
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
BAPTA-based potentiometric polymer sensor: towards sensing inflammations and infections†
Nikol
Janić‡
ab,
Hanna
Zhukouskaya‡
ac,
Peter
Černoch
a,
Jiří
Pánek
a,
Jan
Svoboda
 a,
Milena
Hajná
a,
Alena
Řezníčková
b,
Elena
Tomšík
*a and
Martin
Hrubý
*a
a,
Milena
Hajná
a,
Alena
Řezníčková
b,
Elena
Tomšík
*a and
Martin
Hrubý
*a
aInstitute of Macromolecular Chemistry CAS, Heyrovského nám. 2, 162 00 Prague 6, Czech Republic. E-mail: tomsik@imc.cas.cz; mhruby@centrum.cz
bUniversity of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic
cDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Prague 2, Czech Republic
First published on 3rd March 2025
Abstract
Potentiometric ion sensors represent a significant subgroup of electrochemical sensors. In this study, we have developed a potentiometric sensor using an electrically conductive copolymer of 2,2′-bithiophene (BT) and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) for the selective detection of Ca2+ ions in extracellular interstitial fluids. The integration of BAPTA with its highly selective calcium chelating properties into a polymer matrix via electrochemical polymerization results in a sensitive conductive polymer layer that effectively detects the presence of calcium ions. This sensor aims at the early detection of inflammation or infection around implants because local calcium concentration is strongly elevated in interstitial fluid in such pathologies. The potentiometric study proves the incorporation of BAPTA into the polymer matrix was successful and its potential decreased upon calcium binding demonstrating the Nernstian behavior with a slope of approximately 20 ± 0.3 mV per decade in the concentration range from 0.1 mM to 1 mM. Moreover, the selectivity coefficient of –0.4 was measured by SSM and calculated from the Nicolsky–Eisenmann equation, which indicates selectivity towards Ca2+ ions with respect to Mg2+ ions.
1. Introduction
Orthopedic surgeries such as total hip and total knee arthroplasty play a pivotal role in restoring joint function for individuals afflicted with osteoarthritis. Given the rising life expectancy worldwide, the demand for these procedures increases, holding the promise of significantly enhancing the quality of life. Regrettably, orthopedic implants are uniquely susceptible to two major complications: Peri-implant sterile inflammation and microbial infections.1 These complications manifest through symptoms like pain, redness, swelling, and discharge at the surgical site. Addressing these issues often entails prolonged hospitalization and may lead to severe consequences such as osteomyelitis, implant failure, sepsis, multi-organ dysfunction, amputation, or even death. Statistics reveal that prosthetic joint infections occur in approximately 1–2% of primary arthroplasty procedures and up to 4% of revision arthroplasty procedures.2 Early detection of inflammation is paramount for timely intervention and mitigating further complications. Implantable inflammation sensors offer real-time monitoring of inflammatory markers, empowering healthcare providers to promptly identify and manage inflammation.3 Early detection enables healthcare professionals to initiate targeted antibiotic therapy or consider implant removal if necessary, thereby preventing infection spread and minimizing patient morbidity.In bacterial infections, additional markers include pH reduction, defensin production, calprotectin release, iron depletion, and elevated Ca2+ ion concentration, particularly at the bone interface.4–6
Calcium ions are one of the most important and versatile second messengers in cell signaling, playing pivotal roles in transcription, apoptosis, activation, cell adhesion, exocytosis, metabolism, and proliferation.7–9 The dysregulation of mitochondrial Ca2+ signaling is associated with various pathological scenarios, including cancer, neurological defects, and cardiovascular diseases. Furthermore, calcium signaling is crucial for the proper function of immune cells. Mutations in genes encoding the calcium release-activated channel, a key Ca2+ channel in the plasma membrane of many immune cells can disrupt its function, leading to severe combined immunodeficiency in humans.10 Notably, extracellular calcium levels are tightly regulated at 0.8–1.2 mM, but they significantly rise as an “alert signal” early in the inflammatory process.11 Thus, precise detection and measurement of calcium levels are vital for elucidating possible deviations in biological processes.12
Various potentiometric sensors have been developed proving their utility in analytical chemistry. They demonstrate exceptional proficiency in detecting crucial targets, showcasing remarkable analytical capabilities. Present efforts are primarily directed towards advancing in situ formats, facilitating direct extraction of valuable information. The inherent advantages of potentiometric assessment, including technical simplicity, affordability, and minimal space and energy demands, underscore its significance.13 Moreover, potentiometric sensors are uniquely suitable for creating miniaturized versions that maintain satisfactory analytical performance. Leveraging these attributes, the objective is to elevate the practicality and accessibility of potentiometric sensor technology across diverse applications.
