
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive core–shell–shell nanocarriers for implant-directed magnetic drug targeting†
Timo
Herrmann
ad,
Nina
Angrisani
 bd,
Janin
Reifenrath
bd,
Jessica
Meißner
c,
Adrian
Hannebauer
a,
Lukas
Mönkeberg
a,
Valentin
Hagemann
ad,
Irene
Morales
a,
Peter
Behrens‡
a,
Nina
Ehlert
*ad and
Sebastian
Polarz
*a
bd,
Janin
Reifenrath
bd,
Jessica
Meißner
c,
Adrian
Hannebauer
a,
Lukas
Mönkeberg
a,
Valentin
Hagemann
ad,
Irene
Morales
a,
Peter
Behrens‡
a,
Nina
Ehlert
*ad and
Sebastian
Polarz
*a
aInstitute of Inorganic Chemistry, Hannover, Germany. E-mail: nina.ehlert@acb.uni-hannover.de; sebastian.polarz@aca.uni-hannover.de
bClinic for Orthopaedic Surgery, Hannover Medical School, Anna-von-Borries-Straße 1-9, 30625 Hannover, Germany
cDept. of Pharmacology, Toxicology and Pharmacy, University of Veterinary Medicine Hannover Foundation, Bünteweg 17, 30559, Germany
dNIFE – Lower Saxony Center for Biomedical Engineering, Implant Research and Development, Stadtfelddamm 34, 30625 Hannover, Germany
First published on 2nd May 2025
Abstract
The treatment of implant-associated infections is still a major topic in medical-related research. As an evolution of classical systemic therapy, many approaches target local treatment at the infection site. Here, we present an innovative material approach to overcome the challenges of this local drug delivery. As an effective nanocarrier, we chose nanoporous silica, which fulfills the need for a high capacity to load antibacterial drugs. Combined with a magnetic iron oxide core, these core–shell particles provide a sophisticated drug-delivery system that allows targeted drug delivery to the desired tissue via a magnetic field. However, the release profile often reveals the problem of an uncontrolled burst release of the incorporated drug in physiological media, leading to the loss of the cargo en route to the site of infection and resulting in an ineffective treatment of implant-associated infection. A pH-responsive polymer shell can provide an elegant solution, as the acidic pH occurring during an infection (pH 5–6) can trigger the release precisely, preventing an early release of the drug. In this study, we selected poly(2-(diethylamino)ethyl methacrylate) (PDEMA) with a perfect fitting isoelectric point of 6.7 for the establishment of a pH-responsive polymer shell. Furthermore, we addressed the issue of a poorly stable dispersion of particles functionalized with hydrophobic polymers in physiological media by adding a sulfonic acid modification to the inner pore surface of the nanoparticles. This modification influenced the amount of attached polymer and the drug release profiles. It was also useful to increase the incorporated amount of enrofloxacin. In summary, we present innovative and effective core–shell–shell nanocarriers based on magnetic nanoporous silica nanoparticles functionalized with a pH-responsive polymer for the pH-triggered delivery of the antibiotic enrofloxacin and suitable for targeting using a magnetic field.
1. Introduction
Considering the demographic change and the aging of society, there is great current and future demand for the treatment and replacement of dysfunctional parts of the human body.1–3 Implant technology in medicine opened up new treatment possibilities for joint replacement, cardiovascular treatment, and hearing loss. While the benefits of implants are tremendous, the risk of implant-associated infections remains a significant threat to successful treatment outcomes. In a not-to-be-neglected case, contamination with pathogenic bacteria is a critical issue, leading to substantial problems days or even longer after insertion. The consequences of infection can be severe, ranging from prolonged hospitalization of the patient with increased antibacterial treatment to the removal and replacement of the failed implant, and can even result in mortality.4 In 2021, 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 knee and 176000 hip arthroplasties were performed in Germany, of which about 14000 and 17000 required reoperation, respectively. Of those, 15% and 16% were performed due to infection, respectively.5 As soon as the bacteria have formed a biofilm, fighting the infection is aggravated because of the increased resistance against antibiotics.6 Depending on the type of implant, there may be a low blood supply, or the infected sites may be difficult to access. Taking these points into consideration, the importance of research leading to new approaches for protecting implants against bacterial colonization becomes clear.7,8
000 knee and 176000 hip arthroplasties were performed in Germany, of which about 14000 and 17000 required reoperation, respectively. Of those, 15% and 16% were performed due to infection, respectively.5 As soon as the bacteria have formed a biofilm, fighting the infection is aggravated because of the increased resistance against antibiotics.6 Depending on the type of implant, there may be a low blood supply, or the infected sites may be difficult to access. Taking these points into consideration, the importance of research leading to new approaches for protecting implants against bacterial colonization becomes clear.7,8
A promising approach is using functional silica nanoparticles as a drug-delivery system, especially if combined with a magnetic iron oxide core. This combination facilitates the guidance of the drug-loaded nanoparticles. However, this becomes more challenging when the target area is an infection or an implant located deep within the body. Here, the use of magnetic particles together with a magnetizable implant offers the possibility of efficient particle targeting. We have demonstrated this concept in previous publications, confirming its potential for targeted drug delivery.9–11 In brief, the particle system consists of nano-scaled and porous, organically modified silica colloids with a multi-domain architecture and a magnetite nanocrystal as an inner core. If an infection occurs, the implant can be magnetized by applying a magnetic field. The drug-loaded silica nanoparticles can now be injected into the body near the implant site. During their movement through the blood and other body fluids, they will pass the implant, accumulating and releasing the drug. When the treatment is finished, the magnetic field can be turned off, and the nanoparticles will detach from the implant as they show a superparamagnetic behavior.
Positioning of the drug-delivery system is only one problem that has to be addressed. To ensure a high level of the antibiotic drug at the implant site, an early release of the drug should be avoided. One approach is to install a trigger that can start the controlled drug release once the nanoparticles reach the implant. Numerous possibilities have been reported in the literature. The release can be triggered by light,12–14 mechanically,15 or by temperature.16 The latter methodologies are quite effective, but they require an external action performed by the medical personnel. Chemical stimuli17–19 open the possibility for an autonomous activation of the antibacterial functionalities. Therefore, we focussed on protons, specifically pH value, as a trigger to open the pores and allow the release of pharmaceuticals. An inflamed tissue or an infected implant typically lowers the pH within the infected region to 5–6 due to the metabolic activity of the present bacteria.20–22 There are two general approaches found in the literature. (a) The drug molecules are attached to the pore walls via pH-labile linkers, such as hydrazones23–25 or acetals.26,27 (b) For the so-called nano-valve concept, the goal is to establish mechanisms that can open and close the entrances to the pores. Intriguing reports have been published using molecular or nanoparticle species as caps for the pores, which show very good pH sensitivity with almost no drug release at neutral pH and high release at acidic pH, as shown by Beňová et al.28,29 and Li et al.30 However, these systems have two drawbacks. First, the cap molecules are also released from the porous materials preventing reversibility. And second, once the pores are open, they remain like this. An interesting alternative that we also explore in this paper is attaching a pH-responsive polymer to the external surface of the nanoporous silica particles that can open and close the pores. A variety of pH-responsive polymers are available, ranging from polyacids31 to more basic polymers19,32,33 and co-polymers comprising acidic and basic monomers,34 which all show promising properties for pH-triggered drug release. The polymer chains are collapsed in their neutral state and become charged upon protonation, which induces swelling and switching to different properties. For the current study, we have selected poly(2-(diethylamino)ethyl methacrylate) (PDEMA) for opening and closing the pores. Due to its pKa of 6.7,35,36 it switches exactly in the pH region needed (5–6) for the described application.
Regarding colloidal stability in water, which is another highly important factor that drug-delivery systems must achieve, the uncharged character of polymers such as PDEMA at neutral pH is a large disadvantage.36–38 The agglomeration of the particles in a drug-delivery system must be avoided by all means because it can lead to clogging of the veins or arteries. This might lead to a heart attack or cardiac arrest. Therefore, a crucial step for using the PDEMA-functionalized particles in drug delivery applications is to enhance their colloidal stability.
Fig. 1 summarizes the material concept. A magnetic core is surrounded by a shell of a porous silica material. A second shell of PDEMA is attached to the external surface. The particles can deposit at a magnetic surface. If there is a biofilm at an infection site, the polymer chains become protonated, expand, and open the pores, respectively. Antibiotic compounds are released. Sulfonic acid groups present in the silica matrix fulfil two functions. They ensure the colloidal stability of the particles in water regardless of the pH value and attract enrofloxacin by interaction with the surface through electrostatic forces. In this study, the drug delivery performance and colloidal stability of the polymer-functionalized particles were investigated. Furthermore, we explored the ability of the particles to deposit on a magnetic surface and the efficacy of the released drug.
 | ||
| Fig. 1 Schematic representation of the implant-directed magnetic drug targeting with the polymer-functionalized particles releasing enrofloxacin (EFX) once they reach the acidic biofilm; detaching from the implant after the magnetic field is turned off, and degradation of the nanoparticles by e.g. macrophages. | ||
2. Experimental
2.1 Materials and methods
For the syntheses, all chemicals were used without further purification, unless otherwise stated. Iron(II) chloride tetrahydrate (≥99%), iron(III) chloride hexahydrate (99%), oleic acid (90%), chloroform (≥99%), cetyltrimethylammonium bromide (CTAB, ≥98%), ammonium hydroxide solution (≥25% NH3 in water), tetraethyl orthosilicate (TEOS, ≥99%) ethyl acetate (99.8%), (3-aminopropyl)triethoxysilane (APTES, 99%), ethanol (EtOH, absolute EMPLURA), 2-(diethylamino)ethyl methacrylate (DEMA, 99%, contains 1500 ppm MEHQ as inhibitor), sodium citrate tribasic dihydrate (>99%), citric acid (>99.5%), enrofloxacin (EFX, 99%), hydrochloric acid (HCl, 2 M), sodium hydroxide (NaOH, 98%), triethylamine (TEA, ≥99%), rhodamine-b-isothiocyanate (RITC, mixed isomers), typtone soy broth and Mueller-Hinton Broth (MHB) were purchased from Sigma-Aldrich Corporation (München, Germany). 2-Bromoisobutyryl bromide (BIBB, ≥98%), methanol (MeOH, ≥99,8%), N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PDETA, >99%), and copper bromide (CuBr, >98%, was purified by dispersing 200 mg in 5 ml of glacial acetic acid for 4 h, subsequent washing with MeOH and drying under vacuum) were purchased from TCI Europe N.V. Tetrahydrofuran (THF, 99.8%) was purchased from Acros Organics (Geel, Belgium), ammonium nitrate (NH4NO3, ≥99%) was purchased from Honeywell GmbH (Seelze, Germany) and 3(-trihydroxysilyl) propane-1-sulfonic acid (TSPSA, 30–35 wt% in water) was purchased from Fluorochem Ltd (Hadfield, United Kingdom). PBS was prepared from phosphate buffered tablets (Fisher Scientific, Geel, Belgium). Citrate buffer was prepared from sodium citrate tribasic dihydrate and citric acid.Transmission electron microscopy (TEM) was performed using a Hitachi HT7800 instrument with a 120 kV field emission gun. For sample preparation, 400-mesh carbon-coated copper grids (MicrotoNano, Harlem, Netherlands) were used. The samples were dispersed in ethanol, dropped onto the grid, and dried. The particle size was determined using NIH ImageJ. Fourier-transform infrared spectroscopy (FT-IR) was performed using an ATR set-up on a Spectrum Two FT-IR spectrometer from PerkinElmer (Waltham, United States). Argon physisorption was performed on 3Flex (Micromeritics Instrument Corporation, Norcross, United States) after outgassing the sample for 24 h at 110 °C. Surface areas, pore sizes, and pore volumes were calculated using the associated software for the machine. Thermogravimetric analysis (TGA) was performed with a Netzsch STA 449 F5 Jupiter (Selb, Germany). The samples were heated to 1000 °C in an Al2O3 crucible with a rate of 5 °C min−1 in an atmosphere of N2 and O2 (80% N2, 20% O2). Hydrodynamic size and zeta potential were measured by dynamic light scattering (DLS) with a Zetasizer Nano ZSP (Malvern Panalytical, Malvern, United Kingdom), whereby the sample was dispersed in water. UV/VIS was measured using a Spark 10M (Tecan Trading AG, Männedorf, Switzerland) for which 200 μl of the sample was pipetted into a 96-well plate and the absorbance was measured at 270 nm three times. The optical density was measured using an Infinite® 200 Pro (Tecan Trading AG, Männedorf, Switzerland). Magnetic characterization was performed using a SQUID MPMS 3 magnetometer obtained from Quantum Design (San Diego, United States). The measurements were performed with the samples in powder form in polypropylene sample holders (QDC-4096-388). Magnetization vs. field curves at 5 T were measured in VSM mode at 300 K (San Diego, United States). Fluorescence spectroscopy was performed using a Synergy 2 (BioTek Instruments Inc., Winooski, United States) for which 200 μl of the sample was pipetted into a 96-well plate, and the fluorescence was measured at 590 nm.
2.2 Synthesis of amino-functionalized and amino-sulfonic acid functionalized magnetic core–shell silica nanoparticles
The magnetic core–shell silica nanoparticles were synthesized as previously reported with some minor changes.10,11,39,40 In the case of the first single-modified particles developed, the structure-directing agent CTAB was removed using calcination at 550 °C for 5 h before the particles were amino-modified. Therefore, 100 mg of nanoparticles were dispersed in 5 ml of ethanol, and APTES (95 μl, 0.46 mmol) was added to the dispersion. The dispersion was then stirred for 1 h at 50 °C.41 Afterwards, the particles were centrifuged, washed with ethanol twice, and dried under vacuum. These particles are denoted as amino-sulfo0. For the dual-modified, amino- and sulfonic acid-modified nanoparticles (amino-sulfoX NPs), the preparation procedure had to be adjusted. To achieve a selective amino modification on the outer surface of the nanoparticles, the amino modification was carried out with CTAB still present in the pores, using the same procedure as described above but with 410 mg of nanoparticles. After drying, CTAB was removed using an alcoholic solution of ammonium nitrate (NH4NO3). For this, the obtained amino-modified particles were dispersed in a mixture of 45 ml of ethanol and 977 mg of NH4NO3. The dispersion was then heated to reflux for two hours. After washing with ethanol once and drying under vacuum, extracted amino-modified particles were obtained.The additional modification of the nanoparticles was carried out using 3(-trihydroxysilyl)propane-1-sulfonic acid (TSPSA). Therefore, 120 mg of the amino-modified particles were dispersed in 40 ml of THF and the dispersion was cooled in an ice bath for 30 min. Next, the corresponding amount of the sulfonic acid was added and the dispersion was stirred for another 30 min in an ice bath to allow the sulfonic acid to diffuse into the pores of the particles. The amount of the added sulfonic acid TSPSA ranged from 0, 0.5, 1, 5, 20, and 50 to 100 equivalents (more details can be found in Table S1, ESI†). The particles are denoted as amino-sulfoX nanoparticles, where X corresponds to the equivalents of the organo-sulfonic acid (TSPSA) used for modification. The ice bath was then removed and the particle dispersion was heated to 65 °C for 1 h to start the condensation reaction between the sulfonic acid and the silanol groups. After washing with ethanol once, the particles were placed in an oven at 60 °C overnight.
2.3 Attachment of the polymer shell by polymerization
The polymer shell was attached in the second step. To serve as a starting molecule for the polymerization carried out later, first 2-bromoisobutyryl bromide (BIBB) was attached to the particles.38 In a typical procedure, 100 mg of amino-sulfoX particles were dispersed in 5.6 ml of THF, and 140 μl of triethylamine (TEA) was added. The dispersion was stirred in an ice bath for 30 min. Next, 120 μl of BIBB were dissolved in 3 ml of THF and slowly added dropwise to the particle dispersion. After the complete addition, the solution was further stirred for 3 h in an ice bath and overnight at room temperature. The bromoalkyl amide-modified particles were washed with ethanol twice and dried under vacuum at 40 °C for 2 h. The polymerization reaction was carried out directly after drying. As it was observed in the course of the experiments that initiator-modified particles stored for a longer period showed poorer properties later in the drug release experiments, these particles were used for the polymerization reaction directly after their production. For the typical adapted polymerization procedure, 100 mg of BIBB-modified particles were dispersed in 9 ml of methanol and the dispersion was degassed by one freeze–pump–thaw cycle. Next, 66.9 μl of N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PDETA), 47 mg of copper bromide (CuBr), and 1.91 ml of 2-(diethylamino)ethyl methacrylate (DEMA) were added and the dispersion was again degassed using one cycle. Then the polymerization was allowed to proceed for 24 h at 50 °C under nitrogen. After 24 h, the flask was opened to the atmosphere to stop the polymerization. The particles were separated by centrifugation (6150g, 5 min) from the deep blue solution. They were washed with ethanol twice to remove unreacted monomer and other organic compounds. Next, they were washed twice with an acidic citrate buffer (pH 5) to remove any water-soluble copper compounds. After another washing step with water, the particles were dried under vacuum at room temperature. The samples are labeled amino-sulfoX-PDEMA, with X referring to the equivalents of organosulfonic acid (TSPSA) used for sulfonic acid modification. For clarity, only the samples with X = 0.5, 20, and 50 are presented in the main manuscript, and the residual data (X = 0.5, 1, and 100) are shown in overall comparison in the ESI.†2.4 Drug loading and release procedure
To load the synthesized nanoparticles with the drug, a solution of enrofloxacin (EFX) with a concentration of 10 mg ml−1 was prepared by dissolving EFX in a water/hydrochloric acid (HCl) mixture in the way that an equimolar amount of HCl to EFX was used. 20 mg of the polymer-functionalized particles were added to 3 ml of the EFX solution along with 50 μl of 0.5 M HCl, to ensure an opening of the polymer chains. The dispersion was shaken for 24 h at room temperature. After this, the particles were centrifuged and washed twice with a NaOH solution (pH 9). After drying under vacuum, the EFX-loaded polymer-functionalized particles are obtained.For the release of EFX, 5 mg of the corresponding particles were dispersed in either 2 ml of a PBS buffer (0.1 M, pH 7.4) or 2 ml of a citrate buffer (0.1 M, pH 5) and stored at 37 °C. After different time periods, the particles were centrifuged and the supernatant was removed and stored until the UV-vis measurement. Subsequently, new buffer was added and the particles were again stored at 37 °C until the next measurement point. The release of EFX from the functionalized particles was carried out over 24 h. To guarantee that any surface adsorbed EFX is removed, the release point after 30 min was evaluated as an additional washing step. As no significant amounts of EFX were released after 6 h, only the release up to 6 h is shown in the resulting release profiles.
2.5 Fluorescence RITC labeling and magnetic immobilization procedure
For the fluorescence labeling with rhodamine-b-isothiocyanate (RITC), 30 mg of particles were dispersed in 10 ml of EtOH. Next, 5 mg of RITC were dissolved in 1 ml of EtOH and added to the dispersion, followed by stirring at 50 °C for 24 h in the dark. After the reaction was finished, the particles were centrifuged and washed with EtOH until no reddish discoloration of the supernatant was observable.For the magnetic accumulation experiment, a 3 mg ml−1 dispersion of the corresponding particle was prepared in a 0.9% saline solution (B. Braun SE). Next, 0.4 ml of this particle dispersion was mixed with 4.6 ml of a 0.9% saline solution to obtain a solution with a concentration of 240 μg ml−1. The aperture was filled with this particle dispersion and it was pumped through the system for 3 min with a flow rate of 5.2 ml min−1. The magnet was transferred to a vial, 1 ml of 0.9% NaCl was added and the particles were removed using an ultrasonic bath in two-minute sections.
2.6 Testing of antibacterial effectiveness by microdilution
Enrofloxacin (EFX) supernatants (in PBS or citrate buffer) were collected after 1 h, 2 h, and 3 h, respectively, frozen and stored at −20 °C until further testing of antibacterial effectiveness. For this purpose, a microdilution setup with S. aureus JSNZ-WT (received from S. Holtfreter, Greifswald, Germany, described by Holtfreter et al.42) was used. Bacteria were first thawed and cultured overnight (37 °C) in sterile tryptone soy broth. Before applying on the well plates, bacteria were washed twice and centrifuged in 0.9% saline solution and diluted until an optical density (photometric measurement, 600 nm) of approximately 0.02 was reached. 20 μl of bacteria were applied in each well of a 96-well microtiter plate for testing antimicrobial effectiveness. Different concentrations of the EFX supernatants in sterile Mueller-Hinton broth (MHB, 21 g l−1) were tested (180 μl each well), starting with 1:2 dilution. 1:10 dilution in MHB followed, which was further diluted in a series of 1:2 until no supernatants were present. As blank controls, the same concentrations of supernatants in MHB were tested with 20 μl of sterile saline instead of bacteria. Additionally, pure MHB with and without bacteria was evaluated as positive and negative controls, respectively. Three replicates of dilution series were pipetted in each well plate and three technical independent replicates were performed. Well plates were cultured for 24 h at 37 °C and photometric measurement of optical density at 600 nm was used to detect bacterial growth.
Additionally, PBS and citrate buffer alone were tested equivalent to the supernatants to verify the influence of buffer media.
3. Results and discussion
Fig. 2 summarizes the synthesis routes of the two differently functionalized nanoparticles compared in this study (data concerning the unmodified core–shell nanoparticles can be found in the ESI,† Fig. S17). The silica nanoparticles with an integrated magnetic core10,11 were either modified only with amino or simultaneously with amino and sulfonic acid groups (detailed information on the synthesis and characterization of these nanoparticles can be found in the ESI†). Afterwards, the pH-sensitive polymer PDEMA was attached to the surface to enable a pH-triggered drug delivery.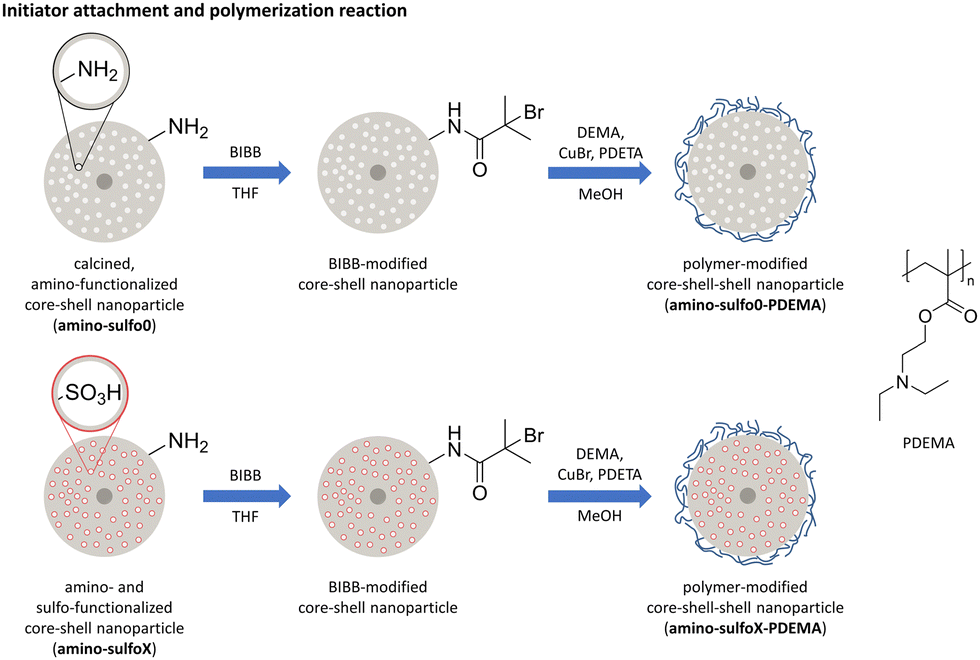 | ||
| Fig. 2 Schematic overview of the synthesis of polymer-functionalized silica core–shell nanoparticles with either a single amino-modification (amino-sulfo0-PDEMA) or a dual amino- and sulfonic acid-modification (amino-sulfoX-PDEMA; X = 5,20,50). X indicates the amounts (equivalents) of sulfonic acid-silane used during particle preparation (data for samples with X = 0.5, 1, and 100 can be found in the ESI†). | ||
3.1 Initiator attachment
Before the polymerization was performed, the initiator 2-bromoisobutyryl bromide (BIBB) was attached to the particles using condensation between the acetyl bromide group of the initiator molecule and the amino groups present on the particle surface. As proof of successful attachment, infrared spectra (IR spectra) for the initiator-modified particles were recorded, as shown in Fig. S6 (ESI†). Here, new vibration bands are visible after the functionalization with the radical starter molecule BIBB, these additional vibration bands were observable at 1650 cm−1 and 1530 cm−1, which belong to the amide(I) and amide(II) vibration of the amide group formed due to the condensation reaction.433.2 Characterization of polymer-functionalized silica core–shell nanoparticles
After the initiator was attached, the polymer was directly polymerized on the nanoparticle using surface-initiated atom transfer radical polymerization (SI-ATRP), which is a well-established method for attaching polymers to e.g. silica surfaces.44–46 In an SI-ATRP reaction, the initiator is anchored to the nanoparticle surface, enabling controlled polymerization, resulting in a well-defined thickness of the polymer shell. In addition, polymerization can only take place where the initiator is present. This prevents the formation of polymer chains that are not attached to the particle. The polymerization is typically performed using copper halide salts or light. Although the usage of copper might be disadvantageous with regard to the application in a biomedical field, as larger amounts of copper ions are toxic for cells,47–49 the usage of light to attach the polymer provides a worse switch between the open and closed form.37 After polymerization, new vibration bands are visible at 1726 cm−1 in the IR spectra (Fig. 3a) belonging to the carbonyl group of the attached polymer. In cases where the organosilyl sulfonic acid TSPSA (for clarity only the samples amino-sulfoX-PDEMA with X = 0.5, 20, and 50 are presented in the main manuscript, the residual data with X = 0.5, 1, and 100 are shown in overall comparison in the ESI†) is used for sulfonic acid group attachment, the intensity of this signal decreases with increasing equivalents used, indicating a lower amount of polymer present on the particles at a higher sulfonic acid group density on the surface (Fig. 3a and Fig. S7a, ESI†). This assumption was further confirmed in the thermogravimetric measurements, as presented in Fig. 3b. The use of higher equivalents of TSPSA in the synthesis results in a lower mass loss and thus organic/polymer content, with a total mass loss of 52% for amino-sulfo5-PDEMA and 34% for amino-sulfo50-PDEMA. Taking into account the mass loss caused by previous amino-sulfo modification, the mass loss of amino-sulfo5-PDEMA and amino-sulfo50-PDEMA caused by the polymer can be estimated to be 41% and 9%, respectively. Hence, the degree of functionalization with sulfonic acid groups influences the amount of attached polymer on the final particle (compare Fig. S2 and Table S2 of the ESI† for results concerning sulfonic acid group content). This could be caused by two reasons. On one hand, the capability of initiator attachment and with that the attachment of the desired polymer could be decreased by the sulfonic acid groups. The higher charge of the particles with higher equivalents of the sulfonic acid used (Fig. S10, ESI†) for modification might lower the amount and the density of the attached initiator. However, this should only have a low impact on the final polymer grafting density, as the initiator efficiency increases for more negatively charged surfaces resulting in similar dense grafted material.50 On the other hand, the sulfonic acid groups or their negative charge respectively could interfere with the polymerization reaction itself. This influence has already been observed for different polymeric nanomaterials and was attributed to the negative charge of the polymeric nanoparticles.50–53 The positively charged catalytic copper-ligand-complex accumulates at the surface of the particles, due to charge equalization. This leads to inefficient polymer chain growth, which is an important reaction step, resulting in polymer chains with lower molecular weight.50 This provides an appropriate explanation to our finding that the mass loss and consecutively polymer content is decreasing with increasing equivalents of TSPSA. For the solely amino-modified nanoparticles, amino-sulfo0-PDEMA, a denser surface coverage with the initiator is expected, due to the high presence of amino groups (concluded from the high organic content caused only by amino modification determined by TG, Fig. S2f and Table S2, ESI†). However, a lower polymer mass is attached, which is probably caused by the lack of negative charge and the associated lack of increased initiator activity. | ||
| Fig. 3 Characterization of amino-sulfoX-PDEMA (X = 0.5, 20, and 50) nanoparticles. (a) IR spectra of polymer-functionalized nanoparticles with a marked carbonyl vibration band, (b) thermogravimetric curves, (c) TEM image of amino-sulfo50-PDEMA (∅ = 83 ± 10 nm). (d) Ar physisorption isotherms (Ar@87 K), filled symbols represent the adsorption branch, and empty symbols represent the desorption branch. Samples: amino-sulfoX-PDEMA grey (◀): X = 0, red (▲): X = 5, violet (▼): X = 20, blue (♦): X = 50. X indicates the amounts (equivalents) of sulfonic acid-silane used during particle preparation. | ||
The same conclusion concerning the correlation of polymer content and degree of sulfonic acid modification is supported by gas physisorption experiments. Due to the blocking of the surface by the attached polymer, only very small specific surface areas of the less polymer-coated samples could be determined from the Ar physisorption isotherms, shown in Fig. 3d. Here, the highest BET surface with 19 m2 g−1 was determined for amino-sulfo50-PDEMA. The main reason behind the higher surface area for this sample is the lower amount of polymer attached to the particles as explained earlier. The samples with higher polymer content (compare also Fig. S7 and Table S3, ESI†) exhibit no porosity. Furthermore, confirmation of the effect of sulfonic acid modification is provided by TEM investigation. The sample amino-sulfo50-PDEMA exhibits no visible polymer shell (Fig. 3c) in comparison to the modified particles before polymerization (Fig. S4 and S5, ESI†). For the lower amounts of used sulfonic acid TSPSA, a material with a brighter contrast is visible around the particles and the particle agglomerates on the grid, as shown in Fig. S8 and S9 (ESI†). These are presumably polymer moieties, which are visible in these samples due to their higher mass fraction. One more important finding was the fact that the sample micrographs revealed no visible damage to the particles, which the polymerization could have caused. So, the spherical shape and the size of the particles were preserved. As further support for successful modification and attachment of the polymer of the amino-sulfo50-PDEMA samples, X-ray photoelectron spectroscopy was carried out in comparison to an unmodified sample. Here, nitrogen and sulphur were only detectable on the polymer-functionalized sample (Fig. S18 and Table S4, ESI†).
3.3 Colloidal stability of polymer-functionalized silica core–shell nanoparticles
The colloidal stability of nanoparticles, which shall be used for drug delivery applications, is indispensable in certain pH ranges, as agglomeration needs to be avoided. Therefore, the properties of our newly synthesized amino-sulfoX-PDEMA particles were investigated concerning their colloidal stability using zeta potential measurements in a water/HCl/NaOH mixture in a pH range from pH 2 to pH 8. This pH range covers the important area for the application in the body. In order to have a comparison, hydrodynamic size and zeta potential were recorded for polymer-functionalized particles with no sulfonic acid groups, amino-sulfo0-PDEMA. The obtained curves are depicted in Fig. 4a. The sample amino-sulfo0-PDEMA generally showed a stable colloidal dispersion in the pH range from approx. 2 to 5 with a size of around 200 nm. With further pH rise, a large increase in the hydrodynamic size occurred, indicating the colloids' agglomeration. The explanation for the observed process is as follows. The amine groups of the polymer shell are protonated below pH 4.5, also documented by the high zeta potential of approx. 35 mV. These positively charged amine groups lead to extended polymer chains due to charge repulsion. As a consequence, the particles are electrostatically stabilized. The deprotonation at a higher pH value is accompanied by discharging and polymer collapse. At a pH of 6, the isoelectric point (IEP) is reached. The colloidal stability is reduced and coagulation takes place, visible by increased particle sizes. The literature on PDEMA-functionalized particles by Sun et al.38 and Su et al.37 confirms the restricted stability at neutral to alkaline pH values. In case of our newly synthesized amino- and sulfonic acid-modified polymer-functionalized nanoparticles, distinct differences were observable. Here, a decrease in the hydrodynamic size was observable over the same pH range for all samples (Fig. 4a for selected – or Fig. S11a for all samples, ESI†). | ||
| Fig. 4 Colloidal stability investigations of amino-sulfoX-PDEMA. (a) pH-dependent hydrodynamic diameter, (b) pH-dependent zeta potential, (c) zeta potential, and (d) hydrodynamic diameter in PBS and citrate buffer grey (▼): X = 0, red (▲): X = 5, violet (♦): X = 20, blue (◀): X = 50. X indicates the amounts (equivalents) of sulfonic acid-silane used during particle preparation. | ||
With the sulfonic acid-modified nanoparticles, we observed the expected course of the hydrodynamic size. With higher pH values, the protonation of the amine groups decreased leading to more entangled polymer chains and smaller particle sizes. In comparison to amino-sulfo0-PDEMA, no significant size increase was observed, which could indicate the formation of large agglomerates. However, for certain samples, agglomeration was observable at the terminal measuring point at pH 8, which appeared to be random. The size decrease was most prominent for the amino-sulfo50-PDEMA, where it decreased from 490 nm at pH 2 to 133 nm at pH 8 (Fig. 4a, blue). This decrease in the hydrodynamic size was again attributed to the behavior of the attached pH-sensitive polymer. In the zeta potential curves (Fig. 4b or Fig. S11b, ESI†), a positive zeta potential was observable over the whole pH range for the sulfo-modified samples amino-sulfo0.5-PDEMA to amino-sulfo20-PDEMA with 35 mV to 15 mV, respectively. It decreased by about 10 mV over the measured pH range.
This positive zeta potential is, again, caused by the protonation of the tertiary amine present within the polymer chains, which persists even at neutral or slightly basic pH to some degree. A negative zeta potential would be expected if all amine groups are neutral in charge because in that case, the charge of the sulfonic acid group would have a stronger effect on the charge of the particle. Accordingly, the consistently positive zeta potential is caused by the large amount of polymer attached to the particle. If the polymer amount and with that the number of amino groups is significantly larger than the number of sulfonic acid groups, then the negative charge caused by the sulfonic acid groups is overcompensated by the positive charged amine groups, resulting in positive zeta potential. This was the case for the samples from amino-sulfo0.5-PDEMA to amino-sulfo20-PDEMA where also a large amount of polymer is attached. This was also true for amino-sulfo0-PDEMA, where a positive zeta potential is observable until agglomeration. When a lower amount of polymer is attached, the number of amino groups and with that the positive charge is not sufficient enough to compensate the negative charge over the whole pH range. As for amino-sulfo50-PDEMA, a steady decrease of the zeta potential was visible, which ends in the negative range with −5.3 mV, now caused by the sulfonic acid groups. Noticeably, no agglomeration of these particles was observed, despite the low zeta potential, indicating stabilization by steric effects. Overall, it could be seen that with the additional functionalization of the particles with sulfonic acid groups by the reaction pathway presented in this work, the stability of the polymer-functionalized particles has improved significantly compared to the single-modified polymer-functionalized amino-sulfo0-PDEMA. Although the zeta potential was not as fine-tuneable for the polymer-coated particles as it is for the dual-functionalized particles without polymers (Fig. S10, ESI†), all polymer-functionalized particles showed better colloidal stability than the comparison sample without sulfonic acid groups. The IEPs for most samples are not in the pH range under consideration. In the only sample with an IEP, it was shifted to a more basic pH of 7.5 and no agglomeration was visible, making all samples suitable candidates for our desired application.
To get a better insight into the colloidal stability of our newly synthesized polymer-functionalized particles during the release procedure, not only the stability of the dispersion in a water/HCl/NaOH system needs to be considered, but the stability in the used buffers is also important. These media differ from the previously observed ones, as they have different ionic strengths and different ions dissolved that could influence the stability of the polymer-coated particles. Therefore, we measured the size and zeta potential of our polymer-functionalized particles in the buffer used for drug release, which are 0.1 M PBS and 0.1 M citrate buffer with a pH of 7.4 and 5.0, respectively. The properties of the particles in these buffers differ from the properties obtained in the simpler water/HCl/NaOH system. The size of the particles, shown in Fig. 4c, corresponds approximately to the size in the water/HCl/NaOH system at the given pH. For most samples, a larger size was determined in the acidic buffer, caused by the open form of the polymer. This situation was different for the zeta potential of the particles, as shown in Fig. 4d (whole data set in Fig. S11, ESI†). Here, a similar zeta potential for all samples up to amino-sulfo20-PDEMA was observed with 9 to 6 mV, which was much lower than the values determined in non-buffered media. So, it becomes clear that the ionic strength of the liquid influences the zeta potential of the particles, resulting in a similar zeta potential in both buffers. When using higher equivalents of TSPSA, the zeta potential decreased, caused by the lower amount of polymer as explained earlier. Despite the low zeta potential, no agglomeration of the particles in the respective buffer was observable for 10 min, as shown in Fig. S12 (ESI†).
3.4 Drug-delivery properties of polymer-modified core–shell particles using enrofloxacin
The release curve of the only amino-modified polymer-functionalized particles amino-sulfo0-PDEMA (Fig. S13a, ESI†) showed a continuous release of enrofloxacin (EFX) with a pH dependency. In the neutral PBS buffer, the polymer chains are present in their closed state, preventing the drug inside the pores from diffusing out. Nevertheless, a small amount of 1.1 μg of EFX per mg of particle was released for amino-sulfo0-PDEMA. This release may result from the incorporation of the polymer chains. However, in the acidic buffer, where the polymer chains are opened up, a higher release with 3.2 μg mg−1 was observed, which represents a 190% higher release in the acidic media, as visualized in Fig. 5b. This confirms the findings by Sun et al.38 and Su et al.,37 where they also received a higher release of their target molecule in acidic media. | ||
Fig. 5 (a) EFX-release curves of amino-sulfo50-PDEMA, filled symbols: release in PBS buffer (0.1 M, pH 7.4), empty symbols: release in citrate buffer (0.1 M, pH 5.0). (b) Proportion of higher EFX released in the acidic citrate buffer and (c) accumulated release after 6 h in the corresponding buffer. Samples: amino-sulfoX-PDEMA (▼): X = 0, (■): X = 0.5, (●): X = 1, (▲): X = 5, (♦): X = 20, (◀): X = 50, (![[hexagon filled, flat-side down]](https://www.rsc.org/images/entities/char_e039.gif) ): X = 100. X indicates the amounts (equivalents) of sulfonic acid-silane used during particle preparation. ): X = 100. X indicates the amounts (equivalents) of sulfonic acid-silane used during particle preparation. | ||
For the presented enhanced polymer-coated particles amino-sulfoX-PDEMA (X ≠ 0), which exhibit higher stability in aqueous solution, also a pH dependency in their release behavior and therefore a higher release in the acidic buffer was observable. In Fig. 5b and c, the differences between the amounts released in neutral PBS and acidic citrate buffer are visualized. It can be stated that the total amount of EFX released was similar for the samples up to amino-sulfo5-PDEMA, which showed a slightly higher release as the only amino-modified polymer-functionalized sample amino-sulfo0-PDEMA with 5.3 μg mg−1 in the acidic buffer. Despite this slight increase, no improvement was achieved in terms of a higher release portion in acidic buffer (46–72%) in comparison to amino-sulfo0-PDEMA (190%) as depicted in Fig. 5b. A possible reason might be the amount of attached polymer because these samples showed a similar, if not higher amount of attached polymer than amino-sulfo0-PDEMA. The amount of attached polymer is too high, so a switching of the polymer between open and closed form will be more difficult, as more functional groups need to be protonated to do so. Also, a high polymer load might lead to clogging of pores, lowering the incorporation and release of EFX. In addition to the polymer load, the added charge to the particles might also influence the switching behavior of the polymer. An improvement in the pH-dependent delivery of EFX was achieved with the samples amino-sulfo50-PDEMA and amino-sulfo100-PDEMA, of which the release curve of amino-sulfo50-PDEMA is shown exemplarily in Fig. 5a. With a release of 51.1 μg mg−1 for amino-sulfo50-PDEMA (and 59.1 μg mg−1 for amino-sulfo100-PDEMA, Fig. S13f, ESI†) in the acidic buffer, these released amounts are significantly higher than the released amounts of all other samples. A reasonable explanation takes the larger amount of sulfonate groups into account. As the EFX is present in a protonated form during the incorporation, an attraction between the positively charged EFX and the negatively charged sulfonic acid groups takes place. This leads to a higher amount of EFX which can be incorporated into the particles.54 As more EFX is incorporated, more EFX can be released during the release experiments. Likewise, the amount of attached polymer is lower, so switching between the open and closed form is easier. Nevertheless, it can be seen that with the samples amino-sulfo50-PDEMA and amino-sulfo100-PDEMA, the same behavior as of amino-sulfo0-PDEMA is achieved, showing a 193% and 149% higher release in the acidic buffer. Since this value is the highest for amino-sulfo50-PDEMA, a higher amount of TSPSA added to the particles does not automatically result in a higher release of EFX. This might be explained by EFX being repeatedly in interaction with the larger number of sulfonic acid groups slowing down or inhibiting a full release of the molecule. Additionally, the sulfonic acid groups may occupy the necessary pore space, limiting further incorporation of EFX.
With these present results, we consider amino-sulfo50-PDEMA the most promising candidate for our application. It showed the best properties from physical characterization and a stable dispersion at our considered pH values. Also, it reached the best release profile with a 193% higher release in the acidic buffer and a high total release of EFX with 51.1 μg mg−1.
3.5 Accumulation onto magnetic implants
For our desired application, the magnetization of our material is another relevant property. As our nanoparticles consist of a magnetic core surrounded by a non-magnetic shell which increases the total mass, it is expected that the magnetization per unit mass of the material (total mass) will be lower than the one of the pure core material (Fig. S16b, ESI†). However, the magnetization should still be high enough, so that the nanoparticles are able to accumulate onto a magnetic implant. For initial tests, we chose the most promising sample, which was amino-sulfo50-PDEMA. Its magnetization was measured using SQUID, and the hysteresis cycle up to 5 T at 300 K is shown in Fig. 6a. The nanoparticles are in the superparamagnetic regime at room temperature, as they show neither remanence nor coercive field. They showcase a saturation magnetization of 0.9 emu gtot−1 with a slight diamagnetic contribution at higher fields, likely caused by the attached polymer.55 As we have observed in our previous studies, this small magnetization of around 1 emu gtot−1 is enough to assure sufficient agglomeration.10,11 The accumulation behavior of the particles was tested using a setup, in which a dispersion of fluorescent-labeled particles could flow past a magnet. The scheme of the experimental setup is shown in Fig. 6b. After the experiment, the particles were removed from the magnet and quantified using fluorescence spectroscopy. During the experiment, the accumulation of the pink nanoparticles onto the magnet was observable with the naked eye, as shown in Fig. 6c. Mostly, the particles were accumulated around the edges of the magnet, where they first came into contact. | ||
| Fig. 6 (a) Magnetization curve of amino-sulfo50-PDEMA, (b) schematic representation of the setup for the particle accumulation, (c) image of a magnet taken directly after the experiment, and (d) accumulated particle concentration after particle removal from the magnet. | ||
This is mainly caused by the edge effect, as the magnetic field is stronger on the edge of a magnet.56,57 The amount of removed particles from the magnet is depicted in Fig. 6d. It turned out that after an ultrasonic treatment of two minutes, almost all particles were removed from the magnet. In the additional washing steps of the magnet, only small amounts of particles were removed, whereby after 6 min the determined particle concentration matched the used particle concentration in the experiment. This means that all particles used for the experiment had accumulated onto the magnet within the duration of the experiment. This proved that the magnetization of the polymer-functionalized particles was still high enough to ensure a comparable accumulation rate to our previously published particles, allowing a similar performance in the desired drug-targeting application.
3.6 Antibacterial effectiveness of the released amounts of EFX
The neutral and acidic supernatants of the EFX release were tested for their antibacterial effectiveness in a dilution series with S. aureus (JSNZ-WT). Therefore, a second release experiment with amino-sulfo50-PDEMA was performed, as shown in Fig. S14a (ESI†). After the release, different amounts of the respective supernatants were mixed with MHB, and an equal amount of the bacteria suspension was added. After incubation of these obtained dilutions with the bacteria, the optical density was measured to assess bacterial growth inhibition. Higher concentrations of EFX allow for greater dilution while still reducing or inhibiting bacterial growth. In Fig. S14c (ESI†), the bactericidal ability of the citrate buffer itself on the bacteria is shown. This ability of the citrate ion is known58 but is no longer observable beyond the dilution ratio of 1 to 40. Thus, starting at this dilution ratio, no effect from the citrate ion is expected, and only the EFX concentration has an impact on bacteria growth. In Fig. 7a, the obtained optical densities (OD) for the different dilution ratios of the supernatants are displayed.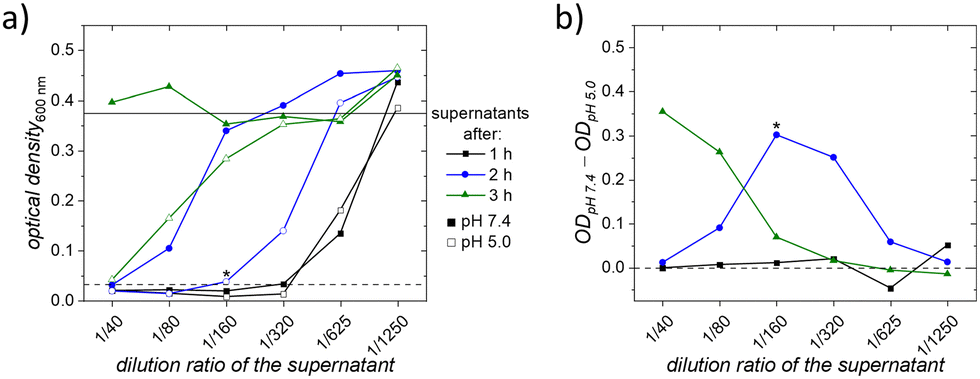 | ||
| Fig. 7 (a) Optical densities of the bacterial dispersion at 600 nm from the diluted supernatants with different pH values; dashed lines: estimated minimal values to inhibit bacterial growth; solid lines: estimated complete bacterial growth, or no reduced bacterial growth and (b) difference in optical density of different supernatants at respective dilution ratios; values above zero indicate a better performance of the acidic supernatant. *Marked values only have n = 2 instead of n = 3. The connection between the data points is just a guide for the eye. | ||
For visualization, two lines are added to Fig. 7a to indicate effectiveness thresholds: if the data point is below the dashed line, it is estimated that the EFX concentration is high enough to inhibit bacterial growth, whereas if the data point is above or at the solid line, it is estimated that no effect on reduced bacteria growth has taken place. The amounts released after one hour in both supernatants perform identically in the test and growth is inhibited even at a high dilution ratio of 1 to 320. This similar performance is also visible in Fig. 7b which visualizes the difference between the ODs of the respective supernatants. If the value in Fig. 7b is positive, the acidic supernatant has a better effect on reducing bacteria growth than the neutral one. At the dilution ratio of 1 to 320, the concentration of EFX was 0.21 μg ml−1 (neutral supernatant) and 0.25 μg ml−1 (acidic supernatant). These results of the minimum inhibition concentrations (MIC) align with the reported values for inhibiting the growth of S. aureus.59,60 With the investigation of the supernatants taken after two and three hours, it became clear that the release in the acidic environment provided higher amounts of EFX, as lower ODs were obtained for the acidic supernatant. Consequently, after three hours, there was still a high EFX release from the particles in an acidic environment. The bacterial growth was still reduced or even inhibited, which was not observed for the neutral supernatant. These results confirm that the released EFX from our core–shell–shell nanocarriers effectively inhibited the growth of bacteria and match the obtained results from our release experiments. Higher amounts of EFX were released in the acidic buffer, where the polymeric chains are open, leading to a more pronounced reduction in bacterial growth. To optimize the required EFX dosage for specific applications, the amount of EFX can be adjusted by varying the number of particles used for the patients’ treatment. Beside the effectiveness of the release EFX, also the biocompatibility of the amino-sulfo50-PDEMA was tested using a cell viability assay. In these tests, the particles showed high biocompatibility and are thus safe to use in the desired application. The complete results can be found in the ESI† in Fig. S18.
Conclusions
We synthesized dual-functionalized particles showing an amino modification on the outside and a sulfonic acid modification on the inside of our magnetic nanoporous silica core–shell nanoparticles. This dual modification allowed us to adjust the zeta potential of the nanoparticles by the amounts of sulfonic acid silane added to the particles during synthesis. In addition, we were able to further attach a pH-responsive polymer to the particles. Thereby, it was observed that the polymer content of the nanoparticles decreased with increasing equivalents of sulfonic acid silane used. As a reason for this, the lower deactivation rate during the polymerization caused by the negatively charged particles was suggested. Our dual-modified, polymer-functionalized nanoparticles showed improved stability in aqueous solution compared to the single-modified nanoparticles. In the release experiment with our model drug EFX, for all samples, a pH-dependent release was observable, where the released amount in the acidic buffer was higher. The most promising sample here was amino-sulfo50-PDEMA, which is why we tested the accumulation behavior of these nanoparticles onto a magnet as a test implant. A complete accumulation of the nanoparticles onto the magnet was observable with the naked eye and verified using fluorescence spectroscopy. It was thus shown that the magnetization of our polymer-coated particles is sufficient to adhere to a magnetic test implant, making these particles promising candidates for their use in magnetic drug targeting. Investigations of the ability to reduce bacterial growth were performed with the supernatants of amino-sulfo50-PDEMA. Here, the results match the findings of the release, and a more pronounced reduction in bacterial growth was observed when using an acidic environment for the release. In addition, the developed nanocarriers exhibit good biocompatibility.Author contributions
T. H.: data generation, particle synthesis and functionalization, investigations, formal analysis, visualization, writing – original draft. N. A.: project administration, funding acquisition, bacteria investigation. J. R.: project administration, funding acquisition, bacteria investigation. J. M.: project administration, funding acquisition, cytotoxicity test, A. H.: physisorption investigation. L. M.: thermogravimetric investigation. P. B.: project administration, funding acquisition, V. H.: data generation, I. M.: SQUID investigation and analysis. N. E.: supervision, validation, project administration, writing – editing. S. P.: supervision, writing – editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the project “Implant-Directed Magnetic Drug Targeting” (ID-MDT) (Project number 280642759) and supported by the SFB/TRR-298-SIIRI-Project 426335750. The authors are thankful for the financial support from the DFG in acquiring the SQUID magnetometer (INST 187/782-1). I. M. acknowledges the support of the Caroline Herschel Program from the Hochschulbüro für Chancenvielfalt, Leibniz University Hannover, and the Cluster of Excellence PhoenixD (EXC 2122, Project ID 390833453).Notes and references
- J. W. Wastesson, L. Morin, E. C. K. Tan and K. Johnell, Expert Opin. Drug Saf., 2018, 17, 1185–1196 CrossRef PubMed.
- H. W. Francis, N. Chee, J. Yeagle, A. Cheng and J. K. Niparko, Laryngoscope, 2002, 112, 1482–1488 CrossRef.
- J. F. Konopka, Y. Lee, E. P. Su and A. S. McLawhorn, JBJS Open Access, 2018, 3, e0007 CrossRef PubMed.
- M. S. Khan, J. Ayub Med. Coll. Abbottabad, 2008, 20, 23–25 Search PubMed.
- The German Arthroplasty Registry (EPRD), Annu. Rep., 2022, 1–172 Search PubMed.
- W. Zimmerli, J. Intern. Med., 2014, 276, 111–119 CrossRef CAS PubMed.
- W. Gao, S. Thamphiwatana, P. Angsantikul and L. Zhang, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2014, 6, 532–547 CAS.
- V. Selvarajan, S. Obuobi and P. L. R. Ee, Front. Chem., 2020, 8, 602 CrossRef CAS PubMed.
- J. Reifenrath, H. C. Janßen, D. P. Warwas, M. Kietzmann, P. Behrens, E. Willbold, M. Fedchenko and N. Angrisani, Nanomedicine, 2020, 30, 102289 CrossRef CAS PubMed.
- H. C. Janßen, N. Angrisani, S. Kalies, F. Hansmann, M. Kietzmann, D. P. Warwas, P. Behrens and J. Reifenrath, J. Nanobiotechnol., 2020, 18, 14 CrossRef.
- H. C. Janßen, D. P. Warwas, D. Dahlhaus, J. Meißner, P. Taptimthong, M. Kietzmann, P. Behrens, J. Reifenrath and N. Angrisani, J. Nanobiotechnol., 2018, 16, 96 CrossRef.
- J. He, Y. He, X. Wu, X. Zhang, R. Hu, B. Z. Tang and Q.-H. Xu, ACS Appl. Bio Mater., 2023, 6, 3433–3440 CrossRef CAS PubMed.
- Y. Yang, B. Velmurugan, X. Liu and B. Xing, Small, 2013, 9, 2937–2944 CrossRef CAS.
- D. Wang and S. Wu, Langmuir, 2016, 32, 632–636 CrossRef CAS PubMed.
- K. Fang, R. Wang, H. Zhang, L. Zhou, T. Xu, Y. Xiao, Y. Zhou, G. Gao, J. Chen, D. Liu, F. Ai and J. Fu, ACS Appl. Mater. Interfaces, 2020, 12, 52307–52318 CrossRef CAS.
- B. Li, M. Criado-Gonzalez, A. Adam, J. Bizeau, C. Mélart, A. Carvalho, S. Bégin, D. Bégin, L. Jierry and D. Mertz, ACS Appl. Nano Mater., 2022, 5, 120–125 CrossRef CAS.
- Y. Li, S. Wang, F. X. Song, L. Zhang, W. Yang, H. X. Wang and Q. L. Chen, Colloids Surf., A, 2020, 590, 124470 CrossRef CAS.
- X. Ma, K. T. Nguyen, P. Borah, C. Y. Ang and Y. Zhao, Adv. Healthcare Mater., 2012, 1, 690–697 CrossRef CAS PubMed.
- H. Fullriede, P. Abendroth, N. Ehlert, K. Doll, J. Schäske, A. Winkel, S. N. Stumpp, M. Stiesch and P. Behrens, Bionanomaterials, 2016, 17, 59–72 CrossRef.
- T. Judl, S. Popelka, E. Tomšík, M. Hrubý, M. Daniel, J. Fojt, P. Melicherčík, I. Landor and D. Jahoda, J. Clin. Med., 2024, 13, 688 CrossRef CAS PubMed.
- G. Hidalgo, A. Burns, E. Herz, A. G. Hay, P. L. Houston, U. Wiesner and L. W. Lion, Appl. Environ. Microbiol., 2009, 75, 7426–7435 CrossRef CAS.
- S. Fulaz, D. Hiebner, C. H. N. Barros, H. Devlin, S. Vitale, L. Quinn and E. Casey, ACS Appl. Mater. Interfaces, 2019, 11, 32679–32688 CrossRef CAS.
- T. Numpilai, T. Witoon, M. Chareonpanich and J. Limtrakul, Appl. Surf. Sci., 2017, 396, 504–514 CrossRef CAS.
- Y. Kuthati, R. K. Kankala, S.-X. Lin, C.-F. Weng and C.-H. Lee, Mol. Pharm., 2015, 12, 2289–2304 CrossRef CAS.
- A. A. Hwang, B. Lee, D. L. Clemens, B. J. Dillon, J. I. Zink and M. A. Horwitz, Small, 2015, 11, 5066–5078 CrossRef CAS PubMed.
- J. Wagner, D. Gößl, N. Ustyanovska, M. Xiong, D. Hauser, O. Zhuzhgova, S. Hočevar, B. Taskoparan, L. Poller, S. Datz, H. Engelke, Y. Daali, T. Bein and C. Bourquin, ACS Nano, 2021, 15, 4450–4466 CrossRef CAS.
- A. Schlossbauer, C. Dohmen, D. Schaffert, E. Wagner and T. Bein, Angew. Chem., Int. Ed., 2011, 50, 6828–6830 CrossRef CAS PubMed.
- E. Beňová, D. Bergé-Lefranc, V. Zeleňák, M. Almáši, V. Huntošová and V. Hornebecq, Appl. Surf. Sci., 2020, 504, 144028 CrossRef.
- E. Beňová, V. Hornebecq, V. Zeleňák, V. Huntošová, M. Almáši, M. Máčajová and D. Bergé-Lefranc, Appl. Surf. Sci., 2021, 561, 150011 CrossRef.
- Z. Li, D. L. Clemens, B.-Y. Lee, B. J. Dillon, M. A. Horwitz and J. I. Zink, ACS Nano, 2015, 9, 10778–10789 CrossRef CAS PubMed.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CrossRef CAS.
- H. Erdemi, H. Sözeri, M. Şenel and A. Baykal, J. Nanopart. Res., 2012, 14, 988 CrossRef.
- S. Niedermayer, V. Weiss, A. Herrmann, A. Schmidt, S. Datz, K. Müller, E. Wagner, T. Bein and C. Bräuchle, Nanoscale, 2015, 7, 7953–7964 RSC.
- M. Yang, H. Wang, Y. Jiang, S. Lai, H. Shang, X. Sun, N. Qiao and X. Zhang, Colloids Surf., A, 2021, 629, 127396 CrossRef CAS.
- L. Salminen, E. Karjalainen, V. Aseyev and H. Tenhu, Langmuir, 2022, 38, 5135–5148 CrossRef CAS.
- X. Chen, D. P. Randall, C. Perruchot, J. F. Watts, T. E. Patten, T. von Werne and S. P. Armes, J. Colloid Interface Sci., 2003, 257, 56–64 CrossRef CAS.
- H.-L. Su, L. Xu, X.-J. Hu, F.-F. Chen, G. Li, Z.-K. Yang, L.-P. Wang and H.-L. Li, Eur. Polym. J., 2021, 148, 110354 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CrossRef CAS.
- A. Polyak, H. Harting, N. Angrisani, T. Herrmann, N. Ehlert, J. Meißner, M. Willmann, S. Al-Bazaz, T. L. Ross, J. P. Bankstahl and J. Reifenrath, J. Nanobiotechnol., 2023, 21, 270 CrossRef CAS PubMed.
- R. Storjohann, B. Gericke, J. Reifenrath, T. Herrmann, P. Behrens, H. Oltmanns and J. Meißner, Int. J. Mol. Sci., 2023, 24, 2565 CrossRef CAS PubMed.
- D. M. Schlipf, S. E. Rankin and B. L. Knutson, Microporous Mesoporous Mater., 2017, 244, 199–207 CrossRef CAS.
- S. Holtfreter, F. J. Radcliff, D. Grumann, H. Read, S. Johnson, S. Monecke, S. Ritchie, F. Clow, C. Goerke, B. M. Bröker, J. D. Fraser and S. Wiles, PLoS One, 2013, 8, e71142 CrossRef CAS.
- B. Jović, M. Panić, N. Radnović, K. Živojević, M. Mladenović, V. Crnojević and N. Knežević, J. Mol. Struct., 2020, 1219, 128562 CrossRef.
- X. Xu, J. He, Y. Zeng, C. Yu and F. Zhang, Eur. Polym. J., 2020, 131, 109724 CrossRef CAS.
- T. Von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409–7410 CrossRef CAS.
- K. Ohno, T. Morinaga, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2005, 38, 2137–2142 CrossRef CAS.
- P. B. Tchounwou, C. Newsome, J. Williams and K. Glass, Met. Ions Biol. Med., 2008, 10, 285–290 Search PubMed.
- S. Puig and D. J. Thiele, Curr. Opin. Chem. Biol., 2002, 6, 171–180 CrossRef CAS.
- S. Shaligram and A. Campbell, Toxicol. In Vitro, 2013, 27, 844–851 CrossRef CAS PubMed.
- J. O. Zoppe, X. Xu, C. Känel, P. Orsolini, G. Siqueira, P. Tingaut, T. Zimmermann and H.-A. Klok, Biomacromolecules, 2016, 17, 1404–1413 CrossRef CAS PubMed.
- J. N. Kizhakkedathu, R. Norris-Jones and D. E. Brooks, Macromolecules, 2004, 37, 734–743 CrossRef CAS.
- J. N. Kizhakkedathu and D. E. Brooks, Macromolecules, 2003, 36, 591–598 CrossRef CAS.
- K. N. Jayachandran, A. Takacs-Cox and D. E. Brooks, Macromolecules, 2002, 35, 4247–4257 CrossRef CAS.
- N. Ehlert, M. Badar, A. Christel, S. J. Lohmeier, T. Luessenhop, M. Stieve, T. Lenarz, P. P. Mueller and P. Behrens, J. Mater. Chem., 2011, 21, 752–760 RSC.
- J. L. Wilson, P. Poddar, N. A. Frey, H. Srikanth, K. Mohomed, J. P. Harmon, S. Kotha and J. Wachsmuth, J. Appl. Phys., 2004, 95, 1439–1443 CrossRef CAS.
- B. A. Belyaev, A. V. Izotov, G. V. Skomorokhov and P. N. Solovev, Mater. Res. Express, 2019, 6, 116105 CrossRef CAS.
- C. Li, Y. Zhou, X. Shi, Z. Liao, J. Du, T. Shen and C. Yao, Mater. Manuf. Processes, 2020, 35, 1040–1050 CrossRef CAS.
- X.-S. Li, J.-Z. Xue, Y. Qi, I. Muhammad, H. Wang, X.-Y. Li, Y.-J. Luo, D.-M. Zhu, Y.-H. Gao, L.-C. Kong and H.-X. Ma, Int. J. Mol. Sci., 2023, 24, 9089 CrossRef CAS PubMed.
- M. C. Lima, M. De Barros, T. M. Scatamburlo, R. C. Polveiro, L. K. De Castro, S. H. S. Guimarães, S. L. Da Costa, M. M. Da Costa and M. A. S. Moreira, BMC Microbiol., 2020, 20, 127 CrossRef CAS.
- L. Oliveira, H. Langoni, C. Hulland and P. L. Ruegg, J. Dairy Sci., 2012, 95, 1913–1920 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00013k |
| ‡ Deceased. Dedicated to the memory of Peter Behrens (†13.01.2023). |
| This journal is © The Royal Society of Chemistry 2025 |