
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making the negative positive – fluorination of indole as an efficient strategy to improve guanidinium-containing gene carriers†
Markus
Kötzsche‡
 a,
Jan
Egger‡
b,
Andreas
Dzierza
b,
Liên Sabrina
Reichel
a,
Ivo
Nischang
acde,
Anja
Traeger
ac,
Dagmar
Fischer
*bcf and
Kalina
Peneva
*ac
a,
Jan
Egger‡
b,
Andreas
Dzierza
b,
Liên Sabrina
Reichel
a,
Ivo
Nischang
acde,
Anja
Traeger
ac,
Dagmar
Fischer
*bcf and
Kalina
Peneva
*ac
aFriedrich Schiller University Jena, Institute of Organic and Macromolecular Chemistry (IOMC), Humboldtstr. 10, Jena, 07743, Germany. E-mail: kalina.peneva@uni-jena.de
bFriedrich-Alexander-Universität Erlangen-Nürnberg, Division of Pharmaceutical Technology and Biopharmacy, Cauerstr. 4, Erlangen, 91058, Germany
cJena Center for Soft Matter (JCSM), Philosophenweg 7, Jena, 07743, Germany
dHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstr. 12-14, Jena, 07743, Germany
eHelmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Hahn-Meitner-Platz 1, Berlin, 14109, Germany
fFAU NeW - Research Center for New Bioactive Compounds, Nikolaus-Fiebiger-Str. 10, Erlangen, 91058, Germany. E-mail: dagmar.fischer@fau.de
First published on 25th March 2025
Abstract
The balance between hydrophilic and hydrophobic components plays an important role in polymeric delivery of nucleic acids. Besides using hydrophobic moieties in the polymer design, fluorination is a promising method to increase the hydrophobicity of polymers. To systematically investigate this effect, N-(2-(1H-indol-3-yl)ethyl)methacrylamide and three fluorinated analogues have been synthesized and copolymerized with 3-guanidinopropyl methacrylamide and 2-hydroxypropyl methacrylamide via an aqueous reversible addition–fragmentation chain transfer (aRAFT) polymerization. A library of eight terpolymers with 5 to 23 mol% of an indole analogue and molar mass about 20 kg mol−1 showed comparably strong DNA binding starting at N/P 2 and formed polyplexes with hydrodynamic diameters around 100 nm. Additionally, no negative impact on biocompatibility was observed. Heparin release studies showed increased DNA binding strength with higher amounts of hydrophobic moieties, while fluorination exhibited similar effects as increasing the indole content. This was also important for pDNA transfection efficiency, where an optimum for DNA binding strength was unveiled. The rapid release and the excessive binding of DNA were identified as factors that negatively impacted transfection efficiency, both influenced by the amount of indole moieties and fluorination. On the other hand, the right degree of hydrophobicity was able to increase the transfection efficiency of the modified polymer by more than threefold. These findings highlight the role of hydrophobic moieties in nucleic acid delivery and provide valuable insights for future polymer design, suggesting that the strategic incorporation of fluorinated monomers can effectively fine-tune DNA interactions.
Introduction
Fluorination has played a crucial role in drug design and has been used to optimize the efficacy and selectivity of therapeutic agents.1 The introduction of fluorine into a drug molecule or peptide can significantly influence its metabolic stability, hydrophobicity, and bioavailability.2–5 Fluorine's unique electronegativity and small size enable the formation of strong, directional bonds, which can improve the binding affinity to biological targets, such as proteins and enzymes. Fluorination has also contributed to the field of gene delivery by enhancing the performance and stability of nucleic acid carriers.6 The incorporation of fluorine atoms into polymeric,7 peptide-based8–10 or lipid-based11,12 delivery systems can improve their chemical stability, resistance to enzymatic degradation, and overall biocompatibility. Fluorinated molecules often exhibit enhanced interactions with cellular membranes, facilitating the uptake of genetic materials like DNA or RNA. This was shown for a broad variety of polymers such as poly(ethylene imine) (PEI),13–20 the commonly used standard for polymeric gene carriers as well as for polyamines,21–23 chitosan24 or polyamidoamine and poly-L-lysine dendrimers.25–29 Additionally, fluorinated carriers can modulate physicochemical properties such as solubility and charge, which are critical for successful transfection.The interplay of charge and polarity is fundamental to the success of gene delivery systems, influencing the efficiency with which nucleic acids are transported into cells.30,31 Charged carriers, commonly cationic polymers or lipids, possess a positive charge that enables the electrostatic association with negatively charged nucleic acids to form the so-called polyplexes.31 This interaction is crucial for the formation of stable complexes, which can protect the genetic material from degradation and facilitate cellular uptake. In our previous study, we could demonstrate that guanidinium-functionalized methacrylamides with guanidinium monomer content equal to or higher than 60%, containing hydrophobic units, such as indoles, are emerging as innovative carriers to enhance gene delivery potential.32–34 These carriers leverage the unique properties of guanidinium groups, which provide a strong positive charge and enable effective electrostatic interactions with negatively charged nucleic acids, forming stable complexes to protect the genetic material from degradation and facilitate its transfection into target cells.35–43 Additionally, incorporation of hydrophobic regions allows the carrier to penetrate the lipid bilayer more efficiently, resulting in enhanced membrane fusion, cellular uptake and endosomal release, a critical step in ensuring that the genetic material reaches the cytoplasm for expression. The hydrophobicity can affect the stability and release profile of the genetic payload, impacting the overall transfection efficiency. Our studies have shown that optimizing the balance between hydrophobicity and hydrophilicity in cationic gene carriers is essential to improve their performance.
In this work, we hypothesize that the insertion of fluorinated indoles as comonomers in guanidinium-functionalized methacrylamides can improve their performance as gene carriers due to the combination of the cationic charge from guanidinium groups and the hydrophobic properties of fluorinated indoles that can optimize the balance between cellular uptake, endosomal release, and controlled release of genetic material. 5- and/or 6-fluoro-substituted indoles are incorporated with different amounts in methacrylamides by aqueous reversible addition–fragmentation chain transfer (aRAFT) polymerization. The resulting polymer library was investigated for the effect on DNA binding, cytocompatibility to human embryonic kidney cells (HEK293T) and the transfection efficiency of pDNA. The more hydrophobic nature of the fluorinated indoles could promote better membrane fusion and improve the cellular uptake, thus allowing more efficient membrane penetration.
Experimental
Monomer synthesis
The N-(2-(1H-indol-3-yl)ethyl)methacrylamide and fluorinated analogues were prepared with the corresponding tryptamines in one step. One molar equivalent of the tryptamine was filled in a Schlenk flask, degassed and dissolved in dry tetrahydrofuran (THF) or dimethylformamide (DMF). Then, one molar equivalent of triethylamine and 1 molar equivalent of N-succinimidyl methacrylate were added. After 3 to 6 h, the solvent was removed and the product purified by a silica column and washed with water to obtain the pure monomer.3-Guanidinopropyl methacrylamide: 1.2 molar equivalents of 3-aminopropyl methacrylamide hydrochloride were dissolved in acetonitrile, water and 3.75 molar equivalents of triethylamine to obtain a 1 M solution. One molar equivalent of N,N′-di-Boc-1H-pyrazole-1-carboxamidine was dissolved in the same amount of acetonitrile and added to the solution. After 1 day at room temperature, the product precipitated and the dispersion was diluted with water. The precipitate was filtered, washed with water and freeze-dried to obtain the Boc-protected 3-guanidinopropyl methacrylamide. Deprotection was done in dichloromethane with trifluoroacetic acid overnight. Afterwards, the solvent was removed and 3-guanidinopropyl methacrylamide obtained as the trifluoroacetic acid salt.
Polymer synthesis
3-Guanidinopropyl methacrylamide and 2-hydroxypropyl methacrylamide were filled in a Schlenk flask, degassed and dissolved in a 1 M acetate buffer at pH 5.2. Chain transfer agent 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (0.1 mg μL−1), initiator 4,4′-azobis(4-cyanovaleric acid) (0.1 mg μL−1) and N-(2-(1H-indol-3-yl)ethyl)methacrylamide analogue (0.5 mg μL−1) were dissolved in dry DMF and added to the monomer solution (600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1, [monomers]:[CTA]:[I]). After 3 freeze–pump–thaw cycles, the mixture was heated for 24 h at 80 °C. The polymerization was terminated with liquid nitrogen and the polymer dialysed at 4 °C against a sodium chloride solution for 1 day and MilliQ water adjusted to pH 4 for 3 days using regenerated cellulose dialysis membranes with a 3.5 kDa molecular weight cut-off. The polymer powder was obtained by freeze-drying. The composition was determined by 1H-NMR spectroscopy and molar mass as well as dispersity by SECDMAc.
:4:1, [monomers]:[CTA]:[I]). After 3 freeze–pump–thaw cycles, the mixture was heated for 24 h at 80 °C. The polymerization was terminated with liquid nitrogen and the polymer dialysed at 4 °C against a sodium chloride solution for 1 day and MilliQ water adjusted to pH 4 for 3 days using regenerated cellulose dialysis membranes with a 3.5 kDa molecular weight cut-off. The polymer powder was obtained by freeze-drying. The composition was determined by 1H-NMR spectroscopy and molar mass as well as dispersity by SECDMAc.
Preparation of polyplexes
Polymers as well as herring testes DNA (Sigma Aldrich, Darmstadt, Germany), pBR322 plasmid DNA (Carl Roth, Karlsruhe, Germany) or pEGFP-N1 DNA were dissolved in 10 mM HEPES ((4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid); Carl Roth) buffered 5% glucose (Sigma Aldrich) pH 7.4 (HBG). Polymer solutions were diluted to different concentrations so that equal volumes of nucleic acid solution and polymer dilution could be mixed to a total volume of 50 μL. Immediately, the mixture was vortexed for 10 s followed by incubation for 10 min before use. Polyplexes with different N/P ratios (ratio of cationic charged nitrogen (N) in the polymer to anionic charged phosphate (P) in the nucleic acid backbone) were obtained. The concentration of DNA was kept to 20 μg mL−1 for all N/P ratios.Nucleic acid binding assay
To quantify uncomplexed DNA, the AccuBlue® High Sensitivity dsDNA Quantification Kit (Biotium, Fremont, USA; purchased via VWR, Darmstadt, Germany) was used according to the manufacturer's protocol. Samples of polyplex dispersions equivalent to 100 ng herring testes DNA were pipetted as triplets in black 96-well plates. Free DNA without the polymer was set as 100% reference, free polymer in the same concentration necessary for N/P 40 was used to exclude any influence of the polymer on the assay, and HBG served as the blank control. The samples were mixed with 200 μL of the working solution (1:100 mixture of a 100X enhancer and quantification solution) per well and incubated under shaking at 200 rpm for 10 min. Fluorescence was measured using a plate reader (Tecan Spark Control; Tecan Group, Männedorf, Switzerland; 485 excitation, 530 nm emission). The results were shown as a percentage of the 100% control.
Horizontal agarose gel electrophoresis
Each polyplex dispersion (50 μL) containing 1 μg pBR322 plasmid DNA was mixed with a loading buffer (1 mM ethylenediaminetetraacetic acid disodium salt (EDTA; Sigma Aldrich), 40 mM tris(hydroxymethyl)aminomethane (TRIS; CarlRoth), and 50% (v/v) glycerol 85% (Carl Roth)) and loaded on a 1% agarose gel containing GelRed® (Biotium) as the fluorescent dye. Free DNA and free polymers were used as controls and bromophenol blue (CarlRoth) as the dye front marker. After electrophoretic separation at 80 V for 1 h in TAE-buffer (40 mM TRIS, 1 mM EDTA and 0.1% acetic acid (SigmaAldrich)), gel photographs were captured under UV transillumination at 312 nm (UV Transilluminator, Intas, Göttingen, Germany) and processed using BioVision software (Vilber, Collégien, France).Stability against enzymatic degradation
Polyplexes (50 μL) containing 1 μg pBR322 plasmid DNA were incubated with 2 μL DNase 1 solution (2.5 U μL−1, Thermo Fisher Scientific, Darmstadt, Germany) for 10 min. Afterwards, all samples were incubated at 37 °C for 45 min in a thermocycler Biometra TOne (Analytic Jena, Jena, Germany) before cooling to 20 °C. The samples were then treated with 2.5 μL EDTA solution (0.1 M) and further incubated at 70 °C for 35 min. After cooling to 20 °C, 10 μL of a heparin solution (2.5 U μL−1, CarlRoth) was added to facilitate the release of DNA. The samples were heated again to 37 °C for 20 min and cooled to 20 °C. Controls containing 1 μg DNA without the polymer were applied (i) untreated, (ii) treated in the same way as described above, and (iii) treated in the same way, except without the addition of DNase. Afterwards, horizontal agarose gel electrophoresis was conducted as described above.Laser light scattering techniques
Hydrodynamic diameters (HD) and polydispersity indices (PDI) were measured via dynamic light scattering (DLS) and the zeta potential (ZP) by laser Doppler anemometry via electrophoretic mobility, using a Zetasizer Ultra (Malvern Panalytical, Kassel, Germany). Polyplexes were formed with herring testes DNA at various N/P ratios. Dispersions with a DNA concentration of 20 μg mL−1 in HBG (refractive index 1.34 and viscosity 1.0204 mPa s) were measured in a high concentration zeta potential cell (Malvern Panalytical) at 25 °C at 633 nm with a scattering angle of 174.8° in three runs per sample. Analysis was performed using ZS Xplorer software (v1.3.1.7, Malvern Panalytical).Heparin release assay
To quantify drug release, a heparin release assay was performed similar to the nucleic acid binding assay described above. Polyplex samples containing 100 ng herring testes DNA were pipetted onto a black 96-well plate, while the same amount of DNA without the polymer served as a 100% reference. HBG was used as the blank. To each sample, 200 μL of the working solution (1:100 mixture of a 100X enhancer and quantification solution) from the AccuBlue® High Sensitivity dsDNA Quantification Kit were added and the plate incubated at 200 rpm for 10 min. Additionally, a solution was prepared in three concentrations, containing 0.025 U μL−1, 0.1 U μL−1 and 2.5 U μL−1 heparin. The first solution (0.025 U μL−1) was used to increase the total heparin content in each well to 1 U in steps of 0.05 U per application. From then on, the total heparin amounts were increased to 2 U in steps of 0.2 U making use of the second solution (0.1 U μL−1), while the last solution (2.5 U μL−1) was used to increase the heparin contents to 16 U per well. After each application, the plate was incubated at 200 rpm for 5 min before the fluorescence was measured using a plate reader. The amount of uncomplexed DNA after each heparin addition was calculated as a percentage of the 100% control. Results were plotted against the logarithmic amount of heparin per well.
Isolation of plasmid DNA
pEGFP-N1 plasmid DNA encoding the enhanced green fluorescent protein (EGFP) for transfection studies was isolated with a Giga Plasmid Kit (Qiagen, Hilden, Germany) from E. coli containing pEGFP-N1 (4.7 kb, Clontech, Mountain View, California). As negative control plasmid, pCMV was purchased from PlasmidFactory (Bielefeld, Germany).Cell culture
The human embryonic kidney cell line HEK293T was cultivated in Dulbecco's modified Eagle's medium (DMEM, Capricorn Scientific GmbH; with 1 g L−1 glucose, CarlRoth), supplemented with 10% (v/v) fetal bovine serum (FBS, Capricorn Scientific GmbH), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (D10, both Capricorn Scientific GmbH) at 37 °C in a humidified 5% (v/v) CO2 atmosphere in TC treated cell culture flasks (Greiner Bio-One International GmbH, Kremsmünster, Österreich and Labsolute, Th. Geyer GmbH & Co. KG). For the determination of metabolic activity (PrestoBlue™ assay), the HEK293T cell line was seeded at a cell concentration of 0.1 × 106 cells per mL in a 96-well plate (TC treated cell culture plates (VWR International GmbH) in a total volume of 100 μL D10 per well. For transfection efficiency studies, the cells were seeded 24 h before the experiment in a 24-well plate (TC treated cell culture plates (VWR International GmbH) at a cell concentration of 0.2 × 106 cells per mL in 500 μL D10.Determination of the metabolic activity (PrestoBlue™ assay)
To evaluate the cytotoxicity of the polymers, the PrestoBlue™ assay (Thermo Fisher Scientific) was performed. 1 h before cell treatment, the old growth medium was changed to 90 μL of fresh D10. In sextuplicates, the cells were treated with 10 μL polymers of different dilutions, ranging from 2 to 250 μg mL−1 and incubated for 24 h. After incubation for 24 h, the medium was replaced by fresh D10 with 10% (v/v) PrestoBlue™ solution, prepared according to the manufacturer's instruction. The cells were further incubated at 37 °C for 45 min and the fluorescence was measured at λEx = 570/λEm = 610 nm using the Tecan infinite M200Pro (Tecan Group). The control cells were treated with buffer on the same plate (100% viability). Values lower than 70% viability were considered cytotoxic. The relative number of viable cells was calculated as follows: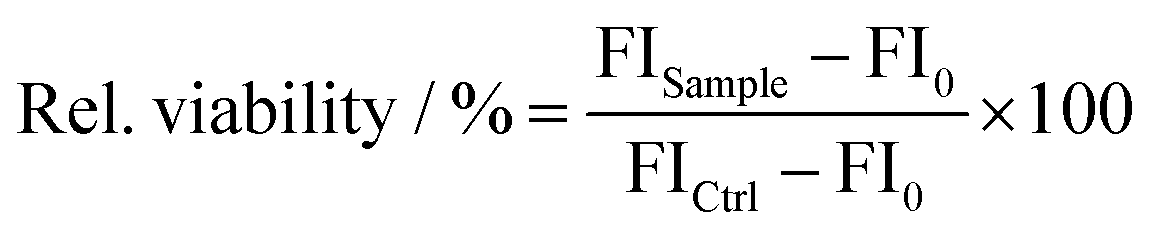 | (1) |
Transfection efficiency
Polyplexes with 4 μg pEGFP-N1 plasmid DNA were used to transfect HEK293T cells. After 24 h of preincubation, media were replaced with 450 μL OptiMEM™ (Thermo Fisher Scientific). OptiMEM™ is a transfection-optimized medium with a reduced serum content. 1 h later, the cells were treated with the polyplexes with different N/P ratios. After 4 h of incubation in OptiMEM™, the medium was changed to 500 μL D10 and the cells were further incubated for 20 h (4 + 20 h). Before harvesting, the supernatant was transferred to a new 96-well plate for the determination of membrane integrity (CytoTox-ONE™ assay). The remaining supernatant was aspirated, and cells were trypsinated (Trypsin in DPBS, Capricorn Scientific GmbH) for 10 min before resuspending in PBS (phosphate-buffered saline, Capricorn Scientific GmbH) and analyzed via flow cytometry (CytoFLEX, Beckmann Coulter, Brea, CA, U.S.). For each experiment, 2 × 104 cells per sample were analyzed using a bandpass detection filter 510 ± 10 nm with signal attenuation (OD1). As the negative control, the cells were treated with polyplexes of pCMV, where no EGPF expression was possible. Linear poly(ethylene imine) (LPEI) with a molar mass of 2500 g mol−1 obtained from Polyscience, Warrington, USA, was used as the positive control.Determination of membrane integrity (CytoTox-ONE™ assay)
To determine the membrane integrity of cells after the exposure of the polyplexes over 24 h, the CytoTox-ONE™ (Promega GmbH, Walldorf, Germany) assay was performed. The assay was conducted in combination with the transfection efficiency assay. After 24 h of incubation with the polyplexes (transfection efficiency assay), an aliquot of the supernatant was transferred to a 96-well plate in triplicate. Following, the solution was equilibrated at room temperature for 20 min. Subsequently, 50 μL of the CytoTox-ONE™ reagent was added to each well and incubated for 10 min at room temperature. To stop the reaction, 25 μL of the stop solution was added to each well. Values lower than 90% viability were considered as cytotoxic. Cells treated with lysis solution were used as a 100% control (100% dead cells). The fluorescence intensity was measured at λEx = 570/λEm = 610 nm using the Tecan infinite M200Pro (Tecan Group) and cytotoxicity was calculated as follows: | (2) |
Results and discussion
Monomer and polymer synthesis
The four hydrophobic monomers N-(2-(1H-indol-3-yl)ethyl)methacrylamide (IEMA), N-(2-(5-fluoro-1H-indol-3-yl)ethyl)methacrylamide (5F-IEMA), N-(2-(6-fluoro-1H-indol-3-yl)ethyl)methacrylamide (6F-IEMA) and N-(2-(5,6-difluoro-1H-indol-3-yl)ethyl)methacrylamide (5,6-di-F-IEMA) were synthesized in one step with a yield of 57%, 42%, 65% and 77%, respectively (Scheme 1). The synthesis of 5,6-di-F-IEMA was performed in dry DMF to ensure the solubility of the more hydrophobic monomer. Both fluorine atoms can be distinguished in 19F-NMR spectroscopy (SI) and can be used as potential probes. The difference in the hydrophobicity of the indole variants was verified by the contact angle measurements of water on spin-coated monomer films on silicon wafers (Fig. S11, ESI†). The non-fluorinated IEMA (52.5 ± 1.1°) was the least hydrophobic, while 5,6-di-F-IEMA (64.0 ± 0.9°) was the most hydrophobic and 6F-IEMA (58.1 ± 0.7°) and 5F-IEMA (62.3 ± 0.6°) exhibited intermediate hydrophobicity. | ||
| Scheme 1 Synthesis of the N-(2-(1H-indol-3-yl)ethyl)methacrylamide analogues and statistical copolymerization with 3-guanidinopropyl methacrylamide (GPMA) and 2-hydroxypropyl methacrylamide (HPMA) via an aqueous RAFT polymerization. Detailed compositions are listed in Table 1. | ||
Statistical terpolymers with HPMA, GPMA and an indole comonomer were prepared in an aqueous RAFT polymerization having 51 to 73 mol% GPMA as cationic moieties to enable the complexation of DNA (Table 1). To adjust the hydrophobicity by fluorination, each of the four indole monomers was once incorporated with 6 mol%. Additionally, the hydrophobicity was varied by increasing the IEMA or 5F-IEMA content to 10 or 20 mol%. The GPMA content was lowered with increasing the IEMA content, because a high content of cationic groups combined with a high amount of hydrophobic groups could negatively affect the toxicity of the terpolymers. The amount of HPMA was kept similar at 24 mol%.
| Abbreviation | Monomer content | M n [g mol−1] | Đ | |||||
|---|---|---|---|---|---|---|---|---|
| HPMA [mol%] | GPMA [mol%] | IEMA [mol%] | 5F-IEMA [mol%] | 6F-IEMA [mol%] | 5,6-di-F-IEMA [mol%] | |||
| Monomer content was determined by 1H-NMR spectroscopy. The molar mass and dispersity were determined by size exclusion chromatography in dimethylacetamide and LiCl with poly(methylmethacrylate) calibration. | ||||||||
| IEMA6 | 24 | 70 | 6 | — | — | — | 21300 |
1.10 |
| IEMA10 | 25 | 65 | 10 | — | — | — | 28600 |
1.17 |
| IEMA20 | 27 | 53 | 20 | — | — | — | 20300 |
1.08 |
| 5F-IEMA6 | 23 | 71 | — | 6 | — | — | 25700 |
1.37 |
| 5F-IEMA10 | 25 | 64 | — | 11 | — | — | 15600 |
1.08 |
| 5F-IEMA20 | 23 | 51 | — | 23 | — | — | 18600 |
1.11 |
| 6F-IEMA6 | 21 | 72 | — | — | 7 | — | 18900 |
1.31 |
| 5,6-di-F-IEMA | 22 | 73 | — | — | — | 5 | 10600 |
1.12 |
The fluorinated monomers were not soluble in the 1 M aqueous acetate buffer at room temperature, but they were dissolved at 80 °C. Purification of the polymers was done by dialysis against water followed by freeze-drying. Due to its increased hydrophobicity, 5,6-di-F-IEMA required additional dialysis against methanol/water to obtain pure water-soluble polymers. The polymerization kinetics were studied using a monomer feed of 20 mol% HPMA, 65 mol% GPMA and 15 mol% 6F-IEMA, revealing an overall statistical incorporation of all comonomers with a slightly slower incorporation of GPMA when compared to HPMA and 6F-IEMA (Fig. S26, ESI†).
The composition of all synthesized polymers was determined by 1H-NMR spectroscopy (SI) and showed a successful polymerization of all four hydrophobic monomers in the desired compositions. The molar mass distributions were characterized by size exclusion chromatography (Fig. S27, ESI†) and the Mn values were around 20 kg mol−1 with a dispersity of 1.1. The higher dispersity of 1.4 for 5F-IEMA6 and 1.3 for 6F-IEMA6 was probably caused by the termination of the polymerization reactions with an initiator to remove the chain-transfer agent, which could lead to chain–chain coupling. The molar mass determined for 5,6-di-F-IEMA was slightly lower than that for the other polymers. This could be related to the lower solubility of the difluorinated monomer affecting the polymerization parameters. To correlate polymer composition and apparent hydrophobicity, we performed a brief chromatographic study in which all polymers were investigated in gradient elution liquid chromatography utilizing a reversed-phase monolith silica column (Fig. S28, ESI†).44 Clearly, in a linear gradient of CH3CN in the aqueous organic mobile phase, retention times increased from IEMA6 to IEMA10, and IEMA20 (Fig. S29, ESI†). This is also observed for the fluorinated compositions 5F-IEMA6 to 5F-IEMA10, and 5F-IEMA20. The difference in elution times among non-fluorinated and fluorinated polymers appears less pronounced under present elution conditions. While IEMA6 and 5F-IEMA6 appear eluting readily similar, differences are seen for IEMA10 and 5F-IEMA10 as well as IEMA 20 and 5F-IEMA20. The smaller difference between fluorinated and non-fluorinated polymers may be attributed to the slightly lower molar masses of the fluorinated ones, which reduce the elution time in reversed-phase liquid chromatography.
DNA binding
The efficiency of DNA binding was evaluated by conducting a fluorescent dye exclusion assay with all synthesized polymers, varying the N/P ratios from 1, 2, 3, 4, 5, 10, 20 to 40 (Fig. S30, ESI†). Regardless of the polymer, virtually no fluorescence signal was detectable starting from an N/P ratio of 2, indicating the complexation of the DNA. This efficient complexation was consistent across all polymers, seemingly not influenced by either the IEMA content or position and degree of fluorination. The results were in good agreement with those from the performed gel electrophoresis assays (Fig. S31, ESI†), where other than for N/P 1 no DNA bands were observable, confirming a complete binding and retardation of the DNA, inhibiting the movement in the gel. These findings also indicate that, when comparing the indole contents from IEMA6 to IEMA10 and to IEMA20, an increase in the content of hydrophobic monomers has no negative effect on the complexation of the DNA. The same can be assumed for the fluorinated variants of 5F-IEMA6, 5F-IEMA10 and 5F-IEMA20. In addition to this, neither the 6-fluorinated nor the di-fluorinated alternatives showed any deviation from the DNA binding profiles of the other polymers, indicating that the position and the degree of fluorination have no negative impact on complexation. To summarize, all tested polymers showed similar binding efficacies, regardless of their structural modifications, instead exhibiting a complete and consistent DNA binding across a wide range of N/P ratios.Physicochemical characterization
Along with the DNA binding studies, all polyplexes were also characterized using dynamic light scattering (DLS) and laser Doppler anemometry (LDA) to determine the hydrodynamic diameter, polydispersity index (PDI), as well as the zeta potential for a variety of different N/P ratios (Fig. 1). Consistently across all polyplexes, hydrodynamic diameters of about 90 to 125 nm were measured, with a tendency towards lower diameters for higher N/P ratios, suggesting more compact polyplexes, possibly due to increasing capacity for condensation of the DNA, the more positive charge is present in the polymer structure. In contrast, PDI values exhibited a trend towards higher polydispersity with increasing N/P ratios. For N/P ratios below 10, typically in the range of what is used in the following biological experiments, the measured PDI for all polyplexes was around 0.2, indicating a uniform distribution of the respective populations. With increasing N/P ratios, PDI values increased to around 0.3 for N/P 40, mostly caused by a higher tendency towards the aggregation of the polyplexes and an increased amount of the free polymer, leading to a broader size distribution. Across all polymers, zeta potential values generally ranged between 40 and 50 mV. A slight trend towards higher zeta potentials was observed for higher N/P ratios, which is to be expected, considering the increase in the surplus of positive charges on the polyplex surface with rising polymer amounts. In general, the DLS measurements confirmed that across all different modifications, the tested polymers form polyplexes of diameters below 150 nm, within an optimal range for cellular uptake, which is one of the many critical factors for an efficient transfection.45,46 The PDI values of around 0.2 to 0.3 indicate a relatively uniform distribution of the polyplexes with a low tendency towards aggregation. The measured zeta potentials of about 40 to 50 mV confirm a sufficiently positive surface charge, critical for strong interactions with cellular membranes, promoting cell uptake and endosomal escape, while also helping to keep the formulation stable. These findings were similar for all tested polymers, once again indicating that neither the amount of hydrophobic monomers, nor the position and amount of fluorination has a negative effect on polyplex formation and formulation stability. | ||
| Fig. 1 N/P dependency of the hydrodynamic diameter, polydispersity index (PDI) and zeta potential, determined via dynamic light scattering (DLS). All measurements were performed as triplicate (mean ± SD). | ||
The main reason for this might be the relatively consistent and high amount of guanidinium comonomer present in all polymers, which imparts them with a constant positive charge, independent of the degree of protonation. Electrostatic interactions are the driving factor for the formation of the polyplexes and therefore also responsible for most of the physicochemical properties, leading to uniform polyplexes with a good DNA complexation above a certain threshold of positive charges, while hydrophobic interactions prove to be important for other interactions discussed later.
Biocompatibility
Hydrophobicity of the polymers often goes hand in hand with increased cellular toxicity and thus a resulting net loss of transfection efficiency. To evaluate this, HEK293T cells were used to perform a PrestoBlue™ assay, determining the number of viable cells by measuring their metabolic capacity to reduce the non-fluorescent dye resazurin into a fluorescent and calorimetric resorufin product (Fig. 2). All polymers exhibited relatively similar biocompatibility profiles independent of their structural modification. Cytotoxic effects were observed starting from polymer concentrations of 8 μg mL−1, whereas a threshold of 50% metabolic activity was reached at around 20 to 30 μg mL−1 for all polymers, revealing a slight increase in cellular toxicity compared to the control polymer LPEI. The selected PEI is acknowledged as the gold standard in gene delivery and was already used in many studies.47–49 Overall, these results indicate that the varying contents of the hydrophobic indole moiety investigated in these measurements did not have a noticeable impact on cytotoxicity, which was also the case for the various fluorinated modifications of the polymers. These findings are in agreement with the determined membrane integrity, which was measured via a CytoTox-ONE™ Assay (Fig. S32, ESI†). | ||
| Fig. 2 PrestoBlue™ assay performed after incubation of HEK293T cells with different concentrations of the polymers for 24 h. Lines represent a DoseRespond fit function (n = 3, mean ± SD). | ||
Stability against enzymatic degradation
Another critical factor for the delivery of genetic material via polyplexes is the protection of the cargo from external influences, more specifically in this case the inhibition of nucleases from degrading the DNA. To quantify how well the genetic material is protected in the polyplexes, a test was performed, measuring the protection against enzymatic degradation (Fig. 3). Overall, this protection and subsequent detection of the intact DNA was effective across all polymers; however, a noticeable trend emerged when analyzing sequences of increasing hydrophobicity in non-fluorinated and fluorinated polymers. The polymer IEMA6 and its fluorinated variants 5F-IEMA6, 6F-IEMA6 and 5,6-di-F-IEMA, as well as IEMA10 and 5F-IEMA10 all displayed clear and intact bands in gel electrophoresis, confirming their adequate protection against degradation by the nuclease. In contrast, for polymers with the highest content of hydrophobic moieties, IEMA20 and 5F-IEMA20, there were no bands visible at all. In these cases, the absence of DNA signals was presumed to be caused by an insufficient release from the polyplexes, rather than an inadequate protection from degradation. These findings suggest that the position and quantity of fluorination on the indole monomer do not exhibit a negative impact on the ability of the polymers to protect the DNA from enzymatic degradation. | ||
| Fig. 3 Qualitative determination of the stability against nucleases determined by horizontal agarose gel electrophoresis conducted after treatment of the polyplexes with DNase 1 and the subsequent release of the intact plasmid DNA. Free polymer (P) and untreated DNA (U) were employed as controls, as well as DNA that was exposed to the same thermal stress, without the addition of DNase (−) and with the addition of DNase (+). | ||
Heparin release assay
To investigate the release of the DNA from the polyplexes, a modified fluorescent dye exclusion assay was conducted. This assay utilized the ability of heparin to affect DNA release from polyplexes by electrostatic interactions.50 By measuring the amount of displaced, non-complexed DNA, it offered insights into polymer–DNA binding affinities (Fig. 4). Supporting previous observations regarding protection from enzymatic degradation, the results showed that among the non-fluorinated polymers, DNA release from IEMA6 was the fastest and most complete, with approximately 90% of uncomplexed DNA after treatment with 16 U heparin. In contrast, when the indole content was increased to 10% in IEMA10, a considerable decline in the kinetics of the DNA release was noted, resulting in approximately 65% of uncomplexed DNA. Further increasing the indole content to 20% in IEMA20 exacerbated this trend, resulting in a markedly delayed release and a final uncomplexed DNA percentage that barely reached 10%, even for very high heparin concentrations. In addition, these results also support the hypothesis regarding fluorination of the aromatic comonomer, which aimed to enhance the hydrophobicity of the modified polymers. A comparison of the non-fluorinated IEMA6 to its fluorinated counterpart 5F-IEMA6 revealed very similar effects to those observed when the indole content was increased from 6% to 10%, highlighting a reduction in the efficiency of DNA release. This effect is observed across all three variations in the indole content, with the fluorinated counterparts consistently showing both delayed and reduced release. The most pronounced example is 5F-IEMA20, which achieves only about 2% uncomplexed DNA. | ||
| Fig. 4 Quantification of uncomplexed herring testes DNA determined by the AccuBlue® High Sensitivity dsDNA Quantification Kit, after treatment with heparin solution (n = 3, mean ± SD). | ||
As previously noted during the physicochemical characterization, the position of the fluorination does not appear to influence the outcome, as 6F-IEMA6 exhibited a release profile similar to that of the 5F-variant. Interestingly, while the di-fluorinated polymer did not reveal any differences in earlier studies, the addition of an extra fluorine atom to the indole monomer appears to considerably affect the release kinetics of the DNA from the respective polymer. Specifically, 5,6-di-F-IEMA displayed a release profile closely resembling that of the non-fluorinated IEMA6, suggesting a reduced DNA binding affinity compared to polymers with similar fluorinated indole contents, like 5F-IEMA6 and 6F-IEMA6.
Overall, these findings suggest that, while the variation of the hydrophobic content did not seem to influence most physicochemical characteristics of the polyplexes, they do indeed affect how tightly the nucleic acids are bound by the polymer, resulting in slower and more incomplete release kinetics from polyplexes with higher contents of hydrophobic moieties in the presence of heparin. In addition to this, fluorination of the polymers appears to be an effective strategy for enhancing their hydrophobicity and improving interactions with DNA.
Transfection efficiency
To evaluate the effect of the different structural modifications of the polymers on their biological activity, pDNA transfection with polyplexes containing pEGFP-N1 as the cargo was performed, using HEK293T cells. The level of transfection was assessed by measuring the fluorescence signal from the transfected cells by flow cytometry (Fig. 5). As mentioned before, the aim behind the fluorination of the polymers was to enhance their hydrophobicity, which would facilitate DNA binding and promote interactions with the hydrophobic components of the cellular membrane, improving uptake into the cells. Indeed, the expression of EGFP positive cells is improved with the fluorinated variant 5F-IEMA6 leading to a more than threefold increase in EGFP positive cells compared to IEMA6. This is comparable to the levels achieved by the gold standard PEI for low N/P ratios, such as N/P 5. Consistent with previous findings, the position of the fluorine atom does not appear to influence these results, with 6F-IEMA6 exhibiting very similar transfection efficiencies to that of 5F-IEMA6. In contrast, the di-fluorinated variant 5,6-di-F-IEMA showed no increase in transfection and revealed comparable efficiency as IEMA6. This aligns with prior observations, as the presence of a second fluorine atom seems to reduce DNA binding affinity. The resulting faster DNA release kinetics for 5,6-di-F-IEMA, which are comparable to that of IEMA6, could explain the similarly low transfection for both polymers. | ||
| Fig. 5 Determination of the transfection efficiency in HEK293T cells over 24 h with 4 μg of pEGFP-N1 DNA. Measurements were conducted in the OptiMEM™ medium with various N/P ratios. As negative controls, polyplexes with pCMV DNA (Ctrl) and pure pEGFP-N1 DNA without the polymer (MM) were used, while LPEI served as a positive control (n = 3, mean ± SD). | ||
When comparing the non-fluorinated polymers, a marked increase in transfection was observed as the indole content was raised from IEMA6 to IEMA10, a boost comparable to the improvement seen with the fluorination of indole. In contrast, when increasing the IEMA content up to 20%, no additional increase in transfection can be achieved. On the contrary, the amount of EGFP positive cells decreases to the level of IEMA6. This outcome suggests that there could be an upper limit for the hydrophobic content, beyond which the presence of excessive hydrophobic moieties becomes detrimental to transfection efficiency. A similar trend was noted among the fluorinated variants, with 5F-IEMA6 exhibiting the highest transfection efficiency, remaining below a critical threshold of hydrophobicity. However, 5F-IEMA10 appeared to surpass this threshold, resulting in a marked decrease in EGFP positive cells. Although DNA could not be released from 5F-IEMA20 with heparin, the polymer showed an increase in transfection compared to 5F-IEMA10, indicating that other effects also influence the delivery to cells. Nevertheless, the amount of transfected cells was still considerably lower than that for 5F-IEMA6.
These findings all correlate to the heparin release measurement and the resulting DNA binding affinities of the polymers. When the binding affinity is too low, as seen for IEMA6 and 5,6-di-F-IEMA, the cargo is not tightly packaged and might be released preemptively, e.g., when the polyplexes encounter the serum with its various charged components, leading to a reduced transfection efficiency.
In similar manner, strong binding affinities likewise negatively affect transfection. This effect can be seen for the polymers IEMA20 and 5F-IEMA20, from which barely any DNA was released when treated with heparin, resulting in reduced amounts of EGFP positive cells. The optimal point seems to be achieved, when the DNA binding affinity is sufficiently high enough, to ensure that the cargo remains bound and protected upon entering the serum, but still low enough to enable the majority of the DNA to disassociate from the polyplex at some point after the particles have been taken up into the cell.
Comparing the data from the heparin release assay with the transfection data, this seems to be achieved for polymers 5F-IEMA6 and 6F-IEMA6, as well as IEMA10, which all cluster around the same amount of released DNA at the endpoint and also specifically show the highest amount of transfection out of all of these polymers. 5F-IEMA10, which shows an increase in DNA binding, seems to be past the optimal point, achieving only low amounts of EGFP positive cells.
Conclusions
aRAFT polymerization was successfully employed to synthesize 8 methacrylamide terpolymers with systematically varying degrees of the indole monomer or fluorinated indole variants to achieve different hydrophobicities. Physicochemical characterization as well as DNA binding studies revealed that all polymers were able to form polyplexes suitable for the delivery of genetic material, with particle sizes below 150 nm and a narrow size distribution, in addition to a complete complexation of the employed DNA. Here, no notable differences were observable between the different polymers, suggesting, that for the physicochemical properties, electrostatic interactions are of greater importance than hydrophobicity. In contrast, hydrophobicity of the employed terpolymers was especially important regarding the binding strength of the complexed DNA and its release kinetics from the polyplexes. With increased hydrophobicity, the release is reduced and thus less DNA could be recovered from the polyplexes. This outcome underpins the improvement of DNA binding affinity by using hydrophobic moieties. In a similar manner, the hydrophobic effect of the fluorination was also observable, with fluorinated polymers showing comparable kinetics to those with overall higher contents of hydrophobic monomers. Interestingly, the position of the fluorine atom had no impact, while a second fluorine atom decreased the DNA binding affinities instead. These results agree with the pEGFP-N1 transfection studies, where an optimal window of hydrophobicity was elucidated. Polymers with 10 mol% indole or 6 mol% mono-fluorinated indole were able to increase transfection. If this hydrophobic threshold was crossed, DNA binding affinities were presumably too high, hindering the release of the DNA from the polyplex.Overall, we could show that the hydrophobic–hydrophilic balance plays an important role in the delivery of nucleic acids via polymeric carriers. Different methods to modify hydrophobicity, like the addition of fluorinated monomers, can be utilized to fine tune interactions with the DNA, impacting future polymer design.
Author contributions
Markus Kötzsche: conceptualization, data curation, investigation, methodology, and writing – original draft; Jan Egger: conceptualization, data curation, investigation, methodology, and writing – original draft; Andreas Dzierza: data curation, investigation, and writing – original draft; Lien Sabrina Reichel: data curation, investigation, and writing – review & editing; Ivo Nischang: conceptualization, data curation, investigation, methodology, and writing – review & editing; Anja Traeger: funding acquisition, supervision, and writing – review & editing; Dagmar Fischer: conceptualization, funding acquisition, project administration, supervision, and writing – review & editing; Kalina Peneva: conceptualization, funding acquisition, project administration, supervision, and writing – review & editing.Data availability
We confirm that the data supporting the findings of our study are fully available within the article and its ESI.† No additional data are available outside of these materials.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 316213987 – SFB 1278 PolyTarget (projects B01, B03 and Z01) for funding. A. T. acknowledges funding by the DFG Heisenberg Programme (514006196). I. N. acknowledges funding by the DFG – 471397362. The expert technical assistance by Martin Klose, Elisabeth Moek and Carolin Kellner is acknowledged for support with the cell experiments and the NMR department at Friedrich Schiller University Jena for their continuous support. The expert technical assistance by Christin Weilandt is acknowledged for support with the HPLC measurements. The authors would like to thank Dr. Purushottam Poudel for supporting the contact angle measurement.Notes and references
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- M. Arias, E. R. Hoffarth, H. Ishida, J. M. Aramini and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1012–1023 CrossRef CAS PubMed.
- J. Shao, B. P. Kuiper, A.-M. W. H. Thunnissen, R. H. Cool, L. Zhou, C. Huang, B. W. Dijkstra and J. Broos, J. Am. Chem. Soc., 2022, 144, 13815–13822 CrossRef CAS PubMed.
- J. Broos, E. Gabellieri, E. Biemans-Oldehinkel and G. B. Strambini, Protein Sci., 2003, 12, 1991–2000 CrossRef CAS PubMed.
- G. D. Galles, D. T. Infield, C. J. Clark, M. L. Hemshorn, S. Manikandan, F. Fazan, A. Rasouli, E. Tajkhorshid, J. D. Galpin, R. B. Cooley, R. A. Mehl and C. A. Ahern, Nat. Commun., 2023, 14, 59 CrossRef CAS PubMed.
- Y. Wan, Y. Yang, M. Wu and S. Feng, Expert Opin. Drug Delivery, 2022, 19, 1435–1448 CrossRef CAS.
- E. C. Tan, J. Lv, J. J. Hu, W. W. Shen, H. Wang and Y. Y. Cheng, J. Mater. Chem. B, 2018, 6, 7230–7238 RSC.
- C. L. Ge, J. D. Yang, S. Z. Duan, Y. Liu, F. H. Meng and L. C. Yin, Nano Lett., 2020, 20, 1738–1746 CrossRef CAS PubMed.
- S. Khemaissa, A. Walrant and S. Sagan, Q. Rev. Biophys., 2022, 55, e10 CrossRef CAS PubMed.
- A. A. Baoum, Plasmid, 2022, 119–120, 102619 CrossRef CAS PubMed.
- H. P. Zhang, C. Y. Meng, X. W. Yi, J. P. Han, J. X. Wang, F. Liu, Q. Ling, H. J. Li and Z. Gu, ACS Nano, 2024, 18, 7825–7836 CrossRef CAS PubMed.
- H. A. Huo, X. D. Cheng, J. X. Xu, J. Q. Lin, N. Chen and X. G. Lu, J. Mater. Chem. B, 2023, 11, 4171–4180 RSC.
- J. Lv, H. Chang, Y. Wang, M. Wang, J. Xiao, Q. Zhang and Y. Cheng, J. Mater. Chem. B, 2015, 3, 642–650 RSC.
- G. Chen, Y. Xu, P. Wu and K. Wang, J. Drug Delivery Sci. Technol., 2020, 55, 101403 CrossRef CAS.
- M. E. Johnson, J. Shon, B. M. Guan, J. P. Patterson, N. J. Oldenhuis, A. C. Eldredge, N. C. Gianneschi and Z. Guan, Bioconjugate Chem., 2016, 27, 1784–1788 CrossRef CAS.
- G. J. Lee and T.-I. Kim, J. Biomed. Mater. Res., Part A, 2019, 107, 2468–2478 CrossRef CAS PubMed.
- Y. Zhang, Z. Yuan, Y. Jin, W. Zhang and W.-E. Yuan, Pharmaceutics, 2021, 13, 2058 CrossRef CAS PubMed.
- Y. P. Xiao, J. Zhang, Y. H. Liu, Z. Huang, B. Wang, Y. M. Zhang and X. Q. Yu, J. Mater. Chem. B, 2017, 5, 8542–8553 RSC.
- Z. C. Deng, W. Gao, F. Kohram, E. H. Li, T. V. Kalin, D. L. Shi and V. V. Kalinichenko, Bioact. Mater., 2024, 31, 1–17 CAS.
- P. K. Wu, X. P. Luo, H. Wu, Q. Y. Zhang, K. K. Wang, M. J. Sun and D. Oupicky, Bioconjugate Chem., 2020, 31, 698–707 CrossRef CAS PubMed.
- G. Chen, K. Wang, Q. Hu, L. Ding, F. Yu, Z. Zhou, Y. Zhou, J. Li, M. Sun and D. Oupický, ACS Appl. Mater. Interfaces, 2017, 9, 4457–4466 CrossRef CAS PubMed.
- Y. Qi, H. Song, H. Xiao, G. Cheng, B. Yu and F.-J. Xu, Small, 2018, 14, 1803061 CrossRef PubMed.
- G. Chen, Y. Wang, A. Ullah, Y. Huai and Y. Xu, Eur. J. Pharm. Sci., 2020, 152, 105433 CrossRef CAS PubMed.
- M. K. Gök, Suleyman Demirel Univ. Fen Bilimleri Enst. Derg., 2019, 23, 885–891 CrossRef.
- X. Cai, R. Jin, J. Wang, D. Yue, Q. Jiang, Y. Wu and Z. Gu, ACS Appl. Mater. Interfaces, 2016, 8, 5821–5832 CrossRef CAS PubMed.
- M. Wang and Y. Cheng, Acta Biomater., 2016, 46, 204–210 CrossRef CAS.
- J. M. Criscione, B. L. Le, E. Stern, M. Brennan, C. Rahner, X. Papademetris and T. M. Fahmy, Biomaterials, 2009, 30, 3946–3955 CrossRef CAS PubMed.
- C. Romani, P. Gagni, M. Sponchioni and A. Volonterio, Bioconjugate Chem., 2023, 34, 1084–1095 CrossRef CAS PubMed.
- M. Wang and Y. Cheng, Biomaterials, 2014, 35, 6603–6613 CrossRef CAS PubMed.
- R. Kumar, C. F. Santa Chalarca, M. R. Bockman, C. Van Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS PubMed.
- C. M. Jogdeo, K. Siddhanta, A. Das, L. Ding, S. Panja, N. Kumari and D. Oupicky, Adv. Mater., 2024, 36 Search PubMed.
- C. Cokca, L. Zartner, I. Tabujew, D. Fischer and K. Peneva, Macromol. Rapid Commun., 2020, 41, 1900668 CrossRef CAS PubMed.
- F. J. Hack, C. Cokca, S. Städter, J. Hülsmann, K. Peneva and D. Fischer, Macromol. Rapid Commun., 2021, 42, 2000580 CrossRef CAS.
- P. P. Mapfumo, L. S. Reichel, K. Leer, J. Egger, A. Dzierza, K. Peneva, D. Fischer and A. Traeger, ACS Macro Lett., 2024, 13, 1000–1007 CrossRef CAS PubMed.
- A. M. Funhoff, C. F. van Nostrum, M. C. Lok, M. M. Fretz, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 2004, 15, 1212–1220 CrossRef CAS PubMed.
- N. J. Treat, D. Smith, C. Teng, J. D. Flores, B. A. Abel, A. W. York, F. Huang and C. L. McCormick, ACS Macro Lett., 2012, 1, 100–104 CrossRef CAS PubMed.
- J. M. Sarapas, C. M. Backlund, B. M. deRonde, L. M. Minter and G. N. Tew, Chem. – Eur. J., 2017, 23, 6858–6863 CrossRef CAS PubMed.
- A. Walrant, A. Bauzá, C. Girardet, I. D. Alves, S. Lecomte, F. Illien, S. Cardon, N. Chaianantakul, M. Pallerla, F. Burlina, A. Frontera and S. Sagan, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183098 CrossRef CAS PubMed.
- H. A. Rydberg, M. Matson, H. L. Åmand, E. K. Esbjörner and B. Nordén, Biochemistry, 2012, 51, 5531–5539 CrossRef CAS PubMed.
- N. Chuard, K. Fujisawa, P. Morelli, J. Saarbach, N. Winssinger, P. Metrangolo, G. Resnati, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 11264–11271 CrossRef CAS PubMed.
- F. A. Jerca, C. Muntean, K. Remaut, V. V. Jerca, K. Raemdonck and R. Hoogenboom, J. Controlled Release, 2023, 364, 687–699 CrossRef CAS PubMed.
- B. M. deRonde, N. D. Posey, R. Otter, L. M. Caffrey, L. M. Minter and G. N. Tew, Biomacromolecules, 2016, 17, 1969–1977 CrossRef CAS PubMed.
- C. R. Hango, C. M. Backlund, H. C. Davis, N. D. Posey, L. M. Minter and G. N. Tew, Biomacromolecules, 2021, 22, 2850–2863 CrossRef CAS PubMed.
- E. Tsarenko, N. E. Göppert, P. Dahlke, M. Behnke, G. Gangapurwala, B. Beringer-Siemers, L. Jaepel, C. Kellner, D. Pretzel, J. A. Czaplewska, A. Vollrath, P. M. Jordan, C. Weber, O. Werz, U. S. Schubert and I. Nischang, J. Mater. Chem. B, 2024, 12, 11926–11938 RSC.
- T. dos Santos, J. Varela, I. Lynch, A. Salvati and K. A. Dawson, Small, 2011, 7, 3341–3349 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- F. J. Hack, C. Cokca, S. Stadter, J. Hulsmann, K. Peneva and D. Fischer, Macromol. Rapid Commun., 2021, 42, e2000580 CrossRef.
- M. Zink, K. Hotzel, U. S. Schubert, T. Heinze and D. Fischer, Macromol. Biosci., 2019, 19, 1900085 CrossRef PubMed.
- S. Ochrimenko, A. Vollrath, L. Tauhardt, K. Kempe, S. Schubert, U. S. Schubert and D. Fischer, Carbohydr. Polym., 2014, 113, 597–606 CrossRef CAS PubMed.
- F. Richter, L. Martin, K. Leer, E. Moek, F. Hausig, J. C. Brendel and A. Traeger, J. Mater. Chem. B, 2020, 8, 5026–5041 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02529f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |