
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArchitectural control of rod-coil block polypeptide thermoresponsive self-assembly via de novo design of coiled-coil orientation†
Bin
Wang
 a,
Weiran
Xie
a,
Tianren
Zhang
ab,
Darrin J.
Pochan
a,
Jeffery G.
Saven
b and
Kristi L.
Kiick
*ac
a,
Weiran
Xie
a,
Tianren
Zhang
ab,
Darrin J.
Pochan
a,
Jeffery G.
Saven
b and
Kristi L.
Kiick
*ac
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE, USA. E-mail: kiick@udel.edu
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA, USA
cDepartment of Biomedical Engineering, University of Delaware, Newark, DE, USA
First published on 14th March 2025
Abstract
The architectural control of the self-assembly of a series of block polypeptides comprising a concatenation of an elastin-like peptide and a coiled-coil, bundle-forming peptide (ELP–BFPs), has been demonstrated. Assembly of the polypeptides is controlled by coacervation of the hydrophobic ELP domain, while the type of coiled-coil assembly of the BFP and the specific placement of short histidine tags significantly tunes assembly behavior. Spectrophotometric analysis of self-assembly demonstrated that the transition temperature of assembly can be controlled by the design of the BFP domain and positioning of the His-tags in the constructs. Cryogenic transmission electron microscopy of assembled polypeptides confirmed distinct morphologies including core–shell particles and multilayer vesicles, depending on the parallel or antiparallel bundle architecture of the block polypeptide. The results have applications in materials design and highlight the potential for controlling multi-stimuli responsiveness and morphologies through fine control of the architectural features of the component polypeptide domains.
Introduction
Self-assembly of biomolecules is critical in controlling the structures and functions of proteins by forming a rich variety of supramolecular structures. Similarly, self-assembly inspired by interactions between biomolecular building blocks is widely regarded as an efficient way to prepare novel functional materials.1 Peptide-mediated assembly can be canonically controlled by formation of secondary structures (e.g., α-helix, β-sheet, random coil), although the contributions of both ordered and disordered domains are important in designing polypeptide sequences that exhibit controlled assembly of complex structures.2–5 Current studies that incorporate ordered protein domains in assembly successfully provide fine control of assembled structures such as nanoscale 2D sheets and 1D fibers.6,7 In addition, exposure of certain residues/sequences in assembled particles can provide selective interactions, such as selective protein-binding peptides and site-specific lysine modification.8,9 The spatial display of functional domains on assembled structures can be used to tune assembly behavior and polypeptide properties.10–12 For disordered peptides or polypeptides (defined by different chain lengths, abbreviated as (poly)peptides), which lack specific secondary or higher-order structures, reversible stimuli-responsiveness can yield hydrogels or assembled structures that can be employed as drug carriers.13,14 By encoding both ordered and disordered domains in block polypeptides, properties from the different building blocks combine to achieve more versatile structures, such as semiflexible chains.2,15 Among different block polypeptides, amphiphilic (poly)peptide-based macromolecules are of particular fundamental and practical interest due to their importance in natural phenomena and tunable assembly.16–18(Poly)peptides, in particular, have been of interest in investigations of both low molecular weight amphiphiles and polymer-based amphiphiles because of their perfectly defined sequences, well-controlled secondary structures, and hierarchical assembly. Lipidated peptides, surfactant-like peptides, and diblock elastin-like polypeptides have been widely investigated.5,19,20 Composed of alternating hydrophilic and hydrophobic residues, surfactant-like peptides commonly adopt β-sheet conformation.20 Due to their short chain length and strict secondary structure requirements, variations in the peptide sequences to introduce more functionality often has deleterious effects on the assembly of the amphiphiles. Lipidated peptides successfully decouple the hydrophilicity of the peptide sequence,19 as they comprise a hydrophobic aliphatic chain and a hydrophilic peptide chain. However, due to the high hydrophilic surface area and low hydrophobic volume, lipidated peptides generally only assemble into spherical micelles or cylindrical rod micelles. Additionally, β-sheet hydrophilic sequences are commonly employed to increase the stability of assemblies. Despite advances in their assembly, lipidated peptides and surfactant-like peptides have strict restrictions on their sequence and can require complicated chemistry with non-natural amino acids. However, elastin-like polypeptides (ELPs), the thermoresponsive polypeptides derived from the extracellular matrix protein elastin, have exhibited high biocompatibility and controllable phase transition behavior, and have therefore been extensively explored as a promising building block for amphiphilic polypeptides.5 Many amphiphilic ELPs have been designed and synthesized to achieve stimuli-responsive assembly.21,22 Nevertheless, the assembly of simple diblock ELPs has been limited largely to micelles due to the long chain lengths normally employed.23
Here, we present a library of de novo-designed polypeptide-based amphiphiles based on disordered ELPs and ordered bundle-forming peptides (BFPs) which can be recombinantly expressed and assembled into controlled nanostructures. Elastin-like polypeptides (ELPs) are employed as the hydrophobic domains of these molecules; ELPs are intrinsically disordered polypeptides (IDPs) that are derived from the hydrophobic domain of tropoelastin, with a pentapeptide repeat unit (Val-Pro-Gly-Xaa-Gly) in which Xaa is a guest residue that can be any amino acid except proline. Since discovered in the 1970s,24 ELP has been a popular material due to its flexibility, tunability, and biocompatibility.5 ELP solutions exhibit reversible LCST-like phase transitions in which the ELPs are soluble at low temperatures and coacervate at high temperatures; these properties have enabled their use in hydrogel preparation, nano-assembly, and inverse transition cycling purification.25,26 While ELPs of high molecular weight (those with more than n = 40 ELP repeats) have been investigated widely over the past few decades, short ELPs remain less understood due to their higher transition temperatures.27 However, short ELPs (those with fewer than n = 10 ELP repeats) can provide a new tool for building biomaterials which can be computationally predicted, with improved insight on the influence and interactions of guest residues in the ELP chain.28,29 The Kiick laboratory has previously reported the fusion of ELPs to collagen-like peptides (CLP), to trigger phase transition of a short ELP (6 repeat units) and to assemble ELP-CLP ‘bilayers’ that exhibit different morphologies depending on the composition and relative lengths of the blocks. These results suggest the possibility of using short ELPs as a building block for a range of (poly)peptide-based assembled systems.2,10
Coiled-coil peptides, consisting of two or more α-helical chains, are common protein structural motifs and not only serve as structural units in natural proteins but also perform functions including accurate chromosome segregation, centriole formation, and DNA recognition and cleavage.30,31 Coiled-coil peptides are characterized by heptad repeat units and formation of coiled-coil bundles which typically present a hydrophobic core and hydrophilic surface.32 Due to the variety of available sequences and their ability to be designed to interact orthogonally, coiled-coil peptides have been applied as ordered nano-assembly modules33 and building blocks for regulating cell signalling.34 The hydrophilic domain of the modular design presented here leverages bundle-forming peptides (BFPs), or “bundlemers”, which are computationally designed α-helical coiled-coil peptides that are thermostable, robust with respect to variation of exterior sequence, exhibit controlled polymerization into nanofibers with terminal functionalization, and assemble via tailored interactions between hydrophilic bundle surfaces.7,35 Forming tetrameric coiled coils, they form a uniform nanoscale cylinder approximately 2 nm in diameter and 4 nm in length. Recent studies have shown that BFPs produced either synthetically or recombinantly can form largely 1D nanostructures by covalent reactions7,36 or physical interactions such as those between resilin-like polypeptides (RLPs).37,38
By fusing ELP, BFP, and His-tag sequences genetically into single sequences, recombinant rod-coil polypeptides employing new ELP and BFP pairs are presented here. The polypeptide amphiphiles were expressed in E. coli, and the successful expression was confirmed by SDS polyacrylamide gel electrophoresis, mass spectrometry, and amino acid analysis. The sequence-dependent phase transition behavior was characterized by spectrophotometric assessment of turbidity, and the influence of pH and salt on the phase transition was also assessed. Circular dichroism spectroscopy confirmed the secondary structure of ELP–BFP constructs at various temperatures. Transmission electron microscopy (TEM and cryogenic-TEM) revealed the morphology of assembled nanostructures, and simulation was performed to suggest the molecular details that guide assembly. Compared to our previously reported RLP-BFP designs that exhibit 1D assembly and UCST (upper critical solution transition)-like phase transitions,38 the polypeptide designs here yield both distinct LCST thermo-responsiveness that is sensitive to the molecular architecture as well as nanoparticle assemblies with layered structures.
Results and discussion
Tuning the LCST-like transition of high-molecular-weight ELPs has been demonstrated to manipulate coacervation and assembly,39 and, in previous studies, the Kiick group has also shown that chemical attachment of short ELPs to an oligomerization unit (collagen-like peptide, or CLP) triggers LCST-like behavior at significantly reduced temperatures due to an increase in the local ELP concentration.29 These results suggest the possibility of building a thermally responsive polypeptide-based amphiphile system by fusing ELP to different oligomerization units. Hence, in this study, ELPs were employed as the thermally responsive, hydrophobic, disordered building block in recombinant polypeptides, while BFP domains served as the hydrophilic oligomerization unit. Three different guest residues were selected for the ELP sequences to test the possibility of controlling the thermal response (Table S1, ESI†).The de novo-designed coiled-coil peptides comprised either an antiparallel bundle-forming peptide (aBFP) or a parallel bundle-forming peptide (pBFP) (previously reported as 44597 and Prll_1a40 respectively) for homotetramer formation; both aBFP (DEEIRRMAEEIRQMAERIQQMAEQIQQEA) and pBFP (DEIIKYLDRIIRQLERIIRQLEEIIKQL) have been shown to form thermally stable coiled-coil helix bundles, but assemble into bundlemer coiled-coil particles with different orientations of the peptide chains (antiparallel vs parallel, Fig. 1), thus offering potential alternatives to finely tune their interactions and assembly when fused with thermoresponsive ELPs (abbreviated as Xn in which X is the guest residue and n is the number of ELP repeat units). The effect of His-tag placement in such systems is largely unexplored, especially with regard to their presentation at the end of unstructured and structured domains of ELP–BFP constructs. His-tags at the ends of unstructured ELP-containing domains may facilitate association of the peptide assemblies; the unstructured domain here comprises the His-tag, an enterokinase recognition site (SSGHIDDDDK), and the ELP sequence (Fig. 1). A useful comparison is provided by a second class of constructs where the His-tag is positioned adjacent to the well-structured bundle at the C-terminus and hence much less mobile. Both BFPs are genetically fused with either (1) a 10-histidine-long sequence, an enterokinase digestion site, and an ELP sequence at the N-terminus (His-ELP–BFP), or (2) an ELP sequence on the N-terminus of the BFP and a 6-histidine repeat at the C-terminus of the construct (ELP–BFP-His) (Fig. 1). A cysteine residue is located between ELP and BFP in the His-V8-pBFP sequence (which does not impact the stability of folded BFPs) and is adapted from previous work describing the design of the parallel sequences.40 The cysteine also offers opportunities for attachment of chromophores and for crosslinking of assembled structures, although such modifications are not considered in the scope of this initial report. Due to these design differences, the ELP–aBFP presents two ELP chains on each end of coiled-coil bundle but the ELP–pBFP presents four ELP chains on one end of the coiled-coil bundle (Fig. 1).
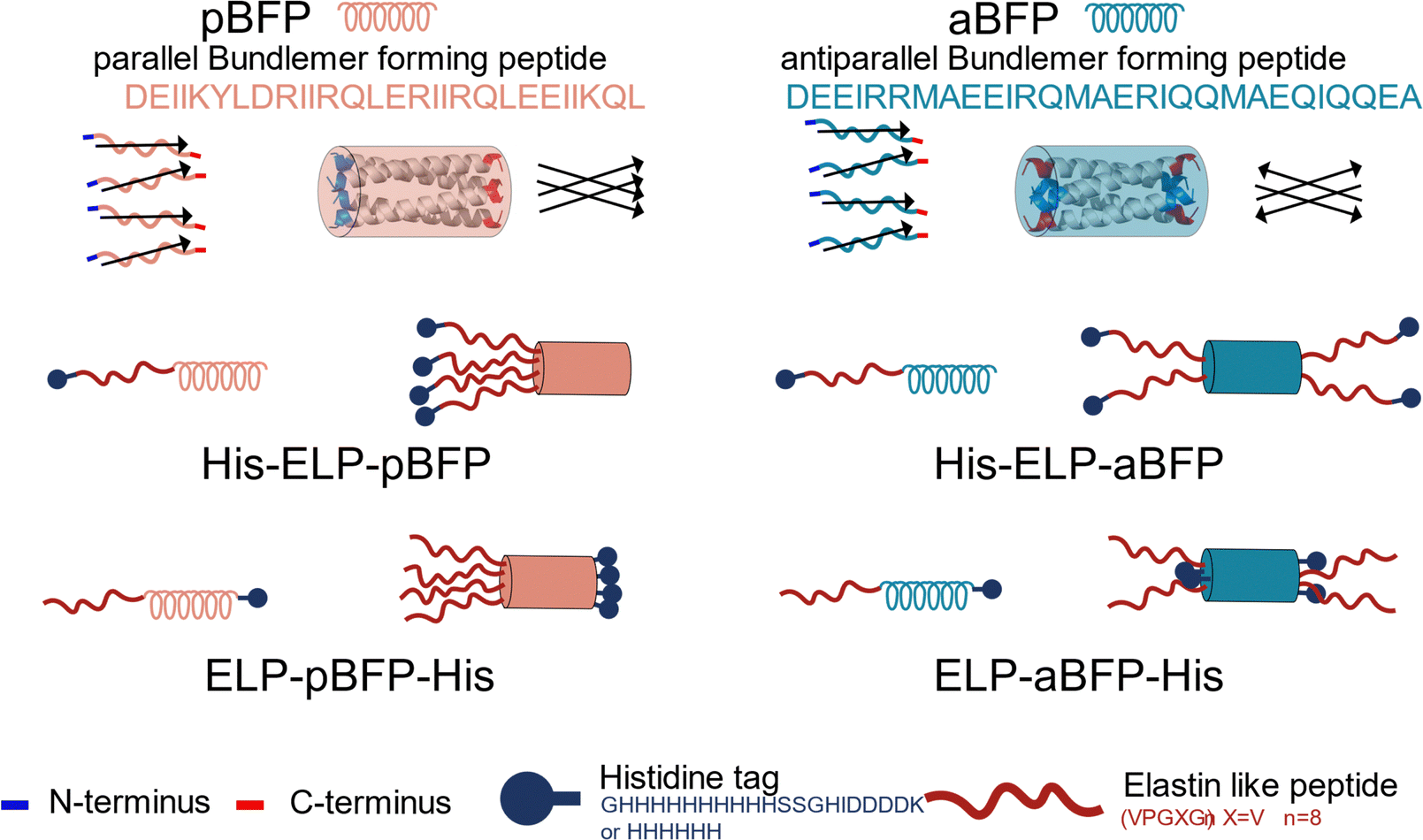 | ||
| Fig. 1 Design of ELP–BFP amphiphiles. The black arrow represents BFP peptide sequences from N-terminus to C-terminus; simplified pBFP tetramers and aBFP tetramers are indicated by these arrows. | ||
The ELP–BFP constructs were recombinantly expressed with IPTG induction and purified by immobilized metal affinity chromatography (IMAC). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was conducted to confirm the production and suggested purity of the desired polypeptide (Fig. S3, ESI†). The expected molecular weights for all constructs were confirmed by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS) and the experimental mass for all constructs was within 0.3% of the theoretical values (Fig. S4, ESI†). Amino acid analysis of all expressed V8-BFPs (which were employed for self-assembly, see below) confirms that the composition determined experimentally is within 5% of the expected values (Table S2, ESI†). Overall, these results indicate the successful expression of a variety of ELP–BFPs via recombinant methods with yields sufficient for comprehensive characterization (Table S3, ESI†).
The purified polypeptides (His-V8-aBFP, His-V8-pBFP, V8-aBFP-His, and V8-pBFP-His) were dissolved in a citric acid-Na2HPO4 buffer (pH 5.6) at 200 μM polypeptide concentration and turbidity tests were conducted by measurement of turbidity at a wavelength of 350 nm as a function of temperature; results are presented in Fig. 2. The ELP–BFP solutions exhibit LCST-like phase transition behavior marked by an increase in turbidity, consistent with association of the ELP domains (Fig. 2(A)). The transition temperatures of His-V8-aBFP, His-V8-pBFP, V8-aBFP-His, and V8-pBFP-His determined from the data in Fig. 2(A) were 35.0 °C ± 2.0 °C, 28.3 °C ± 0.7 °C, 61.0 °C ± 3.2 °C, and 48.7 °C ± 1.2 °C respectively, indicating that the BFP sequence and placement of the His tag both impacted the transition temperature. The transition temperature of the four different polypeptides at different concentrations was also tested (Fig. 2(B) and Fig. S5–S8, ESI†). All sequences showed decreased transition temperatures with increased concentration, as expected. The transition temperature of ELP–BFPs that contain alanine (His-A8-aBFP, His-A16-pBFP) and phenylalanine (His-F6-aBFP and His-F6-pBFP) as ELP guest residues were also tested to assess their possible assembly at experimentally accessible temperatures. ELP–BFPs with alanine residues remain soluble at 85 °C while ELP–BFPs with phenylalanine are insoluble at 5 °C (Table S1, ESI†), so these molecules were not characterized further.
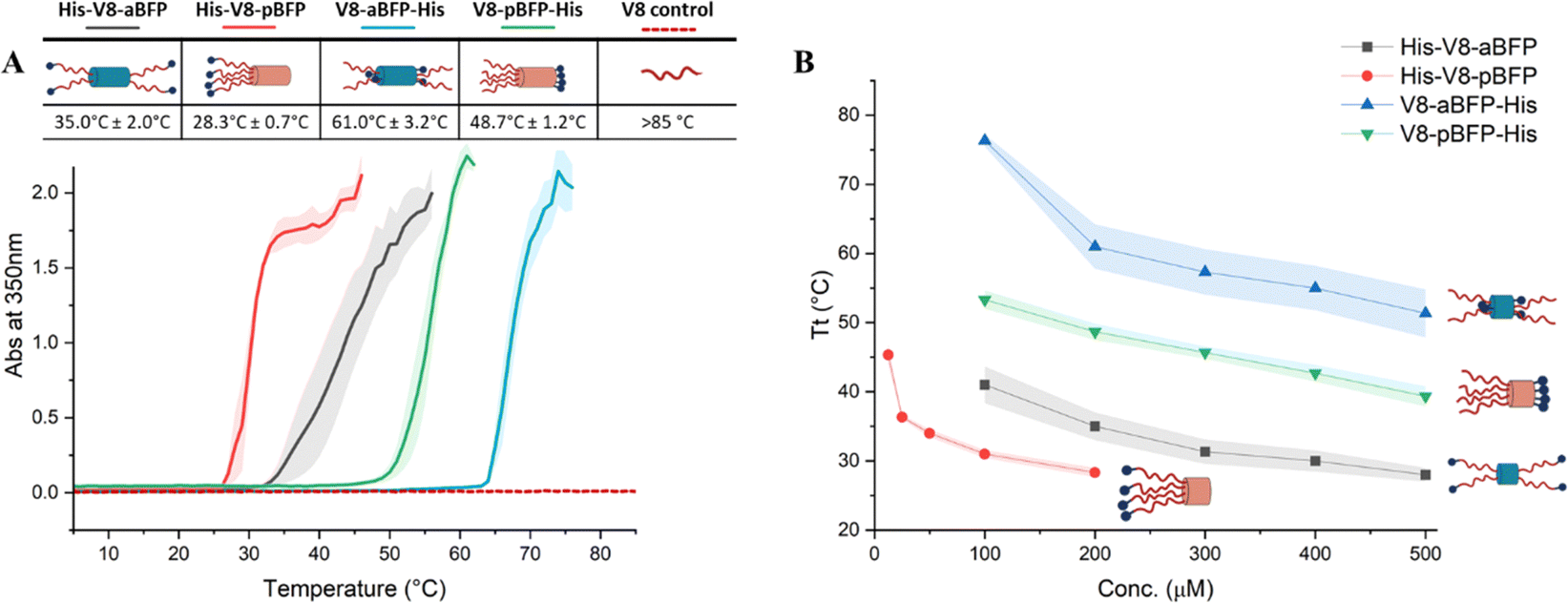 | ||
| Fig. 2 Transition temperature measurements for V8-BFP polypeptides. (A) Different transition temperatures with different BFP designs and histidine tag placement and turbidity (at 350 nm) of different amphiphile designs at 200 μM concentration in pH = 5.6 citric acid–sodium phosphate buffer. (B) For the same ELP–BFP construct, the transition temperature remains stable except in the low concentration range. Detailed turbidity results are shown in Fig. S5–S8 (ESI†). | ||
All four V8-containing polypeptides showed LCST-like transition temperatures lower than that of the isolated (VPGVG)8 sequence, underscoring the importance of the oligomerization of the BFP unit. (The Tt of the isolated (VPGVG)8 sequence was measured as >85 °C for the isolated peptide and is indicated by the red dashed line in Fig. 2(A); the theoretically calculated value is estimated to be 99 °C at 200 mM41). In general, the ELP–pBFP constructs showed lower Tt than ELP–aBFP polypeptides; these observations indicate the sensitivity of the LCST-like behavior of these molecules to the architecture of the ELP–BFP and the possibility of tuning the Tt of ELP–BFP systems by changing the order of the sequence without changing building blocks. The increased local concentration of ELP at one end of the ELP–pBFP (four ELP chains) relative to the two ELPs on each end of the ELP–aBFP constructs is sufficient to induce a lower Tt in the former, consistent with our reports for ELP-CLP constructs.10,29,42 In addition, constructs with an N-terminal His-tag have lower onset transition temperatures (Tt = 29–36 °C) than those where the tag is at the C-terminus (Tt = 49–61 °C) (Fig. 2(A)). The origin of this difference is likely due to the structural differences at the N- and C-termini. At the flexible N-terminus, histidine residues are available for potential intra- and inter-molecule aromatic interactions in ELP domains. At the C-terminus, the fixed proximity of the His-tag to the well-structured bundle may impede such interactions. At the N-terminus, variation of the length of the His-tag (6 and 10 residues) had little effect on the transition temperature, yielding differences of only 4–6 °C (Fig. S9, ESI†). Previous studies have shown that histidine–histidine interactions can improve the stability of polypeptides in both simulation43 and experiment.44 Differential placement of the His-tag (e.g., N- or C-terminal) has been applied in previous studies as a design element to tune the phase transition behavior and assembled morphology of ELP-based sequences. For example, an N-terminal His tag has been reported to reduce Tt slightly (3 °C) for long ELPs.45 Similar results (a 5 °C reduction in Tt) were also observed for highly charged ELPs near their isoelectric point,46 and only minor effects on assembly were reported for mCherry-ELP molecules with N- and C-terminal placement of a His-tag.47 To our knowledge, the placement of the His tag has had little influence on the Tt of ELP-containing constructs in prior studies, whereas for the ELP–BFP constructs considered herein, repositioning of the His-tag from the unstructured N-terminus to the C-terminus of helical segments in the bundle can lead to shifts in Tt by as much as 15 °C.
Circular dichroism (CD) spectroscopy measurements were conducted to determine the secondary structure of the polypeptides at low concentrations at different temperatures (Fig. 3(A), (B) and Fig. S10, ESI†). All molecules showed two minima (208 nm and 222 nm) at 25 °C suggesting the formation of the coiled-coil structure, which was stable at temperatures up to 85 °C. Given the stability of the BFP domains at 85 °C (Fig. 3(A)), the decrease in the molar ellipticity at higher temperatures in the ELP–BFP constructs (Fig. 3(B)) is most likely attributable to the phase transition which decreases the concentration of soluble polypeptides in the aqueous phase. The differences in signal strength between the sequences in Fig. 3(B) suggest that there might also be interactions between ELP and BFP that alter the stability of the BFP coiled-coil structure. Based on assembly results (below), the weaker signal of His-V8-pBFP and V8-aBFP-His may also be a result of stronger particle aggregation and precipitation which decreases the ELP–BFP concentration in solution. In summary, the BFPs in the reported molecules formed stable coiled-coil structures at all temperatures investigated, consistent with our previous reports in which a stable coiled-coil structure was also maintained when resilin-like polypeptides were fused to the termini of BFPs.38
 | ||
| Fig. 3 CD spectra (molar ellipticities) of individual sequences and different constructs. (A) CD spectra for aBFP, pBFP, ELP control peptides. (B) CD spectra for His-V8-aBFP, His-V8-pBFP, V8-aBFP-His, V8-pBFP-His at indicated temperatures, confirming that the BFPs retain their coiled-coil structure in at the temperatures investigated. | ||
Next, we sought to determine if the placement of the His tag and the anti-parallel or parallel orientation of the coiled-coil BFP domains would affect resulting morphologies upon thermally-triggered assembly above the Tt of the ELP domain. Transmission electron microscopy (TEM) revealed the design-dependence of the resulting nanostructures. Both His-V8-aBFP (Fig. 4(A) and Fig. S11A, S12C, ESI†) and V8-aBFP-His (Fig. 4(C) and Fig. S11B, S12D, ESI†) formed droplets ranging from approximately 200 nm to 500 nm in diameter; these observations suggest the possibility that because ELP is located on both ends of the aBFP, it colocalized with the aBFP to form mixed nanoparticles rather than to form a more regular assembled structure with unique BFP and ELP domains.
 | ||
| Fig. 4 Cast-film, negatively stained TEM and simulation results for ELP–aBFP constructs: His-V8-aBFP (A) & (B); His-V8-aBFP (C) & (D) TEM of assemblies collected from samples prepared above Tt: (A) His-V8-aBFP; (C) V8-aBFP-His. Representative configurations sampled from simulation: (B) His-V8-aBFP; (D) V8-aBFP-His. Peptide segments are colored dark blue, dark red, and blue (cyan) represent the sequences of His-tag, ELP, and aBFP, respectively (same coloring as in Fig. 1). Additional cast-film TEM images are shown in Fig. S12 (ESI†). Additional information about molecular structures sampled during the simulation are shown in Fig. S14 (ESI†). | ||
To provide insight into this association process, His-V8-aBFP and V8-aBFP-His were simulated using a MARTINI Coarse-Grained (CG) approach. The MARTINI CG representation and force field efficiently address association and assembly when compared to all-atom simulations and have been previously applied to the simulation of ELP and RLP constructs.38,48 Simulation results (Fig. 4(B) and (D)) suggest that the V8-aBFP constructs aggregate in a manner consistent with the liquid–liquid phase transition associated with ELP when heated above its Tt. This phase separation leads to nanodroplets during the preparation of TEM samples, rather than more specific assemblies with regular nanostructures. The simulation results suggest colocalization of ELP, aBFP, and the His tag together within the aggregated structure for both His-V8-aBFP and V8-aBFP-His (Fig. S15A and B, ESI†), while the pair distribution function (Fig. S16B, ESI†) corroborates the proximity of the BFP and the His-tag in neighboring V8-aBFP-His tetrameric units. For the His-V8-aBFP sequence, the His-tag is in proximity to ELP of distinct neighboring tetramers (Fig. S16E, ESI†). The N-terminal His-tag participates in aromatic and pi–cation interactions with ELP units during assembly, suggesting a potential origin of the lower transition temperature of the His-V8-aBFP relative to V8-aBFP-His. The RLP-aBFP polypeptides we previously reported assemble into linear nanofibers.38 The current observations suggest, however, that the combination of the different chain lengths, different amino acid compositions, and guest residue (valine in ELP and tryptophan in RLP), lead to LLPS-like phase separation in the V8-aBFP constructs instead of assembly into rod-like structures.
Interestingly, for the ELP–pBFP constructs in which ELP domains are localized on one end of the BFP, the His-V8-pBFP (Fig. 5(A) and (B)) formed nanoassemblies with an apparent peptide shell and a heterogeneous interior, while V8-pBFP-His (Fig. 5(D) and (E)) formed assemblies with an onion-like, nanolamellar morphology. These well-defined assemblies likely arise from the restriction of the hydrophobic ELP to one end of the pBFP bundle, supporting association into a shell comprising ELP with BFP on the surface for His-V8-pBFP or into a bilayer for the V8-pBFP-His, respectively. Evaluation of the cryo-TEM images for the well-assembled layers in V8-pBFP-His indicated that the ELP layer (which was presumed to be darker in the image owing to its expected higher scattering cross-section upon association) had a thickness of approximately 4.7 nm while the pBFP layer was approximately 10 nm thick (Fig. 5(D)). Statistical analysis of the thickness of each layer indicates the uniformity of the layers (Fig. S13, ESI†), and the measured thickness for the individual layers is consistent with the expected dimensions of each domain. Specifically, the length of an individual pBFP bundle is 4 nm,49 so the observed thickness of the pBFP layer (10 nm) is consistent with the association of two pBFP-His domains, where their associated His tags are located between the two pBFP bundles. A similar ELP–BFP bilayer structure was also observed at the surface of His-V8-pBFP shell-covered nanoparticles. The 4.7 nm, collapsed hydrophobic ELP layer is discernible in the TEM owing to its greater contrast (Fig. 5(A)), although the surface of the particles are expected to be rich in BFP owing to its hydrophilicity. Characterization via confocal microscopy of V8-pBFP-His, in the presence of the hydrophobic dye AZ488 (Fig. S18, ESI†), supports that the assembled nanostructures have sufficient hydrophobic character to concentrate the hydrophobic dye, consistent with localization of the hydrophobic ELP within the assembled nanostructures.
 | ||
| Fig. 5 Cryo-TEM, cast-film TEM, and simulation results for pBFP-containing constructs. (A) cryoTEM, (B) cast-film TEM, and (C) simulation of His-V8-pBFP assembly above Tt and (D) cryoTEM, (E) cast-film TEM, and (F) simulation of V8-pBFP-His assembly above Tt. The different placement of the His-tag in the polypeptides leads to distinct morphologies in the assembled nanoparticles. The white arrows indicate a ELP layer. Simulation results of the assembly of (C) His-V8-pBFP and (F) V8-pBFP-His above Tt. The simulations corroborate the experimentally observed morphologies. Dark blue, dark red, and pink-red chains in simulation represent the sequence of His-tag, ELP, and pBFP, as indicated in the schematic in Fig. 1. Extra cast-film TEM images are shown in Fig. S12 (ESI†). Details of the molecular structures during the simulation process can be found in Fig. S14 (ESI†). | ||
Simulations of both His-V8-pBFP (Fig. 5(C)) and V8-pBFP-His (Fig. 5(F)) were conducted to shed light on the differences in molecular organization and assembly associated with the His tag placement. The simulation results indicated the formation of an amphiphile bilayer with an ELP core and BFP surface for both designs (Fig. S15C and D, ESI†), and both His-V8-pBFP and V8-pBFP-His form a stable bilayer in the simulation, in qualitative agreement with the experimental observations. The thicknesses of the ELP layer obtained by simulation are approximately 6 nm (His-V8-pBFP) and 8 nm (V8-pBFP-His). The slightly larger thickness of the ELP layer in the simulation (6–8 nm) versus that observed in the cryo-TEM studies (4.7 nm ± 1.1 nm for His-V8-pBFP and 4.7 nm ± 0.9 nm for V8-pBFP-His) is attributed to limitations in accurately capturing the conformations of the intrinsically disordered protein (IDP) domain with the Martini force field. The simulation results indicated close proximity of the His-tags with one another for both His-V8-pBFP and V8-pBFP-His, consistent with aromatic interactions between the His-tags (Fig. S17F, ESI†). These simulation results support that both the His-tag/ELP contacts and the resulting structures are sensitive to their relative placement within the peptide sequence (Fig. S17E, ESI†). As a result of the association involving His and ELP, the Tt for His-V8-pBFP is expected to be the lowest of all of the V8-BFP constructs, which is also corroborated by the large amplitude in the inter-tetrameric ELP-ELP and His-ELP pairs at short distances (Fig. S17E, ESI†). For the V8-pBFP-His, the strong propensity for His tags on neighboring tetramers to be in proximity of the pBFP layer (Fig. 5(D), the 10 nm thick lighter layer) drives the close packing of the V8-pBFP-His into multilayers. In contrast, no regular packing is observed for His-V8-pBFP, resulting in a less-ordered particle interior.
Based on the morphologies observed in cryo TEM and on the simulation data, potential assembly principles, based on the positioning of the His tag, emerge for His-V8-pBFP (Fig. 6(A)) and V8-pBFP-His (Fig. 6(B)). In the simulations of His-V8-pBFP, the His-ELP chains (Fig. 1) necessarily splay, in some cases to the point that His-V8 chains bend to allow His residues to form persistent contacts with the pBFP bundle exteriors (Fig. 5(C)). This looping of the His-ELP unit such that it contacts the exterior of the pBFP bundle distorts the cylindrical shape and impedes the alignment of bundles within the pBFP layer. On the surface of the His-V8-pBFP droplet, the His-ELP chains are largely directed toward the interior of the bundle, partly due to the hydrophobicity of ELP; pBFP bundles can be aligned and ordered to form a layer at the droplet's surface. This surface layer organization is consistent with the cryo TEM observations (Fig. 5(A)). Interior to the His-V8-pBFP droplet, layers do not persist, and the ELP–pBFP bilayer does not adopt aligned packing of bundles. The frequent appearance of unaligned amphiphile bilayers and formation of micelles in simulation are consistent with the cryo TEM observations, where nanodroplets have surface shell layers but lack well-defined interior layers. For V8-pBFP-His, in contrast, the ELP chains exhibit few persistent contacts with the exteriors of the pBFP units. The pBFP bundles align due to their cylindrical bundle structure, the juxtaposition of His tags to the well-structured C-termini of the bundles, and the contacts between His-tag domains at the interface between leaflets in the bilayer (Fig. 1 and 6(B)). These ordered layers persist despite the experimentally observed curvature of ELP–BFP bilayer (Fig. 5(D) and Fig. S17B, ESI†). As a result, V8-pBFP-His assembles into a multilamellar assembly in which each layer is packed closely (Fig. 5(D) and 6(B)).
 | ||
| Fig. 6 The proposed assembly mechanisms for (A) His-V8-pBFP and (B) V8-pBFP-His. | ||
The combination of (cryo-)TEM and simulation reveals the importance that the location of the His tag has in specifying the properties and assembly of these ELP–BFP constructs. His-tag-containing amphiphiles have been previously shown to assemble into multilayer vesicles,50 micelles, and nanofibers51 when the His-tag is fused to hydrophobic polymers such as oligostyrene50 or aliphatic chains.51 The metal-binding properties of the His-tag have also been widely used in metal–organic framework assembly.52 However, there have been no reports to our knowledge indicating the significant impact that His-tag positioning has on controlling polypeptide assembly. Our results thus suggest a novel approach to control assembly with appropriate positioning of a commonly used purification tag.
Discussion
Coiled-coil peptides have long been regarded as powerful tools for controlled peptide assembly, although most designs focus on using individual helical bundles, coiled-coil peptides as linkers between polymer chains, or functional proteins.3,53,54 Helical bundles have also been applied as a reversible link between hydrophilic fluorescent proteins and hydrophobic ELPs to build enzyme-based amphiphiles.3,54 Other protein oligomerization units have some disadvantages for controlling assembly, such as necessarily long peptide lengths (e.g., SpyTag/SpyCatcher system55), uncontrolled numbers of peptide units in the assembly (e.g. silk-like peptides56), and low stability with a lack of sequence variety (e.g. collagen-like peptides2). Coiled-coil peptides, in contrast, provide an oligomerization unit with short but highly chemically versatile sequences, well-structured oligomeric assemblies, and tunable stimuli-dependent responses. The exploitation of other physicochemical and structural properties of the coiled-coil domains—such as their hydrophilicity, rod-like structure, and thermally sensitive structural transitions—provides a powerful route for designing new peptide materials, especially in constructs with the short ELP and BFP domains presented herein. Taking advantage of these design features, especially the placement of the His-tag and use of antiparallel and parallel bundle-forming sequences, permits tuning of BFP organization from random distributed layers to well-aligned multilayers.In this work, we report differences in ELP–BFP design that impact observed transition temperatures and assembly. We note that previous reports of ELP-coiled-coil constructs based on leucine zipper coiled-coils have examined formation of hydrogels and bilayers,53,57–60 and hydrophilic globular proteins such as mCherry and GFP can be incorporated to help drive bilayer formation.3,61,62 Herein, simpler systems are considered, where ELP is conjugated with designed BFPs that control of the positioning unstructured and helical segments via bundle folding and tetramerization. These constructs can also be used to modulate display of His-tags for exploring sequence-and structured-based determinants of assembly (Fig. 2).
The ELP–pBFP system also provides a unique chance to bridge the properties of lipid bilayers and polymeric bilayer systems. Both ELP–pBFP and phospholipids comprise a hydrophilic head and multiple hydrophobic chains, and the increased molecular weight and rigidity of the domains of the ELP–pBFP supports the formation of bi- and multi-layered structures of greater dimensions. The increased dimensions and stability may afford distinct advantages for application. For example, compared to liposomes, ELP–pBFP vesicles may be able to sequester larger amounts of drug and achieve a longer sustained release based on their greater stability. Similar to polymeric bilayers, the ELP–pBFP system can form layers based on molecules of high molecular weight relative to the lipid systems, but with molecular chain lengths that have been successfully reduced from hundreds of repeat units to 80 repeat units.63 Chain lengths necessary to produce stable bilayers can be further reduced by controlling interactions between ELP. In addition, the incorporation of ELP provides adjustable stimuli-responsiveness which could support different therapeutic regimens such as the burst release of cargo under hypothermal conditions, different salt concentrations, or sudden pH change; indeed, the transition temperatures of the ELP–BFP polypeptides herein are preliminarily indicated to be sensitive to pH and salt concentrations (see ESI,† Fig. S19).
In simulations, understanding the self-assembled structures that result from the interactions of intrinsically disordered polypeptides (IDPs) remains a challenge as those polypeptides lack stable tertiary structures and have significant compositional and structural heterogeneity that has limited computational approaches to polypeptide design. This study demonstrates the correspondence between experimentally observed morphology and simulation results, which reveals that the assembly of the ELP–BFP constructs can be qualitatively understood via computational methods. These findings support the possibility for computational-guided system prediction and design of an ELP–BFP sequence with specific morphology and transition temperature by combining computational peptide design, molecular dynamic simulations, and machine learning techniques, which would simplify and accelerate peptide design and investigation.
Conclusions
In conclusion, we have designed a series of new polypeptide amphiphiles, based on ELP and BFP domains, which can be recombinantly expressed in bacterial hosts. ELP–BFP amphiphiles showed sequence-dependent thermally responsive behavior influenced by the relative position of each peptide domain in the overall molecular design, and preliminary studies suggest that pH-triggered phase-transition behavior should also be possible (Fig. S19, ESI†). These molecules assemble into distinct nanostructures such as nanodroplets or bilayer vesicles based on the design of the amphiphile. The ELP–BFP design can be further modified by changing ELP and BFP separately and forming a library of polypeptide amphiphiles with controlled morphology and phase transition behavior. These amphiphiles have the potential to be applied in multiple biomedical applications such as pH-responsive drug delivery carriers or thermal-responsive hydrogels. The ELP–BFP system also provides a unique chance to predict, study, and design assembled structures computationally as they are shorter in length and amenable to computational simulation. Although design modifications to our reported ELP–BFP molecules will be required to achieve assembly and stability under physiologically relevant conditions, the establishment of an ELP–BFP library will provide novel peptide-based materials with tuneable responsiveness, high stability, versatility, and biocompatibility.Materials and methods
Materials
Plasmid DNA (pET19b for sequence with N-terminus histidine tag and pET30a(+) for sequence with C-terminus histidine tag) encoding ELP and BFP sequences were purchased from Genscript Corporation (Piscataway, NJ). The chemically competent E.coli strains BL21(DE3) and SIG10 were purchased from MilliporeSigma (Burlington, MA) for construction and expression of recombinant plasmid encoding ELP–BFP sequences. The chemicals for preparing media used in the plasmid construction and expression was purchased from Fisher Scientific (Hampton, NH). Isopropyl β-D-1-thiogalactopyranoside (IPTG) for induction of polypeptide expression was purchased from Gold Bio (St. Louis, MO). RNase A for polypeptide purification was purchased from Qiagen (Germantown, MD). All amino acids, resins, and activators for solid-phase peptide synthesis were purchased from ChemPep (Wellington, FL) and CEM (Matthews, NC) corporation and used as received. MSP 96 target ground steel BC plate and Protein Standard I for MALDI-TOF MS were purchased from Bruker (Billerica, MA). Water for buffers or media was deionized and filtered using either a ThermoFisher Barnstead NANOpure diamond water purifier or a Milli-Q Synergy water purification system. All other reagents were purchased and used as received from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Hampton, NH) unless indicated otherwise.Peptide synthesis and purification
Short peptide control sequences were prepared using standard solid phase peptide synthesis (SPPS) on a Liberty Blue automated microwave peptide synthesizer (CEM corporation) at 0.10 mmol scale. Fmoc-based protocols with a Rink amide resin (ChemPep), which yields an amidated C-terminus after cleavage, were used. The Fmoc was deprotected with 20% piperidine (Sigma) in dimethyl formamide (DMF) (Fisher) at 75 °C for 3 min, along with 5 washes with DM F after deprotection. Subsequent coupling steps were performed at 90 °C for 5 min with 4 eq. of the appropriate protected amino acid (0.2 mM, ChemPep), 4 eq. ethyl (hydroxyimino)cyanoacetate (Oxyma, 1 mM, CEM), and 4 eq. N,N′-diisopropylcarbodiimide (DIC, 1 mM, ChemImpex). To increase the yield of full-length peptide, all amino acids were coupled twice. After synthesis, the peptides were washed with DMF and dichloromethane (DCM) (Fisher) 3 times, then cleaved in 10 mL Reagent K cleavage solution (82.5% trifluoroacetic acid (Sigma), 5% phenol (Thermo Scientific), 5% water, 5% thiolanisole (Sigma) and 2.5% 1,2-ethanedithiol (Sigma)) by shaking at room temperature for 2 hours. The cleaved peptide solutions were precipitated using cold ethyl-ether and centrifuged at 4000 rpm for 5 min and washed with cold ethyl-ether three times. The precipitates were dried overnight and re-dissolved in deionized 95% Milli-Q water and 5% ACN. The solutions were further purified via high-performance liquid chromatography (HPLC, Quaternary Gradient Module (Waters 2545), Waters Corporation, Milford, MA) using a reverse-phase BEH130 Prep C18 10 μm column (XBridge, Waters Corporation). Pure fractions of the peptides were combined and lyophilized. Their purity and molecular weight were confirmed via analytical ultra-high performance mass spectrometry (UPLC-MS). (Waters Xevo G2-S QTof, Waters Corporation)Cloning of ELP–BFP constructs
The designed DNA sequences are shown in Fig. S1 (ESI†) and polypeptide sequences are shown in Fig. S2 (ESI†). For the construction of the His-ELP–BFP vector, pET19b-ELP plasmid DNA and pET19b-BFP plasmid DNA were first digested with Xho1 at 37 °C for 1 hour. The Xho1 digested dephosphorylated pET19b-ELP vector and BFP sequence were separated and purified by agarose gel extraction. The BFP sequences were then ligated into Xho1 digested and dephosphorylated pET19b-ELP vector via treatment with T4 DNA ligase at 16 °C overnight. 4 μl ligation mixture was then mixed with 40 μl SIG10 chemically competent cells (MilliporeSigma) and incubated in ice for 30 min. The mixture was then heat shocked at 42 °C water bath for 45 seconds and chilled in ice for 2 min before adding 960 μl recovery media provided with the competent cell stock. After adding the recovery media, the mixture was incubated at 37 °C in an incubator with a 225 rpm shaking speed for 1.5 hour. An aliquot of 200 μL transformation mixture was then plated on a LB-agar plate with ampicillin or kanamycin and incubated in a 37 °C oven for 14 hours. Three individual colonies were selected as for a seed culture and were amplified in a culture tube containing 11 mL LB media at 37 °C overnight. A 1 mL aliquote of the overnight culture was mixed with 50% glycerol to create cell stock and the remaining 10 mL overnight culture was purified by QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) to acquire pure plasmid for DNA sequencing and transformation to an expression strain. Sanger DNA sequencing (University of Delaware Sequencing and Genotyping Center) using center-stocked T7p and T7t primers was then performed to confirm the successful construction of a given His-ELP–BFP construct. The plasmid was then transformed into BL21(DE3) chemically competent E. coli cells (MilliporeSigma) following standard protocol to generate the expression host that was employed for polypeptide expression. All sequences were confirmed by DNA sequencing before use. The construction of ELP–BFP-His sequences followed the same protocol except for the use of a pET30a(+)-ELP vector.Polypeptide expression and purification
Protocols for expression and purification of ELP–BFP constructs were developed based on the QIAexpress system (Qiagen, Valencia, CA).64,65 The desired concentration of antibiotics (100 mg L−1 ampicillin for pET19b plasmid and 50 mg L−1 kanamycin for pET30a(+)) was maintained during expression. An overnight starter culture comprising a single colony of transformed cells in 150 mL sterile lysogeny broth (LB) was used to inoculate 6 flasks, each containing 750 mL of sterile 2xYT media (16 g L−1 tryptone, 10 g L−1 yeast, 5 g L−1 sodium chloride). The culture was shaken at 37 °C until the OD600 reached 0.6–0.8. 1 mM IPTG was then used to induce the polypeptide expression and the culture was shaken at 25 °C overnight after induction. The cells were harvested by centrifugation (4284 RCF, 15 min, 4 °C) after expression and were stored at −20 °C. The frozen cell pellets were resuspended overnight in the cold room (4 °C) in 200 mL native lysis buffer (50 mM sodium phosphate monobasic monohydrate, 300 mM sodium chloride, 10 mM imidazole, pH 8.0) containing 1.8 mg mL−1 lysozyme from chicken egg white. A Fisher Scientific model 500 Sonic Dismembrator with a 10 mm tapered horn was used to sonicate the cell lysate for 3 min on ice, with a 10 s recovery time, in order to further disrupt cells. To reduce lysate viscosity, the lysate was next incubated with RNase A (10 μg mL−1) and DNase I (5 μg mL−1) on ice for 1 hour. The purification was then performed under denaturing conditions (8M urea). The pH of buffer B (100 mM sodium phosphate monobasic monohydrate, 10 mM tris base, 8 M urea, pH = 8.0), buffer C (100 mM sodium phosphate monobasic monohydrate, 10 mM tris base, 8 M urea, pH = 6.1), buffer D (100 mM sodium phosphate monobasic monohydrate, 10 mM tris base, 8 M urea, pH = 5.9), and buffer E (100 mM sodium phosphate monobasic monohydrate, 10 mM tris base, 8 M urea, pH = 4.5) was adjusted immediately before use due to the dissociation of urea. An equal volume of lysis buffer B was added to the lysed and sonicated cell product and the mixture was stirred for 1 h. Centrifugation for 15 min at 4 °C, 47808 RCF was used to remove insoluble impurities. The supernatant was incubated with 25 mL of HisPurTM Ni-NTA resin (Thermofisher) with gentle shaking to prevent settling of the resin. The polypeptide-loaded resin was packed into a gravitational flow column and washed with 250 mL wash buffer C and 150 mL of elution buffer D before elution of the target polypeptide with 100 mL elution buffer E. Buffer E fractions containing purified polypeptide were dialyzed against a 50-fold excess of deionized water at 4 °C (MWCO 3.5 kDa) with at least eight changes of water over 3 days. The desalted polypeptide was then lyophilized and stored at −20 °C until further use. The yield of each sequence is shown in Table S2 (ESI†).General characterization of ELP–BFP
Matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (Bruker MicroFlex MALDI-TOF, Billerica, MA) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were performed to confirm the purity and molecular weight of each purified polypeptide construct. Amino acid analysis was performed by the Molecular Structure Facility at the University of California, Davis (Davis, CA) using a Hitachi L-8800 sodium citrate-based amino acid analyzer (Tokyo, Japan) as a check to confirm the composition of the V8-BFP polypeptides, which were employed for assembly studies.Circular dichroism spectroscopy
CD experiments were utilized to confirm the formation of coiled-coils in the designed polypeptides. All CD experiments were performed using a Jasco J-1500 CD spectropolarimeter (Jasco, Easton, MD) with a 0.1 cm path length quartz. The ELP–BFP sample was dissolved in water at 200 μM concentration and sonicated at room temperature in a bath sonicator (Branson 2510-DTH) for 30 min. The sonicated sample was then stored at 4 °C overnight to resolubilize the particles formed by heating during sonication. The sample was then diluted to 50 μM with water. 400 μL of polypeptide solution was added to the quartz cuvette and cooled down to 5 °C in the CD instrument. Temperature-dependent CD experiments were performed in the temperature range from 5 °C to 85 °C at a heating rate of 1 °C min−1. All wavelength scans were acquired as an average of three scans from 190 to 250 nm with data collection at a 1 nm data pitch, a 1 nm bandwidth, a 4-second response time, and a scanning rate of 50 nm min−1. The ellipticity data were subsequently converted to the mean residue ellipticity [θMRE] using the following equation: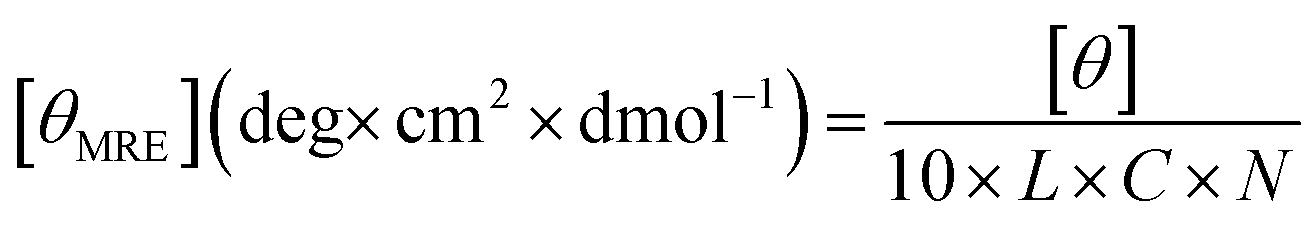 | (1) |
Turbidimetry measurements
The phase transition temperatures for the ELP–BFP constructs were determined via UV-Vis assessments of turbidity for polypeptides under various buffer conditions. Citric acid–monosodium phosphate (Na2HPO4) buffers at different pH were prepared by mixing 0.2 M citric acid and 0.1 M monosodium phosphate and the pH was determined (Accumet™ Basic AB15 equipped with an Accumet™ Liquid-Filled pH/ATC electrodes). ELP–BFP polypeptides were dissolved in citric acid–Na2HPO4 buffer with different pH at different concentrations (500 μM, 400 μM, 300 μM, 200 μM, 100 μM). The phase transition behavior of different constructs was characterized by temperature-dependent measurements of their optical density at 350 nm through ultraviolet (UV)-visible (Vis) spectroscopy with a 10 mm path length quartz cuvette. A Cary 60 UV-Vis spectrophotometer (Agilent, Santa Clara, CA) equipped with a Peltier holder t2 Sport/Cary60 (Quantum Northwest, Liberty Lake, WA) and house air to constantly purge the cuvettes of any condensing water vapor was used to acquire turbidity data while heating from 5 °C to 85 °C at a rate of 2 °C min−1. The baseline was normalized to 0, and the transition temperature was defined as the temperature where the OD350 reached 0.03 units above the baseline. Dynamic light scattering (DLS) measurements were also conducted and a similar transition temperature was observed. The dynamic heterogeneity of the assembled structures, however, complicated the use of DLS to estimate particle sizes. Additional TEM data are presented in the ESI,† illustrating the large dispersity of the nonmicellar structures.Transmission electron microscopy (TEM)
TEM imaging was performed on a FEI TALOS F200C microscope with 200 kV accelerating voltage. All the images were recorded via a FEI Ceta 16M camera. To prepare the sample grids, carbon-coated 200 mesh copper grids (CF400-Cu; Electron Microscopy Sciences, Inc.) were freshly treated by glow discharge using a glow discharge cleaning system (Pelco EasiGlow; Ted Pella, Inc.). All TEM sample preparation was performed in an 80 °C oven; sample solutions at protein concentrations of 500 μM (His-V8-aBFP, V8-His-aBFP, and V8-pBFP-His) or 200 μM (His-V8-pBFP) were pre-incubated for 30 minutes or overnight, with no significant difference in the assembled morphologies observed via TEM for the different incubation times. Stain solutions, sample grids, and pipette tips were incubated in the same oven prior to sample grid preparation. An aliquot of 5 μL of the sample solution was applied to plasma-treated grids for about 30–60 seconds, then the remaining liquid was blotted from the edge of the grids using filter papers. To negatively stain the grids, 5 μL of an aqueous solution of phosphotungstic acid (2% w/v; pH 5.6 citric acid–Na2HPO4 buffer) was applied to the cast-film grids and incubated for about 20–40 seconds. The grids were then blotted with filter paper to remove the remaining staining solution. The stained grids were allowed to sit for at least 5 min under ambient conditions before TEM observation.Cryogenic transmission electron microscopy (Cryo-TEM)
Cryo TEM imaging was performed on a FEI TALOS F200C microscope with 200 kV accelerating voltage. The images were recorded via a FEI Ceta 16M camera. Lacey carbon grids (300 mesh, Ted Pella Inc.) were plasma treated by a plasma cleaner (Pelco EasiGlow; Ted Pella, Inc.) before sample grid preparation. Vitrified grids were prepared using the FEI Vitrobot Mark IV, an automated plunge-freezing device. The climate chamber on the Vitrobot was set to the maximum temperature (60 °C) and 60% humidity before the preparation of the sample grids. Sample solution, lacey carbon grids, and pipette tips were preheated in a thermomixer (set at 80 °C) or the climate chamber on the Vitrobot. A droplet of 4 μL sample solution at 500 μM (His-V8-aBFP, V8-His-aBFP, and V8-pBFP-His) or 200 μM (His-V8-pBFP) was deposited onto the plasma-treated lacey grid. (The temperature for assembly was chosen to be higher than sample Tt. The Tt values of each sample were: 500 μM HisV8aBFP: 28.0 ± 1.0 °C, 200 μM HisV8pBFP: 28.3 ± 0.7 °C, 500 μM V8aBFPHis: 51.3 ± 3.4 °C, 500 μM HisV8aBFP: 39.3 ± 1.4 °C) Then the grid was blotted with blotting paper and rapidly plunged into liquid ethane. Grids were stored in liquid nitrogen until transferred to the electron microscope for imaging.Fiji (ImageJ) was used for the precise determination of the layer thickness of the ELP–BFP polypeptides.67 One hundred data points were meticulously measured for each polypeptide structure, selected from multiple structures obtained from a minimum of two cryo-EM images. Subsequently, the collected data was imported into Origin (OriginLab Co. Northampton, MA) for further analysis.
Simulation
ELP–BFP polypeptides were simulated using a coarse-grained (CG) approach with the MARTINI 3.0 force field combined with an elastic network model.68,69 His-V8-aBFP, V8-aBFP-His, His-V8-pBFP, and V8-pBFP-His, the BFP with a (VPGVG)8 sequence and an unprotonated Histidine tag were simulated in this study, respectively. The N-terminal ELP sequences were added, to a computationally-generated, all-atom model of BFP, using PyMol and Alphafold2. The His-tag sequence was included accordingly. The CG representation was generated by the transformation of atomistic structures via the Martinize2 script.68 During the CG simulation, the tetrahelical structure of the coiled-coil bundlemer was maintained by an elastic network, constructed by adding the elastic bonds among the backbone atoms of the helical bundle. The elastic bonds were applied between pairs of CG atoms if the distance was between 0.5 and 0.9 nm, with a bond force constant of 700 kJ mol−1 nm−2. The ELP segments were unconstrained, i.e., treated as “random coil.”The initial configurations of the CG systems for His-V8-aBFP and V8-aBFP-His were generated by randomly inserting 15 copies of non-overlapping CG HisV8aBFP and V8aBFPHis into a CG water box with dimensions of 30 × 30 × 30 nm3, respectively. For the His-V8-pBFP and V8-pBFP-His, an anisotropic simulation box with size of 15 × 15 × 40 nm3 was constructed, and 18 copies of nonoverlapping CG His-V8-pBFP and V8-pBFP-His were randomly inserted, respectively. Sodium and chloride ions were added to achieve 150 mM sodium chloride and no net charge with each system. After constructing the initial configuration, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of steepest descent energy minimization were performed, then the systems simulated for 10 ns with an NVT ensemble at selected temperatures. Subsequently, the production run in the NPT ensemble was executed for 4 μs using the integration timestep of 0.02 ps at a temperature 330 K and a pressure of 1 bar. The configurations were collected every 1000 ps for analysis purposes. During the NPT production, a v-rescale algorithm70 with a time constant of 1.0 ps, and Parrinello–Rahman barostat71 with a coupling constant of 12.0 ps was applied to maintain the constant temperature and pressure respectively. The leapfrog algorithm was used to update the positions of atoms. Gromacs72 and MARTINI 3.0 force field were used to conduct all the simulations. PyMol was used for visualization purposes.73 The MDAnalysis toolkit was utilized to analyze the data, including the pair distribution functions (PDF) and density calculations.74
000 steps of steepest descent energy minimization were performed, then the systems simulated for 10 ns with an NVT ensemble at selected temperatures. Subsequently, the production run in the NPT ensemble was executed for 4 μs using the integration timestep of 0.02 ps at a temperature 330 K and a pressure of 1 bar. The configurations were collected every 1000 ps for analysis purposes. During the NPT production, a v-rescale algorithm70 with a time constant of 1.0 ps, and Parrinello–Rahman barostat71 with a coupling constant of 12.0 ps was applied to maintain the constant temperature and pressure respectively. The leapfrog algorithm was used to update the positions of atoms. Gromacs72 and MARTINI 3.0 force field were used to conduct all the simulations. PyMol was used for visualization purposes.73 The MDAnalysis toolkit was utilized to analyze the data, including the pair distribution functions (PDF) and density calculations.74
Author contributions
Bin Wang: conceptualization, methodology, investigation, visualization, formal analysis, writing – original draft, writing – review & editing Weiran Xie: investigation, visualization, writing – review & editing Tianren Zhang: software, investigation, formal analysis, visualization, writing – review & editing Darrin J. Pochan: conceptualization, methodology, writing – review & editing Jeffery G. Saven: methodology; writing–review & editing Kristi L. Kiick: conceptualization, methodology, writing – review & editing.Data availability
All data needed to evaluate the conclusions in the paper are presented in the paper and/or the ESI.† Plasmid, materials, and additional data related to this paper may be requested from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was primarily supported by National Science Foundation through the University of Delaware CHARM Materials Research Science and Engineering Center, DMR-2011824. Additional partial support for the reported studies was also provided by NSF (DMR-1609544, DMR-2004796, DMR-0239744, DMR-2004890, CBET-2023668). Microscopy equipment employed in the studies was acquired with shared instrumentation grants (S10 RR027273 and S10 OD016361) and access was supported by the NIH-NIGMS (P20 GM103446), the NIGMS (P20 GM139760), and the State of Delaware. This work used Stampede 2 through allocation TG-CHE110041 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) and XSEDE programs, which is supported by the National Science Foundation grants #1548562, #2138259, #2138286, #2138307, #2137603, and #2138296. Data storage was supported by the University of Delaware Center for Bioinformatics and Computational Biology Core Facility [RRID:SCR_017696] was made possible by support from an NIH Shared Instrumentation Grant (NIH S10OD028725), Delaware INBRE (NIH P20GM103446), and the Delaware Biotechnology Institute. The views expressed here are the responsibility of the authors and do not necessarily reflect the position of the funding agencies. The authors would like to thank Yao Tang from the Pochan group for providing the SPPS control of aBFP, Zihan Zhang from the Pochan group for providing the SPPS control for pBFP, and Akshay Patel and Victoria Thompson from the Kiick group for assistance with the recombinant synthesis of replicate samples.References
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS PubMed.
- J. Qin, J. D. Sloppy and K. L. Kiick, Sci. Adv., 2020, 6, eabd3033 CrossRef CAS PubMed.
- W. M. Park and J. A. Champion, J. Am. Chem. Soc., 2014, 136, 17906–17909 CrossRef CAS PubMed.
- Y. Mizuguchi, Y. Mashimo, M. Mie and E. Kobatake, Biomacromolecules, 2020, 21, 1126–1135 CrossRef CAS PubMed.
- S. Saha, S. Banskota, S. Roberts, N. Kirmani and A. Chilkoti, Adv. Ther., 2020, 3, 1900164 CrossRef PubMed.
- C. S. Thomas, M. J. Glassman and B. D. Olsen, ACS Nano, 2011, 5, 5697–5707 CrossRef CAS PubMed.
- D. Wu, N. Sinha, J. Lee, B. P. Sutherland, N. I. Halaszynski, Y. Tian, J. Caplan, H. V. Zhang, J. G. Saven, C. J. Kloxin and D. J. Pochan, Nature, 2019, 574, 658–662 CrossRef CAS PubMed.
- L. Cao, B. Coventry, I. Goreshnik, B. Huang, W. Sheffler, J. S. Park, K. M. Jude, I. Marković, R. U. Kadam, K. H. G. Verschueren, K. Verstraete, S. T. R. Walsh, N. Bennett, A. Phal, A. Yang, L. Kozodoy, M. DeWitt, L. Picton, L. Miller, E.-M. Strauch, N. D. DeBouver, A. Pires, A. K. Bera, S. Halabiya, B. Hammerson, W. Yang, S. Bernard, L. Stewart, I. A. Wilson, H. Ruohola-Baker, J. Schlessinger, S. Lee, S. N. Savvides, K. C. Garcia and D. Baker, Nature, 2022, 605, 551–560 CrossRef CAS PubMed.
- A. W. Jacobitz, A. B. Dykstra, C. Spahr and N. J. Agrawal, J. Pharm. Sci., 2020, 109, 293–300 CrossRef CAS PubMed.
- T. Luo and K. L. Kiick, J. Am. Chem. Soc., 2015, 137, 15362–15365 CrossRef CAS PubMed.
- D. Britton, L. F. Christians, C. Liu, J. Legocki, Y. Xiao, M. Meleties, L. Yang, M. Cammer, S. Jia, Z. Zhang, F. Mahmoudinobar, Z. Kowalski, P. D. Renfrew, R. Bonneau, D. J. Pochan, A. J. Pak and J. K. Montclare, Biomacromolecules, 2024, 25, 258–271 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS PubMed.
- C. Garcia Garcia, S. S. Patkar, N. Jovic, J. Mittal and K. L. Kiick, ACS Biomater. Sci. Eng., 2021, 7, 4244–4257 CrossRef CAS PubMed.
- Y. Ji, D. Liu, H. Zhu, L. Bao, R. Chang, X. Gao and J. Yin, Acta Biomater., 2023, 164, 74–93 CrossRef CAS PubMed.
- D. R. Dautel, W. T. Heller and J. A. Champion, Biomacromolecules, 2022, 23, 3678–3687 CrossRef CAS PubMed.
- M. H. Sangji, H. Sai, S. M. Chin, S. R. Lee, I. R. Sasselli, L. C. Palmer and S. I. Stupp, Nano Lett., 2021, 21, 6146–6155 CrossRef CAS PubMed.
- M. M. Rvachev, Biophys. Rev. Lett., 2010, 05, 73–88 CrossRef CAS.
- S. Kansız and Y. M. Elçin, Adv. Colloid Interface Sci., 2023, 317, 102930 CrossRef PubMed.
- V. Castelletto, C. J. C. Edwards-Gayle, F. Greco, I. W. Hamley, J. Seitsonen and J. Ruokolainen, ACS Appl. Mater. Interfaces, 2019, 11, 33573–33580 CrossRef CAS PubMed.
- J. Li, J. Wang, Y. Zhao, P. Zhou, J. Carter, Z. Li, T. A. Waigh, J. R. Lu and H. Xu, Coord. Chem. Rev., 2020, 421, 213418 CrossRef CAS.
- J.-W. Choi, S.-H. Choi and J.-I. Won, Biomacromolecules, 2021, 22, 2604–2613 CrossRef CAS PubMed.
- D. H. T. Le, V. Ibrahimova, S. A. H. van den Wildenberg, H. Wu, A. Fonseca, T. Torres, E. Garanger, W. P. J. Leenders, R. Brock, S. Lecommandoux and J. C. M. van Hest, Angew. Chem., Int. Ed., 2023, 62, e202300511 CrossRef CAS PubMed.
- W. Hassouneh, E. B. Zhulina, A. Chilkoti and M. Rubinstein, Macromolecules, 2015, 48, 4183–4195 CrossRef CAS PubMed.
- D. W. Urry, M. M. Long, B. A. Cox, T. Ohnishi, L. W. Mitchell and M. Jacobs, Biochim. Biophys. Acta, Protein Struct., 1974, 371, 597–602 CrossRef CAS PubMed.
- A. K. Varanko, J. C. Su and A. Chilkoti, Annu. Rev. Biomed. Eng., 2020, 22, 343–369 CrossRef CAS PubMed.
- B. Wang, S. S. Patkar and K. L. Kiick, Macromol. Biosci., 2021, 21, 2100129 CrossRef CAS PubMed.
- T. Christensen, W. Hassouneh, K. Trabbic-Carlson and A. Chilkoti, Biomacromolecules, 2013, 14, 1514–1519 CrossRef CAS PubMed.
- B. Zhao, N. K. Li, Y. G. Yingling and C. K. Hall, Biomacromolecules, 2016, 17, 111–118 CrossRef CAS PubMed.
- P. A. Taylor, H. Huang, K. L. Kiick and A. Jayaraman, Mol. Syst. Des. Eng., 2020, 5, 1239–1254 RSC.
- L. Truebestein and T. A. Leonard, BioEssays, 2016, 38, 903–916 CrossRef CAS PubMed.
- Y. Wu and J. H. Collier, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1424 Search PubMed.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- J. M. Fletcher, R. L. Harniman, F. R. H. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Science, 2013, 340, 595–599 CrossRef CAS PubMed.
- T. Lebar, D. Lainšček, E. Merljak, J. Aupič and R. Jerala, Nat. Chem. Biol., 2020, 16, 513–519 CrossRef CAS PubMed.
- H. V. Zhang, F. Polzer, M. J. Haider, Y. Tian, J. A. Villegas, K. L. Kiick, D. J. Pochan and J. G. Saven, Sci. Adv., 2016, 2, e1600307 CrossRef PubMed.
- N. J. Sinha, C. J. Kloxin, J. G. Saven, G. V. Jensen, Z. Kelman and D. J. Pochan, J. Biotechnol., 2021, 330, 57–60 CrossRef CAS PubMed.
- M. J. Haider, H. V. Zhang, N. Sinha, J. A. Fagan, K. L. Kiick, J. G. Saven and D. J. Pochan, Soft Matter, 2018, 14, 5488–5496 RSC.
- S. S. Patkar, Y. Tang, A. M. Bisram, T. Zhang, J. G. Saven, D. J. Pochan and K. L. Kiick, Angew. Chem., Int. Ed., 2023, 135, e202301331 CrossRef.
- F. Aladini, C. Araman and C. F. W. Becker, J. Pept. Sci., 2016, 22, 334–342 CrossRef CAS PubMed.
- Z. Zhang, J. E. Blum, R. Guo, C. J. Kloxin, J. G. Saven and D. J. Pochan, ACS Macro Lett., 2024, 13, 1591–1597 CrossRef CAS PubMed.
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2004, 5, 846–851 CrossRef CAS PubMed.
- A. Prhashanna, P. A. Taylor, J. Qin, K. L. Kiick and A. Jayaraman, Biomacromolecules, 2019, 20, 1178–1189 CrossRef CAS PubMed.
- J. Heyda, P. E. Mason and P. Jungwirth, J. Phys. Chem. B, 2010, 114, 8744–8749 CrossRef CAS PubMed.
- K. Haghani, K. Khajeh, H. Naderi-Manesh and B. Ranjbar, Int. J. Biol. Macromol., 2012, 50, 1040–1047 CrossRef CAS PubMed.
- K. Trabbic-Carlson, D. E. Meyer, L. Liu, R. Piervincenzi, N. Nath, T. LaBean and A. Chilkoti, Protein Eng., Des. Sel., 2004, 17, 57–66 CrossRef CAS PubMed.
- C.-Y. Lin and J. C. Liu, J. Mater. Chem. B, 2019, 7, 5245–5256 RSC.
- G. Qin, P. M. Perez, C. E. Mills and B. D. Olsen, Biomacromolecules, 2016, 17, 928–934 CrossRef CAS PubMed.
- T. Mabuchi, J. Kijima, Y. Yamashita, E. Miura and T. Muraoka, Macromolecules, 2023, 56, 794–805 CrossRef CAS.
- Z. Zhang, J. E. Blum, R. Guo, C. J. Kloxin, J. G. Saven and D. J. Pochan, ACS Macro Lett., 2024, 13, 1591–1597 CrossRef CAS PubMed.
- A. S. Knight, J. Larsson, J. M. Ren, R. Bou Zerdan, S. Seguin, R. Vrahas, J. Liu, G. Ren and C. J. Hawker, J. Am. Chem. Soc., 2018, 140, 1409–1414 CrossRef CAS PubMed.
- T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed.
- R. Röder, T. Preiß, P. Hirschle, B. Steinborn, A. Zimpel, M. Höhn, J. O. Rädler, T. Bein, E. Wagner, S. Wuttke and U. Lächelt, J. Am. Chem. Soc., 2017, 139, 2359–2368 CrossRef PubMed.
- A. J. Olsen, P. Katyal, J. S. Haghpanah, M. B. Kubilius, R. Li, N. L. Schnabel, S. C. O’Neill, Y. Wang, M. Dai, N. Singh, R. S. Tu and J. K. Montclare, Biomacromolecules, 2018, 19, 1552–1561 CrossRef CAS PubMed.
- Y. Li and J. A. Champion, Adv. Healthcare Mater., 2021, 10, 2001810 CrossRef CAS PubMed.
- A. R. Sutherland, Md. K. Alam and C. R. Geyer, ChemBioChem, 2019, 20, 319–328 CrossRef CAS PubMed.
- R. Chakraborty, J. Fan, C. C. Lai, P. V. Raghuvamsi, P. X. Chee, G. S. Anand and D. Yang, Int. J. Mol. Sci., 2020, 21, 4466 CrossRef CAS PubMed.
- J. Hume, J. Sun, R. Jacquet, P. D. Renfrew, J. A. Martin, R. Bonneau, M. L. Gilchrist and J. K. Montclare, Biomacromolecules, 2014, 15, 3503–3510 CrossRef CAS PubMed.
- M. Dai, J. Haghpanah, N. Singh, E. W. Roth, A. Liang, R. S. Tu and J. K. Montclare, Biomacromolecules, 2011, 12, 4240–4246 CrossRef CAS PubMed.
- J. Kronenberg, Y. Jung, J. Chen, M. J. Kulapurathazhe, D. Britton, S. Kim, X. Chen, R. S. Tu and J. K. Montclare, ACS Appl. Bio Mater., 2024, 7, 3714–3720 CrossRef CAS PubMed.
- M. K. Sing, W. R. Burghardt and B. D. Olsen, Macromolecules, 2018, 51, 2951–2960 CrossRef CAS.
- A. Fernández-Colino, F. J. Arias, M. Alonso and J. C. Rodríguez-Cabello, Biomacromolecules, 2015, 16, 3389–3398 CrossRef PubMed.
- D. R. Dautel and J. A. Champion, Biomacromolecules, 2021, 22, 116–125 CrossRef CAS PubMed.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- The QIAexpressionist – (EN) – QIAGEN, https://www.qiagen.com/us/resources/resourcedetail?id=79ca2f7d-42fe-4d62-8676-4cfa948c9435&lang=en, (accessed October 23, 2023).
- C. Garcia Garcia and K. L. Kiick, Acta Biomater., 2019, 84, 34–48 CrossRef CAS PubMed.
- N. J. Greenfield, in Protein-Protein Interactions: Methods and Applications, ed. H. Fu, Humana Press, Totowa, NJ, 2004, pp. 55–77 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domański, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, X. Periole, D. P. Tieleman, A. H. de Vries and S. J. Marrink, Nat. Methods, 2021, 18, 382–388 CrossRef CAS PubMed.
- X. Periole, M. Cavalli, S. J. Marrink and M. A. Ceruso, J. Chem. Theory Comput., 2009, 5, 2531–2543 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 9901 CrossRef PubMed.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- E. Lilkova, P. Petkov, N. Ilieva and L. Litov, The PyMOL Molecular Graphics System, Version 2.6, Schrödinger, LLC, 2015, preprint.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, D. L. Dotson, J. Domanski, S. Buchoux, I. M. Kenney and O. Beckstein, in MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations, ed. S. Benthall and S. Rostrup, Proceedings of the 15th Python in Science Conference, Austin, TX, SciPy, 2016, pp. 98–105 DOI:10.25080/majora-629e541a-00e.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02420f |
| This journal is © The Royal Society of Chemistry 2025 |