A variety of sensors have been engineered to detect the concentration of Ca2+ ions, employing diverse principles and materials to ensure high sensitivity and selectivity.14 For instance, a highly stable calcium ion-selective sensor based on poly(3,4-ethylenedioxythiophene)-poly(styrene-sulfonate) (PEDOT-PSS) polymer layer was prepared by electrochemical polymerization using the potentiodynamic cyclic voltammetry. The prepared Ca2+-specific electrode showed a high sensitivity of 37.7 mV per decade over the calcium ion concentration range of 10−4 to 10−1 M with a rapid response time of less than 20 seconds. Additionally, the polymer layer demonstrated negligible interference from other ions commonly found in human biofluids, and it had a reduced potential drift, indicating high stability and suitability for long-term monitoring.15
To prepare novel ion-sensitive field-effect transistor devices suitable for Ca2+ detection in liquids, mixed Langmuir–Blodgett monolayers containing carboxylic ionophores were used. Rochefeuille et al. reported that such devices exhibited not just sensibility to electrical parameters, but were also sensitive to the surrounding ions, demonstrating a linear response with a slope of 19 mV pNa−1 in the range of 10−6 to 10−2 M and a slope of 23 mV pCa−1 in the range of 10−4 to 10−2 M.16
A study by Arunkumar et al. described the efficacy of chemically prepared bichromophoric squaraine dye foldamer sensor for the selective detection of calcium ions. In order to investigate the sensitivity of those sensors towards Ca2+ detection, spectroscopic methods were used, including UV-vis absorption spectroscopy and fluorometry. The sensor utilized a bichromophoric system designed to specifically bind calcium ions, demonstrating significant selectivity and sensitivity in the presence of other cations, such as Na+, K+, and Mg2+.17
Another approach to the preparation of highly sensitive and selective Ca2+ sensors was developed by Park et al. A terthiophene dendron-based sensor, modified with tetraethylene glycol, was able to detect calcium ions at concentrations as low as 10−8 M, thus demonstrating a strong interaction between the ions and chelating groups on the surface of the sensor.18
Ishiwari et al. prepared a solid-state fluorescent calcium ion sensor for the extra-cellular Ca2+ imaging. It performed exceptionally well at high Ca2+ concentrations, even amidst competing alkali and alkaline-earth metal ions, sugars, and amino acids.19
The research of Qin et al. presented a significant advancement in the design of plasticizer-free ion-selective sensors, based on methyl methacrylate-co-decyl methacrylate polymer matrix, with potential applications extending to in vivo sensing and other areas requiring high biocompatibility and stability.20
Potentiometric ion sensors, also known as ion-selective electrodes (ISEs), represent a significant subgroup of electrochemical sensors. These sensors, which most often rely on polymeric membranes containing neutral or charged carriers (ionophores), offer a versatile means of determining numerous inorganic and organic ions. Of particular interest are solid-type ion sensors, which find application in industrial and medical settings as substitutes for traditional liquid inner contact ion-selective electrodes.21–24 The appeal of these sensors lies in their potential for easy miniaturization and cost reduction. Typically, they feature a polymer ion-selective membrane, commonly poly(vinyl chloride) (PVC) with a specific plasticizer and ionophore, deposited onto a solid inner electrode. Many solid-type ion sensors utilizing conductive polymers employ PVC as the membrane polymer matrix. However, a prevalent issue with these sensors involves water penetration through the membrane, leading to the formation of thin aqueous layers or pools of water at the electrode interfaces. This phenomenon can result in potential instability and inadequate adhesion of the membrane to the solid contact, representing a primary cause of sensor failure. Moreover, the gradual leak of the ionophore, which is not covalently bound in the membrane, strongly limits the lifetime of such sensors.25
Conducting polymers serve as invaluable ion-to-electron transducers in solid-state ISEs for several compelling reasons. Firstly, they possess electronic conductivity, establishing ohmic contact with materials possessing high work functions like carbon or gold. Secondly, conducting polymers can be conveniently electrodeposited onto electronic conductors through the electrochemical polymerization of a diverse range of monomers.26 Alternatively, several conducting polymers are soluble, enabling their deposition from solution. Thirdly, conducting polymers exhibit electroactivity coupled with mixed electronic and ionic conductivity. This unique characteristic allows them to efficiently transduce an ionic signal into an electronic one within the solid state.27 The multifaceted nature of conducting polymers thus renders them exceptionally well-suited for ion-to-electron transduction in solid-state applications. Among such polymers, polythiophenes are highly promising materials for the creation of conductive layers in ISEs. Polythiophenes consist of covalently linked electron-rich heterocyclic monomer units, which create a system of conjugated double bonds. This enables the electric conductivity of the resulting polymer and provides other useful properties, including superior biocompatibility, environmental stability, and tunable fluorescence. Moreover, the chemical structure of polythiophenes can be easily modified in order to change their physical properties, which is why they are a great choice for sensor fabrication.28
Ion selectivity can be achieved through two distinct methods. One approach involves utilizing the conducting polymer solely as an ion-to-electron transducer (solid contact) in conjunction with traditional ion-selective membranes. Alternatively, selectivity can be induced by directly incorporating ion-recognition sites into the conducting polymer matrix. This can be accomplished by doping the conjugated polymer with counter ions containing ion-complexing groups or by covalently binding ion-recognition sites to the conjugated polymer chain.29 This latter approach necessitates precise control over both the electronic and ionic transport properties of the membrane to optimize ion recognition and transduction processes. The primary challenge lies in achieving a selective ionic response while minimizing redox interference. However, the potential benefits are substantial, as the covalent binding of ion-recognition sites to the conducting polymer backbone enables the integration of ion-recognition sites and ion-to-electron transducers within the same macromolecule.
In this study, we have developed a potentiometric sensor using an electrically conductive copolymer of 2,2′-bithiophene (BT) and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetracarboxylic acid (BAPTA) for the selective detection of Ca2+ ions in extracellular interstitial fluids. This sensor aims at the early detection of inflammation or infection around implants because BAPTA is a selective chelator of calcium cations. An increase in the electrical potential of such a sensor due to elevated concentration of calcium in adjacent tissues would be a clear sign of ongoing infection. To address the solubility issues of BAPTA, its tetramethyl ester dihydrochloride (TMeBAPTA) was used instead for the polymer preparation, which was followed by the hydrolytic deprotection of chelating carboxyl groups on the electrode surface. As observed in this study, the electron-rich aromatic ring moieties (N,N,O-trisubstituted o-aminophenol) present in the structure of BAPTA and TMeBAPTA are susceptible to oxidative polymerization, and TMeBAPTA readily electropolymerized with BT, forming a sensitive conductive polymer layer with BAPTA chelator (after removal of protecting methyl ester groups), directly bound to the polymer chains. The deprotection of carboxyl groups in the copolymer layer was done by methyl ester saponification with sodium hydroxide solution after the polymerization. The conjugated polymer chain served as an electron-withdrawing group, allowing adjustment of calcium sensitivity to bio-relevant millimolar levels.
The working hypothesis is that it is possible to construct a polymer layer electrode-based sensor, which would not need sample preparation for in situ measurement, is easily miniaturizable as a chip, is inexpensive, does not need additional large instrumentation, has minimum power consumption, and does not leak ionophore as the calcium chelator is covalently bound to the conductive polymer electrode.
2. Materials and methods
2.1. Reagents and materials
Sodium chloride, acetonitrile, methanol, diethyl ether, ethanol, acetone, and sodium hydroxide were obtained from Lach-ner Ltd (Neratovice, Czech Republic). 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), thionyl chloride, 2,2′-bithiophene, tetrabutylammonium hexafluorophosphate and calcium chloride were purchased from Sigma-Aldrich Ltd (Burlington, MA, USA). Graphite pencil leads (4190 HB, ø 2 mm) were produced by Koh-i-Noor Hardtmuth (České Budějovice, Czech Republic). Aqueous solutions of NaCl and CaCl2 were prepared from Milli-Q water (Millipore Corp.), resistivity of 18.2 MΩ cm at 25 °C.2.2. Liquid-state 1H nuclear magnetic resonance (NMR) spectroscopy
Proton NMR spectra were recorded on Bruker NEO 400 spectrometer (Bruker; Billerica, MA, USA) operating at 400 MHz, which allowed to determine the chemical structure and composition of the analyzed compounds as well as their reaction yields.2.3. Fourier-transform infrared spectroscopy with attenuated total reflectance (ATR-FTIR)
The ATR-FTIR measurements of the samples were performed using a Nexus 870 FTIR spectrometer (Thermo Nicolet; Madison, WI, USA) equipped with a mercury cadmium telluride detector and a universal ATR accessory with a diamond prism. The spectra were averaged over 256 scans at a resolution of 4 cm−1. This method was used to determine the chemical structure of compounds.2.4. Mass spectrometry (MS)
The molecular mass of compounds was determined on an LCQ Fleet mass analyzer with electrospray ionization (Thermo Fisher Scientific; Waltham, MA, USA).2.5. Cyclic voltammetry (CV) deposition
All depositions were carried out using a Metrohm® AUTOLAB Potentiostat/Galvanostat PGSTAT302N with a frequency analyzer FRA32M module controlled by the software NOVA 2.1.4. Three-electrode cell configurations were used for all electrochemical measurements: working electrodes (carbon rods), counter-electrode (Pt), and reference electrode Ag/AgCl. The deposition was done at room temperature. A Faraday cage was used to reduce electrical noise. Al least 10 electrodes were obtained from one deposition mixture, and the reproducibility of the deposition was followed by comparing the CV curves.2.6. Potentiometry study
All measurements were performed using a 6-channel high input impedance voltmeter with an input impedance of 1010 Ω (Lawson Laboratories, Malvern, PA, USA). The 0.15 M NaCl was used as electrolyte and all measurements were performed at room temperature. The 5 electrodes simultaneously were connected with one reference electrode, and potentiometric read out was measured for each concentration point. The error bars in the graph indicates the standard deviation of the measurement.2.7. X-ray photoelectron spectroscopy (XPS)
The XPS spectra were measured on a K-Alpha+ spectrometer (ThermoFisher Scientific; East Grinstead, UK) operating at a base pressure of 1 × 10−6 Pa. All samples were analyzed using a microfocused, monochromated Al Kα X-ray radiation (400 μm spot size) at an angle of incidence of 30° (measured from the surface) and an emission angle normal to the surface. The kinetic energy of the electrons was measured using a 180° hemispherical energy analyzer operated in the constant analyzer energy mode at 200 eV and 50 eV pass energy for the survey and high-resolution spectra respectively. The data acquisition and processing were performed using the Thermo Avantage software. All spectra were referenced to the C 1s peak of hydrocarbons at 285.0 eV.2.8. Synthesis of TMeBAPTA
The glassware was dried overnight in an oven at 120 °C. BAPTA tetramethyl ester di-hydrochloride (Scheme 1) was synthesized by a modified procedure according to ref. 30. | ||
| Scheme 1 Synthesis of TMeBAPTA. | ||
Into a 100 mL glass vessel with a magnetic stirrer, BAPTA (200 mg, 0.42 mmol, 1 eq.) was placed and under the flow of argon 10 mL of methanol was added through a septum. The suspension was cooled in an ice bath and within 8 minutes 1.5 mL of thionyl chloride (21 mmol, 50 eq.) was dropwise added from a syringe during 10 minutes. The mixture was stirred in the ice bath for the next 2 h, and after that, the reaction continued at room temperature (RT) for an additional 20 h. The obtained mixture was refluxed for the next 5 h and an excess of methanol and thionyl chloride was subsequently removed under vacuum (200–100 mbar) at 50 °C. The viscous residuum was washed with diethyl ether three times and dried in vacuo, resulting in a yield of 248 mg (97.6%).
1H NMR (400 MHz, DMSO): δ 12.40 (s, 4H), 6.96 (m, 2H), 6.84 (m, 4H), 6.74 (m, 2H), 4.23 (s, 4H), 4.03 (s, 8.00H). 1H NMR of TMeBAPTA (400 MHz, DMSO): δ 9.19 (s, 2H), 6.94 (m, 2H), 6.83 (m, 4H), 6.71 (m, 2H), 4.17 (s, 4H), 4.10 (d, 8H), 3.47 (d, 12H).
FT-IR: ν(O–H) 3150–2891 cm−1, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1693 cm−1, ν(aromatic ring) 1595–1504 cm−1, ν(–CH2– in dioxyethylene) 1483–1429 cm−1, ν(tertiary amine) 1398–1375 cm−1, ν(C–O–C) 1237–1205 cm−1, ν(C–O) 1124–1038 cm−1, δ(hydrogen bonds) 972–868 cm−1, δ(C–H) 750–716 cm−1. FT-IR of TMeBAPTA: ν(O–H) 2951 cm−1 (traces of methanol), ν(CO) 1736 cm−1, ν(aromatic ring) 1595–1502 cm−1, ν(–CH3 in ester) 1435 cm−1, ν(C–O–C) 1122 cm−1, ν(C–O) 1043 cm−1, δ(C–H) 993–744 cm−1.
O) 1693 cm−1, ν(aromatic ring) 1595–1504 cm−1, ν(–CH2– in dioxyethylene) 1483–1429 cm−1, ν(tertiary amine) 1398–1375 cm−1, ν(C–O–C) 1237–1205 cm−1, ν(C–O) 1124–1038 cm−1, δ(hydrogen bonds) 972–868 cm−1, δ(C–H) 750–716 cm−1. FT-IR of TMeBAPTA: ν(O–H) 2951 cm−1 (traces of methanol), ν(CO) 1736 cm−1, ν(aromatic ring) 1595–1502 cm−1, ν(–CH3 in ester) 1435 cm−1, ν(C–O–C) 1122 cm−1, ν(C–O) 1043 cm−1, δ(C–H) 993–744 cm−1.
The MS spectra of TMeBAPTA are given in the ESI† (Fig. S1).
2.9. Electrochemical deposition of the sensing layer and potentiometric testing in the presence of Ca2+
The PBT/BAPTA polymer (Scheme 2) was prepared by electrochemical polymerization using potentiodynamic cyclic voltammetry of bithiophene and TMeBAPTA in the presence of tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting salt in acetonitrile as an electrolyte. The TBAPF6 not only enhanced the electric conductivity of the acetonitrile but also compensated for the electric charge of the developing conductive polymer layer. The reaction was performed as follows: TBAPF6 (80 mg, 0.2 mmol, 10 eq.) and bithiophene (49.9 mg, 0.3 mmol, 15 eq.) were thoroughly dissolved in 5 mL of acetonitrile each, while TMeBAPTA (10.7 mg, 0.02 mmol, 1 eq.) was dissolved in 1 mL of MeOH with further addition of 10 mL of acetonitrile. The prepared solutions were mixed and poured into a three-electrode cell consisting of graphite as a working electrode, previously cleaned in an ultrasonic bath using acetone, ethanol, and water; Pt as a counter electrode, and Ag/AgCl as a reference electrode. The electrochemical polymerization was carried out in the potential window from 0 V to +1.5 V at a scan rate of 100 mV s−1 for 10 scans. After finishing the electrochemical deposition of PBT/BAPTA, the working graphite electrodes were washed with distilled water and dried on air. To remove the protective methyl groups from the PBT/BAPTA layer, the electrode coated with the polymer was soaked in 0.1 M solution of NaOH for 24 h, washed with distilled water, and dried. | ||
| Scheme 2 Synthesis of PBT/BAPTA copolymer, representing two possible ways of inclusion of the TMeBAPTA monomer units into the polymer chain. | ||
The graphite electrodes, covered with a layer of deprotected PBT/BAPTA were immersed in 100 mL of 0.15 M NaCl solution for 1 h at RT in order to reach the ion exchange equilibrium. Aliquot volumes of 0.01 M CaCl2 solution were added into the cell in 5 min intervals and the values of electric potential was recorded. The total volume of added CaCl2 solution reached 5 mL (with 200 μL aliquots).
3. Results and discussion
Each of the currently used methods for calcium determination possesses some drawbacks. Some need expensive large instrumentation not compatible with miniaturization and may also require addition of potentially toxic chemicals (spectrophotometry/fluorimetry with chromogenic/fluorogenic chelators, chelatometric titration). Additionally, they cannot be miniaturized for in situ measurement without sample processing (all abovementioned methods). The current calcium ion-selective electrodes are also membrane-type with ionophores noncovalently dissolved in the polymer film and therefore suffering from due to leakage of the ionophore, which limits shelf-life and the released ionophore can also be toxic. Our sensor would not need sample preparation for in situ measurement, is easily miniaturizable, inexpensive, not needing additional large instrumentation, with minimum power consumption, and does not leak ionophore as the calcium chelator is covalently bound to the conductive polymer electrode.The BAPTA is a unique chelating agent, which is primarily used for its ability to bind calcium ions and is often utilized in soluble calcium-sensing fluorescent probes. Due to its properties such as high calcium affinity, rapid binding kinetics, pH stability, and especially selectivity towards other cations of biogenic elements such as Mg2+,31 we hypothesize that BAPTA-based films can be effectively utilized as sensors for Ca2+ ions. The preparation of TMeBAPTA monomer involved the methylation of carboxyl groups yielding the corresponding tetramethyl ester as salt with hydrochloric acid. It can be seen from the 1H NMR spectra (Fig. 1) that BAPTA has been successfully converted to TMeBAPTA. This structural transformation involves the replacement of the carboxyl hydrogens with methyl groups, which is proved by the disappearance of a peak at δ 12.40 and the appearance of a new peak at δ 3.47.
 | ||
| Fig. 1 1H NMR spectra of BAPTA (left) and TMeBAPTA (right). | ||
The infrared spectra shown in Fig. 2 also confirm that BAPTA was transformed into its tetramethyl ester due to the change in peaks at 1693 and 3150–2891 cm−1 after the reaction, attributed to the CO and –OH stretching of the carboxylic group bonds, respectively. In the case of TMeBAPTA, its spectrum displayed new peaks, where the main bands associated with ester groups were at 1736, 1435, and 1122 cm−1.
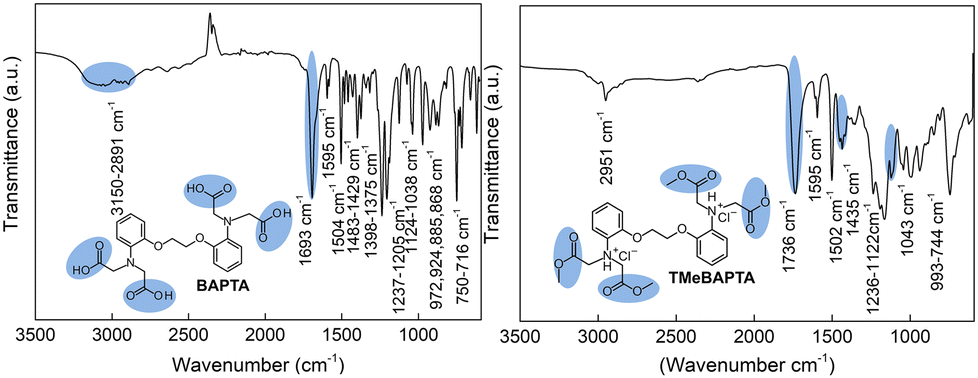 | ||
| Fig. 2 ATR-FTIR spectra of BAPTA (left) and TMeBAPTA (right). | ||
To incorporate TMeBAPTA into the polymer matrix, we utilized the presence of two electron-rich aromatic rings able to oxidatively (co)polymerized into a conjugated polymer.
