
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlutaraldehyde functionalized reduced graphene oxide based resistive sensors for detection of PCA3
Bolivia
Konthoujam
a,
Nikita
Bhandari
b,
Shridhar C.
Ghagane
ce,
Rajendra B.
Nerli
de,
Sudhanshu
Shukla
b and
Ruma
Ghosh
 *a
*a
aDepartment of Electrical, Electronics and Communication Engineering, Indian Institute of Technology Dharwad, Karnataka – 580011, India. E-mail: rumaghosh@iitdh.ac.in
bDepartment of Biosciences and Bioengineering, Indian Institute of Technology Dharwad, Karnataka – 580011, India
cKAHER's Dr Prabhakar Kore Basic Science Research Centre, 3rd Floor, KLES V. K. Institute of Dental Science Campus, Belagavi, Karnataka – 590010, India
dDepartment of Urology, JN Medical College, KLE Academy of Higher Education and Research, Belagavi, Karnataka – 590010, India
eUrinary Biomarkers Research Centre, KLES Dr Prabhakar Kore Hospital & Medical Research, Belagavi, Karnataka – 590010, India
First published on 19th December 2024
Abstract
Prostate cancer antigen 3 (PCA3) has emerged as a critical biomarker for the early detection of prostate cancer, complementing the traditional prostate-specific antigen (PSA) testing. This research presents a novel resistive sensor based on reduced graphene oxide (RGO) functionalized with glutaraldehyde (GA)/complementary single-stranded DNA (ss-DNA) for the detection of the PCA3 RNA. The device was meticulously characterized at each fabrication step to confirm the successful integration of the various layers on the sensor device, utilizing atomic force microscopy (AFM) which confirmed the increase in the thickness of the sensor from ∼1.4 nm (only RGO) to ∼25 nm for the RGO–GA/ss-DNA/PCA3 device. Field emission scanning electron microscopy exhibited a change in the surface morphology after each step of device fabrication and testing. Fourier-transform infrared (FTIR) spectroscopy was conducted to confirm the presence of functional groups in each component of the sensor. The fabricated resistive sensor demonstrated mean response ranging from 1.203 to 59.44% for 0.1 to 100 ng mL−1 PCA3 RNA. Notably, the device exhibited stability over a period of two weeks and displayed high selectivity for PCA3. The developed RGO–GA/ss-DNA/PCA3 sensor was tested with RNA extracted from multiple prostate cancer cell lines and other cancer cell lines and demonstrated response only to RNA extracted from LNCaP as it was the only cell line expression of PCA3. The findings from the developed sensor were cross-validated with the observations of a semi quantitative polymerase chain reaction (qPCR), and were found to be closely matched.
1. Introduction
The global burden of cancer is increasing at an alarming rate, with prostate cancer ranking as the second most prevalent cancer among men worldwide.1,2 According to a recent report from the World Health Organization (WHO), approximately 1.4 million new cases and nearly 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths from prostate cancer were documented in 2022.3 While age remains the predominant risk factor, early detection is crucial, as timely intervention can significantly improve treatment outcomes.
000 deaths from prostate cancer were documented in 2022.3 While age remains the predominant risk factor, early detection is crucial, as timely intervention can significantly improve treatment outcomes.
The current screening methods primarily include digital rectal examination (DRE) and the measurement of serum prostate-specific antigen (PSA) levels. The identification of PSA as a biomarker has revolutionized the diagnosis and prognosis of prostate cancer. PSA is a protein secreted by prostate epithelial cells, and its elevated serum levels (greater than 4 ng mL−1) have been strongly correlated with malignancy.4 However, despite its utility, PSA lacks specificity, as elevated levels may also occur in various non-malignant conditions, including benign prostatic hyperplasia (BPH), urinary tract infections (UTIs), and prostatitis. Furthermore, in some cases, the growth of prostate tumors is slow and it remains indolent, resulting in PSA levels that may not rise above the diagnostic threshold.5 This complicates the clinical decision-making. These limitations of PSA screening contribute to a substantial number of unnecessary biopsies, which are invasive and painful. Additionally, the risk of false negatives can delay the diagnosis of clinically significant cancers and subsequently, the commencement of treatment, underscoring the pressing need for more reliable biomarkers.
Prostate cancer antigen 3 (PCA3) is a long noncoding ribonucleic acid (lncRNA) consisting of 3922 nucleotides that is significantly overexpressed in prostate cancer tissue.6,7 Notably, PCA3 demonstrates a high degree of specificity, showing no correlation with other prostate related conditions like prostatitis or BPH. This specificity is particularly advantageous, as it reduces the likelihood of false positives. Moreover, PCA3 can be detected in urine, allowing for non-invasive sampling, which is both convenient for patients and enhances compliance with screening protocols.8,9 Recent studies have suggested that PCA3 may also provide additional insights into tumor aggressiveness and may be used in conjunction with PSA levels to improve risk stratification. The integration of PCA3 testing into clinical practice could lead to more informed decision-making regarding the necessity of biopsies and the management of prostate cancer.
The expression of RNAs is commonly assessed using polymerase chain reaction (PCR)-based methodologies due to their high sensitivity, making them widely utilized in clinical settings for RNA and DNA detection.10 In addition, techniques like in situ hybridization, northern blot (NB) analysis, and mass spectrometry (MS) are also employed to evaluate RNA expression levels.11,12 Most studies investigating RNA or DNA as a biomarker rely on these established methods for quantifying the nucleic acid expression. While these techniques demonstrate considerable sensitivity, they typically necessitate advanced laboratory equipment, intricate protocols, and specialized expertise for sample handling, characterization, and analysis. Additionally, they often require substantial sample volumes (in the range of hundreds of microliters) and can take several hours to yield results. These limitations may discourage patients from undergoing testing during the early manifestation of symptoms, thereby impeding timely diagnosis.
The development of portable sensors that are user-friendly, require smaller sample volumes, provide rapid results, and are cost-effective could significantly alleviate these challenges. Electrochemical sensors have emerged as a promising alternative to conventional detection methods for RNA and nucleic acids, primarily due to their portability and potential for real-time monitoring.4,13,14 For example, Soares et al. have developed an electrochemical and impedance-based biosensors for the detection of PCA3.8 The authors employed a PCA3-complementary single-stranded DNA (ss-DNA) probe, immobilized on a layer of chitosan and carbon nanotubes to detect as low as 0.128 nM L−1 PCA3 RNA. Though electrochemical sensors are highly accurate, they suffer from poor shelf-lives. In addition to electrochemical sensors, field-effect transistor (FET)-based sensors represent an emerging alternative to traditional RNA detection methods.15 Tian et al. developed a graphene-based FET sensor incorporating 1-pyrenebutanoic acid succinimidyl ester (PBASE) for RNA detection.16 Similarly, Wang et al. created graphene-based solution-gated FETs (G-SGFETs) for DNA detection.17 These FET sensors exploit the high electron mobility of graphene, allowing for rapid and sensitive detection of nucleic acids. While FET sensors offer high sensitivity and compactness, their fabrication often requires sophisticated and costly cleanroom facilities.
Many of the aforementioned methods rely on optical and electronic signals generated from the hybridization of ss-DNA and RNA. The alteration in charge distribution associated with ss-DNA/RNA hybridization can be quantitatively measured using resistive sensors, which operate by inducing changes in electrical resistance of the sensors resulting from bio-reactive interactions between ss-DNA and RNA.18 These sensors can be fabricated using simpler and more cost-effective methods, possess longer shelf lives, and can be made portable. Each device typically comprises three essential components: (a) a bioreceptor, which in the case of RNAs, is carefully designed ss-DNA, (b) a transducer layer, which serves to anchor the ss-DNA without inhibiting its bio-reactivity and converts the biological interactions into electronic signals, and (c) a signal measurement unit.
The transducer layer plays a crucial role and can be made from materials with high carrier mobility to effectively transfer changes in charge density, as well as possessing ample functional groups for ss-DNA anchoring. Reduced graphene oxide (RGO) is a two-dimensional carbon nanomaterial known for its exceptional electronic properties, with functional groups present on its basal planes and edges, allowing for effective interaction with biomolecules.19 RGO has been successfully utilized as a transducer layer in various biosensor applications, including resistive sensors, owing to its promising characteristics, such as high surface area, higher carrier mobilities, and conductivity. However, the functional groups on RGO may sometimes be insufficient to anchor an adequate number of probes. One potential solution is the incorporation of gold nanoparticles, which are biocompatible and can facilitate ss-DNA anchoring through coordination chemistry, enabling stronger interactions with the ss-DNA probes.18 Nevertheless, the cost of gold nanoparticles may limit their widespread application in low-resource settings. As an alternative, organic compounds featuring the desired functional groups could serve as a more economical option compared to gold nanoparticles. For instance, compounds with carboxyl, amine, or thiol groups can provide effective anchoring sites for ss-DNA, thereby enhancing sensor performance while maintaining cost-effectiveness. Recent advancements in hybrid composites of nanomaterials combining RGO with other nanostructures have shown promise in improving sensor sensitivity and specificity. These materials can enhance electron transfer rates and increase the overall binding capacity for target nucleic acids, further advancing the field of portable nucleic acid detection.
This research reports the development of 2-port resistive sensor devices using RGO and glutaraldehyde (GA) for detection of a wide range of concentrations (0.1 to 100 ng mL−1) of PCA3. GA is an aldehyde which covalently binds with amine.17,20,21 A judiciously designed ss-DNA probe terminated with an amine group was used as the receptor to bind with PCA3 RNA. The developed sensors were tested with RNA extracted from different cancer cell lines. The detailed findings are discussed in the subsequent sections.
2. Materials and methods
2.1. Material synthesis
Indium tin oxide (ITO)-coated glasses with sheet resistivity 30–60 per sq. were purchased from Sigma Aldrich (703184). HPLC water and all the other chemicals were purchased from Sigma Aldrich and were used without any further purification. Graphene oxide (GO) was synthesized using modified Hummers’ method as per one of our earlier reports.222.2. Fabrication of the biosensor
ITO-coated glasses were used as substrates for the resistive sensors. The conducting ITO electrodes were defined on the two edges of the substrate by selectively etching ∼1 mm wide ITO from the middle of the substrate using a mixture comprising 36% hydrochloric acid (HCl) and deionized (DI) water in a 4:1 ratio. Next, the exposed ITO/glass surfaces were coated with GO–GA and left to dry at 200 °C for variable durations, optimizing the reduction of GO–GA such that the baseline resistance of the device ranges within 10 ± 3 kΩ. GA functioned as binding sites for single-stranded DNA (ssDNA). The ss-DNA ([AmC6] TTTTTTTCCCAGGGATCTCTGTGCTTCC) was designed and synthesized at Eurofins Genomics India labs. 5 μL volume of 9 ng mL−1 concentration of the designed ss-DNA probe was coated onto the RGO–GA, followed by heating the device at 50 °C for varying durations (10/30/45/60 minutes). After the initial binding, the samples were washed with HPLC grade water to remove the unbound ss-DNA molecules. For the biomarker detection, the hybridization of RNA/DNA was facilitated by dropping 5 μL of the required concentration of PCA3 (GGAAGCACAGAGATCCCTGGG, synthesized at Eurofins Genomics India) on the RGO–GA/ss-DNA sensors. Subsequently, the device was heated at 50 °C for varied durations (10/30/45 minutes), followed by washing with HPLC water to eliminate any unbound PCA3 RNA. This step minimizes the non-specific signals emerging from the sensors. The binding temperature was chosen the same as the melting temperature of PCA3 (50 °C). The steps of the biosensor fabrication are shown schematically in Fig. 1.
 | ||
| Fig. 1 Fabrication steps and working of the PCA3 RNA biosensor. | ||
2.3. Material and device characterization
The surface morphologies of the samples were analysed using a Carl Zeiss Gemini 300 field emission scanning electron microscope (FESEM). The presence of RGO–GA, probe and target were confirmed through an Oxford energy dispersive spectrometer (EDS), which was integrated with the FESEM. The binding of various elements within the sensing layer of the device was cross-validated using an Agilent Cary 5000 UV-vis-NIR spectrophotometer. A Park System NX10 atomic force microscope (AFM) was employed to obtain the thickness profile of the GO flakes, and efforts were made to capture images of the immobilized probe and target which determine the thickness of the layers using AFM. A Keithley Data Acquisition Unit (DAQ6510) was used to measure the change in sensor resistances.2.4. RNA extraction and analysis using semi-quantitative RT-PCR
The expression of PCA3 in five cancer cell lines (LNCaP, PC3, T47D, DU145 and A549) was analysed using semi-quantitative RT-PCR. RNA was extracted from the cell lines using RNAiso Plus (Takara, #9108Q) following the manufacturer's protocol. Cells in RNAiso Plus were treated with 100 μL of chloroform, mixed vigorously, and incubated for 5 minutes. The samples were then centrifuged at 13000g for 15 minutes, and the top aqueous layer was carefully transferred to a new tube. Next, 250 μL of isopropanol was added, mixed, and incubated for 10 minutes. The samples were centrifuged for 10 minutes, and the RNA pellet was washed with 75% ethanol which was then air dried.
After quantifying the RNA, reverse transcription was carried out using the Prime Script™ RT reagent Kit (Takara, #RR037A), and the resulting cDNA was diluted to a concentration of 10 ng μL−1. Semi-quantitative RT-PCR was then performed using Emerald Amp GT PCR Master Mix (Takara, #RR310). A thermal cycler was used in synthesis and conditions were set as follows: 95 °C for 3 minutes, 60 °C for 1 minute, and 72 °C for 30 seconds. After 32 cycles for PCA3 amplification and 25 cycles for TPT1, the reactions were incubated at 72 °C for 10 minutes, followed by cooling at 4 °C.
2.5. Urine sample collection and processing
The urine samples of the patients reporting with preliminary symptoms and histologically confirmed prostate cancer at KLE's Dr Prabhakar Kore Hospital & Medical Research Centre, Belagavi, India were collected after acquiring the informed consents from the patients. Voided urine specimens were collected from 2 subjects undergoing treatment for castrate resistant prostate cancer (CRPCa). Urine specimens (30–50 mL) were centrifuged at 700g for 10 min and urinary sediments were washed with ice-cold PBS buffer before being centrifuged again under the same conditions. The supernatant collected was subject to biosensor testing.23 The collected samples were directly drop casted on RGO–GA/ss-DNA devices and their PCA3 concentrations were evaluated. The PCA3 levels as found using the resistive sensors were validated using semi quantitative PCR. For semiquantitative PCR, all the steps mentioned in Section 2.4 were followed.3. Results and discussion
3.1. Material characterization
The morphologies of the sensor device at each step were observed using FESEM. The RGO/GA-coated device when analysed under the electron microscope, revealed irregular, large, crumbled, and wrinkled sheets of RGO with lateral dimensions reaching a few microns and clusters of bead-like GA, confirming the presence of both RGO and GA (Fig. 2(a)). The EDS spectrum of RGO–GA demonstrates the presence of only carbon and oxygen which are attributed to sp2 hybridized carbon atoms and the associated functional groups from RGO and also to the organic compound (Fig. 2(b)). In the RGO–GA/ss-DNA coated device, thicker crumpled sheets of ss-DNA above RGO–GA were visible (Fig. 2(c)). This image was captured following multiple washing steps to ensure the proper immobilization of the ss-DNA on the base transport layer. Also, no spherical GA could be seen in RGO–GA/ssDNA devices or the devices acquired in subsequent steps. This is because the ssDNA anchored on GA covering them completely. The ss-DNA was modified with an amine group, which was confirmed by the EDS spectrum of the RGO–GA/ss-DNA sample which shows the coexistence of carbon and oxygen from RGO, GA, and ss-DNA and nitrogen from the amine-terminated ss-DNA (Fig. 2(d)). Notably, the nitrogen peak was absent in the RGO–GA sample. Not only, nitrogen was captured in the RGO–GA/ss-DNA but also the carbon and oxygen content increased slightly in Fig. 2(d), perhaps due to the introduction of ssDNA in the device. The nitrogen weightage in the EDS spectrum of the RGO–GA/ss-DNA/PCA3 device increased to 2.5% from 1.8% confirming the presence of N in the RNA too (Fig. 2(f)). As GO is a 2D material and was synthesized following a top-down approach, it was necessary to ascertain the efficacy of the exfoliation. This was done by assessing the thickness of RGO–GA using AFM. The thickness of RGO–GA was found to range between 1–2 nm indicating the presence of mono to a few layers of RGO–GA (Fig. 2(g)). The thickness of the RGO–GA/ss-DNA was found to range from 15–20 nm confirming the attachment of ss-DNA to the transducer layer (Fig. 2(h)). Furthermore, the thickness of the RGO–GA/ss-DNA/PCA3 was found further increase by ∼25 nm (Fig. 2(i)). This ensured the hybridization of the probe with the target. | ||
| Fig. 2 FESEM images of (a) RGO–GA coated on ITO coated glass, (c) RGO–GA/ssDNA, and (e) RGO–GA/ssDNA/PCA3 device; EDS spectra of (b) RGO, (d) RGO–GA/ssDNA, and (f) RGO–GA/ssDNA/PCA3; and (g) AFM image and thickness profile of RGO–GA, (h) AFM image and thickness profile of RGO–GA/ssDNA, and (i) AFM image and thickness profile of RGO–GA/ss-DNA/PCA3. | ||
The absorption characteristics of the device at different stages were studied using UV-visible absorption spectroscopy. The sharp absorption peak seen at 226 nm in Fig. 3(a) shows π–π* transition of aromatic C–C bonds in GO with a shoulder peak at 310 nm due to n–π* transition C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The π–π* transition remains the same in all other samples. Also, absorption peaks around 307 nm and at 342 nm could be seen in the case of the RGO–GA/ss-DNA sample with a broader and stronger band (Fig. 3(a)).
O. The π–π* transition remains the same in all other samples. Also, absorption peaks around 307 nm and at 342 nm could be seen in the case of the RGO–GA/ss-DNA sample with a broader and stronger band (Fig. 3(a)).
 | ||
| Fig. 3 (a) UV-visible spectra of GO, RGO–GA, RGO–GA/ss-DNA probe and RGO–GA/ssDNA probe/PCA3 RNA (b) and (c) FTIR spectra of (b) GO and RGO–GA (c) RGO–GA/ss-DNA and RGO–GA/ss-DNA/PCA3. | ||
The absorption intensities increased significantly after the probe/target hybridization. FTIR spectra of the sensors at different stages of fabrication were acquired to ensure the presence of each added material in the developed sensor. The green plot in Fig. 3(b) represents the dominant peaks present in GO at 3400, 2927, 1720, 1619, 1224 and 1080 cm−1 indicating the presence of hydroxyl, carbonyl, carboxyl, and epoxide in the synthesized GO. In the RGO–GA spectrum, similar peaks to those observed for GO were observed (Fig. 3(b)) signifying the presence of the same peaks. The N–H peak intensity was observed to increase after the PCA3 target bound to the sensor device, as shown in Fig. 3(c).
3.2. Biosensor characterization
To evaluate the capabilities of the developed biosensors, they were to be tested with various concentrations of PCA3 RNA. The resistance across the RGO–GA/ss-DNA device was measured and recorded as the baseline resistance, Rss-DNA. As soon as a specific concentration of PCA3 was introduced to the sensor the resistance of the device was observed to increase to RPCA3. The sensor's response was calculated using the formula in eqn (1) | (1) |
To the best of the authors’ knowledge, RGO–GA has never been employed as a transducer layer for detection of PCA3. Hence, before the biosensors could be tested with different concentrations of PCA3, it was necessary to optimize multiple parameters of the material to get the highest response to the target RNA. Firstly, to optimize and achieve the efficient immobilization of ss-DNA on RGO–GA, various concentrations of GA were mixed with RGO ranging from 0.2–2% while maintaining a constant ss-DNA concentration of 9 ng mL−1. This exercise aimed at determining the optimal concentration of glutaraldehyde (GA) required to be present at the sensor surface, ensuring anchoring of adequate ss-DNA to respond to the highest concentrations of PCA3. Fig. 4(a) illustrates the comparative response of multiple sensor devices to 50 ng mL−1 PCA3. It was noted that the percentage of GA had an impact on the performance of the biosensors and the response of the device was found to be highest for 1% GA (Fig. 4(a)). This perhaps happened because with an increase in the concentration of GA, the attachment site increased thereby, anchoring more ss-DNA till 1% GA. When the concentration of the organic compound increased further, perhaps it started agglomerating owing to a very high concentration. This would have led to loss in the available attachment sites for the probes which subsequently resulted into a poorer response to the RNA. Another reason that could have possibly led to a decrease in the response of RGO–GA/ss-DNA devices to PCA3 with an increase in GA% beyond 1% is GA is insulating in nature. The change in the charge due to hybridization of ss-DNA and RNA needs to be transferred to the measuring device. This task is accomplished by the semiconducting RGO present in the transducer layer. However, with a high concentration of GA (2%), the continuity of the RGO layer may have been disturbed which might have prohibited the carrier mobility of the transport layer ultimately leading to a reduced response. The next parameter to be optimized was the binding temperature of the ss-DNA of RGO–GA. This was done by following all the fabrication steps but by varying the incubation temperature from 25 (room temperature) to 70 °C and testing the devices for 50 ng mL−1 PCA3.
 | ||
| Fig. 4 (a) Response of RGO–GA/ssDNA sensors (3 devices each) to 50 ng mL−1 PCA3 for varying percentage of GA, (b) by varying the temperature of incubation from 25 °C to 70 °C, and (c) change in resistance of the RGO–GA/ss-DNA devices after heating the same at 50 °C for four different durations (10–60 minutes). (d) Response of RGO–GA/ss-DNA sensors for 50 ng mL−1 PCA3 bound on the devices for 10, 30 and 45 minutes. | ||
It was found that heating the probe at 50 °C results in the best immobilization of the probe as the response of the biosensors was found to be highest for the devices fabricated by heating the probes on RGO–GA at that temperature (Fig. 4(b)). The next key parameters to optimize were the binding times of ss-DNA and the RNA. For binding resulting into the highest response towards PCA3, three devices for each duration were heated at 50 °C for 10–60 minutes. It was observed that the device's response reduced when heated for longer than 45 minutes (Fig. 4(c)). Similarly, DNA/RNA hybridization was performed by heating the sensors at 50 °C for 10, 30, and 45 minutes, with both sets of experiments yielding nearly highest responses for 45 minutes (Fig. 4(d)). As a result, subsequent device fabrication involved heating them for 45 minutes each after introducing ss-DNA and PCA3 onto the devices.
After the different parameters related to the transducer layer and process steps were optimized, the developed RGO–GA/ss-DNA devices were tested over a wide range of concentrations (0.1–100 ng mL−1). For each concentration, multiple devices (3 to 5) were assessed, showing mean responses from 1.203% to 59.44% for PCA3 levels of 0.1 ng mL−1 to 100 ng mL−1 (Fig. 5(a)). The limit of detection of the device was calculated using the formula below
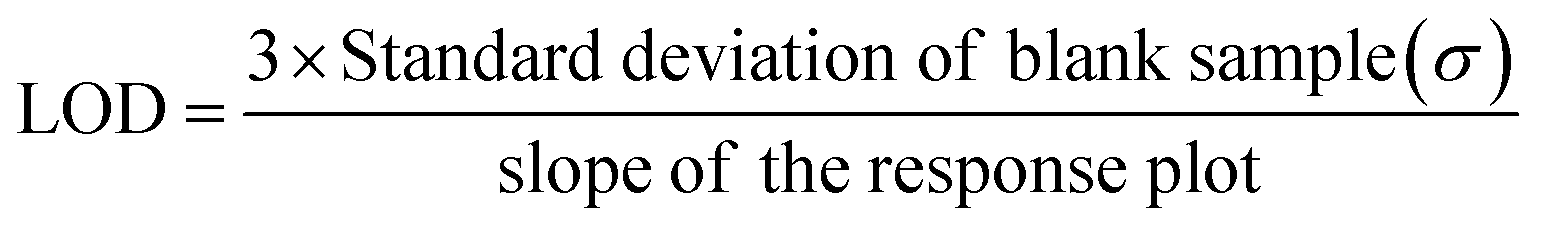
 | ||
| Fig. 5 (a) Response of RGO–GA/ssDNA sensors (3 devices each) for different concentrations varying from 0.1–100 ng mL−1 PCA3, (b) exponentially fitted curve of the response plot, and (c) stability of RGO–GA/ssDNA sensors tested up to the 2nd week using 50 ng mL−1 PCA3. (d) Selectivity test results of RGO–GA/ssDNA sensors. (e) Semi quantitative PCR results using different cancer cell lines. | ||
The LOD of the sensors was found to be 0.014 ng mL−1, where the standard deviation (σ) value was 0.51316 and the slope (m) was 72.60345 as seen in Fig. 5(b). Fig. 5(b) also indicates that the responses of the developed sensors were found to increase linearly as the concentration of the RNA increased. As it is not possible to fabricate the device afresh before testing the samples in the real-time, it was necessary to find the lifetime of the developed devices. This was ascertained by fabricating multiple devices and testing them across weeks. A total of 9 devices were fabricated and 3 of them were tested with 50 ng mL−1 on the same day they were fabricated and the remaining devices were stored at room temperature in an evacuated desiccator. These devices were tested in the subsequent two weeks and the responses of the devices were recorded. The mean response of the fresh set of devices was found to be ∼24% while the responses of the sensors in the subsequent two weeks were found to slightly reduce to ∼20% indicating good stability of the device (Fig. 5(c)). For practical application, the sensors would need to directly interact with RNA present in patients' urine or serum. Given that human urine is highly saline and both urine and serum contain multitudes of RNAs, ensuring the sensor's selectivity for PCA3 becomes essential. To evaluate this, the RGO–GA/ss-DNA sensors were tested with various samples, including PCA3 negative control (NC, CTAATGTCCTTCCCTCACAAGCG), RNA extracted from a lung cancer cell line (A549 LUAD), a breast cancer cell line (T47D), and three prostate cancer cell lines LNCaP, PC3, and DU145. Among these, only the LNCaP cell line expresses PCA3 RNA, and hence, no PCA3 signal was expected from the other samples. As shown in Fig. 5(d), the sensors exhibited the highest response (∼20%) to RNA extracted from LNCaP, successfully indicating the presence of 50 ng mL−1 PCA3 in the cancer cell. For other cell line and the NC sample, the mean responses of the RGO–GA/ss-DNA sensor were found to be very low. This confirms the sensor's high selectivity for PCA3. The expressions of PCA3 in five tested cancer cell lines were also evaluated using semi quantitative PCR (qPCR), a widely used commercial technique for RNA/DNA detection. As shown in Fig. 5(e), the LNCaP cell line exhibited the highest PCA3 expression compared to A549 LUAD, T47D, DU145, and PC3 as was expected. These results aligned perfectly with the findings from the developed portable, label-free, and easy-to-use resistive sensor. Not only the developed resistive sensors were found to be highly sensitive and selective towards PCA3, the performance of the devices was also found to be comparable and, in some cases, better than the recently reported works on PCA3 detection (Table 1).
Finally, the developed RGO–GA/ss-DNA sensors were tested with urine samples of two patients and the device expressed the presence of greater than 50 ng mL−1 PCA3 in one case and a concentration of 50 ng mL−1 of another sample, as shown in Fig. 6(a). The findings with the developed sensors were cross validated with qPCR in which the expression of PCA3 in the standard 50 ng mL−1 PCA3 sample (labelled as positive control) and in the clinical samples was checked (Fig. 6(b)). The presence of PCA3 was clearly indicated in the PCR results as well which aligned with the observations with the developed sensors greatly.
 | ||
| Fig. 6 (a) Response of RGO–GA/ssDNA sensors for urine samples (b) and semi quantitative PCR results with urine samples and 50 ng mL−1 PCA3 positive control. | ||
| S. no. | Type of sensor | Materials used | Dynamic range | Limit of detection (LOD) | Ref. |
|---|---|---|---|---|---|
| 1 | Electrochemical and impedance sensor | CHT/CNT/ssDNA/PCA3 | 10−6 to 10−16 mol L−1. | 0.128 nmol L−1 and 1.42 nmol L−1 | 8 |
| 2 | Electrochemical sensor | Cu-MOFs nano sheet/BiVO4 nano rod | 0.1 pM to nM | 0.02 fM | 24 |
| 3 | DNA-EGFET sensor | Au/PANI/ssDNA | 1 pmol L−1 to 1 mol L−1 | 9.77 pmol L−1 | 25 |
| 4 | G-FET sensor | Graphene/PBASE/probe DNA/RNA | 100 aM to 1 pM for DNA probe-modified G-FET | 100 aM | 26 |
| 5 | MoS2-FET sensor | SiO2/MoS2/PASE/PMO probe/DNA | 10 fM to 1 nM | 6 fM | 27 |
| 6 | Resistive biosensor | RGO/AuNPs/ssDNA/PCA3 | 0.1–100 ng mL−1 | 18 | |
| 7 | RNA biosensor | RGO/GA/ssDNA/PCA3 | 0.1–100 ng mL−1 (15.37 pM–15.37 nM) | 0.014 ng mL−1 (2.172 pM) | This work |
3.4. Sensing mechanism
RGO is a p-type semiconductor, and acted as the base transport layer of the developed sensor devices.19 GA is an organic molecule with two aldehyde groups that allow it to cross-link biomolecules such as proteins, nucleic acids, or other biological recognition elements onto various base layers, in this case, RGO.17 The aldehydes present in GA form stable covalent bonds with amine groups (–NH2) with which the ss-DNA probe was terminated, enabling the latter's immobilization on the sensor surface. The ss-DNA is a charged biomolecule which imparts electrons to the p-type RGO thereby resulting in a resistance increase of the semiconducting carbon nanomaterial.18 Next, when the PCA3 RNA interacts with the bioreceptor, in this case, the ss-DNA, the overall charge density changes due to the hybridization of RNA/DNA. Mostly, the hybridization results in transferring more negative charges to the base layer which increases the resistance of RGO even further as seen in a schematic diagram of the sensing mechanism in Fig. 7. This change in the resistance of the base transport layer is proportional to the amount of PCA3 interacting with the bioreceptor. Hence, the extent of increase in RGO resistance as recorded by the DAQ indicates precisely the concentration of the PCA3 bound to the sensor device. | ||
| Fig. 7 Schematic diagram of the sensing mechanism. | ||
4. Conclusion
This work presents a simple and highly selective PCA3 RNA sensor. The RGO–GA/ss-DNA (9 ng mL−1) sensors were tested for PCA3 concentrations from 0.1 to 100 ng mL−1, showing a mean response ranging from 1.203% to 59.44%. The wt% of GA was optimized by preparing the sensors with GA varying from 0.2 to 2%. The devices with 1% GA in the RGO–GA composite were found to exhibit the highest response as compared to PCA3. Similarly, the binding temperature of ss-DNA and the hybridization temperature of PCA3 on ss-DNA were optimized by heating the devices at three temperatures ranging from room temperature to 70 °C and found to have best binding/hybridization at 50 °C. Next, the binding times were also optimized and found to be of 45 minutes. The sensor exhibited excellent selectivity for PCA3 RNA, distinguishing it from negative controls and RNA samples extracted from not only lung and breast cancer cell lines but also from PC3 and DU145, which are prostate cancer cell lines but do not strongly express PCA3 RNA. These results were validated with the results of qPCR, a standard clinical method for RNA detection. The limit of detection of the developed sensor was found to be 0.014 ng mL−1. It is believed that this investigation would lead to a simpler, portable, and efficient method for detecting PCA3 which in turn is expected to lead to improved and early diagnosis of prostate cancer, potentially minimizing unnecessary prostate biopsies.Data availability
All the data generated in this research are presented in Fig. 2 to 6 of the main manuscript. The personal details of the patients are not disclosed due to ethical reasons.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ruma Ghosh and Sudhanshu Shukla acknowledge the partial support of Science and Engineering Research Board (SERB), India through the Core Research Grant (CRG) scheme (CRG/2022/003365). The authors are also grateful to sophisticated central instrumentation facility and department of Chemistry, IIT Dharwad for helping us with the material characterization.References
- X. Ma and H. Yu, Yale J. Biol. Med., 2006, 79, 85–94 Search PubMed.
- H. Nagai and Y. H. Kim, J. Thorac. Dis., 2017, 9, 448–451 CrossRef PubMed.
- L. Wang, B. Lu, M. He, Y. Wang, Z. Wang and L. Du, Front. Public Health, 2022, 10, 811044 CrossRef PubMed.
- L. H. Pan, S. H. Kuo, T. Y. Lin, C. W. Lin, P. Y. Fang and H. W. Yang, Biosens. Bioelectron., 2017, 89, 598–605 CrossRef CAS PubMed.
- S. Ma, X. Li, Y. K. Lee and A. Zhang, Biosens. Bioelectron., 2018, 117, 276–282 CrossRef CAS PubMed.
- V. Yamkamon, K. Phyu Pyar Htoo, S. Yainoy, T. Suksrichavalit, T. Tangchaikeeree and W. Eiamphungporn, EXCLI J., 2020, 19, 501–513 Search PubMed.
- S. Takita, A. Nabok, M. Mussa, M. Kitchen, A. Lishchuk and D. Smith, Biosens. Bioelectron.: X, 2024, 18, 100462 CAS.
- J. C. Soares, A. C. Soares, V. C. Rodrigues, M. E. Melendez, A. C. Santos, E. F. Faria, R. M. Reis, A. L. Carvalho and O. N. Oliveira, ACS Appl. Mater. Interfaces, 2019, 11, 46645–46650 CrossRef CAS PubMed.
- D. Lee, S. R. Shim, S. T. Ahn, M. M. Oh, D. G. Moon, H. S. Park, J. Cheon and J. W. Kim, Clin. Genitourin. Cancer, 2020, 18, 402–408.e5 CrossRef PubMed.
- O. Khorkova, J. Stahl, A. Joji, C. H. Volmar and C. Wahlestedt, Nat. Rev. Drug Discovery, 2023, 22, 539–561 CrossRef CAS PubMed.
- W. Ahmad, B. Gull, J. Baby and F. Mustafa, Curr. Issues Mol. Biol., 2021, 43, 457–484 CrossRef CAS PubMed.
- F. Chen, J. Xue, L. Zhou, S. Wu and Z. Chen, Anal. Bioanal. Chem., 2011, 401, 1899–1904 CrossRef CAS PubMed.
- M. Kushwah, R. Yadav, A. N. Berlina, K. Gaur and M. S. Gaur, J. Solid State Electrochem., 2023, 27, 559–574 CrossRef CAS.
- H. Xu, L. Wang, H. Ye, L. Yu, X. Zhu, Z. Lin, G. Wu, X. Li, X. Liu and G. Chen, Chem. Commun., 2012, 48, 6390–6392 RSC.
- C. S. Lee, S. Kyu Kim and M. Kim, Sensors, 2009, 9, 7111–7131 CrossRef CAS PubMed.
- M. Tian, S. Xu, J. Zhang, X. Wang, Z. Li, H. Liu, R. Song, Z. Yu and J. Wang, Adv. Condens. Matter Phys., 2018, 2018, 8146765 Search PubMed.
- Z. Wang and Y. Jia, Carbon, 2018, 130, 758–767 CrossRef CAS.
- S. Kumar, N. Bhandari, S. Shukla and R. Ghosh, Biosens. Bioelectron.: X, 2024, 18, 100481 CAS.
- B. Konthoujam, N. Bhandari, M. P. Kamal, P. N. Srinivas, B. Thati, P. Bondugula, P. Reddy, R. C. Antaratan, N. Kadayinti, S. Shukla and R. Ghosh, Biosens. Bioelectron.: X, 2024, 20, 100530 CAS.
- E. B. Aydın, M. Aydın and M. K. Sezgintürk, Sens. Actuators, B, 2023, 378, 133208 CrossRef.
- Q. Wang, B. Zhang, X. Lin and W. Weng, Sens. Actuators, B, 2011, 156, 599–605 CrossRef CAS.
- S. Joshi, G. Guruprasad, S. Kulkarni and R. Ghosh, IEEE Sens. J., 2022, 22, 1138–1145 CAS.
- R. B. Nerli, S. C. Ghagane, S. R. Bidi, M. L. Thakur and L. Gomella, Cytojournal, 2021, 12(26) DOI:10.25259/Cytojournal_76_2020.
- X. Zhang, P. Wang, Z. Liang, W. Zhong and Q. Ma, Talanta, 2024, 266, 124952 CrossRef CAS.
- H. J. N. P. D. Mello, B. Bachour Junior and M. Mulato, Sens. Actuators, A, 2021, 318, 112481 CrossRef CAS.
- M. Tian, M. Qiao, C. Shen, F. Meng, L. A. Frank, V. V. Krasitskaya, T. Wang, X. Zhang, R. Song, Y. Li, J. Liu, S. Xu and J. Wang, Appl. Surf. Sci., 2020, 527, 146839 CrossRef CAS.
- J. Mei, Y. Li, H. Zhang, M. Xiao, Y. Ning and Z. Zhang, Biosens. Bioelectron., 2018, 110, 71–77 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |