated surfaces and surfaces with immobilized FITC-MCMs were imaged immediately following preparation to quantify initial fluorescence intensity (Fig. 6, Day 1). After a 10-day incubation at 4 °C in water in the dark, both surfaces were again imaged and their fluorescence intensity was measured (Fig. 6, Day 10). The mean fluorescence intensity did not change significantly over time was observed, suggesting that in aqueous environments at 4 °C, the MCMs remained stably attached to the surfaces and preserved their architecture.
 | ||
| Fig. 6 Stability of surface-immobilized MCMs. CLSM images recorded on day 1 and day 10 of a bare surface (left panels) and a surface with immobilized FITC-MCMs (right panels). Scale bars = 20 μm. The mean fluorescence intensity of a 200 × 200 μm area was recorded on day 1 and compared to a corresponding area on day 10 (far right). Two-way ANOVA, followed by Tukey's post hoc test was used for comparison of mean fluorescence intensity measured on surfaces with immobilized FITC-MCMs. n.s. (not significant), * (p < 0.05), ** (p < 0.005), *** (p < 0.0005). | ||
3.3 Antibacterial properties of surfaces with immobilized RIF-MCMs
Initially, we evaluated the antibacterial effect of RIF-MCM decorated surfaces using CLSM with live-dead staining of Bacillus subtilis, as the efficacy of RIF against this strain is well established.55 While bacterial viability was significantly reduced by surface-adsorbed particles and, to a lesser extent, by MCM-decorated surfaces (Fig. S8, ESI†), CLSM served only for a qualitative screening due to its limitations. These include potential imaging bias, limited visualization of bacterial populations across large surfaces, and challenges in obtaining quantitative data.To further assess the antimicrobial potency of RIF-MCM decorated surfaces, we evaluated their ability to inhibit bacterial growth using resazurin reduction and colony count assays. Resazurin conversion to fluorescent resorufin serves as an indicator of bacterial metabolic activity and is widely used for antimicrobial testing.56 To analyze bacterial adhesion and proliferation, Staphylococcus aureus (S. aureus), another Gram-positive bacterium, was directly applied to RIF-MCM-modified, MCM-modified or unmodified surfaces and incubated for 24 h at 37 °C. Bacterial viability was quantified by measuring resorufin production at λ = 588 nm, in response to bacterial metabolic activity in samples collected from the different surfaces (Fig. 7A). In comparison to unmodified glass surfaces (control), incubation of S. aureus on RIF-MCM surfaces led to a 98 ± 0.3% reduction in viable bacteria. S. aureus is known forms biofilms that hinder antibiotic penetration and enable bacterial persistence, necessitating prolonged antimicrobial activity at infection sites.57 RIF exerts its bactericidal activity by binding to the β-subunit of bacterial DNA-dependent RNA polymerase (RNAP), inhibiting RNA synthesis and preventing bacterial replication.58 This transcriptional disruption, combined with the need for prolonged antimicrobial activity, has driven extensive research toward enhancing RIF's clinical efficacy through controlled-release formulations, such as poly(lactic-co-glycolic acid) (PLGA) microspheres59,60 and mesoporous silica nanoparticles.61–63 The RIF-MCM surface, with its high local RIF concentration and prolonged exposure due to sustained release profile, effectively prevented bacterial adhesion and proliferation, making it particularly suitable for implant-associated and prosthetic joint infections. In contrast, bacterial survival on surfaces coated with MCMs lacking rifampicin (102 ± 17% viability) was comparable to the survival on the control surfaces. Although the (HR)3(WL)6 peptide itself features some characteristics typically associated with antimicrobial peptides, i.e., hydrophobic tryptophane-rich regions combined with cationic regions,64,65 it is noteworthy that after assembly, unloaded MCMs showed no antimicrobial activity under the conditions tested. However, this finding does not entirely rule out any antimicrobial potential of the MCMs themselves, as the ratio of MCMs to bacteria, the disassembly state of the MCMs or their interactions with medium components could affect their ability to kill bacteria.
 | ||
| Fig. 7 Antimicrobial activity of MCM-modified surfaces against S. aureus after 24 hours. (A) Resazurin metabolic assay reporting S. aureus viability as a percentage relative to viability on unmodified surfaces. (B) Antibacterial activity assessed by estimating live bacteria based on counting colonies on agar plates. Colony count was determined following sampling of bacteria containing solutions from the corresponding surfaces, serial dilution, and plating. All surfaces were carefully rinsed prior to adding S. aureus and running antimicrobial activity assays, and tested in quadruplicate. | ||
In addition to the resazurin assay, a plate count assay was performed to quantify bacterial colonies in accordance with ISO 22196 standards.45 RIF-MCM modified surfaces showed strong antimicrobial activity, with colony counts reduced by over three orders of magnitude, reaching only 187 colonies after 24 h, compared to 962500 for untreated surfaces (Fig. 7B and Fig. S9, ESI†). The large reduction of colonies is in good agreement with data obtained by the resazurin assay. Interestingly, in colony count experiments where bacteria for plating were collected from the medium above the glass surface, MCM-modified surfaces led to a reduction in colony counts, with less than half (418333 colonies) the number of colonies observed on plates grown from samples collected from unmodified glass surfaces (Fig. 7B and Fig. S9, ESI†). This reduction in colony formation is attributed to the enhanced surface roughness resulting from the heterogeneous surface modification by the MCMs, which hinders bacterial adhesion and biofilm formation via passive antibacterial mechanisms. Further support for a passive mechanism is provided by the fact that at 37 °C, MCMs tend to dissociate, resulting in changes to the surface topography, which in turn limit bacterial attachment and growth. Thus, while the resazurin assay did not show a direct reduction in bacterial viability attached to surface, the colony count from the medium above the glass surface indicates that the surface roughness and structural features of both RIF-MCMs and unloaded MCMs play a role in reducing bacteria.
3.4 Biocompatibility of surfaces with high antibacterial activity
To determine whether our active surfaces with high antibacterial efficacy are safe for human cells, we evaluated the cytotoxicity of the MCMs used for surface modification toward eukaryotic cells by an MTS assay. A549 lung cancer cells were exposed to various concentrations of RIF-MCMs (Dose I, Dose II, Dose III), while unloaded MCMs and free RIF were investigated at equivalent concentrations for comparison (Fig. 8). Dose I corresponded to the RIF-MCM concentration needed for full surface coverage, Dose II was double this concentration, and Dose III was triple. In the case of unloaded MCMs, the peptide concentration was matched to that of RIF-MCMs, while the free RIF dosages used to treat A549 cells were equivalent to the amount of RIF entrapped in RIF-MCMs. Control cells were incubated under identical conditions but without treatment. After 24 h of treatment, neither MCMs nor RIF-MCMs exhibited significant cytotoxicity at any of the concentrations tested (Fig. 8). In contrast, free RIF resulted in a notable reduction in cell viability, with values of 86 ± 5%, 77 ± 6%, and 61 ± 1.5% observed for dose I, dose II, and dose III, respectively (Fig. 8). These findings demonstrate that peptide MCMs are not cytotoxic, whereas free rifampicin exerts toxicity, particularly at higher doses. The encapsulation of RIF within MCMs, followed by its sustained release proved advantageous in shielding cells from the direct exposure to high-dose of RIF, thereby reducing toxic effects and enhancing biocompatibility. A similar trend was observed after 48 h of exposure, with slightly more pronounced cytotoxic effects at higher dosages (Fig. S10, ESI†). This indicates that the use of peptide MCMs for antibiotic delivery can reduce undesired cytotoxicity towards human cells while preserving antimicrobial efficacy.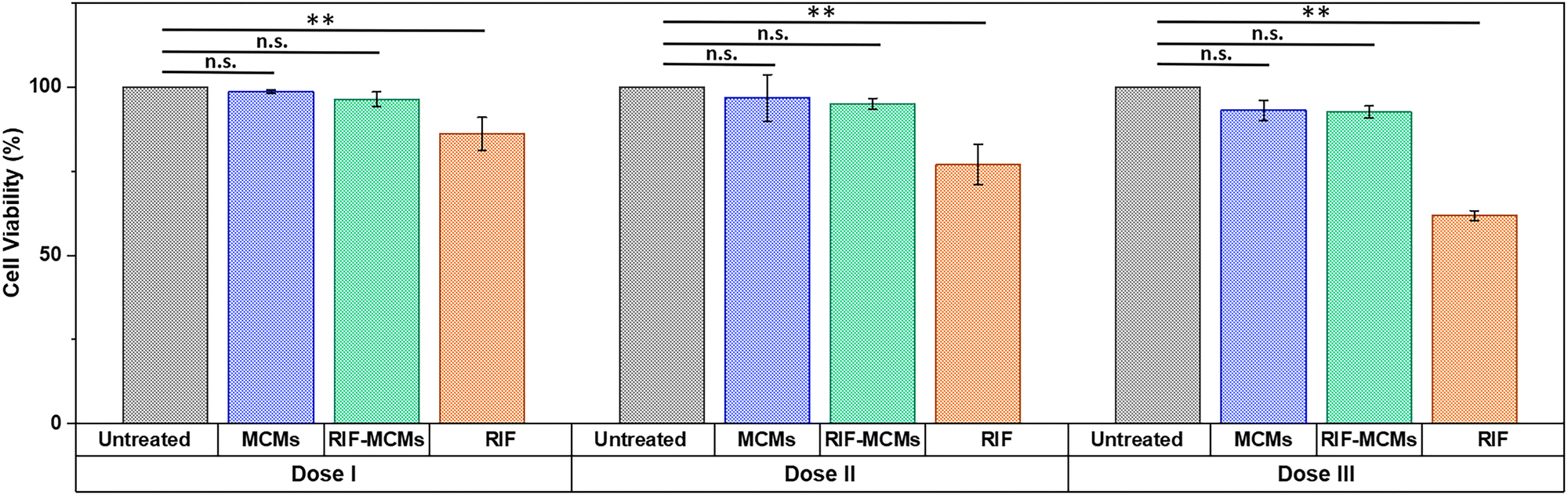 | ||
| Fig. 8 Effect of MCMs and RIF on human cell viability. The proliferation of A549 cells after 24 h of treatment with Dose I, Dose II, and Dose III at 37 °C was assessed using MTS assays. For RIF-MCMs, dose I corresponds to the concentration applied for full surface coverage with RIF-MCMs, dose II is double this concentration, and dose III triple. For unloaded MCMs, doses I to III have peptide concentrations equivalent to those used for RIF-MCM treatment. Doses I, II and III of free RIF correspond to the amount of RIF entrapped in the respective doses of RIF-MCMs. Control cells were incubated correspondingly but without treatment. Error bars represent standard deviation of triplicate measurements. Statistical comparisons were conducted using two-way ANOVA followed by Tukey's HSD (honestly significant difference); p > 0.05 (n.s., not significant), p < 0.05 (*), and p ≤ 0.01(**). | ||
4. Conclusion
This study presents a novel approach using surface-immobilized rifampicin-loaded multicompartment micelles (RIF-MCMs), combining sustained antibiotic release (active) with contact-based bactericidal activity (passive). Following the systematic optimization of self-assembled RIF-MCMs, their immobilization on surfaces resulted in prolonged antimicrobial effects, crucial for preventing bacterial colonization and biofilm formation. These are key challenges in infections associated with medical implants, where the synergy between sustained antibiotic release and passive bactericidal activity is essential to enhance localized therapeutic outcomes. Our in vitro antimicrobial assays demonstrate that RIF-MCM-modified surfaces reduce bacterial viability by over 98% and decrease colony counts by over three orders of magnitude. Furthermore, the surfaces exhibit minimal toxicity to human cells, indicating their biocompatibility and suitability for clinical applications. This dual-functionality approach represents a step forward in overcoming the limitations of conventional antibiotics in clinical practice, aligning with current trends in nanomedicine to improve antimicrobial therapies. By incorporating these nanoparticles into medical device coatings, this strategy reduces the need for systemic antibiotics, potentially decreasing the risk of antibiotic resistance. To validate their broad potential in healthcare, future studies will focus on evaluating the clinical efficacy of these surfaces in more complex infection models, exploring different solid supports for coating and expanding the range of bacterial targets.Author contributions
S. T. and C. G. P. performed conceptualization. S. T., S. N. A., J. K. M. B. and C.-A. S. performed the methodology. S. T., S. N. A., J. K. performed the investigation. S. T., S. N. A., M. B. and C.-A. S. performed visualization. C. G. P. performed funding acquisition. S. T. and M. B. wrote the original manuscript, S. T., N. A., C.-A. S. and C. G. P. reviewed and edited the manuscript.Data availability
The data that support the findings of this study are openly available in Zenodo at https://doi.org/10.5281/zenodo.14710264.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The authors gratefully acknowledge financial support from the University of Basel, the National Centre of Competence in Research Molecular Systems Engineering (NCCR-MSE, grant no. 51NF-40-205608), and the Swiss National Science Foundation (SNSF, grant no. 207383). The authors thank Susanne Erpel (University of Basel) for TEM measurements, and Dr Mohamed Chami and Carola Alampi (Biozentrum) for cryo-TEM. The authors gratefully acknowledge Dr Mirela Malekovic (University of Basel) for her support in sample preparation for cryo-TEM and FTIR.References
- L. Kadirvelu, S. S. Sivaramalingam, D. Jothivel, D. D. Chithiraiselvan, D. Karaiyagowder Govindarajan and K. Kandaswamy, Curr. Res. Microb. Sci., 2024, 6, 100231 CAS
.
- F. Jahanmard, M. Croes, M. Castilho, A. Majed, M. J. Steenbergen, K. Lietaert, H. C. Vogely, B. C. H. van der Wal, D. A. C. Stapels, J. Malda, T. Vermonden and S. Amin Yavari, J. Controlled Release, 2020, 326, 38–52 CrossRef CAS PubMed
- M. Villegas, F. Bayat, T. Kramer, E. Schwarz, D. Wilson, Z. Hosseinidoust and T. F. Didar, Small, 2024, 20, 2404351 CrossRef CAS PubMed
- S. Zhang, H. Yang, M. Wang, D. Mantovani, K. Yang, F. Witte, L. Tan, B. Yue and X. Qu, The Innovation, 2023, 4, 100503 CrossRef CAS PubMed
- A. Uneputty, A. Dávila-Lezama, D. Garibo, A. Oknianska, N. Bogdanchikova, J. F. Hernández-Sánchez and A. Susarrey-Arce, Colloid Interface Sci. Commun., 2022, 46, 100560 CrossRef CAS
- H. Cao, S. Qiao, H. Qin and K. D. Jandt, J. Funct. Biomater., 2022, 13, 86 CrossRef CAS PubMed
- S. Akay and A. Yaghmur, Molecules, 2024, 29, 1172 CrossRef CAS PubMed
- V. M. Vijayan, M. Walker, J. J. Morris and V. Thomas, Curr. Opin. Biomed. Eng., 2022, 22, 100377 CrossRef CAS
- S. M. Imani, L. Ladouceur, T. Marshall, R. Maclachlan, L. Soleymani and T. F. Didar, ACS Nano, 2020, 14, 12341–12369 CrossRef CAS PubMed
- M. F. Kunrath, G. Rubensam, F. V. F. Rodrigues, D. R. Marinowic, P. Sesterheim, S. D. de Oliveira, E. R. Teixeira and R. Hubler, Colloids Surf., B, 2023, 228, 113417 CrossRef CAS PubMed
- T. Wei, Y. Qu, Y. Zou, Y. Zhang and Q. Yu, Curr. Opin. Chem. Eng., 2021, 34, 100727 CrossRef
- M. Cloutier, D. Mantovani and F. Rosei, Trends Biotechnol., 2015, 33, 637–652 CrossRef CAS PubMed
- Z. Li, X. Yang, H. Liu, X. Yang, Y. Shan, X. Xu, S. Shang and Z. Song, Chem. Eng. J., 2019, 374, 564–575 CrossRef CAS
- S. Rigo, C. Cai, G. Gunkel-Grabole, L. Maurizi, X. Zhang, J. Xu and C. G. Palivan, Adv. Sci., 2018, 5, 1700892 CrossRef PubMed
- Y. Huang, X. Guo, Y. Wu, X. Chen, L. Feng, N. Xie and G. Shen, Signal Transduction Targeted Ther., 2024, 9, 1–50 CrossRef PubMed
- X. Bai, J. Yu, J. Xiao, Y. Wang, Z. Li and H. Wang, Front. Bioeng. Biotechnol., 2023, 10, 1016001 CrossRef PubMed
- J. Sahoo, S. Sarkhel, N. Mukherjee and A. Jaiswal, ACS Omega, 2022, 7, 45962–45980 CrossRef CAS PubMed
- M. Bina, J. P. Coats, M. Skowicki, M. Malekovic, V. Mihali and C. G. Palivan, Langmuir, 2024, 40, 23178–23188 CrossRef CAS PubMed
- J. He, M. Hong, W. Xie, Z. Chen, D. Chen and S. Xie, J. Controlled Release, 2022, 351, 301–323 CrossRef CAS PubMed
- O. M. Eggenberger, P. Jaśko, S. Tarvirdipour, C.-A. Schoenenberger and C. G. Palivan, Helv. Chim. Acta, 2023, 106, e202200164 CrossRef CAS
- S. K. Mondal, S. Chakraborty, S. Manna and S. M. Mandal, RSC Pharm., 2024, 1, 388–402 RSC
- L. Caselli, E. Parra-Ortiz, S. Micciulla, M. W. A. Skoda, S. M. Häffner, E. M. Nielsen, M. J. A. van der Plas and M. Malmsten, Small, 2024, 20, 2309496 CrossRef CAS PubMed
-
J. Jampílek and K. Král’ová, in Nanostructures for Antimicrobial Therapy, ed. A. Ficai and A. M. Grumezescu, Elsevier, 2017, pp. 23–54 Search PubMed
- Y. Jiang, W. Zheng, K. Tran, E. Kamilar, J. Bariwal, H. Ma and H. Liang, Nat. Commun., 2022, 13, 197 CrossRef CAS PubMed
- G. Mi, D. Shi, M. Wang and T. J. Webster, Adv. Healthcare Mater., 2018, 7, 1800103 CrossRef PubMed
- I. Qayoom, A. K. Teotia, A. Panjla, S. Verma and A. Kumar, ACS Infect. Dis., 2020, 6, 2938–2949 CrossRef CAS PubMed
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227–1249 CrossRef CAS PubMed
- D. Kışla, G. G. Gökmen, G. Akdemir Evrendilek, T. Akan, T. Vlčko, P. Kulawik, A. Režek Jambrak and F. Ozogul, Trends Food Sci. Technol., 2023, 135, 144–172 CrossRef
- X. Zhu, Q. Tang, X. Zhou and M. R. Momeni, Microb. Pathog., 2024, 193, 106741 CrossRef CAS PubMed
- V. K. Pal and S. Roy, ACS Appl. Nano Mater., 2022, 5, 12019–12034 CrossRef CAS
- M. Abbas, M. Ovais, A. Atiq, T. M. Ansari, R. Xing, E. Spruijt and X. Yan, Coord. Chem. Rev., 2022, 460, 214481 CrossRef CAS
- S. Tarvirdipour, C.-A. Schoenenberger, Y. Benenson and C. G. Palivan, Soft Matter, 2020, 16, 1678–1691 RSC
- S. J. Sigg, V. Postupalenko, J. T. Duskey, C. G. Palivan and W. Meier, Biomacromolecules, 2016, 17, 935–945 CrossRef CAS PubMed
- L. Abad, J. Josse, J. Tasse, S. Lustig, T. Ferry, A. Diot, F. Laurent and F. Valour, J. Antimicrob. Chemother., 2020, 75, 1466–1473 CrossRef CAS PubMed
- L. Ferreira, E. Pos, D. R. Nogueira, F. P. Ferreira, R. Sousa and M. A. Abreu, Front. Microbiol., 2024, 15, 1435720 CrossRef PubMed
- G. Tan, J. Xu, W. M. Chirume, J. Zhang, H. Zhang and X. Hu, Coatings, 2021, 11, 1401 CrossRef CAS
- P. A. Aristoff, G. A. Garcia, P. D. Kirchhoff and H. D. Hollis Showalter, Tuberculosis, 2010, 90, 94–118 CrossRef CAS PubMed
- B. Jacob, O. Makarewicz, A. Hartung, S. Brodt, E. Roehner and G. Matziolis, Sci. Rep., 2021, 11, 23425 CrossRef CAS PubMed
- P. Li, J. Luan, L. Jiang, Y. Zhang, W. Liu, Z. Yan and X. Ke, J. Mater. Chem. A, 2024, 12, 6014–6027 RSC
- M. Motiei, L. Pleno de Gouveia, T. Šopík, R. Vícha, D. Škoda, J. Císař, R. Khalili, E. Domincová Bergerová, L. Münster, H. Fei, V. Sedlařík and P. Sáha, Molecules, 2021, 26, 2067 CrossRef CAS PubMed
- H. Singh, R. Bhandari and I. P. Kaur, Int. J. Pharm., 2013, 446, 106–111 CrossRef CAS PubMed
- W. Xi, V. Hegde, S. D. Zoller, H. Y. Park, C. M. Hart, T. Kondo, C. D. Hamad, Y. Hu, A. H. Loftin, D. O. Johansen, Z. Burke, S. Clarkson, C. Ishmael, K. Hori, Z. Mamouei, H. Okawa, I. Nishimura, N. M. Bernthal and T. Segura, Nat. Commun., 2021, 12, 5473 CrossRef CAS PubMed
- I. Ul Haq and K. Krukiewicz, Appl. Surf. Sci. Adv., 2023, 18, 100532 CrossRef
- Y.-C. Yeh, T.-H. Huang, S.-C. Yang, C.-C. Chen and J.-Y. Fang, Front. Chem., 2020, 8, 286 CrossRef CAS PubMed
- ISO 22196:2011(en), Measurement of antibacterial activity on plastics and other non-porous surfaces, https://www.iso.org/obp/ui/#iso:std:iso:22196:ed-2:v1:en, (accessed December 5, 2024).
- JISZ2801, https://microbe-investigations.com/jisz2801/, (accessed December 5, 2024).
- T. Rawal, R. Parmar, R. K. Tyagi and S. Butani, Colloids Surf., B, 2017, 154, 321–330 CrossRef CAS PubMed
- D. Ge, Y. Li, L. Yang, Z. Fan, C. Liu and X. Zhang, Thin Solid Films, 2011, 519, 5203–5207 CrossRef CAS
- T. N. Murakami, Y. Fukushima, Y. Hirano, Y. Tokuoka, M. Takahashi and N. Kawashima, Colloids Surf., B, 2003, 29, 171–179 CrossRef CAS
- J. R. Vig, J. Vac. Sci. Technol., A, 1985, 3, 1027–1034 CrossRef CAS
- S. H. Behrens and D. G. Grier, J. Chem. Phys., 2001, 115, 6716–6721 CrossRef CAS
-
W. L. W. Hau, in Encyclopedia of Microfluidics and Nanofluidics, ed. D. Li, Springer US, Boston, MA, 2008, pp. 1911–1914 Search PubMed
- F. Baino and E. Verné, Biomed. Glasses, 2017, 3, 1–17 Search PubMed
- I. Georgakopoulos-Soares, E. L. Papazoglou, P. Karmiris-Obratański, N. E. Karkalos and A. P. Markopoulos, Colloids Surf., B, 2023, 231, 113584 CrossRef CAS PubMed
- J. E. Bandow, H. Brötz and M. Hecker, J. Bacteriol., 2002, 184, 459–467 CrossRef CAS PubMed
- M. Králová, S. Patakyová, M. Veselá, M. Baudys, J. Viktorová, J. Krýsa, M. Veselý and P. Dzik, J. Photochem. Photobiol., A, 2024, 455, 115769 CrossRef
- F. F. Tuon, P. H. Suss, J. P. Telles, L. R. Dantas, N. H. Borges and V. S. T. Ribeiro, Antibiotics, 2023, 12, 87 CrossRef CAS PubMed
- J. Pupaibool, Antibiotics, 2024, 13, 1223 CrossRef CAS PubMed
- E. Snejdrova, J. Loskot, J. Martiska, T. Soukup, L. Prokes, V. Frolov and T. Kucera, J. Drug Delivery Sci. Technol., 2022, 73, 103435 CrossRef CAS
- J. Reinbold, T. Hierlemann, L. Urich, A.-K. Uhde, I. Müller, T. Weindl, U. Vogel, C. Schlensak, H. P. Wendel and S. Krajewski, Drug Des., Dev. Ther., 2017, 11, 2753–2762 CrossRef CAS PubMed
- F. Rizzi, E. Fanizza, M. Giancaspro, N. Depalo, M. L. Curri, B. González, M. Colilla, I. Izquierdo-Barba and M. Vallet-Regí, Microporous Mesoporous Mater., 2025, 383, 113414 CrossRef CAS
- M. Vallet-Regí, F. Schüth, D. Lozano, M. Colilla and M. Manzano, Chem. Soc. Rev., 2022, 51, 5365–5451 RSC
- B. Xu, S. Li, R. Shi and H. Liu, Signal Transduction Targeted Ther., 2023, 8, 1–28 CrossRef PubMed
- S. Omardien, S. Brul and S. A. J. Zaat, Front. Cell Dev. Biol., 2016, 4, 111 Search PubMed
- L. Caselli, T. Traini, S. Micciulla, F. Sebastiani, S. Köhler, E. M. Nielsen, R. G. Diedrichsen, M. W. A. Skoda and M. Malmsten, Adv. Funct. Mater., 2024, 34, 2405047 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00246j |
| This journal is © The Royal Society of Chemistry 2025 |