This approach brings the conjugated electrically conductive chain to the proximity of the chelating unit, a charge of which is changed upon calcium binding causing a potentiometric response. The copolymer with bithiophene was selected as the sensing layer material as TMeBAPTA homopolymer is water-soluble. Polymerization must be done in organic solvent due to the solubility of bithiophene (Scheme 1, it is worth mentioning that the position on BAPTA to which the polymer chain is attached is only hypothesized and may also occur in the para position to the nitrogen atom of the BAPTA moiety. However, no effect on the chelating ability of BAPTA was observed). However, BAPTA is not soluble in such an environment, which was solved by its esterification and then deprotection after polymerization.
The cyclic voltammogram of PBT/TMeBAPTA (Fig. 3a) showed that the current started to increase significantly around +0.8 V, suggesting the beginning of the electrochemical deposition of the material onto the working electrode. The optimal potential range was between +0.8 and +1.2 V, which means that the monomers effectively undergo redox reactions to form the polymer chain, despite the oxidation potential not being particularly high (for details see Fig. 3b). The cyclic voltammetry curves of the polymer deposition have only one oxidation and one reduction peak, which means that polymerization proceeded without any intermediate steps or side reactions. The consistent peaks in the multiple cycles suggested that the polymer undergoes repeated redox processes without degradation, and the progressive increase of the currents with the number of scans proves that such electrochemical deposition is suitable for the sensing film preparation. Moreover, all these factors indicate that PBT/TMeBAPTA is suitable for applications requiring stable and reversible electrochemical behavior to be used as sensors. It was also tested that films deposited with 10 cycles exhibited optimal sensitivity and selectivity, compared to the thicker films formed with 20 cycles or thin ones with 5 cycles, making them more suitable for further experiments.
 | ||
| Fig. 3 Cyclic voltammograms of the deposition of PBT/TMeBAPTA film recorded in ACN/MeOH solution with the scan rate of 100 mV s−1. The reference electrode was Ag/AgCl/3 M KCl. (a) demonstrates the whole voltammogram and deposition behavior of the polymer, and (b) displays multiple cycles of the deposition process on a narrow potential window, highlighting changes over time. | ||
The conclusion was made based on the potentiometric responses of sensing films towards Ca2+ ions. That is why further investigations were done only for the sensing film obtained by 10 CV cycles.
According to XPS measurements, we confirmed the presence of both the BAPTA (nitrogen) and BT (sulfur) monomeric units in the final sensing film (Fig. 4).
 | ||
| Fig. 4 High-resolution S 2p, N 1s, and Ca 2p XPS spectra of PBT/BAPTA film after immobilization of Ca2+ ions. | ||
The BAPTA monomer contains two electron-rich aromatic rings from which only one or both can be embedded into the polymer film backbone, the suggested routes are depicted in Scheme 2. As there is only one peak recorded for nitrogen (singlet at 399.9 eV) N 1s, we assume that the BAPTA moiety is symmetrically embedded by both its aromatic rings. Also, the high-resolution XPS spectra of sulfur (S 2p) doublet located at 164.1 and 165.3 eV correspond to the non-charged sulfur in the BT ring.
To evaluate the efficiency of the PBT/BAPTA layer as a potentiometric polymer sensor, its electrical response to calcium chelation was studied in the presence of an excessive amount of other physiological ions (in Fig. 5). The 5 electrodes were attached to the holder and connected to the high input impedance voltmeter. In the middle of the holder was placed the Ag/AgCl (3 M KCl) reference electrode that was connected to the same voltmeter. The 0.15 M NaCl was used as an electrolyte and all measurements were performed at room temperature. The electrodes were left to equilibrate at least up to 1 h to give the constant potentiometric response. The electric potential of neat graphite electrodes and the ones coated only with PBT film was examined to confirm that both (support and support with PBT film only) are not sensitive to Ca2+ ions (Fig. 5a and b) to compare it to the potential of electrodes covered with PBT/BAPTA film. At least 5 deposited films were investigated for every case as replicates (standard deviations among them are represented by error bars). For a divalent Ca2+ ion, the Nernstian slope at RT should be 29.58 mV per decade change in concentration (often rounded to 30 mV per decade for practical purposes).32 However, in some cases experimental data revealed a slope, that exceeded this value, indicating a super-Nernstian behavior that could be possibly explained by material properties, membrane interactions, and environmental conditions.33 A slope of approximately 20 ± 0.3 mV per decade was observed in the current work. The deviation from the ideal Nernstian response 30 mV per decade for divalent ions can be given by polymer film thickness, ion transport limitations, polymer properties, electrostatic proximity effects as the chelator moieties are close to each other, etc. The potentiometric response curve exhibited a linear region in the range of logarithmic calcium cation concentrations from 10−4 to 10−3.4 mol L−1 (see Table 1 for head-to-head comparison with literature data). Chelation has led to the formation of a complex between Ca2+ and multiple donor atoms of the functional groups of BAPTA, namely nitrogen and oxygen, which have surrounded the calcium cation. This process shifted the electron density from the benzene rings in the conjugated polymer chain, thus the binding of Ca2+ has resulted in a decrease in the pKa of the remaining carboxyl groups, meaning that they are more likely to deprotonate, which is similar to the behavior of EDTA during its chelation of metal cations.34 On the other hand, the examined neat electrodes and the ones coated only with PBT (Fig. 5b and c) did not show any change in electric potential in the studied concentration range of Ca2+ ions, which confirms the chelating ability of the prepared PBT/BAPTA films.
 | ||
| Fig. 5 Potentiometric detection of Ca2+ for (a) clean electrode, (b) poly(bithiophene), (c) PBT/BAPTA sensor films in 0.15 M NaCl, (d) potentiometric detection of Mg2+ by PBT/BAPTA sensor films and (e) potentiometric detection of Ca2+ ions by PBT/BAPTA sensor films in the presence of interfering ions Zn2+ and Mg2+. | ||
| Type of sensing layer | Detection method | Linear range (M) | Time of detect. | Ref. |
|---|---|---|---|---|
| nr – not relevant. | ||||
| PEDOT-PSS | 37.7 mV per decade | 10−4 to 10−1 | <20 s | 15 |
| LB monolayers with ionophore | 23 mV pCa−1 | 10−4 to 10−2 | — | 16 |
| Terthiophene dendron-based sensor | Fluorescent | 8 × 10−8 to 12 × 10−7 | — | 18 |
| Polypyrrole-calcion | 29.1 mV pCa−1 | 10−1 – 10−4 | nr | 35 |
| Chemically-crosslinked polyacrylic acid gel | Fluorescent | 0.1 – 1.1 × 10−3 | nr | 36 |
| Manganese-based MRI contrast agent | Magnetic resonance imaging | 0–100 × 10−6 | nr | 37 |
| Ca-selective dye, BEEF-CP | Fluorescent | 10 × 10−9 to 1 × 10−6 | nr | 38 |
| BAPTA | Fluorescent | — | nr | 39 |
| Calcium ion-selective liquid membrane | Microelectrode technique, 25 mV per decade | 10−5 – 10−2 | nr | 40 |
| Ca2+ dyes | Fluorescent | In the cell | nr | 41 |
| Physiologically relevant Ca2+ concentration 0.8–1 × 10−3 | ||||
| PBT/BAPTA copolymer | 20 mV per decade | 0.1 × 10−3 to 1 × 10−3 | 60 s | This work |
Additionally, the high-resolution XPS spectra of PBT/BAPTA film after immobilization of Ca2+ (Fig. 4) ions confirm the presence of Ca 2p doublet at 347.4 and 351.0 eV (Fig. 4). It proves that constructed sensing polymer film can be used for Ca2+ detection. The Survey XPS spectra of PBT/BAPTA are presented in ESI† in Fig. S2.
The mechanism underlying the detection of Ca2+ ions has not been fully established yet.42,43 Based on the measured data, the following explanation for the mechanism of the developed polymer sensor membrane is proposed: XPS measurements confirm the detection of Ca2+ ions, implying that Ca2+ ions were incorporated during the potentiometric measurements. According to the mechanism illustrated in Fig. 5c, Ca2+ ions are bound to BAPTA. The Mg2+ and Zn2+ could interfere with the PBT/BAPTA sensing film, which is why the potentiometric response of the film in the presence of individual ions was studied, and the results are shown in Fig. 5d for Mg2+ ions. The relevant concentration of free Mg2+ ions in the body is in the range from 2.06 mmol L−1 to 2.54 mmol L−1. In our research, we decided to check the response of the PBT/BAPTA film sensing film towards Mg2+ ions in the range from 0.1 mmol L−1 to 3 mmol L−1. It is obvious from the Fig. 5d that the film is sensitive towards Mg2+ ions and has a linear response in the concentration range from 0.2 mmol L−1 to 0.5 mmol L−1 with a response slope of 30.6 mV per decade. It must be emphasized that the measured potential drops substantially when the PBT/BAPTA sensing film interacts with Mg2+ ions, and it's in the range from 35 mV vs. Ag/AgCl reference electrode. The measured potential for individual Ca2+ ions is in the range from 80 mV to 96 mV vs. Ag/AgCl reference electrode (see Fig. 5c). The PBT/BAPTA sensing film does not respond towards Zn2+ ions.
The potentiometric response of the PBT/BAPTA sensing film towards Ca2+ ions in the presence of 15 μmol L−1 of Zn2+ and 2 mmol L−1 Mg2+ ions was recorded and shown in Fig. 5e. The response slope is 17.5 mV per decade in a similar concentration range as for the test where no interfering ions were added. However, the measured potential drops compared to the individual Ca2+ ions detection and it is similar to the one measured for the Mg2+ ions detection. To prove that our sensing film can detect Ca2+ ions in the presence of the interfering ions, the XPS analysis was conducted (on the sensing film after potentiometric measurements) to study the nature of ions that were interacting with the PBT/BAPTA sensing film, and the results are presented in ESI† Fig. S3 (Survey XPS for 2 electrodes) and Fig. S4 for Ca 2p, Mg 2p and Zn 2p high-resolution spectra. Both Ca2+ and Mg2+ ions are detected in the PBT/BAPTA sensing film. The high-resolution of Zn 2p spectrum is noisy and could not be considered reliable. Based on the XPS data it is concluded that PBT/BAPTA sensing film could detect Ca2+ ions even in the presence of interfering Mg2+ and Zn2+ ions. To confirm it the selectivity coefficient was calculated.
The selectivity study was performed to determine the Ca2+ ions sensor's ability to select the primary ions over the interference ions. The selectivity study was carried out using the separate solution method (SSM) to determine the sensor's logarithm selectivity coefficient via the Nicolsky–Eisenmann equation below:
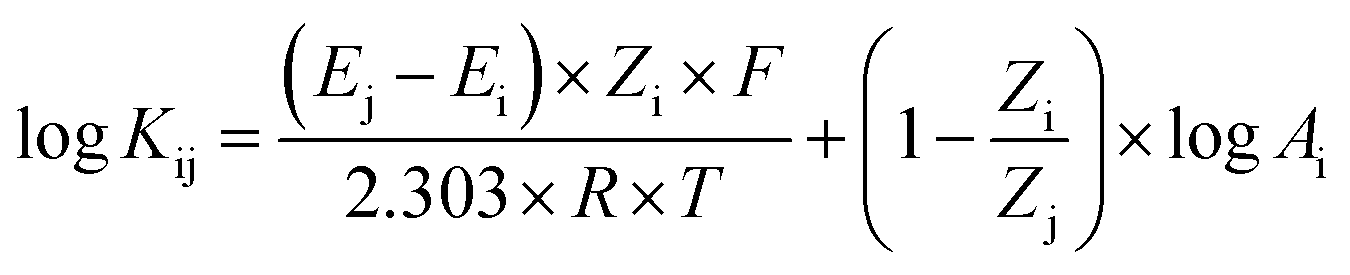
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kij = −0.4. It can be seen from this value that the PBT/BAPTA sensing film possesses selectivity towards Ca2+ ions with respect to Mg2+ ions.43
Kij = −0.4. It can be seen from this value that the PBT/BAPTA sensing film possesses selectivity towards Ca2+ ions with respect to Mg2+ ions.43
In the current research, the analytical properties of the developed sensor were studied in the presence of excessive amounts of other physiological ions, particularly 0.15 M NaCl. The results of this study confirm the fact that the PBT/BAPTA sensing film is suitable for the detection of Ca2+ ions even in the presence of the interfering ions (Mg2+), see Table 1. However, we summarized the data reported in the literature for other types of films for Ca2+ detection either by potentiometric or fluorescent methods (as the most investigated). Particularly, the fluorescent method is one of the most investigated,18,36,38,39,41 but this type of detection is not suitable for integration into implants. The magnetic resonance imaging37 and microelectrode technique40 are methods of detection that could be applied to study the cells. Generally, it can be summarized that specific application of Ca2+ detection requires the development of tailored polymer sensing films that work for particular purposes only.
4. Conclusion
We successfully developed a potentiometric sensor utilizing an electrically conductive copolymer of BT and BAPTA. We clearly demonstrated the superior potential of our PBT/BAPTA sensor to selectively detect Ca2+ ions in extracellular interstitial fluids-relevant concentrations to facilitate early detection of inflammation or infection around implants. The integration of BAPTA and its highly selective chelating properties into a polymer matrix via electrochemical polymerization results in a sensitive conductive polymer layer that effectively binds calcium ions. Our study demonstrates the incorporation of BAPTA into the polymer matrix and its functionality as a calcium sensor through an increase in electric potential upon calcium binding. The observed increase demonstrates a response slope of approximately 20 ± 0.3 mV per decade, deviating from the theoretical Nernstian value of ∼30 mV per decade. This phenomenon may be influenced by various conditions. Moreover, the selectivity coefficient of −0.4 was measured by SSM and calculated from the Nicolsky–Eisenmann equation, which indicates selectivity towards Ca2+ ions with respect to Mg2+ ions. The initial results are indeed promising, further research could see this technology becoming a vital part of post-operative care and an advance in medical diagnostics.The practical deployment of the sensor in clinical settings will include integration to implants such as hip and knee replacements (with suitable electronics for wireless signal transfer to external reader) and development of compact portable user friendly devices enabling real-time measurement at orthopaedic operating rooms, general practitioners’ offices as well as clinical analytical laboratories.
Author contributions
Nikol Janić – investigation, data curation, writing – original draft; Hanna Zhukouskaya – investigation, data curation, writing – original draft; Peter Černoch – investigation, data curation, visualization; Jiří Pánek – supervision, formal analysis, writing – review & editing; Jan Svoboda – investigation, data curation, visualization, writing – original draft; Milena Hajná – investigation; Alena Řezníčková – supervision, formal analysis; Elena Tomšík – conceptualization, visualization, writing – review & editing, supervision, data curation; Martin Hrubý – conceptualization, supervision, visualization, writing – review & editing, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank for financial support to the Ministry of Education, Youth and Sports of the Czech Republic (grant # EATRIS CZ LM2023053). The study was co-funded by the project New Technologies for Trans-lational Research in Pharmaceutical Sciences/NETPHARM, project ID CZ.02.01.01/00/22_008/0004607, co-funded by the European Union. Financial support from the Ministry of Education, Youth and Sports of the Czech Republic (grant INTER-EXCELLENCE II # LUAUS24272) is also acknowledged.References
- H. Jacobs, G. H. Seeber, K. Allers and F. Hoffmann, BMC Musculoskeletal Disord., 2021, 22, 711 CrossRef PubMed.
- I. Potapova, Diagnostics, 2013, 3, 356–371 CrossRef PubMed.
- C. R. Arciola, S. Ravaioli, R. Mirzaei, P. Dolzani, L. Montanaro, M. Daglia and D. Campoccia, Int. J. Mol. Sci., 2023, 24, 16669 CrossRef CAS PubMed.
- T. G. Nakashige, B. Zhang, C. Krebs and E. M. Nolan, Nat. Chem. Biol., 2015, 11, 765–771 CrossRef CAS PubMed.
- M. K. Yilmaz, A. Abbaszadeh, S. Tarabichi, I. Azboy and J. Parvizi, Antibiotics, 2023, 12, 1054 CrossRef CAS PubMed.
- Q. Y. Zhang, Z. B. Yan, Y. M. Meng, X. Y. Hong, G. Shao, J. J. Ma, X. R. Cheng, J. Liu, J. Kang and C. Y. Fu, Mil. Med. Res., 2021, 8, 48 CAS.
- E. C. David, Cell, 2007, 131, 1047–1058 CrossRef PubMed.
- Z. Dong, P. Saikumar and J. M. Weinberg, Annu. Rev. Pathol.: Mech. Dis., 2006, 1, 405–434 CrossRef CAS.
- K. Thurley, A. Skupin, R. Thul and M. Falcke, Biochim. Biophys. Acta, 2012, 1820, 1185–1194 CrossRef CAS PubMed.
- A. B. Parekh, Nat. Rev. Drug Discovery, 2010, 9, 399–410 CrossRef CAS PubMed.
- D. K. Atchison and W. H. Beierwaltes, Pflugers Arch. – Eur. J. Physiol., 2013, 465, 59–69 CrossRef CAS PubMed.
- L. Modesti, A. Danese, V. Angela Maria Vitto, D. Ramaccini, G. Aguiari, R. Gafà and P. Pinton, Cells, 2021, 10, 1317 CrossRef CAS PubMed.
- N. Lenar, R. Piech, C. Wardak and B. Paczosa-Bator, Membranes, 2023, 13, 876 CrossRef CAS PubMed.
- J. Bobacka, A. Ivaska and A. Lewenstam, Chem. Rev., 2008, 108, 329–351 CrossRef CAS PubMed.
- C. Park, H. Yoon, M. A. Zahed, S. Zhang, S. Yoon, D. Kim, D. Kim and J. Park, IEEE Sens. J., 2021, 99, 1 Search PubMed.
- S. Rochefeuille, C. Jimenez, S. Tingry, P. Seta and J. P. Desfours, Mater. Sci. Eng., C, 2002, 21, 43–46 CrossRef.
- E. Arunkumar, A. Ajayaghosh and J. Daub, J. Am. Chem. Soc., 2005, 127, 3156–3164 CrossRef CAS PubMed.
- Y. Park, D. C. Apodaca, J. Pullen and R. C. Advincula, J. Phys. Chem. B, 2010, 114, 13084–13094 CrossRef CAS PubMed.
- F. Ishiwari, H. Hasebe, S. Matsumura, F. Hajjaj, N. Horii-Hayashi, M. Nishi, T. Someya and T. Fukushima, Sci. Rep., 2016, 6, 24275 CrossRef CAS PubMed.
- Y. Qin, S. Peper, A. Radu, A. Ceresa and E. Bakker, Anal. Chem., 2003, 75, 3038–3045 CrossRef CAS PubMed.
- M. A. Zayed, W. H. Mahmoud, A. A. Abbas, A. E. Ali and G. G. Mohamed, RSC Adv., 2020, 10, 17552–17560 RSC.
- I. Bedlechowicz-Sliwakowska, P. Lingenfelter, T. Sokalski, A. Lewenstam and M. Maj-Zurawska, Anal. Bioanal. Chem., 2006, 385, 1477–1482 CrossRef CAS PubMed.
- E. Wang, H. Chen, H. Patel, I. Sadaragani and C. Romero, Anal. Chim. Acta, 1999, 397, 287–294 CrossRef CAS.
- M. H. Lee, C. L. Yoo, J. S. Lee, I.-S. Cho, B. H. Kim, G. S. Cha and H. Nam, Anal. Chem., 2002, 74, 2603–2607 CrossRef CAS PubMed.
- S. G. Choi, S. H. Kang, J. Y. Lee, J. H. Park and S. K. Kang, Front. Bioeng. Biotechnol., 2023, 11, 1335188 CrossRef PubMed.
- P. Zhang, B. Zhu, P. Du and J. Travas-Sejdic, Chem. Rev., 2024, 124, 722–767 Search PubMed.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Conjugated polymer-based chemical sensors, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS.
- V. Sethumadhavan, S. Rudd, E. Switalska, K. Zuber, P. Teasdale and D. Evans, BMC Mat., 2019, 1, 4 CrossRef.
- J. Charton, M. Gauriot, J. Totobenazara, N. Hennuyer, J. Dumont, D. Bosc, X. Marechal, J. Elbakali, A. Herledan, X. Wen, C. Ronco, H. Gras-Masse, A. Heninot, V. Pottiez, V. Landry, B. Staels, W. G. Liang, F. Leroux, W. J. Tang, B. Deprez and R. Deprez-Poulain, Eur. J. Med. Chem., 2015, 90, 547–567 CrossRef CAS PubMed.
- K. A. Christensen, J. T. Myers and J. A. Swanson, J. Cell Sci., 2002, 115, 599–607 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, ed. Bard, A. J. and Faulkner, L. R., Wiley, New York, NY, USA, 2nd edn, 2001, 200–250 Search PubMed.
- L. Manjakkal, S. Dervin and R. Dahiya, RSC Adv., 2020, 10, 8594–8617 RSC.
- M. N. Leiske, J. A. Walker, A. Zia, N. L. Fletcher, K. J. Thurecht, T. P. Davis and K. Kempe, Polym. Chem., 2020, 11, 2799–2810 RSC.
- T. Blaz, J. Migdalski and A. Lewenstam, Talanta, 2000, 52, 319–328 CrossRef CAS.
- F. Ishiwari, H. Hasebe, S. Matsumura, F. Hajjaj, N. H. Hayashi, M. Nishi, T. Someya and T. Fukushima, Sci. Rep., 2016, 6, 24275 CrossRef CAS.
- A. Barandov, B. B. Bartelle, C. G. Williamson, E. S. Loucks, S. J. Lippard and A. Jasanoff, Nat. Commun., 2019, 10, 897 CrossRef PubMed.
- M. Domokos, S. Gergely, C. Levente, K. Zoltán, P. Bernadett, F. Gábor, K. Noémi, F. Anna, S. Áron, H. Polett, C. Attila, Z. Ákos, K. Kristóf, K. Gergely, M. Zoltán, J. R. Balázs and K. Ervin, Cell. Mol. Biol. Lett., 2024, 29, 105 Search PubMed.
- Xinqi Zhou, Kayla J. Belavek and Evan W. Miller, Biochemistry, 2021, 60(46), 3547–3554 Search PubMed; A. Csomos, B. Kontra, A. Jancsó, G. Galbács, R. Deme, Z. Kele, B. J. Rózsa, E. Kovács and Z. Mucsi, Eur. J. Org. Chem., 2021, 5248–5261 CrossRef CAS.
- C. E. M. Berger, B. R. Horrocks and H. K. Datta, Electrochem. Acta, 1991, 44, 2677–2683 CrossRef.
- J. T. Lock, I. Parker and I. F. Smith, Cell Calcium, 2015, 58, 638–648 CrossRef CAS PubMed.
- E. Bakker, P. Buhlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS PubMed.
- K. N. Mikhelson, Ion-selective electrodes, Springer, 2013 DOI:10.1007/978-3-642-36886-8; E. Lindner and Y. Umezawa, Pure App. Chem., 2008, 80(1), 85–104 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02586e |
| ‡ N. Janić and H. Zhukouskaya are contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |