
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formulation and evaluation of PVA-based composite hydrogels: physicochemical, leachables, and in vitro immunogenicity studies†
Achmad
Himawan
 ab,
Anna
Korelidou
a,
Ana M.
Pérez-Moreno
cg,
Juan L.
Paris
c,
Juan
Dominguez-Robles
d,
Lalitkumar K.
Vora
a,
Andi Dian
Permana
b,
Eneko
Larrañeta
a,
Robert
Graham
e,
Christopher J.
Scott
f and
Ryan F.
Donnelly
*a
ab,
Anna
Korelidou
a,
Ana M.
Pérez-Moreno
cg,
Juan L.
Paris
c,
Juan
Dominguez-Robles
d,
Lalitkumar K.
Vora
a,
Andi Dian
Permana
b,
Eneko
Larrañeta
a,
Robert
Graham
e,
Christopher J.
Scott
f and
Ryan F.
Donnelly
*a
aSchool of Pharmacy, Queen's University Belfast, Belfast BT9 7AF, UK. E-mail: r.donnelly@qub.ac.id
bDepartment of Pharmaceutical Science and Technology, Faculty of Pharmacy, Universitas Hasanuddin, Makassar 90245, Indonesia
cAllergy Research Group, Instituto de Investigación Biomédica de Málaga y Plataforma en Nanomedicina-IBIMA Plataforma BIONAND, Málaga 29590, Spain
dDepartment of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, University of Seville, Seville 41004, Spain
eSchool of Biological Science, Queen's University Belfast, Belfast BT9 7AF, UK
fPatrick G Johnson Centre for Cancer Research, Queen's University Belfast, Belfast BT97BL, UK
gUniversidad de Málaga, Málaga 29071, Spain
First published on 6th January 2025
Abstract
This study explores the formulation and characterization of poly(vinyl alcohol) (PVA)-based composite hydrogels synthesized through solid-state crosslinking. Comprehensive assessments were conducted on their physicochemical properties, leachables, and immunogenicity. Swelling experiments demonstrated that the incorporation of poly(vinylpyrrolidone) (PVP) enhanced water retention, while chitosan had a minimal effect on swelling behavior. Qualitative analysis of leachables identified water-soluble components, including dehydrated PVA and PVP. Fourier-transform infrared (FTIR) spectroscopy confirmed the formation of ester bonds and indicated increased hydrogen bonding post-crosslinking. Thermal stability was validated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), with decomposition observed at 320–330 °C. X-ray diffraction (XRD) analysis revealed enhanced crystallinity following crosslinking. Solid-state nuclear magnetic resonance (NMR) further confirmed chemical changes consistent with the results from other characterization techniques. In vitro assays using DC2.4 mouse dendritic cells showed that hydrogel extracts inhibited cell proliferation without causing cytotoxicity or triggering significant immune responses. These findings highlight the hydrogels’ biocompatibility and stability, supporting their potential for biomedical applications.
Introduction
Hydrogels are three-dimensional crosslinked networks of insoluble polymers that can absorb and retain water within their structure.1 Due to their high water content, hydrogels provide a medium for the diffusion of water-soluble substances.2 When a hydrogel dries, it transforms into a xerogel, which can swell again upon rehydration.3 A dry hydrogel can also be synthesized through solid-state chemistry, where crosslinking occurs after the polymeric blend has been dehydrated. This type of material is classified as a hydrogel-forming polymer since it swells into a hydrogel when exposed to water.4 Hydrogels have been used extensively in the biomedical and pharmaceutical fields as wound dressing,5 scaffolds for tissue engineering,6 sensors7 and drug delivery.8 Typically, hydrogels are fabricated from biocompatible polymers such as poly(vinyl alcohol) (PVA), poly(vinylpyrrolidone) (PVP) and chitosan.9,10 The polymers can be combined into one composite hydrogel to achieve specific preferred physical, chemical and biological characteristics.11–13 Extra caution is taken when formulating hydrogels that are intended for biological applications, especially if they are meant to make direct contact with biological tissue.14–16 PVA was selected as the primary hydrogel-forming polymer in this study due to its widespread use in pharmaceutical and biomedical applications in various forms such as microneedles,17 injectable hydrogels,18 oral hydrogels,19 and implantable hydrogels.20Every design of hydrogel-based medical devices and therapeutics faces a significant challenge due to the potential leachables.21–24 Hydrogels that are produced by crosslinking polymers using solid-state chemistry, which involves the formation of new chemical bonds via polymerization or macromolecule side group reactions, tend to yield incomplete reactions.25–29 For example, chemically crosslinked hydrogels have been shown to produce hydrogels with a gel fraction less than 100%. This indicates that the hydrogel contains unreacted monomers, polymers, or crosslinkers that are soluble and leach when the hydrogel swells.1,2,25,29–31
Although the majority of hydrogels are made from biocompatible materials, many of the other components, such as crosslinkers or photoionizers, are not.32,33 The use of hydrogels in applications such as implants and microneedles, where they are in direct contact with unprotected living tissue, theoretically, leads to the deposition and accumulation of harmful leachables in the body.34,35 The safety profiles of biocompatible polymers have been extensively researched and reported. Numerous studies have shown that these materials are safe for use in a variety of pharmaceutical preparations.12,36,37 For instance, low molecular weight PVA has been observed to be eliminated from the skin within six days following its topical application as microneedles and has shown no significant toxicity in healthy mice, even with daily insertions over a 160-day period.38 However, there have been several reports of people experiencing anaphylactic shock after ingesting or applying these polymers.39–43 The most common allergic reaction to PVP can occur through oral ingestion or vaginal or dermal application.39–42 PVP is known to trigger type I allergies.39 Allergic reactions to chitosan are rare, but it has been reported that chitosan can boost dendritic cell maturation.43 In addition, although PVA is regarded as nonhemolytic, it can cause minor irritation to the surrounding tissues.44
Dendritic cells (DCs) are in charge of initiating all antigen-specific immune responses. DCs are antigen-presenting cells (APCs) that are responsible for presenting antigens to native T cells.45,46 DCs are crucial in the initiation of allergic reactions.47 Following an allergen encounter, DCs become activated, mature, and differentiate into immunostimulatory DCs capable of effectively presenting antigens to T cells.45 It is known that exposing DCs to biomaterials can modulate DC responses. Depending on the material's properties, this exposure can either accelerate DC maturation or promote DC tolerance.48 The modulation of the DC response toward materials is influenced by their surface chemistry, surface hydrophilicity, surface topography, spatial structure, surface roughness, and surface charge.48
Herein, we report, for the first time, the formulation and characterisation of leachable-free PVA-based composite hydrogels synthesised through solid-state crosslinking reaction. The leachables from the hydrogels were identified and their toxicity toward dendritic cell lines was studied. We also report the response of the dendritic cells after being exposed to hydrogel extracts. DC maturation and activation markers were also analysed.
Materials and methods
Materials
Chemicals and materials used in this study were PVA (87–89% hydrolysed, 85–124 kDa; Sigma Aldrich, Dorset, UK), PVP K-29/32 (Ashland, Kidderminster, UK), chitosan (low molecular weight; Sigma Aldrich, Dorset, UK), OA (Sigma Aldrich, Dorset, UK), RPMI-1640 (Sigma-Aldrich, Madrid, Spain), ELISA Flex Mouse TNF-α kit (Mabtech AB, Nacka Strand, Sweden), FITC anti-mouse CD40 (BioLegend, San Diego, California, USA), APC anti-mouse CD80 (BioLegend, San Diego, California, USA), Zombie Aqua™ fixable viability kit (BioLegend, San Diego, California, USA), PBS tablet (pH 7.4, 10 mM; Sigma-Aldrich, Madrid, Spain), WST-1 reagent (Sigma-Aldrich, Madrid, Spain), Gibco Trypsin EDTA (Sigma-Aldrich, Madrid, Spain), 96-well polystyrene F bottom clear ELISA high binding plate (Merck Life Science S.L.U., Madrid, Spain), and 96-well cell culture plate (Cole-Parmer Instrument Co., Ltd, Eaton Socon, UK). The DC2.4 mouse dendritic cell line was purchased from Merck (Merck Life Science S.L.U., Madrid, Spain). Purified water was obtained from an ELGA Purelab Flex 2® ultrapure water purification system (Vivendi Water System Ltd, Bucks, UK), and Milli-Q water was obtained using Millipore Q-POD® (Merck Life Science S.L.U., Madrid, Spain) equipped with a Biopak® polisher (Merck Life Science S.L.U., Madrid, Spain).Hydrogel synthesis
The solvent casting process was employed to manufacture hydrogel films.17 The composition of each formulation is provided in Table 1 and the preparation of the polymeric stock solution is presented in the ESI.† Initially, the polymeric aqueous solutions were weighed and mixed thoroughly in a Falcon® tube. Next, a volume of 250 μL of the mixture was accurately transferred into a silicone mould with dimensions of 12 × 12 × 3.2 mm using a positive displacement pipette. Following centrifugation at 5000 rpm for 15 minutes, the mixture was left to dry at room temperature for the next 48 hours. The dried films were gathered, punched into circular shapes using a paper puncher (∼8 mm in diameter), and thereafter subjected to crosslinking in a preheated oven at a temperature of 130 °C for a duration of one hour.| Composition | Amount (wt%) | ||||
|---|---|---|---|---|---|
| P | PP | PPChi0.125 | PPChi0.25 | PPChi0.625 | |
| PVA 85-124k | 15 | 15 | 15 | 15 | 15 |
| PVP K29/32 | — | 10 | 10 | 10 | 10 |
| Chitosan | — | — | 0.125 | 0.25 | 0.625 |
| OA | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 |
| Acetic acid | — | — | 0.2375 | 0.475 | 1.1875 |
| Purified water | 84.3 | 74.3 | 73.9375 | 73.575 | 72.4875 |
In this paper, uncrosslinked polymeric film formulations are denoted by the prefix ‘u’ (e.g., uP, uPP, uPPChi), crosslinked formulations by ‘c’ (e.g., cP, cPP, cPPChi), and washed or leachable-free formulations by ‘w’ (e.g., wP, wPP, wPPChi).
Swelling experiments
 | (1) |
 | (2) |
 | (3) |
Qualitative leachable experiments
Physicochemical characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μs and 5744 scans, with an overall experimental time of 8 h was employed. Prior to the NMR analysis, the hydrogel films were frozen and pulverized in a mill (Retsch ZM 200, Haan, Germany) using liquid nitrogen and a 0.5 mm output sieve.
μs and 5744 scans, with an overall experimental time of 8 h was employed. Prior to the NMR analysis, the hydrogel films were frozen and pulverized in a mill (Retsch ZM 200, Haan, Germany) using liquid nitrogen and a 0.5 mm output sieve.
In vitro immunogenicity assay
000 cells per well). Two separate plates were prepared. The plates were thereafter placed in a humidified incubator at a temperature of 37 °C, with a CO2 concentration of 5%, overnight. Subsequently, the plate was transferred to a sterile hood. Following the removal of the media by aspiration, 100 μL of pre-warmed media was introduced into each well. Subsequently, a volume of 100 μL of the treatment solution was introduced into the specified wells, following the schematic diagram outlined in the ESI.† The procedure for preparing the treatment solution is also included in the ESI.† The plate was thereafter placed in a humidified incubator with 5% CO2 at a temperature of 37 °C for a duration of 24 hours. Cell viability was evaluated using WST-1 labelling and flow cytometry. The WST-1 staining procedure was carried out in accordance with the methodology provided by the manufacturer. The live and dead cell analysis in flow cytometry was performed by labelling the cells with the Zombie Aqua™ dye, following the instructions provided by the manufacturer.
Statistical analysis
Statistical analyses were performed using GraphPad Prism® software version 10.30.1 for Windows 64-bit (GraphPad Software, Boston, MA, USA). One-way ANOVA with Tukey's multiple comparison test was used for analysis. Normality was assessed prior to testing using the Shapiro-Wilk test. P-Values are reported to indicate statistical significance.Results and discussion
Swelling experiments
All the obtained hydrogels were subjected to swelling kinetic studies in purified water and PBS pH 7.4. The results are shown in Fig. 1(A). The results indicate that the cPP formulation exhibited the highest swelling percentage at t = 48 h, with 448% ± 42.3% in water and 324% ± 6% in PBS pH 7.4. This was followed by the chitosan-containing hydrogels: cPPChi0.125 (390% ± 13% in water and 308% ± 9% in PBS pH 7.4), cPPChi0.25 (394% ± 7% in water and 318% ± 8% in PBS pH 7.4), and cPPChi0.625 (385% ± 8% in water and 317% ± 3% in PBS pH 7.4). The cP formulation showed the lowest swelling, with 175% ± 6% in water and 246% ± 20% in PBS pH 7.4. Compared to the cP formulation, all differences in swelling percentages were statistically significant, with p < 0.0001. The addition of PVP significantly increased the swelling percentage of the PVA-based hydrogel after 48 hours (p < 0.0001), possibly through the formation of semi-IPN networks. PVP did not participate in chemical crosslinking in these hydrogel formulations; rather, the polymer network of PVP was entangled with the PVA network, resulting in a relatively loose network, which in turn increased the capacity of the hydrogel to hold more water.49 Similar behaviour was observed in alginate–gelatine hydrogels, where the alginate–gelatine semi-IPN formulation had a twofold greater degree of swelling than the crosslinked alginate hydrogel.49 The calculations of %EWC and %GF presented in Fig. 1(B) were performed based on the data from the swelling study in water. It was clear that the addition of PVP significantly reduced %GF (p = 0.0007) but increased %EWC (p < 0.0001) due to the formation of semi-IPNs.49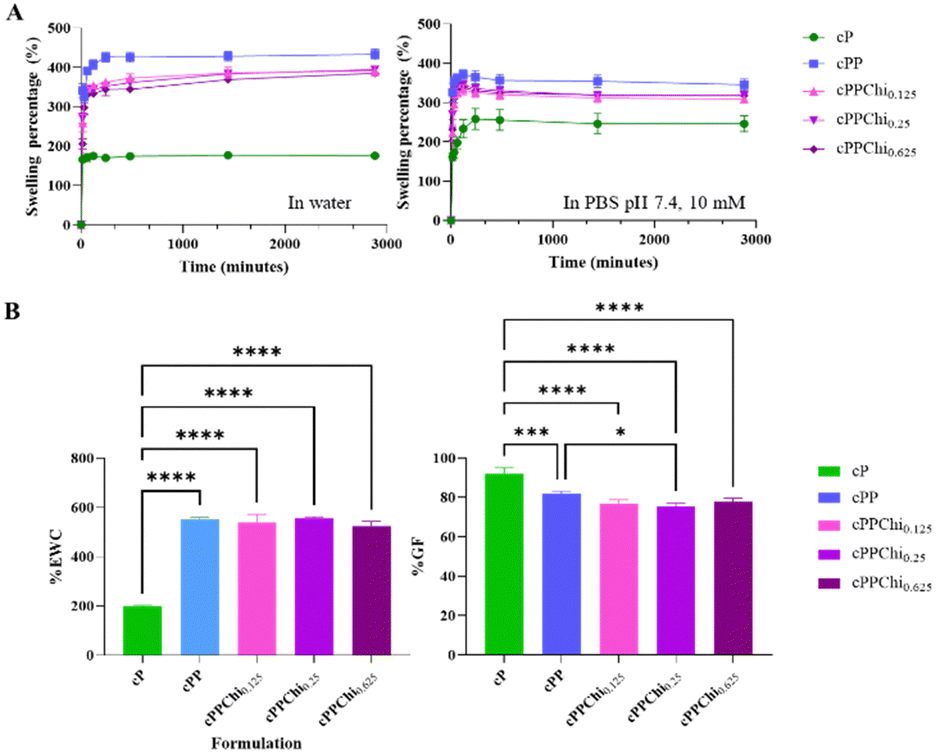 | ||
| Fig. 1 (A) Plots of swelling percentage over time of all five formulations in water and PBS pH 7.4 (means ± SD, n = 3). (B) Comparison of the gel fraction and equilibrium water content for all the formulations (means + SD, n = 3). | ||
The inclusion of chitosan in cPP decreased the swelling percentage of PVA–PVP hydrogels after 48 hours (p = 0.0040, p = 0.0063, and p = 0.0014 for comparisons between cPP and cPPChi0.125, between cPP and cPPChi0.25 and between cPP and cPPChi0.625, respectively). However, the effect was miniscule on the %GF of the chitosan-containing formulation, where the difference was not statistically significant except for the difference in %GF between the cPP and cPPChi0.25 groups (p = 0.0178). The effect of chitosan addition to the hydrogel on %EWC was not statistically significant (p > 0.05). In addition, the concentration of chitosan did not significantly affect the swelling percentage or %GF (p > 0.05). Similar results were obtained when the swelling study was performed using PBS pH 7.4 as the medium, although the difference in swelling percentage between cPP with cPPChi0.25 and cPP with cPPChi0.625 was not statistically significant (p = 0.1331 and p = 0.1208 for cPPChi0.25 and cPPChi0.625, respectively).
A comparison of the swelling study results in water and PBS pH 7.4 revealed that the swelling percentages of cPP, cPPChi0.125, cPPChi0.25 and cPPChi0.625 in water were greater than those in PBS pH 7.4 (p < 0.05). This was due to the salt screening effect that was exerted by the ions in PBS pH 7.4 on the polar/ionisable groups in the hydrogel. Similar results have been reported previously.50,51 On the other hand, the percentage of swollen cP in PBS pH 7.4 was greater than that in the water counterpart (p = 0.0044). A similar phenomenon was observed for ethylene glycol diglycidyl ether (EDGE)-crosslinked PVA-microcrystalline cellulose.52 It is possible that the pH of the medium played a greater role than the dissolved ions. For example, the same swelling behaviour can be observed in a boric acid-crosslinked PVA-chitosan hydrogel, where the swelling ratio in PBS at pH 7.4 was greater than that in PBS at pH 4.0.53,54 Considering that there was a negligible effect on %EWC and %GF after the incorporation of various concentrations of chitosan into the cPP formulation, only the formulation with the highest concentration of chitosan (cPPChi0.625) was characterised for the remaining studies.
Qualitative leachable analysis
Water soluble leachables were evaluated by a simple water extraction step and the results are presented in Fig. 2. The PVP spectra presented in the graph do show a distinct peak but instead a large “mountain” toward the end of the UV spectrum. This profile is consistent with what has been found in the literature.55,56 This band could be assigned to the π → π* transition of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl and tertiary alicyclic amine from the pyrrolidone ring of PVP. Like those of PVA and PVP, the polymeric chains of chitosan bear far-UV chromophore groups, for which the maximum absorbance wavelength is 201 nm, in dilute hydrochloric acid.57 At higher concentrations, both polymers have visible “shoulder” signals in the spectra, but no useful peak can be observed.
O carbonyl and tertiary alicyclic amine from the pyrrolidone ring of PVP. Like those of PVA and PVP, the polymeric chains of chitosan bear far-UV chromophore groups, for which the maximum absorbance wavelength is 201 nm, in dilute hydrochloric acid.57 At higher concentrations, both polymers have visible “shoulder” signals in the spectra, but no useful peak can be observed.
 | ||
| Fig. 2 (A) The evolution of the gel fraction and equilibrium water content of the hydrogels after each washing cycle (means ± SD, n = 3). (B) The appearance of uncrosslinked hydrogels (uP, uPP, uPPChi0.625), crosslinked hydrogels (cP, cPP, cPPChi0.625), and washed hydrogels (wP, wPP, wPPChi0.625). | ||
The absorption spectra of the extraction solution clearly show the difference between crosslinked and washed hydrogels. In the cP hydrogel, only a small absorption shoulder can be observed. The gel fraction calculation indicated that the soluble part of the hydrogel was less than 10%, in addition to the low molar absorptivity of the polymeric solution yielded in that spectrum profile. The wP counterpart of the formulation, however, did not show any peak in the scanning range. This characteristic was also observed for the cPP and cPPChi0.625 hydrogels. Both formulations had gel fractions of approximately 75–80%, which means that >20% of their mass was soluble. Judging from the intensity of the shoulder of the spectrum, some of the PVP constituents were leached to the extraction media. In all crosslinked formulation cases, leachables were detected even after one minute of extraction and became more intense as the extraction time increased. For the washed hydrogels, no characteristic shoulder was observed at any of the time points. The results indicated that the possible leachables were removed during the washing process. This finding is consistent with the gel fraction calculation results discussed before (Fig. 3).
 | ||
| Fig. 3 (top row) Absorbance of PVA, PVP and Chi from 200 to 800 nm. (middle row) The absorbance spectra of crosslinked hydrogel extracts showing the presence of potential leachables in the solvent at different extraction times for up to 30 minutes. (bottom row) The absorption spectra of washed hydrogel extracts showing the absence of potential leachables in the solvent at different extraction times, up to 30 minutes. | ||
The leachables from each crosslinked hydrogel were then isolated by drying through the lyophilization process, followed by vacuum evaporation to remove the remaining water. Less mass was isolated from the cP hydrogel than from the cPP and cPPChi0.625 formulations. This corresponded to the high gel fraction of the cP formulation; hence, less material was leached from the hydrogels. These isolated substances were dissolved in DMSO-d6 and scanned using a 400 MHz 1H-NMR spectrometer. For comparison, pristine and heat-treated water soluble polymers were also scanned. The spectrograms of all the materials are given in Fig. 4.
 | ||
| Fig. 4 1H-NMR spectra of pristine and heat-treated PVA, pristine and heat-treated PVP, as well as cP, cPP and the cPPChi0.625 water extracts. | ||
The 1H-NMR spectra of pristine and heat-treated PVA were identical, except for the lower intensity of the water peak at ∼3.4 ppm due to water removal after heating.58,59 The NMR spectra of both polymers showed the characteristics of partially hydrolysed PVA. The methine (Cα) hydrogens can be found at ∼5.1–4.8 ppm due to the deshielding effect of the neighbouring oxygen. The presence of hydroxyl hydrogens could be found in the range of ∼4.2–4.8 ppm.58,59 There were several peaks assigned to the hydroxyl groups, which indicated different arrangements of the –OH groups in the polymer backbone.58,59 Similarly, the position of the methylene (Cβ) hydrogen atom varied depending on the adjacent group, acetyl (X) or hydroxyl (Y) group, and these peaks were observed between 1.8 and 1.3 ppm. The hydrogen from the acetyl CH3 group was assigned to the broad peak at ∼1.9 ppm.58,59 The other peak assignments for Hδ, Hη, Hε, Hζ and Hγ were 3.8–3.4, 3.3–2.9, 2.2–2.0, 1.8–1.7 and 1.7–1.4 ppm, respectively. This order was ascribed to the proximity of neighbouring electronegative atoms (nitrogen and oxygen).60,61
Due to the decreased amount of the isolated polymer (higher %GF), the signal of the cP extract was weak, and low-intensity peaks were not visible. However, the peaks that corresponded to Hγ (YY), CH3 and Cα protons were still detectable in the spectrogram. The spectra of the cPP and cPPChi0.625 extracts were stronger than those of the other samples, indicating that the former contained a mixture of PVA and PVP polymers. All five peaks of PVP were evident. The Hζ peak from PVP overlapped with the CH3 peak from PVA, but each could still be identified clearly. However, Hβ (XX) and Hβ (XY) could not be identified because they overlapped with the Hγ peak in the spectrum of the PVA molecule. In addition, these peaks also had weaker intensities. Interestingly, all the hydroxyl peaks of PVA cannot be identified from the spectrogram. This result indicated that the majority of the hydroxyl groups in the extracted polymer were removed, which most likely happened through dehydration in acidic environments.62,63 These findings corroborated the hypothesis of the formation of alkene bonds in the PVA hydrogel and explained the desaturation of the yellow colour in the washed hydrogel (compared to that in the crosslinked formulations), as the presence of alkenes bearing PVA is one of the contributing factors to the observed hydrogel discolouration.62,63
Physicochemical characterisation
The complete IR spectra from wavenumbers 4000 cm−1 to 750 cm−1 of the hydrogels and their individual constituents are given in the ESI.† The list of peaks of interest from all constituent materials is also given in the ESI.†. Fig. 5 depicts the breakdowns of some peaks of interest between 1800 and 1500 cm−1, and 4000 and 3000 cm−1, the PVA crystalline peak (∼1140 cm−1) and the chitosan skeletal C–O peak (∼665 cm−1). | ||
| Fig. 5 (A) Overlaid CO peaks of the polymers and crosslinker between wavenumbers 1800 and 1500 cm−1. (B) Overlaid O–H and N–H peaks of the polymers and crosslinker between wavenumbers 4000 and 3000 cm−1. (C) ATR-FTIR spectra of formulations between wavenumbers 1800 and 1500 cm−1. (D) ATR-FTIR spectra of formulations between wavenumbers 4000 and 3000 cm−1. (E) Crystalline peaks (∼1140 cm−1) of PVA, crosslinked, and washed hydrogels. (F) Characteristic C–O chitosan skeletal vibration peak (∼665 cm−1) of chitosan, wP, wPP, and chitosan-containing formulations. | ||
For the formulation containing only PVA and OA, the spectra were very similar to that of neat PVA. All the characteristic peaks of PVA can be observed in the spectra of the formulations. The ratio of saturated to unsaturated CO band intensity exhibited the most noticeable difference. The peak corresponding to saturated CO in neat PVA had a greater intensity than that in the rest of the PVA formulations (uP, cP and wP). When comparing the spectra of the uncrosslinked and crosslinked formulations, it was clear that the saturated CO peak intensity was greater before heating. The thermal dehydration of PVA contributed to this phenomenon. When PVA is heated, CC bonds are expected to form, particularly near the CO group.64,65 Further analysis revealed no differences between these features in the cP and wP hydrogels, confirming that no hydrolysis occurred after one cycle of washing, which agrees with the 100% gel fraction calculation reported in the previous section.
A comparison of the O–H vibrational bands revealed that the corresponding peak was shifted to a lower wavenumber, indicating that hydrogen bond formation increased after casting (νO–H PVA > νO–H uP). Upon heating, the wavenumber increased and did not change after the hydrogel was washed. The subtle difference between the spectra of cP and wP was the PVA crystalline peak at 1141 cm−1. The intensity of this peak increased after crosslinking but then decreased after washing. This trend was also observed for the cPP–wPP and cPPChi0.625–wPPChi0.625 formulations. The disappearance of the peak was an indication of the loss of crystallinity after the swelling–deswelling process.66,67
One unique difference between the uP, cP and wP spectra was the increased intensity of the shoulder peak at 2850 cm−1. In some reports, this peak was assigned to the C–H stretching vibration.68–70 Some other studies mentioned that this band was attributed to C–H stretching related to aldehydes and was often an indicator of successful crosslinking between PVA and glutaraldehyde.66,71,72 As the reaction between carboxylic acid (OA) and alcohol (PVA) will form ester bridges and not acetal bridges, the peak at 2850 cm−1 could be assigned to C–H stretching of the CH group. The increased intensity in cP and wP implies the formation of stable CC bonds after crosslinking reactions.73
In the PVA–PVP–OA formulation, the formation of new unsaturated ester bonds was also observed, which was less obvious than that in the PVA-containing formulations. The presence of PVP was confirmed by the intense amide I band at 1651 cm−1 in all the PVA–PVP–OA formulations. Like the νO–H peak in the spectrum of the PVA-containing formulations, the νO–H peak in the spectrum of the PVA–PVP composite formulations also shifted to a higher wavelength after the crosslinking reaction but remained the same after the swelling–deswelling step. A comparable trend was observed for the PVA–PVP–Chi–OA composite hydrogel, where the formation of new unsaturated CO esters was confirmed along with the shift in the νO–H band after crosslinking. The only noticeable chitosan peak that can be found across all chitosan-containing formulations was a weak band at 665 cm−1, which is a characteristic peak of the saccharide structure of chitosan.74
All the formulations and starting materials were subjected to DSC analysis, and the results are shown in Fig. 6. The PVA DSC thermogram revealed several thermal events between 25 and 400 °C. The first was the glass transition temperature (Tg), indicated by the shift in the baseline which was at 92.9 °C. The first broad endothermic peak at 191.0 °C can be assigned to the melting of PVA. The second endothermic peak at 324.9 °C was attributed to the decomposition of PVA. The first very broad peaks at 98.9 °C for PVP and 88.1 °C for chitosan were attributed to the loss of moisture during the heating process. A valley was formed at >350 °C in the PVP thermogram indicating the onset of decomposition. According to the literature, the PVP decomposition peak appears at ∼420 °C in the DSC thermogram.75 For chitosan, an exothermic decomposition peak can be observed at 302.9 °C. The DSC thermogram of OA showed a characteristic thermal profile for a crystalline material. The first peak that appeared at 86.2° can be assigned to the removal of water molecules.76 The sharp endothermic peak at 193.2 °C could be attributed to a melting event followed by decomposition. This was confirmed by the 100% weight loss in the TGA thermogram. OA decomposes to form water, carbon monoxide and carbon dioxide molecules, leaving no char at the end of the reaction.76
 | ||
| Fig. 6 DSC thermograms of the polymers, crosslinker, and formulations. | ||
In the PVA–OA formulation, a peak at approximately 165–170 °C was observed. This peak corresponded to the melting/relaxation point of the formulation. The intensity of this peak increased after crosslinking. This can be attributed to the change in the crystallinity of the material.77,78 Further examination revealed that all the formulated hydrogels had very similar thermograms. The first broad peak could be attributed to the removal of bound water from the formulations, which occurred between 90 and 100 °C for the PVA–OA formulation, 103 and 120 °C for the PVA–PVP–OA formulation, and 103 and 115 °C for the PVA–PVP–Chi–OA formulation. The peak that appeared at ∼320–330 °C in all formulations was assigned to decomposition. The thermogram shows that the energy required to remove water from the material depends on the composition and the state of the hydrogel.
All the formulations and starting materials were subjected to DSC analysis, and the results are shown in Fig. 7. From the graph, it can be seen that OA decomposes completely at around 180 °C while PVA and chitosan decompose at around 250 and 220 °C, respectively, leaving 40% char at the end of the analysis (400 °C). On the other hand, PVP did not decompose within the scanning range. For all starting materials, the first step of weight loss (<100 °C) corresponded to the removal of moisture or bound water and the second weight loss (not observed in PVP) corresponded to the decomposition. According to the literature, the PVP decomposition peak appears at ∼420 °C in the DSC thermogram.75 All other formulations showed similar steps in weight loss.
 | ||
| Fig. 7 TGA thermograms of the polymers, crosslinker, and formulations. | ||
Several peaks corresponding to both monoclinic and orthorhombic PVA were observed in the diffractogram depicted in Fig. 8. The strong broad peak at 19.7° corresponded to the orthorhombic lattice at the (2 0 2) plane. The other three halo peaks at 11.6°, 22.9° and 40.7° can be attributed to the monoclinic lattice in the (2 0 0), (0 0 2), and (3 1 ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ) directions, respectively.79,80 However, all these peaks were very broad, indicating that the pristine PVA polymer used in this study had low crystallinity.17 The PVP diffractogram showed typical halo peaks at approximately 11.3 and 21.8° 2θ, corresponding to the amorphous form of the PVP polymer.81 The diffractogram of pure chitosan showed broad peaks at 10.2° and 20.2°, which can be assigned to the (0 2 0) and (1 1 0) planes of non-deacetylated chitin, respectively. In chitosan, these peaks become broader and less intense due to the disruption of intermolecular hydrogen bonds after the removal of acetyl groups from the polymer backbone.82,83 The diffractogram of OAs indicated that the type of OA used in this work was the α form of the anhydrous crystal.84 This form was characterized by strong major peaks at 22.9°, 31.0° and 37.4° 2θ corresponding to the (1 1 1), (2 0 1) and (0 2 2) planes of the orthorhombic lattice of the material (data retrieved from the Open Crystallography Database using Difrac.Eva® software).84
) directions, respectively.79,80 However, all these peaks were very broad, indicating that the pristine PVA polymer used in this study had low crystallinity.17 The PVP diffractogram showed typical halo peaks at approximately 11.3 and 21.8° 2θ, corresponding to the amorphous form of the PVP polymer.81 The diffractogram of pure chitosan showed broad peaks at 10.2° and 20.2°, which can be assigned to the (0 2 0) and (1 1 0) planes of non-deacetylated chitin, respectively. In chitosan, these peaks become broader and less intense due to the disruption of intermolecular hydrogen bonds after the removal of acetyl groups from the polymer backbone.82,83 The diffractogram of OAs indicated that the type of OA used in this work was the α form of the anhydrous crystal.84 This form was characterized by strong major peaks at 22.9°, 31.0° and 37.4° 2θ corresponding to the (1 1 1), (2 0 1) and (0 2 2) planes of the orthorhombic lattice of the material (data retrieved from the Open Crystallography Database using Difrac.Eva® software).84
 | ||
| Fig. 8 X-ray diffractograms of polymers, crosslinkers, and all hydrogel formulations. | ||
All hydrogel film formulations showed similar diffraction patterns. All the samples exhibited a characteristic halo peak at 19.5°, which can be assigned to the (2 0 2) plane of orthorhombic PVA. The results showed that all crosslinked formulations (cP, cPP, and cPPChi0.625) had sharper peaks at this diffraction angle, possibly indicating a greater degree of crystallinity. This property could be attributed to the removal of residual water during the crosslinking process and the abundance of energy introduced by the heating process.85–89 Another feature that needs to be highlighted is the disappearance of the peaks at 14.8° and 30.3° in the formulation after crosslinking. These peaks could be attributed to the (2 0 0) and (−1 1 1) planes of the OA dihydrate form (not shown in the graph).84,90 After the polymeric solution dried, OA could have recrystallised. According to the thermal analysis results, the melting and decomposition of OA did not occur at the temperature used for the crosslinking process, so the peak disappearance could not be attributed to either melting or decomposition. One possible explanation is that OA was consumed by the crosslinking reaction, so no further OA peaks were detected after the crosslinking process. The disappearance of the crosslinker peak in the formulation after crosslinking has also been reported for suberic acid-crosslinked PVA hydrogels.91
Fig. 9 displays the methylene signal at 45 ppm and methine carbon resonances at approximately 70 ppm (PVA-II) and 65 ppm (PVA-III), along with a weak signal at 76 ppm (PVA-I). The peaks for PVA-I and PVA-II were assigned to the isotactic triad with two intramolecular hydrogen bonds and the heterotactic triads with one intramolecular hydrogen bond, respectively. The PVA-III peak was attributed to syndiotactic triads without any intervening intramolecular hydrogen bonds.92 The peak at the highest field was linked to the CH3 group of the residual acetate in PVA, while the peak at the highest frequency was associated with the carbonyl carbon.93 The PVP spectrum revealed methylene peaks at 42 ppm, 32 ppm, and 18 ppm. The peaks at 18 ppm and 32 ppm were assigned to carbon atoms nearest to the carbonyl group of PVP, and the peak at the lowest field was attributed to the carbonyl carbon.94
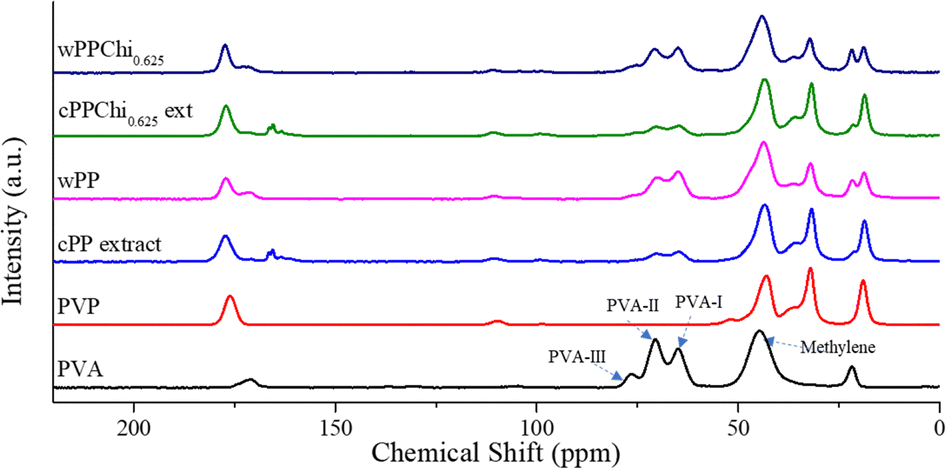 | ||
| Fig. 9 CP-MAS NMR spectra of PVA, PVP, hydrogel extracts and clean hydrogels. | ||
The CH3 peak from PVA and the methylene peak of PVP at 18 ppm are well-resolved in the spectrograms of all hydrogel extracts, whereas in the hydrogels, the CH3 peak appears as a shoulder peak. Similarly, the carbonyl peak of PVA also appears as a shoulder in the hydrogels’ spectrograms. Notably, in the extracts’ spectrograms, weak, split peaks are observed at 166 ppm and 165 ppm, likely due to leached oxalic acid molecules during the hydrogel extraction process.95 The PVA methylene peaks are not clearly visible in either the clean hydrogels or the hydrogel extracts; only the methine peaks for PVA-II and PVA-III are observed in all samples, suggesting the conversion of C–C to CC after the crosslinking process. Additionally, the intensity of the PVA-II and PVA-III peaks is less pronounced in the extracts, indicating that most leachables were unbounded PVP. The ratio between PVA-II and PVA-III also changes, reflecting a shift in the tacticity of the PVA hydrogel backbones. These findings are consistent with the FTIR and liquid H-NMR results previously discussed.
In vitro immunogenicity assay
The number of viable cells in each treatment group was determined using WST-1 staining. The cytotoxicity of the pristine polymers, undiluted hydrogel extracts, and 1:103 and 1:106 dilutions of each extract was tested, and the results are shown in Fig. 10(A). The results indicate that all the pristine polymers did not cause cytotoxicity to DC2.4 cells at a concentration of 1 μg per well. For comparison, a study on the cytotoxicity of PVA and PVP against primary murine DCs reported a modest reduction in cell viability. The viability of these cells was not dose dependent when the concentrations were tested across three orders of magnitude, which were 1 μg per well and 1 ng per well.96 Another study reported that PVA is nearly nontoxic, as reflected by the small reduction in immature human dendritic cell viability measured using the MTT assay.97 There are no reports on the cytotoxicity of chitosan against DCs. However, several studies have reported that chitosan is nontoxic to several other healthy cells, such as the BJ dermis cell line98 and the HT29-MTX-E12 intestinal cell line.12
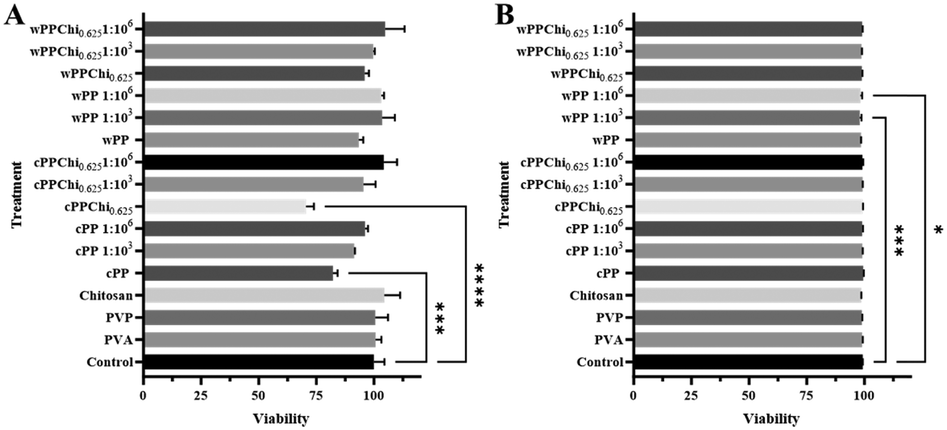 | ||
| Fig. 10 (A) Graphs showing cell viability/proliferation after treatment with polymer solution and hydrogel extracts measured using WST-1 analysis. (B) Graphs showing cell viability after treatment with polymer solution and hydrogel extracts measured using flow cytometry. | ||
Among the formulations, the cPP and cPPChi0.625 hydrogel extracts produced a significantly lower number of viable cells than did the control (p < 0.001). This reduction in viable cells, however, did not occur at higher dilutions. The undiluted cPP and cPPChi0.625 hydrogel extracts exhibit lower pH values than the fresh RPMI-1640 media due to the release of the acidic leachable component. A lower pH is known to inhibit cell growth in cell culture.99 Hence, it is possible that due to the nonideal conditions of the expansion medium, the proliferation of cells in the undiluted cPP and cPPChi0.625 treatment groups was slower than that in the control group, yielding lower cell counts. In general, the hydrogels used in this study can also be considered noncytotoxic toward DC2.4 cells.
Several studies on hydrogels containing PVA, PVP, and chitosan (separately or in combination) have reported the in vitro cytotoxicity of hydrogel extracts to several other cell lines. A study reported that the gelatine/PVA/chitosan hydrogel extract is nontoxic to the HT29-MTX-E12 cell line.100 Other studies reported that PVP/chitosan/glycerol and poly(2-hydroxyethyl acrylate)/itaconic acid/PVP hydrogels were nontoxic toward murine L929 fibroblasts.101,102 PVA/clay hydrogels have also been proven nontoxic to the K562 cell line.103 In addition, a study reported the cytocompatibility of PVP/carboxymethylcellulose hydrogels with MG63 human osteosarcoma cells and murine 929 cells.104
Live and dead cell analysis was also performed using a flow cytometer after cell staining with the Zombie Aqua™ dye, and the results are presented in Fig. 11(B). As shown in the graph, none of the polymers or hydrogels tested were cytotoxic at the test concentration. These results complemented the findings from the WST-1 study and confirmed that the reduction in the cell's absorbance in the undiluted cPP and cPPChi0.625 treatment groups relative to the control was due to inhibition of the cell's proliferation process rather than killing of the cells. These results once again confirmed the cytocompatibility of the material used in the hydrogel formulation, specifically with DCs.
 | ||
| Fig. 11 (A) Normalised single-parameter flow cytometry histogram of CD40+ cell analysis. (B) Graphs showing the percentage of CD40+ cells after treatment with polymer solutions and hydrogel extracts. | ||
The expression of the CD40 protein on the surface of the DCs was measured via flow cytometry. The numbers of CD40+ cells in all the control and treatment groups are depicted in Fig. 11. From the graphs, it is evident that an increase in CD40+ cells was observed in the LPS-treated cells but not in the other treatment groups. A study with primary DCs revealed that PVA and PVP at 1 g per well and 1 ng per well induced a slight increase in the CD40+ cell count. However, compared to that in LPS-treated cells in the study, the increase in these parameters was much lower.96 The increase in CD40 expression was greater at lower polymer doses, which could indicate the occurrence of feedback mechanisms during encounters with high or persistent polymer doses.96 In this study, we observed slight downregulation of surface CD40 expression in the cPP and wPPChi0.625 treatment groups, but this downregulation was irrelevant to the activation and maturation of DCs. CD40 is a surface transmembrane glycoprotein receptor that belongs to the tumour necrosis factor receptor (TNFR) superfamily. CD40 is a biomarker of DC activation and maturation. CD40 signalling induces changes in DCs, which increase the effectiveness of APCs.105 Through the engagement of toll-like receptor 4 (TLR4), LPS induces DC activation. DC activation boosts the expression of costimulatory molecules such as CD40, CD80, and CD86.106 These results indicate that, while stimulation with LPS induced a strong activation of the dendritic cells (marked increase in CD40+ DC2.4 cells), none of the studied experimental groups showed any significant dendritic cell activation.
Flow cytometry was used to assess the expression of CD80 on the surface of the DCs and the results are presented in Fig. 12. The graphs show that the basal expression of CD80 is high in this cell line. However, an increase in CD80 expression was still observed in LPS-treated cells. Similarly, undiluted cPP and cPPChi0.625 extract-treated cells exhibited a detectable increase in the transmembrane protein. This phenomenon could be attributed to the slightly acidic expansion medium pH in those treatment groups. CD80 expression on the surface of DCs indicates cell maturity.107 It was reported that exposing DCs to acidic media could trigger their maturation.108,109 In addition, modestly downregulated expression of CD80 was found in the chitosan treatment groups as well as in the high dilutions (106-fold) of the hydrogel treatment groups, except for the cPP hydrogel. A slight decrease in CD80 expression is irrelevant to cell maturation, and no explanation can be provided for this phenomenon. CD80 is a maturation biomarker for DCs. Along with CD86, CD80 is a shared ligand for CD28 and CD152.110 Mature dendritic cells express high levels of the costimulatory molecules CD80 and CD86, which provide the signal needed for triggering T-cell activation, expansion, and differentiation via interaction with CD28.111 Strong immunogenic molecules such as LPS activate DCs and increase the expression of common costimulatory proteins such as CD40, CD80 and CD86.106
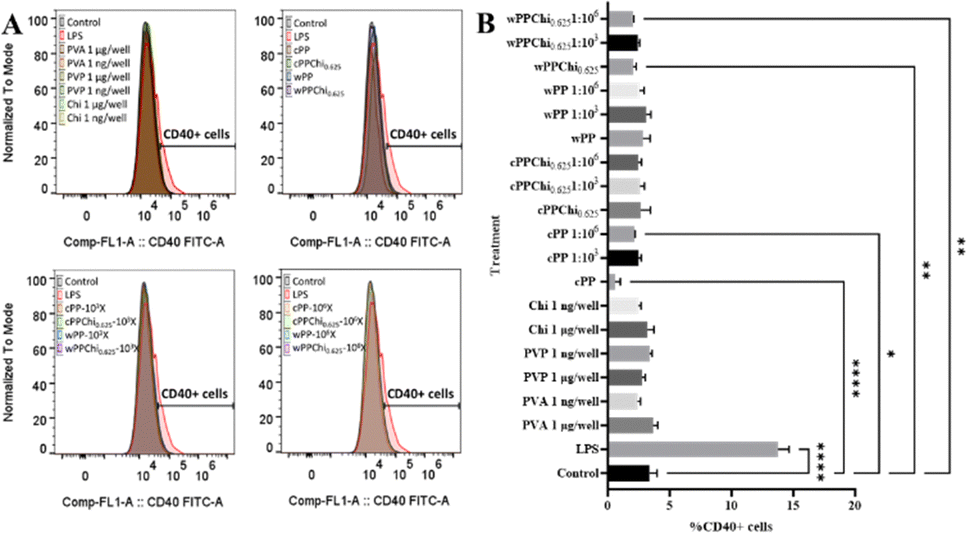 | ||
| Fig. 12 (A) Normalised single-parameter flow cytometry histogram of CD80+ cell analysis. (B) Graphs showing the percentage of CD80+ cells after treatment with polymer solutions and hydrogel extracts. | ||
The quantification of TNF-α secretion by DCs was accomplished through ELISA analysis of the cytokine concentration in the expansion medium subsequent to incubation. The TNF-α concentrations in each of the treatment and control groups are presented in Fig. 13. A statistical analysis was conducted to identify differences in the secretion of TNF-α compared to that in the control group. The graphs provide clear evidence of an increase in TNF-α secretion in the group that was treated with LPS. Furthermore, cells treated with the cPPChi0.625 extract also exhibited a slight increase in TNF-α production. In contrast, there were no notable differences in TNF-α secretion among the remaining groups.
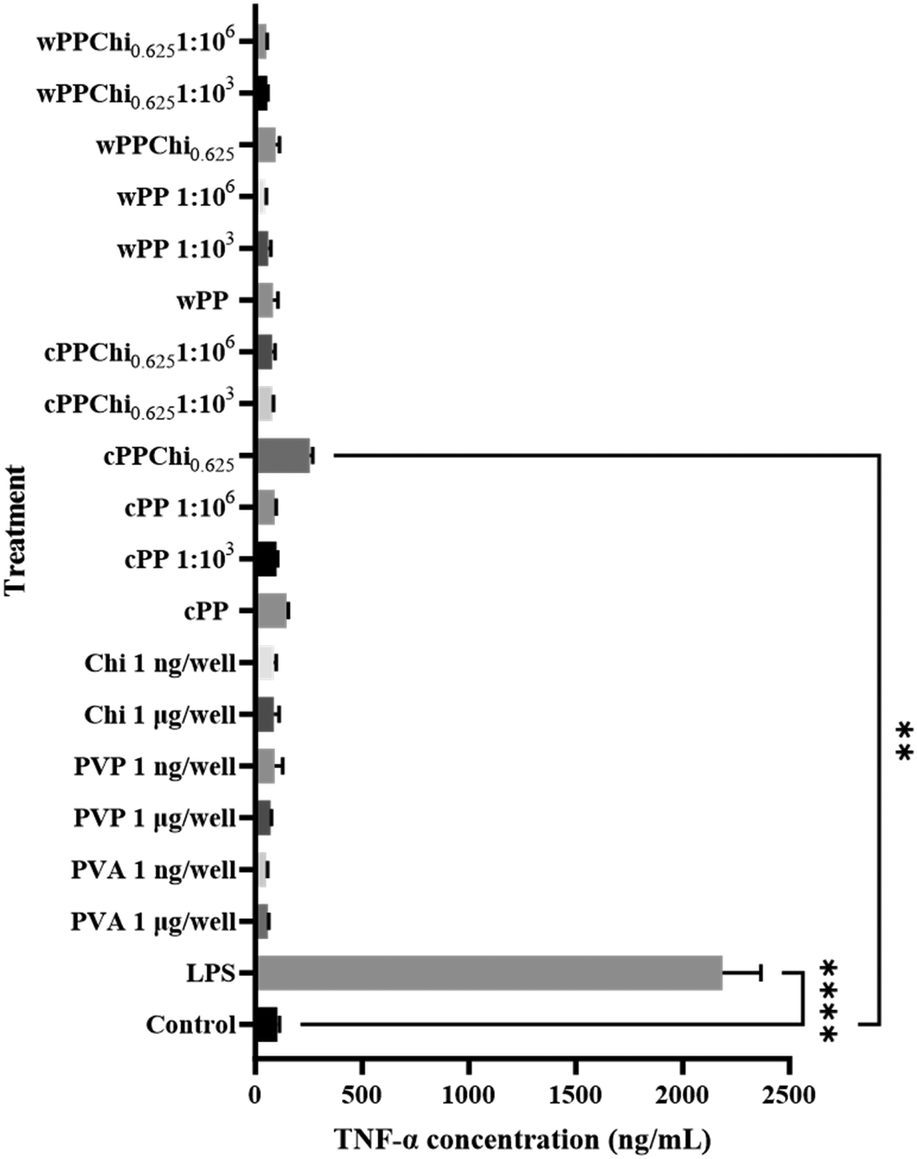 | ||
| Fig. 13 Graphs indicating the level of TNF-α in the complete culture media after cells’ treatment with polymer solutions and hydrogel extracts. | ||
TNF-α, an important pleiotropic cytokine, has been linked to the development of numerous inflammatory disorders, including allergies.112 TNF-α production can be induced in DCs through stimulation with LPS.113 The burst release of TNF-α by DCs signifies the activation of the cells via the TLR pathway.114 In untreated cells, the basal concentration of TNF-α was 103.4 ± 9.4 pg mL−1 after 24 hours of incubation. It has been mentioned that immature murine DCs exhibit increased amounts of TNF-α.115 The modest increase in TNF-α production in the cPPChi0.625 hydrogel extract (254.6 ± 13.6 pg mL−1) could be linked to cell maturation due to exposure to an acidic environment, as discussed in the previous section. This is evident as a similar increase (although not statistically significant) was observed in the cPP group (147.1 ± 8.3 pg mL−1). The results obtained in this study indicated that the hydrogel extracts and the pristine polymers did not activate DCs through the TLR pathway.
Conclusions
This study details the properties of clean PVA-based composite hydrogels produced using the solid-state crosslinking process. The hydrogels maintain their properties during the swelling–deswelling process, with little alterations in thermal properties and crystallinity. The ATR-FTIR analysis indicated that the final product contains a higher number of CC bonds, which can be attributed to the thermal dehydration of the PVA chain during the crosslinking process. The leachables obtained from the swelling and deswelling process were identified as a combination of PVA and PVP. The effect of the crosslinked and washed hydrogel extracts was evaluated in DC2.4 cells. Although the hydrogel extracts of both cPP and cPPChi0.625 hindered cell growth, they did not cause any harmful effects on the DC2.4 cell line, as was definitively proven. Furthermore, the wPP and wPPChi0.625 hydrogel extracts had minimal impact on the expression of several markers. From the production of the proinflammatory cytokine TNF-α and the unique expression patterns of CD40 and CD80, it is clear that all hydrogel formulations did not possess immunogenicity against the DC2.4 cell line.
This study addresses the potential biocompatibility concerns related to the immunogenicity of the crosslinked hydrogels. It provides a strong foundation for extending the claim of biocompatibility and non-immunogenicity to the hydrogel formulations produced from the individual polymers used in this study—PVA, PVP, and chitosan—despite some evidence of chemical changes, such as the formation of diene bonds, after crosslinking. As an immediate follow-up to this finding, future work will focus on utilizing non-immunogenic, leachable-free formulations for biomedical applications. These formulations could be employed in the fabrication of microneedle devices for drug delivery and interstitial fluid sampling, transmucosal patches, or implantable hydrogel-based devices. However, the potential applications are not limited to microneedles and may extend to other pharmaceutical and biomedical purposes, such as inserts, implants, and devices based on PVA, PVP, and chitosan hydrogels.
Author contributions
Achmad Himawan: conceptualisation, investigation, data curation, methodology, formal analysis, project administration, visualisation, writing – original draft; Anna Korelidou: methodology, investigation, writing – original draft; Ana M. Pérez-Moreno: investigation: writing – original draft; Juan Luis Paris: conceptualisation, methodology, investigation, formal analysis, data curation, resources, writing – review & editing; Juan Dominguez-Robles: methodology, investigation, formal analysis, data curation, writing – review & editing; Lalitkumar K. Vora: conceptualisation, methodology, writing – review & editing; Andi Dian Permana: methodology, writing – review & editing; Eneko Larraneta: conceptualisation, methodology, writing – review & editing; Robert Graham: methodology, formal analysis; Christopher J. Scott: conceptualisation, supervision, writing – review & editing; Ryan F. Donnelly: conceptualisation, project administration, resources, supervision, validation, writing – review & editing.Data availability
Data supporting this article can be found throughout or in the ESI.†Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Ryan Donnelly is an inventor of patents that have been licenced to companies developing microneedle-based products and is a paid advisor to companies developing microneedle-based products. The resulting potential conflict of interest has been disclosed and is managed by Queen's University Belfast. The companies had no role in the design of the present studies, in the collection, analyses or interpretation of the data, in the writing of the manuscript or in the decision to publish the work. Other all authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
AH and AK thankfully acknowledge the financial support of the CITI-GENS programme at Queen's University Belfast, co-funded with the EU Horizon 2020 scheme under the Marie Sklodowska-Curie Action grant agreement number 945231 – CITI-GENS. JLP acknowledges grant RYC2021-034536-I funded by MCIN/AEI/10.13039/501100011033 and by European Union NextGenerationEU/PRTR. APM acknowledges PhD fellowship PREP2022-000567 (associated to grant PID2022-142781OA-I00) funded by MCIN/AEI/10.13039/501100011033 and by ESF+. JDR acknowledges financial support from the Ramón y Cajal grant RYC-2021–034357-I funded by MCIN/AEI/10.13039/501100011033 and by the “European Union NextGenerationEU/PRTR”.References
- T. R. Raj Singh, P. A. McCarron, A. D. Woolfson and R. F. Donnelly, Eur. Polym. J., 2009, 45, 1239–1249 CrossRef CAS.
- T. R. Raj Singh, A. D. Woolfson and R. F. Donnelly, J. Pharm. Pharmacol., 2010, 62, 829–837 CrossRef PubMed.
- A. Léonard, S. Blacher, M. Crine and W. Jomaa, J. Non-Cryst. Solids, 2008, 354, 831–838 CrossRef.
- R. F. Donnelly, T. R. R. Singh, M. J. Garland, K. Migalska, R. Majithiya, C. M. McCrudden, P. L. Kole, T. M. T. Mahmood, H. O. McCarthy and A. D. Woolfson, Adv. Funct. Mater., 2012, 22, 4879–4890 CrossRef CAS PubMed.
- N. Hasan, C. Jiafu, A. Z. Mustopa, A. Himawan, R. N. Umami, M. Ullah, N. Wathoni and J.-W. Yoo, J. Pharm. Invest., 2023, 53, 781–801 CrossRef CAS.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 RSC.
- J. Tavakoli and Y. Tang, Polymers, 2017, 9, 364 CrossRef PubMed.
- C. A. Dreiss, Curr. Opin. Colloid Interface Sci., 2020, 48, 1–17 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- Y. Zhang and Y. Huang, Front. Chem., 2021, 8, 615665 CrossRef PubMed.
- P. Sánchez-Cid, M. Jiménez-Rosado, A. Romero and V. Pérez-Puyana, Polymers, 2022, 14, 3023 CrossRef PubMed.
- R. Rodríguez-Rodríguez, H. Espinosa-Andrews, C. Velasquillo-Martínez and Z. Y. García-Carvajal, Int. J. Polym. Mater. Polym. Biomater., 2019, 69, 1–20, DOI:10.1080/00914037.2019.1581780.
- W. Liu, H. Du, M. Zhang, K. Liu, H. Liu, H. Xie, X. Zhang and C. Si, ACS Sustainable Chem. Eng., 2020, 8, 7536–7562 CrossRef CAS.
- S. Dugam, R. Tade, R. Dhole and S. Nangare, Future J. Pharm. Sci., 2021, 7, 19 CrossRef.
- S. A. Meenach, K. W. Anderson and J. Z. Hilt, in Safety of Nanoparticles: From Manufacturing to Medical Applications, ed. T. J. Webster, Springer, New York, NY, 2009, pp. 131–157 Search PubMed.
- R. Ansar, S. Saqib, A. Mukhtar, M. B. K. Niazi, M. Shahid, Z. Jahan, S. J. Kakar, B. Uzair, M. Mubashir, S. Ullah, K. S. Khoo, H. R. Lim and P. L. Show, Chemosphere, 2022, 287, 131956 CrossRef CAS PubMed.
- A. Himawan, Q. K. Anjani, U. Detamornrat, L. K. Vora, A. D. Permana, R. Ghanma, Y. Naser, D. Rahmawanty, C. J. Scott and R. F. Donnelly, Eur. Polym. J., 2023, 186, 111836 CrossRef CAS.
- A. A. El-Fattah, M. N. Hassan, A. Rashad, M. Marei and S. Kandil, Int. J. Polym. Anal. Charact., 2020, 25(5), 362–373 CrossRef.
- Y. Hu, S. Zhang, Z. Wen, H. Fu, J. Hu, X. Ye, L. Kang, X. Li and X. Yang, Int. J. Biol. Macromol., 2022, 221, 806–820 CrossRef CAS PubMed.
- W. Lan, M. Xu, M. Qin, Y. Cheng, Y. Zhao, D. Huang, X. Wei, Y. Guo and W. Chen, Mater. Des., 2021, 204, 109652 CrossRef CAS.
- R. Li, L. Zhang, X. Jiang, L. Li, S. Wu, X. Yuan, H. Cheng, X. Jiang and M. Gou, J. Controlled Release, 2022, 350, 933–948 CrossRef CAS PubMed.
- M. Wancura, M. Talanker, S. Toubbeh, A. Bryan and E. Cosgriff-Hernandez, J. Mater. Chem. B, 2020, 8, 4289–4298 RSC.
- F. A. Maulvi, T. G. Soni and D. O. Shah, J. Biomater. Sci., Polym. Ed., 2015, 26, 1035–1050 CrossRef CAS PubMed.
- Ø. Øvrebø, G. Perale, J. P. Wojciechowski, C. Echalier, J. R. T. Jeffers, M. M. Stevens, H. J. Haugen and F. Rossi, Bioeng. Transl. Med., 2022, 7, e10295 CrossRef PubMed.
- E. Larrañeta, R. E. M. Lutton, A. J. Brady, E. M. Vicente-Pérez, A. D. Woolfson, R. R. S. Thakur and R. F. Donnelly, Macromol. Mater. Eng., 2015, 300, 586–595 CrossRef PubMed.
- E. Larrañeta, R. E. M. Lutton, A. D. Woolfson and R. F. Donnelly, Mater. Sci. Eng., R, 2016, 104, 1–32 CrossRef.
- J. G. Hardy, E. Larrañeta, R. F. Donnelly, N. McGoldrick, K. Migalska, M. T. C. McCrudden, N. J. Irwin, L. Donnelly and C. P. McCoy, Mol. Pharmaceutics, 2016, 13, 907–914 CrossRef CAS PubMed.
- J. G. Turner, L. R. White, P. Estrela and H. S. Leese, Macromol. Biosci., 2021, 21, 2000307 CrossRef CAS PubMed.
- S. Sharifi, A. A. Saei, H. Gharibi, N. N. Mahmoud, S. Harkins, N. Dararatana, E. M. Lisabeth, V. Serpooshan, Á. Végvári, A. Moore and M. Mahmoudi, ACS Appl. Bio Mater., 2022, 5, 2643–2663 CrossRef CAS PubMed.
- I. A. Tekko, G. Chen, J. Domínguez-Robles, R. R. S. Thakur, I. M. N. Hamdan, L. Vora, E. Larrañeta, J. C. McElnay, H. O. McCarthy, M. Rooney and R. F. Donnelly, Int. J. Pharm., 2020, 586, 119580 CrossRef CAS PubMed.
- R. F. Donnelly, M. T. C. McCrudden, A. Zaid Alkilani, E. Larrañeta, E. McAlister, A. J. Courtenay, M.-C. Kearney, T. R. R. Singh, H. O. McCarthy, V. L. Kett, E. Caffarel-Salvador, S. Al-Zahrani and A. D. Woolfson, PLoS One, 2014, 9, e111547 CrossRef PubMed.
- E. Zeiger, B. Gollapudi and P. Spencer, Mutat. Res., 2005, 589, 136–151 CAS.
- B. Zeng, Z. Cai, J. Lalevée, Q. Yang, H. Lai, P. Xiao, J. Liu and F. Xing, Toxicol. In Vitro, 2021, 72, 105103 CrossRef CAS PubMed.
- K. Ahmed Saeed AL-Japairai, S. Mahmood, S. Hamed Almurisi, J. Reddy Venugopal, A. Rebhi Hilles, M. Azmana and S. Raman, Int. J. Pharm., 2020, 587, 119673 CrossRef CAS PubMed.
- J. Premkumar, K. SonicaSree and T. Sudhakar, in Handbook of Polymer and Ceramic Nanotechnology, ed. C. M. Hussain and S. Thomas, Springer International Publishing, Cham, 2021, pp. 1329–1355 Search PubMed.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923–943 CrossRef CAS.
- V. Balan and L. Verestiuc, Eur. Polym. J., 2014, 53, 171–188 CrossRef CAS.
- X. P. Zhang, B. B. Wang, W. X. Li, W. M. Fei, Y. Cui and X. D. Guo, Eur. J. Pharm. Biopharm., 2021, 160, 1–8 CrossRef CAS PubMed.
- A. Adachi, A. Fukunaga, K. Hayashi, M. Kunisada and T. Horikawa, Contact Dermatitis, 2003, 48, 133–136 CrossRef PubMed.
- K. Yoshida, Y. Sakurai, S. Kawahara, T. Takeda, T. Ishikawa, T. Murakami and A. Yoshioka, Int. Arch. Allergy Immunol., 2008, 146, 169–173 CrossRef PubMed.
- T. Itazawa, Y. Adachi, Y. Okabe, Y. S. Adachi, M. Nakabayashi, T. Harai, K. Miya, K. Uese and T. Miyawaki, J. Allergy Clin. Immunol., 2006, 117, S134 CrossRef.
- M. A. Bruusgaard-Mouritsen, C. Mortz, L. Winther and L. H. Garvey, Ann. Allergy, Asthma, Immunol., 2021, 126, 598–600 CrossRef CAS PubMed.
- Y.-C. Lin, P.-J. Lou and T.-H. Young, Biomaterials, 2014, 35, 8867–8875 CrossRef CAS PubMed.
- N. Alexandre, J. Ribeiro, A. Gärtner, T. Pereira, I. Amorim, J. Fragoso, A. Lopes, J. Fernandes, E. Costa, A. Santos-Silva, M. Rodrigues, J. D. Santos, A. C. Maurício and A. L. Luís, J. Biomed. Mater. Res., Part A, 2014, 102, 4262–4275 Search PubMed.
- M. J. Toebak, S. Gibbs, D. P. Bruynzeel, R. J. Scheper and T. Rustemeyer, Contact Dermatitis, 2009, 60, 2–20 CrossRef CAS PubMed.
- M. Collin and V. Bigley, Immunology, 2018, 154, 3–20 CrossRef CAS PubMed.
- P. Humeniuk, P. Dubiela and K. Hoffmann-Sommergruber, Int. J. Mol. Sci., 2017, 18, 1491 CrossRef PubMed.
- S. Wang, Y. Chen, Z. Ling, J. Li, J. Hu, F. He and Q. Chen, Int. J. Oral Sci., 2022, 14, 1–15 CrossRef PubMed.
- X. Hu, L. Lu, C. Xu and X. Li, Int. J. Biol. Macromol., 2015, 72, 403–409 CrossRef CAS PubMed.
- Z. Li, G. He, J. Hua, M. Wu, W. Guo, J. Gong, J. Zhang and C. Qiao, RSC Adv., 2017, 7, 11085–11093 RSC.
- Y. Shi, D. Xiong, Y. Liu, N. Wang and X. Zhao, Mater. Sci. Eng., C, 2016, 65, 172–180 CrossRef CAS PubMed.
- S. D. K. Seera, D. Kundu and T. Banerjee, Cellulose, 2020, 27, 6521–6535 CrossRef CAS.
- M. A. Abureesh, A. A. Oladipo and M. Gazi, Int. J. Biol. Macromol., 2016, 90, 75–80 CrossRef CAS PubMed.
- M. Wiśniewska, V. Bogatyrov, I. Ostolska, K. Szewczuk-Karpisz, K. Terpiłowski and A. Nosal-Wiercińska, Adsorption, 2016, 22, 417–423 CrossRef.
- A. M. Khalil, M. L. Hassan and A. A. Ward, Carbohydr. Polym., 2017, 157, 503–511 CrossRef CAS PubMed.
- G. Oster and E. H. Immergut, J. Am. Chem. Soc., 1954, 76, 1393–1396 CrossRef CAS.
- D. Liu, H. Ge, M. Song, Y. Jiang, X. Gong, T. You, L. Fu, Z. Fu and Y. Zhang, Carbohydr. Res., 2006, 341, 782–785 CrossRef CAS PubMed.
- O. S. Lezova, D. V. Myasnikov, O. A. Shilova, A. G. Ivanova and S. I. Selivanov, Int. J. Hydrogen Energy, 2022, 47, 4846–4853 Search PubMed.
- B. M. Budhlall, K. Landfester, E. D. Sudol, V. L. Dimonie, A. Klein and M. S. El-Aasser, Macromolecules, 2003, 36, 9477–9484 CrossRef CAS.
- B. Sesta, A. L. Segre, A. D’Aprano and N. Proietti, J. Phys. Chem. B, 1997, 101, 198–204 CrossRef CAS.
- M. I. Loría-Bastarrachea, W. Herrera-Kao, J. V. Cauich-Rodríguez, J. M. Cervantes-Uc, H. Vázquez-Torres and A. Ávila-Ortega, J. Therm. Anal. Calorim., 2011, 104, 737–742 CrossRef.
- K. V. Phung, US Pat., US6046272A, 2000.
- T.-J. Lee, D.-J. Lee, J.-H. Seo, K.-S. Lee, D. Guerin, P. Martinez, M.-K. Lee and J.-Y. Ryu, ACS Sustainable Chem. Eng., 2018, 6, 1702–1707 CrossRef CAS.
- I. Yu Prosanov and A. A. Matvienko, Phys. Solid State, 2010, 52, 2203–2206 CrossRef CAS.
- Full article: Chemical and biology aging of novel green membranes made of PVA and wood flour fibers reinforced with nanosilica manufactured by compression molding process, https://www.tandfonline.com/doi/full/10.1080/1023666X.2017.1404271, (accessed 1 October 2023).
- H. S. Mansur, C. M. Sadahira, A. N. Souza and A. A. P. Mansur, Mater. Sci. Eng., C, 2008, 28, 539–548 CrossRef CAS.
- Dissolution mechanism of semicrystalline poly(vinyl alcohol) in water – Mallapragada – 1996 – Journal of Polymer Science Part B: Polymer Physics – Wiley Online Library, https://onlinelibrary.wiley.com/doi/abs/10.1002/%28SICI%291099-0488%28199605%2934%3A7%3C1339%3A%3AAID-POLB15%3E3.0.CO%3B2-B, (accessed 1 October 2023).
- S. M. Pawde and K. Deshmukh, J. Appl. Polym. Sci., 2008, 109, 3431–3437 CrossRef CAS.
- S. Bonakdar, S. H. Emami, M. A. Shokrgozar, A. Farhadi, S. A. H. Ahmadi and A. Amanzadeh, Mater. Sci. Eng., C, 2010, 30, 636–643 CrossRef CAS.
- O. Akin and F. Temelli, Desalination, 2011, 278, 387–396 CrossRef CAS.
- H. S. Mansur, R. L. Oréfice and A. A. P. Mansur, Polymer, 2004, 45, 7193–7202 CrossRef CAS.
- D. Choi, M. H. Khan and J. Jung, Int. Biodeterior. Biodegrad., 2019, 145, 104788 CrossRef CAS.
- Z. M. O. Rzayev, M. Şimşek, U. Bunyatova and B. Salamov, Colloids Surf., A, 2016, 492, 26–37 CrossRef CAS.
- A. M. Abdelghany, M. S. Meikhail, A. H. Oraby and M. A. Aboelwafa, Polym. Bull., 2023, 80(12), 13279–13298 CrossRef CAS.
- L. S. Usmanova, M. A. Ziganshin, I. T. Rakipov, N. M. Lyadov, A. E. Klimovitskii, T. A. Mukhametzyanov and A. V. Gerasimov, BioMed Res. Int., 2018, 2018, e2412156 Search PubMed.
- K. Terumitsu, S. Ko and I. Akira, J. Phys. Chem., 1987, 91, 2366–2371 CrossRef.
- N. A. Peppas and E. W. Merrill, J. Appl. Polym. Sci., 1976, 20, 1457–1465 CrossRef CAS.
- Y. Liu, L. M. Geever, J. E. Kennedy, C. L. Higginbotham, P. A. Cahill and G. B. McGuinness, J. Mech. Behav. Biomed. Mater., 2010, 3, 203–209 CrossRef PubMed.
- C. W. Bunn, Nature, 1948, 161, 929–930 CrossRef CAS.
- B. G. Colvin, Nature, 1974, 248, 756–759 CrossRef CAS.
- R. Ramadan and A. M. Ismail, J. Inorg. Organomet. Polym. Mater., 2023, 33, 2506–2516 CrossRef CAS.
- G. Hao, Y. Hu, L. Shi, J. Chen, A. Cui, W. Weng and K. Osako, Sci. Rep., 2021, 11, 1646 CrossRef CAS PubMed.
- Y. Zhang, C. Xue, Y. Xue, R. Gao and X. Zhang, Carbohydr. Res., 2005, 340, 1914–1917 CrossRef CAS PubMed.
- F. Colmenero, Phys. Chem. Chem. Phys., 2019, 21, 2673–2690 RSC.
- A. Xiang, C. Lv and H. Zhou, J. Vinyl Addit. Technol., 2020, 26, 613–622 CrossRef CAS.
- M. P. Illa, C. S. Sharma and M. Khandelwal, J. Mater. Sci., 2019, 54, 12024–12035 CrossRef CAS.
- J.-S. Park, H.-A. Kim, J.-B. Choi, H.-J. Gwon, Y.-M. Shin, Y.-M. Lim, M. S. Khil and Y.-C. Nho, Radiat. Phys. Chem., 2012, 81, 857–860 CrossRef CAS.
- N. A. Peppas and E. W. Merrill, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 441–457 CrossRef CAS.
- O. A. Zakurdaeva, S. V. Nesterov, N. A. Shmakova, G. K. Semenova, E. O. Sozontova and V. I. Feldman, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 356–361 CrossRef CAS.
- V. Štejfa, V. Pokorný, J. Rohlíček, M. Fulem and K. Růžička, J. Chem. Thermodyn., 2021, 160, 106488 Search PubMed.
- A. K. Sonker, K. Rathore, R. K. Nagarale and V. Verma, J. Polym. Environ., 2018, 26, 1782–1794 CrossRef CAS.
- S. Lai, M. Casu, G. Saba, A. Lai, I. Husu, G. Masci and V. Crescenzi, Solid State Nucl. Magn. Reson., 2002, 21, 187–196 CrossRef CAS PubMed.
- T. Terao, S. Maeda and A. Saika, Macromolecules, 1983, 16, 1535–1538 CrossRef CAS.
- H. Feng, Z. Feng and L. Shen, Polymer, 1993, 34, 2516–2519 CrossRef CAS.
- R. Koike, K. Higashi, N. Liu, W. Limwikrant, K. Yamamoto and K. Moribe, Cryst. Growth Des., 2014, 14, 4510–4518 CrossRef CAS.
- S. A. Shah, R. S. Oakes, S. M. Kapnick and C. M. Jewell, Front. Immunol., 2022, 13, 843355 CrossRef PubMed.
- M. Riehl, M. Harms, A. Hanefeld, R. B. Baleeiro, P. Walden and K. Mäder, Int. J. Pharm., 2017, 532, 37–46 CrossRef CAS PubMed.
- B. Tyliszczak, A. Drabczyk, S. Kudłacik-Kramarczyk, K. Bialik-Wąs, R. Kijkowska and A. Sobczak-Kupiec, Colloids Surf., B, 2017, 160, 325–330 CrossRef CAS PubMed.
- M. A. M. Vis, K. Ito and S. Hofmann, Front. Bioeng. Biotechnol., 2020, 8, 911 CrossRef PubMed.
- R. Rodríguez-Rodríguez, Z. Y. García-Carvajal, I. Jiménez-Palomar, J. A. Jiménez-Avalos and H. Espinosa-Andrews, J. Appl. Polym. Sci., 2019, 136, 47149 CrossRef.
- X. Yang, K. Yang, S. Wu, X. Chen, F. Yu, J. Li, M. Ma and Z. Zhu, Radiat. Phys. Chem., 2010, 79, 606–611 CrossRef CAS.
- B. D. Krezović, M. G. Miljković, S. T. Stojanović, S. J. Najman, J. M. Filipović and S. Lj Tomić, Chem. Eng. Res. Des., 2017, 121, 368–380 CrossRef.
- M. Sirousazar, M. Kokabi and Z. M. Hassan, J. Biomater. Sci., Polym. Ed., 2011, 22, 1023–1033 CrossRef CAS PubMed.
- N. Saha, R. Shah, P. Gupta, B. B. Mandal, R. Alexandrova, M. D. Sikiric and P. Saha, Mater. Sci. Eng., C, 2019, 95, 440–449 CrossRef CAS PubMed.
- D. Y. Ma and E. A. Clark, Semin. Immunol., 2009, 21, 265–272 CrossRef CAS PubMed.
- L. Biscari, C. D. Kaufman, C. Farré, V. Huhn, M. F. Pacini, C. B. Balbi, K. A. Gómez, A. R. Pérez and A. Alloatti, Front. Cell. Infect. Microbiol., 2022, 12, 897133 CrossRef CAS PubMed.
- W. Huo, X. Yang, B. Wang, L. Cao, Z. Fang, Z. Li, H. Liu, X. Liang, J. Zhang and Y. Jin, Biomaterials, 2022, 288, 121722 CrossRef CAS PubMed.
- D. Martínez, M. Vermeulen, E. von Euw, J. Sabatté, J. Maggíni, A. Ceballos, A. Trevani, K. Nahmod, G. Salamone, M. Barrio, M. Giordano, S. Amigorena and J. Geffner, J. Immunol., 2007, 179, 1950–1959 CrossRef PubMed.
- A. J. Courtenay, A. M. Rodgers, M. T. C. McCrudden, H. O. McCarthy and R. F. Donnelly, Mol. Pharmaceutics, 2019, 16, 118–127 CrossRef CAS PubMed.
- D. M. Sansom, C. N. Manzotti and Y. Zheng, Trends Immunol., 2003, 24, 313–318 CrossRef PubMed.
- J.-G. Li, Y.-M. Du, Z.-D. Yan, J. Yan, Y.-X. Zhuansun, R. Chen, W. Zhang, S.-L. Feng and P.-X. Ran, Exp. Ther. Med., 2016, 11, 878–884 CrossRef CAS PubMed.
- H. T. Idriss and J. H. Naismith, Microsc. Res. Tech., 2000, 50, 184–195 CrossRef CAS PubMed.
- A. Rhule, B. Rase, J. R. Smith and D. M. Shepherd, J. Ethnopharmacol., 2008, 116, 179–186 CrossRef CAS PubMed.
- D. P. Vangasseri, Z. Cui, W. Chen, D. A. Hokey, L. D. Falo and L. Huang, Mol. Membr. Biol., 2006, 23, 385–395 CrossRef CAS PubMed.
- Y.-D. Xu, M. Cheng, P.-P. Shang and Y.-Q. Yang, J. Leukocyte Biol., 2022, 111, 695–709 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02181a |
| This journal is © The Royal Society of Chemistry 2025 |