
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntrinsic fluorescence hydrogels for ON/OFF screening of antidiabetic drugs: assessing α-glucosidase inhibition by acarbose†
F. Javier
Patiño‡
 ab,
Josué
M. Galindo‡
ab,
Alicia
Jiménez
ab,
Yolanda
Alacid
c,
C. Reyes
Mateo
c,
Ana Ma
Sánchez-Migallón
ab,
Ester
Vázquez
ab,
Sonia
Merino
*ab and
M. Antonia
Herrero
*ab
ab,
Josué
M. Galindo‡
ab,
Alicia
Jiménez
ab,
Yolanda
Alacid
c,
C. Reyes
Mateo
c,
Ana Ma
Sánchez-Migallón
ab,
Ester
Vázquez
ab,
Sonia
Merino
*ab and
M. Antonia
Herrero
*ab
aDepartamento de Química Inorgánica, Orgánica y Bioquímica, Facultad de Ciencias y Tecnologías Químicas, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: sonia.merino@uclm.es; Mariaantonia.herrero@uclm.es
bInstituto Regional de Investigación Científica Aplicada (IRICA), UCLM, 13071 Ciudad Real, Spain
cInstituto de Investigación, Desarrollo e Innovación en Biotecnología Sanitaria de Elche (IDiBE), Universidad Miguel Hernández de Elche (UMH), 03202 Elche, Spain
First published on 10th January 2025
Abstract
Diabetes remains one of the most prevalent chronic diseases globally, significantly impacting mortality ratetables. The development of effective treatments for controlling glucose level in blood is critical to improve the quality of life of patients with diabetes. In this sense, smart optical sensors using hydrogels, responsive to external stimuli, have emerged as a revolutionary approach to diabetes care. In this study, changes in the optical properties of a hydrogel are employed for monitoring α-glucosidase activity, a critical enzyme involved in diabetes mellitus type II due to its role in breaking terminal α-glycosidic bonds, releasing α-glucose. The enzyme is encapsulated within a triazine-based hydrogel that exhibits intrinsic blue fluorescence. Upon hydrolysis of the substrate p-nitrophenyl-α-D-glucopyranoside (p-NPG) by α-glucosidase, the fluorescence is quenched due to the release of p-nitrophenol (PNP). However, when exposed to potential antidiabetic drugs, the enzyme's activity is inhibited, and the hydrogel's fluorescence remains intact. This ON/OFF fluorescence-based assay enables rapid screening of drug candidates by evaluating their ability to inhibit α-glucosidase enzymatic activity. Sensor optimization involves conducting swelling studies, fluorescent assays, reusability tests and a trial with a real antidiabetic drug. This innovative approach holds potential for enhancing antidiabetic drug screening and management, offering a more accessible and efficient solution compared to traditional biosensors.
1. Introduction
Diabetes mellitus (DM) is a metabolic disorder characterized by elevated blood glucose levels, known as hyperglycemia, resulting from disruptions in insulin secretion (as seen in type I diabetes) or varying degrees of insulin resistance (commonly seen in type II diabetes).1 Diabetes is a global health concern, affecting more than 400 million individuals worldwide2 and causing 1.5 million deaths annually. Efforts are underway to reduce its prevalence by 2025.3 While there is no definitive cure, management involves diet, lifestyle changes, and pharmacological therapy.4 In this sense, type I DM patients require insulin injections, while type II DM patients are treated with medications like metformin to control blood glucose levels.5Several of these medications, especially in type II DM, focus on inhibiting the action of the enzyme α-glucosidase. This enzyme plays a vital role in carbohydrate digestion as it is responsible for breaking down terminal α-glycosidic bonds, releasing α-glucose into the bloodstream. Inhibiting this enzyme can slow the digestion of complex carbohydrates, resulting in a gradual increase in blood glucose levels following a meal.6 Thus, monitoring the activity of the enzyme α-glucosidase is crucial for the development of new drugs and therapies for type II DM.
In this context, biosensors were born with the final goal of detecting and transforming a biological signal into an analytical data, allowing for the detection of specific biological molecules or activities.7 They are used to detect and measure the presence or concentration of substances such as glucose, enzymes, and antibodies, among others. Typically, biosensors comprise a biological component, such as an enzyme or antibody, and a transducer that translates the biological response into an analytical signal. Transducers may operate based on different principles, including electrochemical, optical, and piezoelectric methods.8,9 Biosensors are crucial for the development of new treatments for different illnesses. Different techniques and biosensors are developed to check the α-glucosidase activity, especially by promoting in situ fluorescent reactions using among others different kinds of nanoparticles, due to the intrinsic nature of fluorescence that usually contributes to a higher sensitivity and specificity.10–12 However, if chromophore nanoparticles are not bound to a support, they may readily aggregate, resulting in reduced stability of the sensor components or properties over time.13,14 This can negatively impact the reusability of the system and result in an inconsistent response.
Hydrogels are presented as new materials in the field of biosensors. They are 3D polymeric networks formed by physical and/or chemical interactions with the ability to swell large amounts of water without dissolving in it. They can interact with different biological molecules or even respond to an external stimulus such as changes in the pH, temperature or light.7,15,16 Besides, due to the porous nature and substantial internal surface area of hydrogels, it becomes feasible to trap and secure molecules within them, while their high water content creates a suitable environment for biological compounds. A diverse range of bioreceptors can be anchored, including antibodies for identifying antigens, nucleic acids for hybridizing with specific DNA sequences of interest, biomimetic receptors, and even entire cells such as bacteria or fungi for assessing toxicity. Nevertheless, the immobilization of enzymes as recognition components garners special attention due to their unique binding specificity and catalytic prowess, which enhances the detection process.17 Several instances of hydrogels as biosensors are found in the literature for measuring cholesterol levels in blood or detecting chemotherapeutic drugs in cancer cells, among other applications.7,16,18–24 Furthermore, different fluorescent hydrogels have been utilized to detect bacterial species through the entrapment of the enzyme α-glucosidase,25–27 and several studies follow a similar pattern by monitoring α-glucosidase activity.28
Thus, knowing the potential of these materials in the field of sensing, a triazine-based hydrogel was used in this work to monitor the activity of the α-glucosidase. This hydrogel has an inherent fluorescence due to an aggregation-induced emission (AIE) process, which gives it the advantage of having a very reproducible fluorescent intensity.29 In contrast to already published hydrogels, in which a fluorescent compound is added into the hydrogel structure, sometimes giving rise to a non-homogeneous structure,30–33 this triazine-based hydrogel emits fluorescence without any extra chromophore, allowing a homogeneous fluorescent emission. In this present work, we found that the hydrogel is a perfect candidate to host the α-glucosidase while keeping its functionality. Furthermore, its emission is completely quenched due to the presence of p-nitrophenol (PNP), which can be produced by the enzyme when breaking the α-o-glucosidic bond present in p-nitrophenyl-α-D-glucopyranose (PNPG) (Fig. 1). This capability aids in monitoring enzyme activity, enabling the identification of molecules that inhibit the enzyme and thereby facilitating the screening of antidiabetic drug candidates.
 | ||
| Fig. 1 Schematic representation of the biosensor working. | ||
2. Materials and methods
2.1. Materials and reagents
Reagents and solvents were used as purchased from commercial sources without further purification. Dimethyl sulfoxide (DMSO), phosphate buffered saline (P-5368, pH = 7.4, 0.138 M NaCl, 0.0027 M KCl) (PBS), p-nitrophenol (PNP), p-nitrophenyl-α-D-glucopyranoside (PNPG), α-glucosidase (E.C. 3.2.1.20, from Saccharomyces cerevisiae, ≥10 unit mg−1), acarbose, acrylamide (AM), oligo(ethylene glycol) methyl ether methacrylate (OEGMA), potassium persulfate (KPS), and poly(ethylene glycol) diacrylate (PEGDA) were obtained from Sigma–Aldrich. Finally, (4-vinylphenyl)-2,4-diamino-1,3,5-triazine (VPhDT) was synthesized following a previous modified method.29,342.2. Synthesis of phenyldiaminotriazine hydrogel (ChDAT)
Phenyldiaminotriazine hydrogels were prepared using 1 mL DMSO as a solvent. Synthesized VPhDT (30 mg, 0.14 mmol) was dissolved in it. Then OEGMA (135 mg, ∼0.14 mmol), and AM (135 mg, 1.90 mmol) were added as comonomers, and PEGDA (1 mg, ∼1.43 × 10−3 mmol) as crosslinker. Finally, KPS (8 mg, 2.96 × 10−2 mmol) was added as radical initiator. The reaction was carried out in a cylinder (1 cm diameter, 0.5 cm high) or cubic (1 × 1 × 1 cm) silicone mould in a stove at 90 °C for 30 min. After that, organogels were placed in a vessel with 250 mL of Milli-Q water, replacing it four times a day for 3 days in order to obtain the final hydrogels. For xerogels, hydrogels were dried at room temperature until their weight was constant.2.3. Characterization of the hydrogels
Swelling studies were carried out by immersing the xerogels in Milli-Q water at room temperature. The samples were weighted at established time intervals, finalizing the measures when hydrogels have a constant weight. In order to avoid measurement errors by water weight, excess water was removed using a paper filter before each measure. The swelling degree (SD) was calculated following the eqn (1): | (1) |
Mechanical tests were carried out using a Mecmesin Multitest 2.5-i dynamic mechanical analyzer at room temperature. Hydrogel disks (×6) were compressed between two plates at a rate of 6 mm min−1 until 40% of strain at their initial swelling degree (1 cm of diameter and 0.5 cm of height). This means that each xerogel is swelled with a specific amount of water to achieve the desired initial swelling degree of the hydrogels (SD = 4). For Young tests, stress–strain curves were analyzed from 2 to 10% of strain.
Absorption spectra were carried out on a Jasco V-530 spectrophotometer.
The porous structure was depicted by scanning electron microscopy (SEM) using a Gemini SEM 500 from Zeiss with a FEI QUANTA 250 apparatus.
Fluorescence properties were measured on a Jasco FP-8300 equipment and, also, in a Photon Technology International, Inc. Spectrofluorimeter. Measures took place in solid state in cylindrical and cube form, without differences between them. No cuvette was used for the measurements, instead the hydrogels were placed in a support for solids.
2.4. Hydrogel loading procedure
Hydrogels have been loaded with different solutions like α-glucosidase enzyme, PNPG and PNP among others. Although details are included in the results and discussion section, the general procedure in all cases was performed as follows: initially, each hydrogel is dried at ambient room temperature until the weight of the dried gel is constant, a process typically spanning 24 hours. After drying, the hydrogel's weight is meticulously recorded. This measurement is crucial for determining the precise volume of solution needed to achieve the desired swelling degree. For instance, if the weight of a dry hydrogel is 100 mg and a SD of 10 is desired, then 1 g of the solution is required for absorption. During the swelling phase, the hydrogel is placed in a hermetically sealed container to ensure complete and uniform absorption of the solution. This absorption process is generally completed over a period of 48 hours.3. Results and discussion
In this study, a chemically crosslinked triazine hydrogel was employed, which we denote as ChDAT hydrogel, to monitor the activity of α-glucosidase, which is confined inside. The hydrogel's emission is completely suppressed in the presence of PNP, a byproduct generated from PNPG through enzymatic action. In the presence of any α-glucosidase inhibitor, the fluorescence emission remains unquenched, enabling the identification of the most promising drug candidates.3.1. ChDAT hydrogel
Triazine-based hydrogels were prepared as previously reported by the group.29,35 VPhDT is a multifunctional monomer because it can take part in numerous noncovalent interactions.36 Initially, an organogel in DMSO is obtained and OEGMA is used as a hydrophilic part in the polymer network in order to offset the intrinsic hydrophobicity of VPhDT, thus, allowing the organogel to exchange the DMSO for water, forming the hydrogel. Finally, AM and PEGDA helps to maintain the consistency of the network through hydrogen bonding and chemical crosslinking, respectively (Scheme 1). | ||
| Scheme 1 Synthesis of chemically crosslinked triazine-based hydrogels (ChDAT). | ||
3.2. Characterization of the ChDAT hydrogel
The swelling degree serves as a crucial parameter to know water absorption capacity of the hydrogel. It can be modulated altering the degree of crosslinking and by introducing hydrophilic or hydrophobic groups. Within the realm of biosensors, this swelling property is harnessed to regulate the diffusion of bioactive molecules into the network.37 However, an excess of swelling ratio can potentially compromise the mechanical integrity of hydrogels. Hence, it is essential to strike a balance between the swelling ratio and the desired mechanical properties.38 In order to characterize this property for ChDAT hydrogel, xerogels were immersed in Milli-Q water and the swelling degree, determined using eqn (1), was obtained after 24 h when the weight of the hydrogels was constant, obtaining a final swelling degree around 16 (Fig. S1, ESI†).Besides, scaffolds must exhibit adequate strength and stiffness to offer structural support and adapt to the mechanical demands of their environment.39 In this context, Young's modulus is commonly employed for comparing the mechanical properties of hydrogels, with values ranging from 0.1 kPa to 1 MPa.40,41 Thus, mechanical compressive tests were performed obtaining the stress–strain curves and Young's modulus (E) was analyzed obtaining 24 kPa (Fig. S2, ESI†), which verifies the structural integrity of the 3D hydrogel network. This Young's modulus means the triazine-based hydrogel is rigid enough to sustain channels without undergoing deformations, which is crucial to allocate the enzyme α-glycosidase and analytes in the hydrogel network42 but enough softness environment that can help the enzyme to keep its activity outside a living being. The three-dimensional network of hydrogels (see SEM images in Fig. S3, ESI†), capable of retaining a substantial amount of water, creates optimal physiological conditions for enzyme activity. The aqueous environment inherent in polymeric hydrogels serves to minimize enzyme denaturation and facilitates enzymatic functions. Consequently, the immobility within the hydrogel polymer matrix offers the prospect of sustaining enzyme activity.43,44
Finally, the predominant feature of this hydrogel is its inherent fluorescence, which arises from the AIE phenomenon previously described by our group (Fig. 2, inset).29 Besides the most effective excitation wavelength (λex) was 386 nm (Fig. S4, ESI†), with the peak emission wavelength measured at 465 nm (Fig. 2).
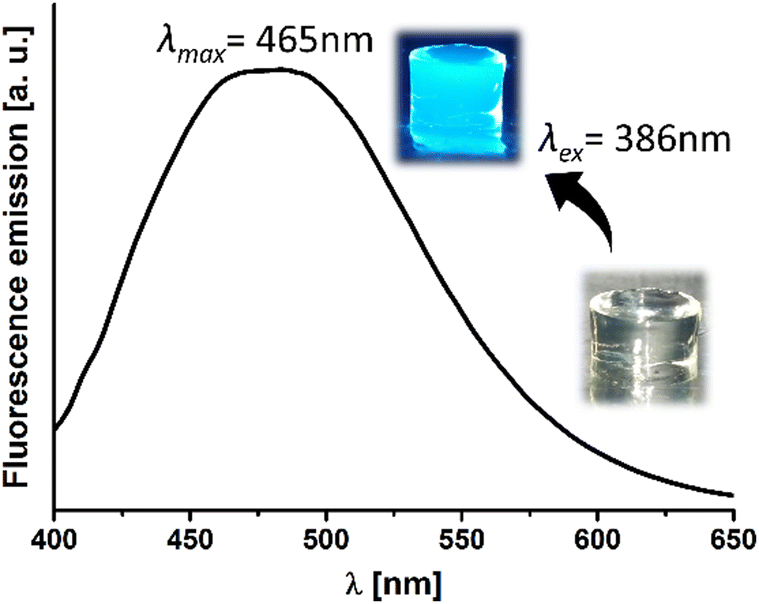 | ||
| Fig. 2 Fluorescence emission spectra of ChDAT hydrogel upon excitation at 386 nm. Inset: Digital image of the hydrogel before and after UV light irradiation, showing its intrinsic fluorescence. | ||
3.3. Effects of biosensor components on fluorescence emission and enzyme encapsulation
As mentioned in the introduction of this study, our final goal is to monitor the activity of the enzyme α-glucosidase. Therefore, PNPG was swelled by the hydrogel as a substrate, since this molecule has an α-o-glycosidic bond that can be hydrolyzed by α-glucosidase releasing α-D-glucose and PNP (Scheme 2).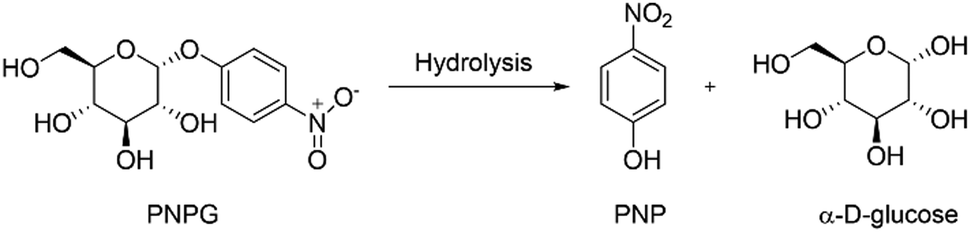 | ||
| Scheme 2 Hydrolysis of PNPG catalyzed by α-glucosidase. | ||
Consequently, PNP could disrupt the AIE within the hydrogel network, leading to quenching of fluorescence and allowing the enzyme activity to be followed through changes in fluorescence intensity. For this, it is imperative to check the influence of PNP and the effect of the inclusion of the enzyme α-glucosidase and the PNPG into the network. Common strategies to achieve effective immobilization of enzymes into a hydrogel network involve physical adsorption, entrapment, covalent bonding, crosslinking, or a combination of these methods.45 Covalent bonding is the most widely employed technique due to the stability it offers. However, it is imperative to carefully monitor the formation of these bonds, as they can potentially interfere with the enzyme's active site, impacting its functionality. In the context of hydrogels, the physical entrapment method is particularly advantageous since it preserves the functionality of the enzyme by securely anchoring it within the hydrogel through interactions like van der Waals forces, salt bridges, or hydrogen bonds. Nonetheless, a drawback is that the immobilization process may be reversible under specific environmental conditions, potentially resulting in the release of the bioreceptor from the hydrogel. Enzyme entrapment within the hydrogel can be achieved either during polymerization (the in situ method) or afterward, following the addition to an already formed hydrogel (the ex situ method).45 In this study, the selected approach involved swelling the enzyme α-glucosidase into the already prepared hydrogel (ex situ method), as it is less aggressive for enzymes.
To investigate the influence of the enzyme, PNPG and PNP on the fluorescence emission, five ChDAT hydrogels were synthesized, dried and subsequently swollen until SD 10 using different solutions:
Hydrogel A: swelled with 0.01 M PBS buffer (pH = 7.4) exclusively.
Hydrogel B: swelled with a 0.8 × 10−6 M solution of the enzyme α-glucosidase.
Hydrogel C: swelled with a 5.26 × 10−3 M solution of PNPG.
Hydrogel D: swelled with a 5.26 × 10−3 M solution of PNP.
Hydrogel E: swelled both with the 5.26 × 10−3 M PNPG and 0.8 × 10−6 M enzyme solutions.
All solutions used to swell the hydrogels were prepared using 0.01 M PBS solution as the solvent to maintain the enzyme's functionality, particularly since the phenol from PNP could influence the hydrogel's pH.
The enzyme concentration (0.8 × 10−6 M) was selected from a previous work11 since for this concentration in solution the time required to hydrolyze micromolar concentrations of PNPG is only a few minutes. However, given that the substrate diffusion in hydrogels is much slower than in solution, the PNPG concentration was increased to millimolar concentrations. Specifically, the optimum substrate concentrations used for these experiments were determined by preliminary studies according to the point at which the fluorescence of the triazine hydrogel was completely quenched by PNP in SD 10 (see Fig. S5, ESI†). Fluorescence quantification was conducted using a spectrofluorometer, and areas under the emission spectra of hydrogels were analyzed. In the initial assessment, it was observed that the enzyme did not influence the fluorescence intensity of the hydrogel. This was clearly demonstrated by the identical behavior of hydrogel A, swollen with PBS buffer, and hydrogel B, swollen with α-glucosidase enzyme. Hydrogel C exhibited a decrease in fluorescence when exposed to PNPG. In this case, there is a significant inner filter effect due to the amount of PNPG added. Nevertheless, complete deactivation was not achieved, in contrast to the impact of PNP, hydrogel D, where the fluorescence was totally quenched. Thus, the notable difference between PNP and PNPG justifies the utilization of this system, particularly since PNPG fails to induce complete fluorescence quenching (Fig. 3).
 | ||
| Fig. 3 Fluorescence emission of the hydrogels loaded. Percentages are given relative to the blank hydrogel. PNPG and PNP solutions are also shown to illustrate the difference in their color, which is also observable on the hydrogels (experiments were performed in triplicate). | ||
It might be presumed that the reduction in fluorescence of hydrogel C (with PNPG) is a result of its spontaneous hydrolysis into PNP, even without the presence of the enzyme. However, this was not the case, as PNP exhibits a distinct yellow coloration (Fig. 3, inset). Hydrogels containing PNP acquire this yellow coloration, which is not observed in hydrogel C. Hence, it can be confidently stated that PNPG diminishes the fluorescence of the system, and the noticeable disparity between PNP and PNPG allows practical utility. In Section 3.5, the wash waters will be analyzed to confirm again that PNPG does not hydrolyze to PNP without the enzyme.
Finally, when examining hydrogels D and E, it became evident that the presence of the enzyme led to a nearly identical quenching of fluorescence, regardless of whether they were swollen with PNPG or PNP. This similarity can be attributed to the fact that the hydrogel with the enzyme and PNPG generates PNP in situ. Moreover, the ability of the enzyme to operate within the hydrogel network was successfully demonstrated. Full spectra emissions of hydrogels C and E can be found in Fig. S5 (ESI†).
3.4. Biosensor working
Once the influence of each component within the hydrogel is established, the next critical step is to determine if it works effectively as a biosensor potentially used for the screening of enzyme inhibitors. In Fig. 1 a schematic interpretation of the biosensor working is shown. To validate this application, acarbose, a commercial competitive α-glucosidase inhibitor, was selected.46The initial assessment involved checking if the inhibitor interfered with the hydrogel's emission. Thus, a dried hydrogel was immersed in a solution of acarbose (1 × 10−3 M) in PBS, and its fluorescence emission was recorded once maximum swelling was archived. The comparation between the fluorescence emission of acarbose-loaded hydrogel and blank hydrogel is shown in Fig. S6 (ESI†). Notably, no significant differences were observed between the hydrogel swollen with PBS buffer and the hydrogel swollen with the inhibitor.
Once it is certified that the inhibitor does not affect the fluorescence, three different experiments were carried out (Fig. 4):
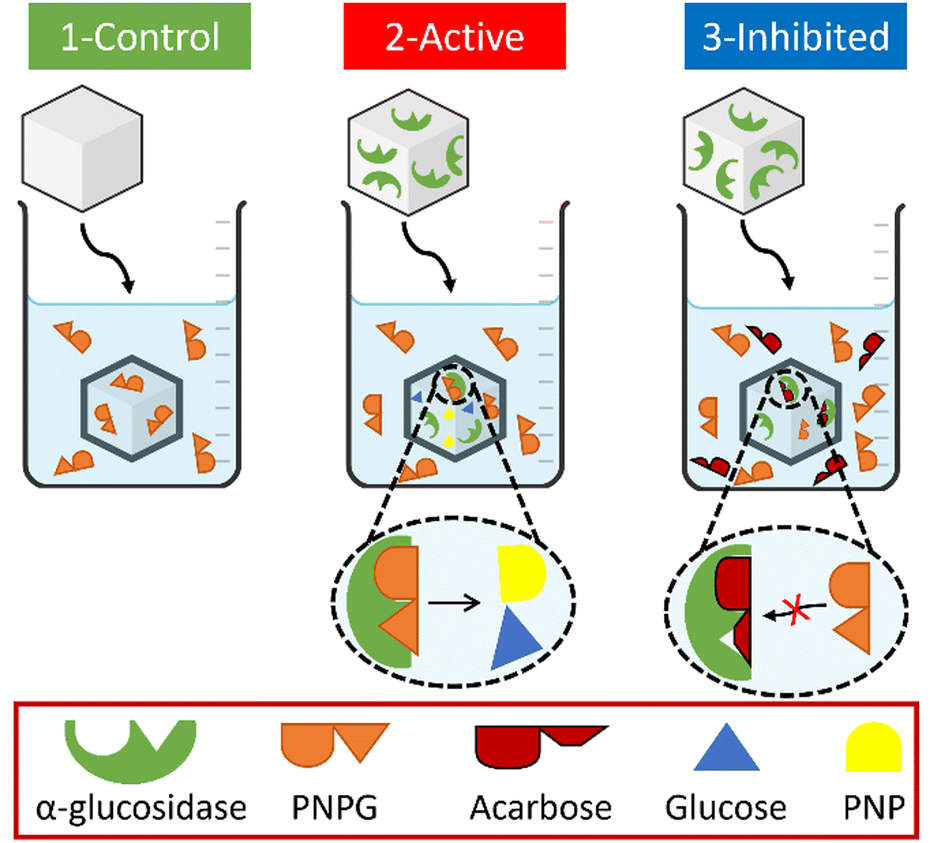 | ||
| Fig. 4 Schematic representation of the processes occurring in each hydrogel. | ||
1. Control experiment: the blank hydrogel was immersed in 20 mL of 5.26 × 10−3 M PNPG solution.
2. Active experiment: the hydrogel loaded with 0.8 × 10−6 M enzyme was immersed in 20 mL of 5.26 × 10−3 M PNPG solution.
3. Inhibited experiment: the hydrogel loaded with 0.8 × 10−6 M enzyme was immersed in 20 mL of 5.26 × 10−3 M PNPG and 1 × 10−3 M of acarbose solution.
The three of them were swelled with PBS buffer until maximum swelling previously to the immersion in the PNPG solution or PNPG-acarbose solution. This process was essential to ensure the complete opening of the hydrogel's porous structure, thereby enhancing the interaction between the substrate and the immobilized enzyme.
The fluorescence of the hydrogel was monitored over time (Fig. 5), revealing a gradual decrease in emission. Notably, the PNPG without the inhibitor quenched the fluorescence much faster than when the inhibitor was present. Additionally, it was observed that even with the inhibitor present, the fluorescence decreased slightly faster than in the absence of the enzyme. This observation aligns with the nature of acarbose as a competitive inhibitor, which means it does not completely block the activity of the enzyme. This is a crucial finding, as drugs of this type do not aim to fully inhibit α-glucosidase but rather to slow it down. In this context, this biosensor could be a valuable tool for selecting the most suitable candidates for further studies.
 | ||
| Fig. 5 (a) Evolution of the fluorescence emission throughout a 180-minute assay. (b) Final fluorescence emission after a 180-minute assay. Percentages are given relative to the initial fluorescence of each hydrogel (Experiments were conducted in triplicate). | ||
3.5. Reusability tests
As explained above, a potential disadvantage of the adsorption of the α-glucosidase enzyme into the hydrogel is that the immobilization process may be reversible under specific environmental conditions, potentially resulting in the release of the mentioned enzyme from the hydrogel. An additional concern regarding reusability pertains to the retention of residual substances such as PNP or PNPG.Thus, to confirm that the enzyme was not released and that the remaining compounds were properly removed, an analysis of the wash waters from these hydrogels was conducted. After testing, the hydrogels were immersed in 100 mL of Milli-Q water for one day, with the process repeated three times. Then, the wash waters were collected and concentrated evaporating the solvent to ensure the reliability of the results. Following this, the concentrated solutions were dissolved in 3 mL of Milli-Q water, their fluorescence and absorbance were measured. Since the α-glucosidase enzyme exhibits fluorescence due to its tryptophan groups, with a maximum fluorescence wavelength at 350 nm,47,48 wash waters were excited at 280 nm, and the fluorescence emission was measured in the range of 290 to 380 nm (Fig. 6). Conclusively, the graph data indicates that the enzyme remains within the hydrogel, as no fluorescence signal is observed in the wash waters at the mentioned wavelength. The enzyme solution used to swell the hydrogels, with a concentration of 0.8 μM, serves as the reference.
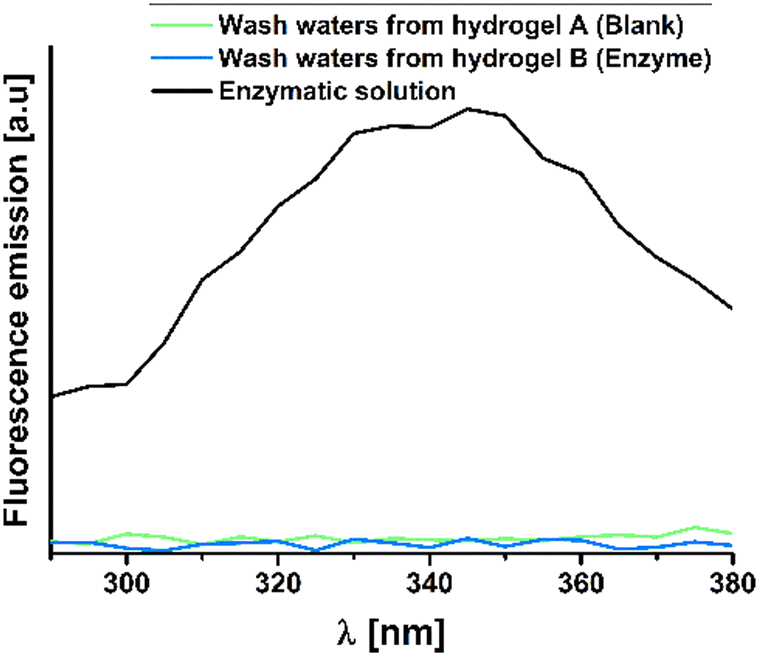 | ||
| Fig. 6 Fluorescence emission of wash waters from a blank hydrogel in comparison with an enzyme-loaded hydrogel and a 0.8 μM enzymatic solution. | ||
Having established that the enzyme remains inside the hydrogel, the next objective was to validate through absorbance experiments that the yellow coloration observed in the wash waters is a result of PNP and that any possible PNPG leftover is no present in the hydrogel. This entails confirming that both PNP and PNPG leach out of the hydrogel during washing (Fig. 7).
 | ||
| Fig. 7 Absorbance spectra of wash waters in PBS buffer from the hydrogels. In black is enzyme and PNPG, in red with PNP, in blue with PNPG, pink and green are PNP and PNPG reference solutions, respectively. | ||
Finally, these washed and empty hydrogels, all while retaining the enzyme were dried at room temperature until the weight of the xerogel were constant and stored in ambient conditions for future use again. Two months later, the hydrogels were reswollen and immersed again in a solution containing PNPG and acarbose, measuring the fluorescence. This washing-using cycle was repeated 3 more times. The maximum and minimum fluorescence of each cycle were recorded (Fig. 8). This indicates that the enzyme remains active after this period and has not been washed out, underscoring the significant advantage of hydrogel reusability and ability to host the enzyme. This is particularly significant considering that, under standard conditions, the enzyme must be stored at −20 °C to maintain its functionality according with the manufacturer Sigma–Aldrich.
 | ||
| Fig. 8 Final fluorescence emission after a 180-minute assay-washing cycles (×4 uses). Percentages are given relative to the initial fluorescence of each hydrogel. Experiments were conducted in triplicate. | ||
4. Conclusions
Leveraging the intrinsic fluorescence of a triazine-based hydrogel, attributed to an aggregation-induced emission (AIE) phenomenon, an α-glucosidase biosensor was successfully developed. In contrast to the majority of previously published fluorescent hydrogel-based biosensors, the innovative design eliminates the need for additional fluorescent molecules in the hydrogel network, enhancing the efficiency, stability and cost-effectiveness of the biosensor. Furthermore, the remarkable swelling capacity and optimal mechanical properties give rise to a robust, transparent matrix that supports the immobilization and activity characterization of the enzyme by monitoring the intrinsic fluorescence of the hydrogel. The changes in fluorescence induced by the hydrolysis of PNPG catalyzed by the enzyme, lead to the release of PNP and the subsequent quenching of the fluorescence, were thoroughly tested.Additionally, the stability of the enzyme within the hydrogel and the possibility of reusing the biosensor multiple times were thoroughly evaluated, demonstrating the reliability of the biosensor. A notable advantage is its potential to function as a portable optical device. The hydrogel can be easily stored, manipulated, and transported in a xerogel state, ready to be swollen with the substrate for rapid and cost-effective measurements.
Finally, as a proof of concept using the commercial drug acarbose, the device has shown its capability to detect α-glucosidase enzyme inhibitors, suggesting potential in identifying compounds that could alter enzyme activity and serve as promising candidates for antidiabetic drugs. This underscores the biosensor's role in advancing the exploration of new antidiabetic treatments, positioning it as an ideal candidate for further research and discoveries in the field.
Author contributions
F. Javier Patiño: investigation, writing – original draft, writing – review & editing. Josué M. Galindo: investigation, writing – original draft, writing – review & editing. Alicia Jiménez: investigation, formal analysis. Yolanda Alacid: investigation. C. Reyes Mateo: writing – review & editing. Ana Mª Sánchez-Migallón: writing – review & editing. Ester Vázquez: writing – review & editing, funding acquisition. Sonia Merino: writing – review & editing, supervision, funding acquisition, conceptualization. M. Antonia Herrero: writing – review & editing, supervision, funding acquisition, conceptualization.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the Spanish Government (projects PID2020-113080RB-I00 and PID2022-138761NB-I00/AEI/10.13039/501100011033), PID2022-138507OB-I00, funded by MICIU/AEI/10.13039/501100011033 and ERDF, EU; the Junta de Comunidades de Castilla-La Mancha (SBPLY/21/180501/000135/1/SBPLY/23/180225/000153) and University of Castilla-La Mancha (2022-GRIN-34076 Consolidado). This study forms part of the Advanced Materials programme and was supported by MCIN with funding from the European Union NextGenerationEU (PRTR-C17.I1) and the Junta de Comunidades de Castilla-La Mancha. F. J. Patiño would also like to express his gratitude to Castilla-La Mancha community board for their pre-PhD contract (2020-PREDCLM-16324). J. M. Galindo acknowledges the Spanish Ministry of Economy and Competitiveness (MINECO) for his FPI Fellowship (FPI-PRE2018-084047).References
- J. E. Reyes-Martínez, J. A. Ruiz-Pacheco, M. A. Flores-Valdéz, M. A. Elsawy, A. A. Vallejo-Cardona and L. A. Castillo-Díaz, J. Tissue Eng. Regener. Med., 2019, 13(8), 1375–1393 CrossRef PubMed.
- B. Zhou, K. Hajifathalian, J. Bentham, M. Di Cesare and G. Danaei, et al., Lancet, 2016, 387(10027), 1513–1530 CrossRef PubMed.
- World Health Organization. https://www.who.int/health-topics/diabetes#tab=tab_1, accessed: April, 2024.
- J. S. Haw, K. I. Galaviz, A. N. Straus, A. J. Kowalski, M. J. Magee, M. B. Weber, J. Wei, K. M. V. Narayan and M. K. Ali, JAMA Intern. Med., 2017, 177(12), 1808–1817 CrossRef.
- R. Caleyachetty, T. M. Barber, N. I. Mohammed, F. P. Cappuccio, R. Hardy, R. Mathur, A. Banerjee and P. Gill, Lancet Diabetes Endocrinol., 2021, 9(7), 419–426 CrossRef.
- Z. Liu and S. Ma, ChemMedChem, 2017, 12(11), 819–829 CrossRef CAS PubMed.
- J. Tavakoli and Y. Tang, Polymers, 2017, 9(8), 364 CrossRef PubMed.
- N. Bhalla, P. Jolly, N. Formisano and P. Estrela, Essays Biochem., 2016, 60(1), 1–8 CrossRef.
- V. Perumal and U. Hashim, J. Appl. Biomed., 2014, 12(1), 1–15 CrossRef.
- F. Liu, Z. Li, G. Kang, Z. Liu, S. Zhu, R. He, C. Zhang, C. Chen and Y. Lu, Microchem. J., 2023, 186, 108352 CrossRef CAS.
- Y. Alacid, M. J. Martínez-Tomé and C. R. Mateo, ACS Appl. Mater. Interfaces, 2021, 13(22), 25624–25634 CrossRef CAS PubMed.
- Q. Zhao, Y. Wang, M. Zhang, D. Wu, J. Sun and X. Yang, Biosens. Bioelectron., 2022, 214, 114504 CrossRef CAS.
- X. Zhang, M. A. Ballem, Z. J. Hu, P. Bergman and K. Uvdal, Angew. Chem., 2011, 123(25), 5847–5851 CrossRef.
- X. Zhang, M. A. Ballem, M. Ahrén, A. Suska, P. Bergman and K. Uvdal, J. Am. Chem. Soc., 2010, 132(30), 10391–10397 CrossRef CAS.
- A. Herrmann, R. Haag and U. Schedler, Adv. Healthcare Mater., 2021, 10(11), 2100062 CrossRef CAS.
- K. Völlmecke, R. Afroz, S. Bierbach, L. J. Brenker, S. Frücht, A. Glass, R. Giebelhaus, A. Hoppe, K. Kanemaru, M. Lazarek, L. Rabbe, L. Song, A. Velasco Suarez, S. Wu, M. Serpe and D. Kuckling, Gels, 2022, 8(12), 768 CrossRef.
- M. Pita, S. Minko and E. Katz, J. Mater. Sci.: Mater. Med., 2009, 20(2), 457–462 CrossRef CAS.
- P. Sokolov, P. Samokhvalov, A. Sukhanova and I. Nabiev, Nanomaterials, 2023, 13(11), 1748 CrossRef CAS PubMed.
- Z. Ma, S. Tang, W. Shen and H. K. Lee, Trends Anal. Chem., 2024, 180, 117907 CrossRef CAS.
- R. Baretta, A. Raucci, S. Cinti and M. Frasconi, Sens. Actuators, B, 2023, 376, 132985 CrossRef CAS.
- O. Gideon, H. S. Samuel and I. A. Okino, Discovery Chem., 2024, 1(1), 34 CrossRef.
- P. Gagni, G. Lodigiani, R. Frigerio, M. Cretich, A. Gori and G. Bergamaschi, Chem. – Eur. J., 2024, 30(46), e202400974 CrossRef CAS PubMed.
- H. Kono, F. Otaka and M. Ozaki, Carbohydr. Polym., 2014, 111, 830–840 CrossRef CAS.
- Y. Alacid, M. J. Martínez-Tomé, R. Esquembre, M. A. Herrero and C. R. Mateo, Int. J. Mol. Sci., 2023, 24(3), 2672 CrossRef CAS PubMed.
- Z. Jia, L. Gwynne, A. C. Sedgwick, M. Müller, G. T. Williams, A. T. A. Jenkins, T. D. James and H. Schönherr, ACS Appl. Bio Mater., 2020, 3(7), 4398–4407 CrossRef CAS PubMed.
- Z. Jia, M. Müller, T. Le Gall, M. Riool, M. Müller, S. A. J. Zaat, T. Montier and H. Schönherr, Bioact. Mater., 2021, 6(12), 4286–4300 CAS.
- M.-M. S. Ebrahimi, M. Laabei, A. T. A. Jenkins and H. Schönherr, Macromol. Rapid Commun., 2015, 36(24), 2123–2128 CrossRef CAS.
- M. Kopjar, I. Ćorković, I. Buljeta, J. Šimunović and A. Pichler, Antioxidants, 2022, 11(8), 1459 CrossRef CAS PubMed.
- J. M. Galindo, J. Leganés, J. Patiño, A. M. Rodríguez, M. A. Herrero, E. Díez-Barra, S. Merino, A. M. Sánchez-Migallón and E. Vázquez, ACS Macro Lett., 2019, 8(10), 1391–1395 CrossRef CAS.
- F. Qiu, Q. Zhu, G. Tong, L. Zhu, D. Wang, D. Yan and X. Zhu, Chem. Commun., 2012, 48(98), 11954–11956 RSC.
- B.-P. Jiang, D.-S. Guo, Y.-C. Liu, K.-P. Wang and Y. Liu, ACS Nano, 2014, 8(2), 1609–1618 CrossRef CAS PubMed.
- Q. Zhao, K. Li, S. Chen, A. Qin, D. Ding, S. Zhang, Y. Liu, B. Liu, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22(30), 15128–15135 RSC.
- X. Guan, D. Zhang, T. Jia, Y. Zhang, L. Meng, Q. Jin, H. Ma, D. Lu, S. Lai and Z. Lei, Ind. Eng. Chem. Res., 2017, 56(14), 3913–3919 CrossRef CAS.
- Á. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, M. del Carmen Mateo, A. Sánchez-Migallón and G. Valiente, New J. Chem., 2004, 28(8), 952–958 RSC.
- J. Leganés, A. Sánchez-Migallón, S. Merino and E. Vázquez, Nanoscale, 2020, 12(13), 7072–7081 RSC.
- T. J. Mooibroek and P. Gamez, Inorg. Chim. Acta, 2007, 360(1), 381–404 CrossRef CAS.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8(5), 607–626 CrossRef CAS.
- F. Wu, Y. Pang and J. Liu, Nat. Commun., 2020, 11(1), 4502 CrossRef CAS PubMed.
- D. Antoni, H. Burckel, E. Josset and G. Noel, Int. J. Mol. Sci., 2015, 16(3), 5517–5527 CrossRef CAS PubMed.
- J. Liu, H. Zheng, P. S. Poh, H.-G. Machens and A. F. Schilling, Int. J. Mol. Sci., 2015, 16(7), 15997–16016 CrossRef CAS PubMed.
- M. S. B. Reddy, D. Ponnamma, R. Choudhary and K. K. Sadasivuni, Polymers, 2021, 13(7), 1105 CrossRef CAS PubMed.
- Y. Ling, J. Rubin, Y. Deng, C. Huang, U. Demirci, J. M. Karp and A. Khademhosseini, Lab Chip, 2007, 7(6), 756–762 RSC.
- C. S. Dawes, H. Konig and C.-C. Lin, J. Biotechnol., 2017, 248, 25–34 CrossRef CAS.
- L. Y. Maroufi, M. Rashidi, M. Tabibiazar, M. Mohammadi, A. Pezeshki and M. Ghorbani, Adv. Pharm. Bull., 2022, 12(2), 309–318 CAS.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef.
- S. R. Dehkordi, N. Pahlavani, M. Nikbaf-Shandiz, R. Bagheri, N. Rasaei, M. Darzi, S. Rastgoo, H. Bahari, F. Shiraseb and O. Asbaghi, J. Diabetes Metab. Disord., 2023, 1–38 Search PubMed.
- C.-P. Pan, P. R. Callis and M. D. Barkley, J. Phys. Chem. B, 2006, 110(13), 7009–7016 CrossRef CAS.
- J. Singh, S. Joshi, S. Mumtaz, N. Maurya, I. Ghosh, S. Khanna, V. T. Natarajan and K. Mukhopadhyay, Sci. Rep., 2016, 6(1), 31492 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02466d |
| ‡ F. Javier Patiño and Josué M. Galindo equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |