
Advances in peptide/polymer antimicrobial assemblies
He
Zhao
,
Jiayi
Sun
,
Yi
Cheng
,
Shuaishuai
Nie
and
Wen
Li
 *
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Qianjin Avenue 2699, Changchun 130012, China. E-mail: wenli@jlu.edu.cn
First published on 12th December 2024
Abstract
Antimicrobial peptides (AMPs) have been extensively exploited as promising drugs to cope with antibiotic-resistant bacteria in clinical treatment. Peptide/polymer assembly provides a particularly important contribution to this topic and has emerged as a new paradigm for the development of nano-antimicrobial systems with previously unattainable outcomes. In this review article, we systematically summarize the recent advances in antimicrobial peptide/polymer assemblies. We describe a brief background and several classified systems based on peptide/polymer assemblies. We discuss the molecular design and the general rules behind the assembled nanostructures and bioactivities. The key role of polymers in improving the antimicrobial activity, stability, cytotoxicity, and bioavailability of peptides is emphasized based on the reported systems. The resulting peptide/polymer assemblies with stimuli-responsiveness, value-added properties and potential applications are demonstrated. The outlook of the antimicrobial peptide/polymer assemblies is also presented from the viewpoint of bio-applications.
1. Introduction
Bacterial infection is one of the greatest public health risks, which poses a significant threat to human health and life safety.1–3 Antibiotics have been proven a reliable weapon in the treatment of bacterial infectious diseases and have saved countless lives.4–6 Unfortunately, the limited number of antibiotics and their overuse have greatly stimulated many bacteria to evolve the ability of antibiotic resistance (AMR),7–10 giving rise to the decline in the reliability of conventional antibiotics. Therefore, one of the urgent tasks is to develop new drugs that are different from the traditional antibiotics in antimicrobial mechanisms.11–13 In an effort to seek that kind of candidate, defensin-inspired antimicrobial peptides (AMPs) have received widespread attention due to their unique membrane-disruption mechanism via ionic bond-driven non-specific electrostatic interactions between peptides and bacteria, and the hydrophobic effect dominated membrane insertion, which can significantly slow or reduce the development of AMR.14–17 Along this line, a lot of natural or artificial long-chain peptides with cationicity and amphiphilicity have been reported.18–21 It was found that long-chain peptides can simultaneously hold many alkaline and hydrophobic amino acid residues in a single sequence, resulting in much higher antimicrobial activity.22,23 Alternatively, AMPs with branched topological structures, such as hyperbranched, dendritic, and star-like peptides were designed and synthesized.24–26 In spite of the high antimicrobial efficacy, the cost-efficiency of long-chain or branched AMPs is poor due to the time-consuming synthesis and low yield. Inversely, linear oligopeptides with relatively short sequences (below 12 amino acid residues) were seldom explored in earlier studies although they can be easily synthesized with a high yield. This fact stems from the poor antimicrobial activity of short peptides.27–30Since the 2000s, peptide self-assembly has been identified as a potent platform for the creation of multivalent nanostructures with tailor-made antimicrobial activities.31–36 The breadth of self-assembled nano-AMPs has been documented by several review articles.37–40 It is noteworthy that most of the self-assembled AMPs need rigorous design in peptide sequences. Recently, chemical conjugation or physical complexation between oligopeptides and diverse objects has emerged as a new paradigm for the development of nano-AMPs.41–45 In this vein, the aqueous assembly of peptide–polymer (including polypeptide–polypeptide) conjugates or the peptide/polymer complexes has gathered particular interest. For the aqueous assembly of amphiphilic peptide–polymer conjugates, the hydrophilic peptide block and the hydrophobic polymer block have a strong propensity to segregate into their distinct subdomain due to the solvation contrast between the dissimilar blocks. The entropic penalties associated with the chain aggregation of the hydrophobic block and interfacial energy enable the peptide–polymer conjugates to form water-dispersed assemblies with a hydrophobic polymer core surrounded by a hydrophilic peptide shell.46,47 In addition, the microphase segregation of the incompatible blocks can further enhance each other in a watery environment, giving rise to the formation of thermodynamically stable nanostructures by adjusting the relative volume fraction between hydrophobic and hydrophilic blocks. In the case of peptide/polymer complexes, ionic bonds together with hydrogen bonds are the primary driving forces to dominate the assembly of peptides and oppositely charged polymers in aqueous solution.48,49 In general, the ionic and hydrogen bonds could be typically depressed in polar aqueous solution owing to the hydration and the screening effect.50,51 However, the introduction of multiple ionic bonds into the co-assembling systems has been proved to greatly increase the association energy and reduce the dissociation probability, leading to stable and effective nano-AMPs.
Up to now, a great deal of effort has been made to develop peptide–polymer conjugates or peptide/polymer complexes. Emphasis has been placed on several aspects, such as antimicrobial efficacy, stability, life span and biocompatibility of the resulting assemblies. In this article, we offer a comprehensive overview of the recent advances in peptide/polymer antimicrobial assemblies. We classified the systems into three types. The first part is the polypeptide–polypeptide conjugates, in which cationic polypeptide blocks are coupled with the hydrophobic or anionic polypeptide blocks. We demonstrate the influence of molecular shape or topology on the assembled nanostructures and bioactivities. The second part focuses on the self-assembly of peptide–polymer conjugates. The volume fraction and hydrophobicity/hydrophilicity of the polymer blocks have been utilized to regulate the structure–property relationship of the resulting assemblies. The third part is the peptide/polymer complexes, where the ionic co-assembly of cationic peptides with a broad range of anionic polymers or polypeptides is illustrated.
2. Polypeptide–polypeptide conjugates
Block polypeptides simultaneously carrying cationic and hydrophobic peptide segments tend to form stable and water dispersed nanostructures, which can greatly improve the hydrolysis stability and the antimicrobial efficacy of the peptides. In an earlier study, a block polypeptide p(Lys)x–p(Ala)y (x + y = 100), consisting of poly-lysine (pLys) and poly-alanine (pAla) was conjugated with 6-arm polyethylene glycol (PEG)-amide succinimidyl glutarate.52 The branched polypeptide exhibited significant antimicrobial activity. Subsequently, star-like polypeptide nanoparticles were designed by S. J. Lam and colleagues. In this case, polypeptide composed of pLys and poly-valine (pVal) was grafted from poly(amido amine) (PAMAM) dendrimers (Fig. 1(a)) via N-carboxylic acid anhydride (NCA) co-polymerization of NCA-Lys and NCA-Val.53 The super-resolution fluorescence imaging using 3D structured illumination microscopy (3D-SIM) demonstrated that the resulting star-like polypeptide nanoparticles (Fig. 1(b)) exhibited enhanced antimicrobial activity against various Gram-negative bacteria due to the highly concentrated charges on the surface of the star-like nanoparticles. Detailed investigations indicated that the polypeptide nanoparticles exerted antibacterial action through a multimodal bacterial death mechanism, including outer membrane destabilization, unregulated ion movement across the cytoplasmic membrane and the apoptosis-like death pathway. In subsequent work, the authors explored the effect of the number and length of arms of star-like polypeptides on antimicrobial activity and biocompatibility.54 It was found that the antimicrobial activity of the polypeptides was proportional to the number and length of the polypeptides, owing to the increased surficial cationicity and α-helix content of the polypeptide nanoparticles. However, the simultaneously enhanced cytotoxicity indicated that therapy is needed to carefully balance the antimicrobial activity and cytotoxicity. The therapeutic index calculations identified the 16-arm and 4-arm polypeptide nanoparticles as the optimal therapeutic agents. Furthermore, the authors assessed the interaction between the polypeptide nanoparticles and different bacteria in complex biological media, such as artificial body fluid and animal serum, in order to stimulate the in vivo antimicrobial activity.55 They argued that divalent cations (such as Ca2+ and Mg2+) in body fluids may bind to polyphosphates in the lipopolysaccharide (LPS) layer of the bacterial outer membrane (OM), stabilize the LPS structure, and reduce the membrane penetration ability of the polypeptide, leading to the decline of antimicrobial activity. This result clearly demonstrates that the in vitro antimicrobial experiments cannot directly reflect the in vivo performance. Harnessing the aggregation-induced emission effect of PAMAM, Zhou and coworkers further explored the real-time monitoring function of the pLys-pVal-PAMAM dendrimers.56 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of star-like polypeptide nanoparticles via ring-opening polymerization of lysine and valine N-carboxyanhydrides (NCAs) initiated from the terminal amines of poly(amido amine) (PAMAM) dendrimers. (b) Optical microscopy experimental 3D-SIM images of Escherichia coli (E. coli) before and after treatment with AF488-tagged star-like polypeptide nanoparticles in MHB. Scale bars, 1 μm. The E. coli cell membrane was stained with FM4-64FX (red) and star-like polypeptide nanoparticles with AF488 (green) in all images. Reproduced with permission from ref. 53. Copyright 2016 Nature Publishing Group. | ||
To balance the antimicrobial activity and cytotoxicity of the peptide–PAMAM conjugates, Zhang et al. decided to improve their selectivity towards bacteria and normal cells by mimicking the structural unit of vitamin U.57 As shown in Fig. 2(a), (b), poly-methionine (pMet) chains carrying partially or completely alkylated sulfonium moieties were conjugated with PAMAM dendrons (G2-PAMAM). The resulting conjugates were denoted as G2-PAMAM-n, where G2-PAMAM means the second generation PAMAM, n (1, 2, and 3) represents the ratio of pMet to G2-PAMAM (50, 25, and 10, respectively). The authors identified that G2-PAMAM-1 with a high proportion of sulfonium moieties exhibited a broad-spectrum antimicrobial activity with an MIC of 4 μg mL−1 against Staphylococcus aureus (S. aureus) and 16 μg mL−1 against E. coli. The antimicrobial mechanisms involved the electrostatic binding and membrane disruption and the reactive oxygen species (ROS) accumulation in the cytoplasm. More importantly, the vitro anti-MRSA experiment and in vivo treatment experiments proved that G2-PAMAM-1 with highly cationic moieties significantly enhanced the hemocompatibility and bactericidal activity. The ratio of HC10 to MIC is 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, which is much higher than that of the reported results. Furthermore, the selective antibacterial activity in a mixed bacterial model of G2-PAMAM-n can be modulated by adjusting the proportion of the sulfonium moieties (Fig. 2(c) and (d)). Products with a high proportion of the sulfonium moieties heightened bactericidal activities against S. aureus, while products with a low proportion of sulfonium moieties exhibited a more targeted response against E. coli.
000, which is much higher than that of the reported results. Furthermore, the selective antibacterial activity in a mixed bacterial model of G2-PAMAM-n can be modulated by adjusting the proportion of the sulfonium moieties (Fig. 2(c) and (d)). Products with a high proportion of the sulfonium moieties heightened bactericidal activities against S. aureus, while products with a low proportion of sulfonium moieties exhibited a more targeted response against E. coli.
 | ||
| Fig. 2 The schematic structures of G2-PAMAM-n carrying partially (a) or completely (b) alkylated sulfonium moieties. Antimicrobial efficiency of mixed bacteria treated with G2-PAMAM-2 carrying partially (c) or completely (d) alkylated sulfonium moieties. Reproduced with permission from ref. 57. Copyright 2023 Wiley-VCH GmbH. | ||
Cheng et al. designed a block polypeptide p(Lys-stat-Phe)–pPhe by conjugating poly-lysine (pLys) with a poly-phenylalanine (pPhe) block.58 As shown in Fig. 3(a), the resulting block peptide can self-assemble into a nanomicelle (diameter ∼45 nm) in an aqueous solution with a cationic surface, which can kill bacteria via destroying the cell membrane. In vivo animal experiments demonstrated that nanomicelles displayed greater advantages than tobramycin when used for the treatment of bacterial keratitis. This kind of block polypeptide p(Lys-stat-Phe) was further grafted on a hyperbranched polymer scaffold to create a hyperbranched block polypeptide with a new “wrapping and penetrating” antimicrobial mechanism.59 As illustrated in Fig. 3(b), the hyperbranched pLys–pPhe has a strong propensity to self-assemble into sheet-like superstructures with a large size, which enables the sheet-like superstructures to contact and wrap bacteria, followed by penetrating into the cell membrane and killing bacteria effectively. This unique “wrapping and penetrating” antimicrobial mechanism led to a very low minimum inhibitory concentration (MIC) against both Gram-negative E. coli and Gram-positive S. aureus (16 μg mL−1). More importantly, sheet-like superstructures had a low surface potential (+6.1 mV) and thus showed low hemolysis and cytotoxicity. This work provides a strategy for solving the contradictory relationship between antimicrobial activity and cytotoxicity.
 | ||
| Fig. 3 (a) Schematic drawing of the chemical structure of block polypeptide p(Lys-stat-Phe)–pPhe and micelle-like assemblies for bacterial keratitis. Reproduced with permission from ref. 58. Copyright 2022 American Chemical Society. (b) Schematic illustration of the sheet-like superstructures of the hyperbranched polypeptide and the “wrapping and penetrating” antibacterial mechanism. Reproduced with permission from ref. 59. Copyright 2016 American Chemical Society. | ||
In another example, Zhu et al. reported a synergistic antimicrobial system by combining chlorin e6 (Ce6) and an arginine (Arg)-rich dendritic peptide with an aliphatic chain (Arg-ADP).60 As illustrated in Fig. 4(a), the amphiphilic peptide dendrimer self-assembled into micelle-like structures with a hydrophobic core, which served as a carrier to load Ce6. The resulting Ce6@Arg-ADP exhibited the ability to treat subcutaneous abscesses and promote wound healing via photodynamic therapy (PDT)-driven NO controllable generation. When Ce6@Arg-ADP penetrated into the biofilm, the PDT behaviour of Ce6 induced the production of H2O2, which further oxidized Arg-ADP to generate NO and citrulline. The NO and 1O2 offered synergistic antimicrobial effects (Fig. 4(b)). After all bacteria at the abscess site were eliminated, Arg-ADP could further generate trace amounts of NO to promote wound healing (Fig. 4(c)).
 | ||
| Fig. 4 Schematic illustration of the preparation of Ce6@Arg-ADP and the related mechanisms of PDT/NO to provide highly efficient synergistic antibacterial effects and promote wound healing. Reproduced with permission from ref. 60. Copyright 2021 Wiley-VCH GmbH. | ||
In an effort to develop a smart antimicrobial system, Xiong et al. reported a class of pH-sensitive di-block polypeptides (pGlu)m-r-(pHGlu-MHH)nvia conjugating polyglutamic acid (pGlu) with poly(γ-6-N-(methyldihexylammonium)hexyl-glutamate) (pHGlu-MHH). As shown in Fig. 5(a), the (pGlu)m-r-(pHGlu-MHH)n adopted a random coil conformation at physiological pH due to the intramolecular electrostatic interactions between the anionic carboxylate and cationic ammonium groups. As a consequence, both the antimicrobial activity and cytotoxicity were low. Under acidic conditions, the conformation of the (pGlu)m-r-(pHGlu-MHH)n transformed from a random coil to a helical one driven by the protonation of the carboxylate groups and the depletion of the side-chain electrostatic interaction. Meanwhile, potent antimicrobial activity was observed against Helicobacter pylori (H. pylori) in the stomach.61 In another work, Gong et al. designed a pH-responsive block polypeptide pLys-POIM-CA by post-polymerization modification of pLys with 1-(propylthio)acetic acid-3-octylimidazolium (POIM) and citraconic anhydride (CA).62 pLys-POIM-CA showed good cytocompatibility and low antimicrobial activity under physiological conditions. Once the polypeptide was placed in an acidic environment, enhanced antimicrobial efficacy was observed because the CA segments were readily hydrolyzed to expose the cationic ε-NH2 (Fig. 5(b)). Yang et al. designed an enzyme-sensitive adhesive AMP by coupling the Phe7-stat-Lys10 block with the poly-tyrosine (pTyr) segment.63 The former block showed excellent antimicrobial activity, while the latter segment was an adhesive precursor, which tended to be catalyzed by tyrosinase into poly-DOPA amino acid (pDOPA), endowing it with excellent adhesion. These synergistic features enabled the polypeptide to be an antibacterial coating on the implant surface via a facile one-step dip coating method. As a result, the polypeptide-based antibacterial coating exhibited excellent surface contact bactericidal activity against planktonic bacteria and desirable performance in biofilm inhibition. By injecting the polypeptide into a mouse subcutaneous implantation model, the polypeptide can in situ transform into an antibacterial coating with the aid of skin tyrosinase to prevent bacterial infection. In view of the above discussion, one can confirm that the assembly of the polypeptides showed the ability to improve their hydrolytic stability and antimicrobial efficacy. Furthermore, hyperbranched or star-like polypeptides with amphiphilic features have unique potential to control the antimicrobial selectivity and solve the contradictory relationship between antimicrobial activity and biocompatibility.
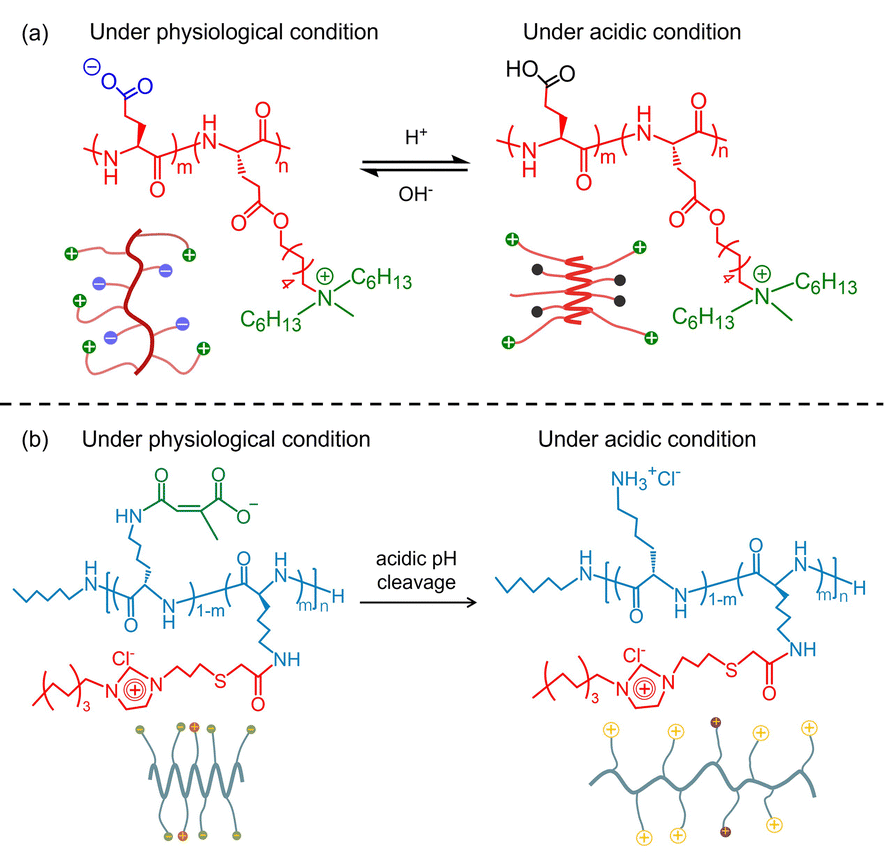 | ||
| Fig. 5 (a) Schematic illustration of the pH-responsive conformation transition of (pGlu)m-r-(pHGlu-MHH)n. Reproduced with permission from ref. 61. Copyright 2017 the National Academy of Sciences of the United States. (b) Schematic illustration of the pH-responsive conformation transition of pLys-POIM-CA. Reproduced with permission from ref. 62. Copyright 2021 Wiley-VCH GmbH. | ||
3. Peptide–polymer conjugates
Apart from the block polypeptide, peptide–polymer conjugates are another class of antimicrobial candidates. Tan et al. designed an amphiphilic peptide–polymer conjugate consisting of an aliphatic block, repeated phenylalanine–proline (Phe–Pro) segment, a polyethylene glycol (PEG) side chain, and a repeated Lys–Pro segment.46 As shown in Fig. 6(a), the aliphatic chain provided hydrophobicity to drive the self-assembly. The repeated Phe–Pro segment enhanced the membrane insertion capability. The authors argued that the Pro residues located at each end of each Phe can inhibit the protease degradation of the peptide chain.64 The repeated Lys–Pro segment provided a rich positive charge to ensure the strong electrostatic binding between the peptide and bacteria. The hydrophilic PEG side chain showed the ability to prevent non-specific protein adsorption and improve protease stability and biocompatibility. As a result, the self-assembled peptide–polymer nanoparticles showed broad-spectrum antibacterial activity, improved stability and good biocompatibility. In a subsequent study, the same group designed a pH-triggered peptide–polymer conjugate by introducing a pH-sensitive oligo-histidine (oligo-His) segment into the polypeptide sequence (Fig. 6(b)).65 The QRKLAAKLT segment encoded into the polypeptide sequence showed specificity and strong affinity to Pseudomonas aeruginosa (P. aeruginosa). Furthermore, the C-terminus of the peptide was aminated to enhance stability. At pH 7.4, the peptide–polymer conjugate self-assembled into nanofibers with a large size but low charge, showing long-term retention and good biocompatibility. Once reaching an acidic environment, the long nanofibers tended to transform into small nanoparticles with enhanced charges, which exhibited good membrane permeability and potent antimicrobial activity. | ||
| Fig. 6 (a) The molecular structure of the amphiphilic peptide–polymer conjugate and the schematic diagram of the peptide–polymer conjugate self-assembly into nanoparticles. Reproduced with permission from ref. 46. Copyright 2022 Wiley-VCH GmbH. (b) The molecular structure of the pH-responsive amphiphilic peptide–polymer conjugate and the schematic diagram of self-assembly and structural transformation of peptide–polymer nanoassemblies. Reproduced with permission from ref.65. Copyright 2023 Wiley-VCH GmbH. | ||
RFRRLRKKWRKRLKKI (labeled as T9W) showed selective antimicrobial activity against P. aeruginosa and low cytotoxicity. However, the strong ionic interactions among the cationic T9W and anionic proteins (albumin, α1-acid glycoprotein, and haptoglobin), and the sensitivity of T9W to protease digestion in serum largely limited its clinical application.66 To reduce the peptide–anion interactions and improve the stability of T9W, Yu and colleagues designed a kind of peptide–polymer conjugate (Fig. 7) by coupling mPEG1000 with T9W at the C-terminus (CT9W1000).67 Compared with the T9W alone, the self-assembled micelles of CT9W1000 exhibited higher stability under salt ion, serum, acid, base, and trypsin environments. A detailed investigation revealed that the CT9W1000 micelles exerted antimicrobial action through synergistic mechanisms, including membrane destruction, ROS accumulation and activation of the apoptosis-like death (ALD) pathway. In addition, the CT9W1000 micelles showed good biocompatibility and a remarkable therapeutic effect on the mouse acute pneumonia model. Similarly, Xu and co-workers demonstrated that the conjugation between AMPs and PEG can greatly reduce the cytotoxicity of the AMPs and improve the antimicrobial activity.68 Cui et al. designed a series of peptide–polymer conjugates via coupling Ac-PESKAIKA(pentenyl)LLKA(pentenyl)VSKERSKRSP-NH2 with linear/star-shaped PEG, varying in arm number and length. The authors found that increasing the arm number and decreasing the length of PEG are beneficial for improving bactericidal activity, lowering hemolysis, and enhancing resistance against proteolytic degradation.69 Instead of controlling the length and arm number of PEG, a ureido-pyrimidinone (UPy) modified linear PEG chain was conjugated with a naturally derived AMP (Lasio III, VNWKKILGKIIKVVK).70 The UPy moiety was able to drive the self-assembly of the polymer–polypeptide conjugate into short nanofibers or nanorods with enhanced antibacterial activity and modulable cytotoxicity.
 | ||
| Fig. 7 The molecular structure of T9W and poly(ethylene glycol) derivatives CT9W1000, Y = mPEG1000-NH2; the molecular weight of mPEG is 1000 Da. Reproduced with permission from ref. 67. Copyright 2023 American Chemical Society. | ||
By coupling PEG with a pH-responsive polypeptide (KLAKLAK)2 protected by 2,3-dimethyl maleic anhydride (DA),71 Hu and colleagues designed a PEG-(KLAKLAK)2–DA conjugate, which can recognize α-cyclodextrin coupled nitric oxide (NO) (α-CD-NO) or Ce6 (α-CD-Ce6) via host–guest interactions, and form stable nanoparticles (α-CD-Ce6-NO-DA) under physiological conditions (Fig. 8(a)). This kind of nanoparticle with negative surfaces showed long-term circulation in the blood. Once the DA moieties were hydrolyzed under acidic biofilm conditions, the nanoparticles underwent charge reversal to expose the positive lysine residues (Fig. 8(b)), which facilitated efficient penetration into the biofilm. Afterwards, overexpressed glutathione (GSH) triggered the rapid release of NO, leading to bacterial eradication (Fig. 8(c)). Simultaneously, the GSH concentration reduced significantly within the biofilm, facilitating photodynamic therapy (PDT). Furthermore, the released NO could react with reactive oxygen species (ROS), leading to the production of reactive nitrogen species (RNS) for improving the PDT efficacy. Following the efficiently penetrating biofilms and depleting GSH mechanism, this pH-responsive nanoparticle significantly improved the PDT efficiency at low photosensitizer doses and laser intensities, minimizing the adverse effects on healthy tissues. In another work, a pH-sensitive polypeptide DDDEEKRWRWRWC was coupled with a PEG chain to explore the antifouling coating on the tooth surface.72 The PEG enhanced the hydrophilicity and biological stability of the polypeptide. The DDDEK fragment of the polypeptide can specifically target towards hydroxyapatite (HA), enabling the conjugate to anchor stably and firmly on the tooth surface in a complex oral environment. The protonated arginine and tryptophan residues encoded in the sequence of the polypeptide endowed the conjugate with pH adaptive properties. This switchable property conferred reduced damage to beneficial flora and maintained the balance of oral microbiota. A polypeptide–PEG conjugate also could be explored as a hydrogel scaffold to improve the stability of AMPs.73 The entanglement of the PEG chain can form the first network, while the second network was established via self-assembly of the β-sheet polypeptide. The resulting double network significantly enhanced the storage modulus of the peptide–PEG hydrogel. Upon encapsulation of AMPs, the hydrogel network retained high antimicrobial activity while showing minimum cytotoxicity toward mammalian cells.
 | ||
| Fig. 8 (a) Schematic diagram of the preparation process of α-CD-Ce6-NO-DA nanocarriers; (b) schematic of acid-activated charge reversal of PEG-(KLAKLAK)2-DA at pH 5.5; and (c) schematic of the mechanisms of the MRSA biofilm associated infection eradication by synergistic effects between ROS and NO produced by α-CD-Ce6-NO-DA nanocarriers. Reproduced with permission from ref. 71. Copyright 2019 American Chemical Society. | ||
Chitosan (CS) is another kind of biocompatible polymer. Zhou and co-workers designed a kind of CS–polypeptide conjugate via coupling poly(Lys11-stat-Phe10) with the CS chain.74 The conjugate can form nanocapsules with enhanced antibacterial activity due to the locally concentrated positive charges. Moreover, the nanocapsule can efficiently encapsulate anticancer (doxorubicin) and antiepileptic (Dilantin) drugs to play a synergistic role. Since biofilms are characterized by water-filled channels and negatively charged extracellular polymeric substance (EPS), CS-PEG-(LeuLys)13 (Fig. 9(a)) has been designed for permeating biofilms.47 As shown in Fig. 9(b), CS-PEG-(LeuLys)13 can self-assemble into neutral nanoparticles (∼100 nm) with the polypeptide (LeuLys)13 located at the core of the sphere and the PEG covered on the surface. After coming into contact with the negatively charged P. aeruginosa biofilm, the neutral nanospheres can spontaneously disassemble to expose the polypeptide segments and combat the bacteria (Fig. 9(c)). The antibacterial efficiency (97.95%) of CS-PEG-(LeuLys)13 was significantly higher than that of individual polypeptides (41.58%). Qi et al. reported an enzyme-responsive chitosan–peptide conjugate composed of a chitosan backbone, an enzyme-cleavable sequence (GPLGVRGC) linked with a poly(ethylene glycol) (PEG, Mw = 2 KDa) terminal and an antimicrobial peptide CGGGKLAKLAKKLAKLAK.75 This molecular design enabled the CS–peptide conjugate to form stable nanoparticles with hydrophilic PEG chains located at the surface of the nanoparticles. When the peptide nanoparticles are placed in a bacterial infection environment, the gelatinase secreted by the bacteria can selectively cleave the GPLGVRGC segment and peel off the PEG protective layer, giving rise to structural transformation from nanoparticles to nanofibers. As a consequence, the α-helical antimicrobial peptide segments were exposed to the bulk solution, causing cell membrane rupture. Importantly, the in situ morphological transformation also increased the accumulation and retention of the CS–peptide conjugate at the site of infection, endowing high antimicrobial activity. Microcin J25 is a potential AMP, which shows excellent antibacterial ability against Gram-negative bacteria and good stability under enzyme and high temperature conditions. However, its application is largely restricted suffering from the narrow-spectrum antimicrobial activity. Yu et al. found that the conjugation of Microcin J25 with CS not only can enhance the antimicrobial efficacy but also achieve broad-spectrum antibacterial activity.76
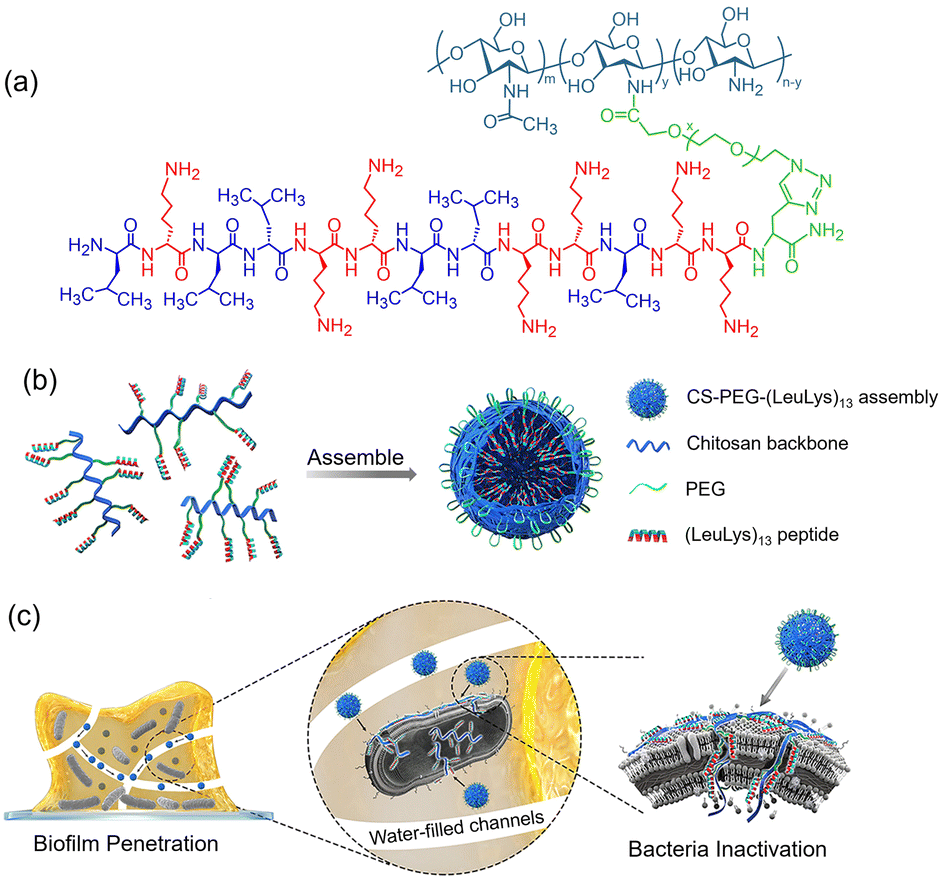 | ||
| Fig. 9 (a) The molecular structure of CS-PEG-(LeuLys)13; (b) schematic illustration of the self-assembled nanoparticles of CS-PEG-(LeuLys)13; (c) schematic illustration of biofilm penetration and bacterial inactivation of CS-PEG-(LeuLys)13 nanoparticles. Reproduced with permission from ref. 47. Copyright 2020 American Chemical Society. | ||
Recently, tri-block conjugates PPGn-(pGLu-Im)m were synthesized by conjugating poly(propylene glycol)(PPG) with imidazolium-modified polyglutamate (pGlu-Im), which can self-assemble into micelles in aqueous solution.77 The antimicrobial activity of the tri-block conjugate is related to the chain length of the cationic pGlu-Im. PPG34-(pGlu-Im)70 with a medium polypeptide chain length exhibited the best antimicrobial activity with a MIC of 25 μg mL−1 and good biocompatibility. Elongating the chain length of pGlu-Im led to increased cytotoxicity. To balance the antimicrobial activity and biocompatibility, a pH-sensitive polymer, maleimide-modified polyacrylic acid (PMAA), was coupled with the C-terminus of a cationic polypeptide (PEPc: NH2-C-VAQLESK-VAQLESK-VSKLESK-VSSLESK-COOH).78 The resulting PEPc–PMAA exhibited negatively charged carboxylate moieties and showed low toxicity at physiological pH. Under acidic conditions, the cationic PEPc block with positive residues can be exposed to bind on the negatively charged liposomes, due to the protonation of the carboxylate groups of PMAA. Meanwhile, the protonated PMAA block transitioned into a hydrophobic state, tending to insert and destroy the cell membranes of bacteria. This antimicrobial property of the pH-triggered peptide–polymer conjugate can only be activated near bacteria, giving rise to low system toxicity.
The conjugation of antimicrobial peptides and hydrophobic polymers is an alternative way to drive the assembly and improve the antimicrobial activity of peptides. M. L. Becker et al. designed a kind of polypeptide–polymer conjugate by coupling tritrpticin with the amphiphilic block copolymer poly(tert-butyl acrylate)-b-polystyrene (PAA-bPS).79 The entropic penalties associated with the aggregation of the hydrophobic PS block drove the polypeptide–polymer conjugate to self-assemble into water-dispersed micellar structures with PS segments as the core surrounded by the hydrophilic tritrpticin shell. Compared with the tritrpticin alone, the micellar assemblies exhibited better antimicrobial activity with a MIC of 13 μg mL−1 against both S. aureus and E. coli. In another work, amphiphilic polypeptide (pLys–pPhe) serving as a side chain was coupled with hydrophobic poly-2-methylallylamine.80 As illustrated in Fig. 10, the resultant polypeptide–polymer can self-assemble into colloidal nanoparticles covered with concentrated lysine groups, which can strongly bind to the anionic cell membrane of bacteria through multivalent electrostatic interactions. As a next step, the hydrophobic pPhe block tended to insert into the membrane, resulting in the death of bacteria. This kind of colloid nanoparticle exhibits broad-spectrum antibacterial efficacy against various bacteria with an MIC of 16 μg mL−1.
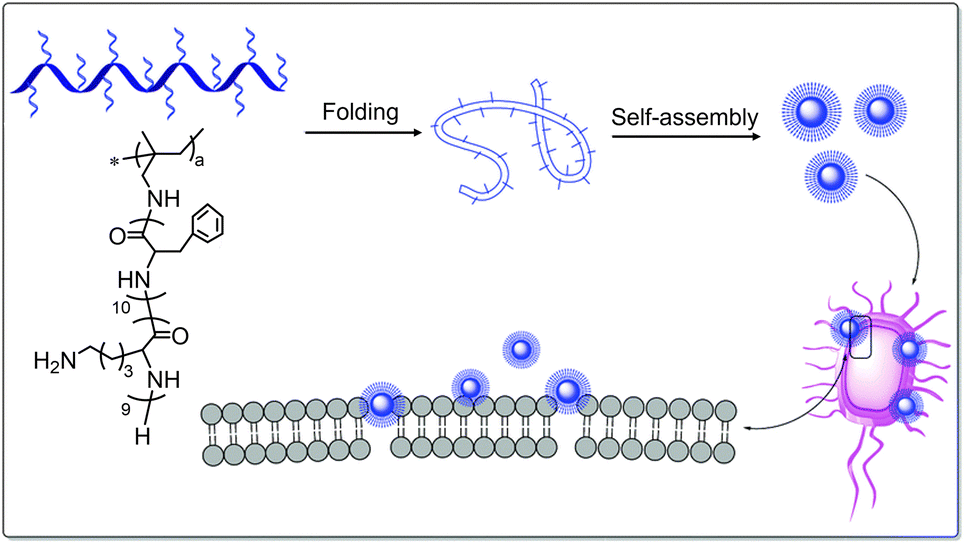 | ||
| Fig. 10 Schematic diagram of the self-assembly process and antibacterial mechanism of amphiphilic peptide-based pectinate polymers with primary amino groups. Reproduced with permission from ref. 80. Copyright 2019 Royal Society of Chemistry. | ||
Poly-ε-caprolactone (PCL) is a kind of biocompatible and biodegradable polymer, which has been typically utilized as biomaterials. In an earlier work, block-poly[phenylalanine-stat-lysine-stat-(lysine-folic acid)] (labeled as p[Phe12-stat-Lys9-stat-(Lys-FA)6]) was coupled with PCL19 to obtain a polypeptide–polymer conjugate PCL19-b-p[Phe12-stat-Lys9-stat-(Lys-FA)6].81 As shown in Fig. 11, the conjugate self-assembled into vesicular structures with the hydrophobic PCL19 block serving as the vesicle membrane while the hydrophilic p[Phe12-stat-Lys9-stat-(Lys-FA)6] block acting as vesicle corona. The vesicle membrane can load antibacterial drugs to inhibit bacteria, while the hydrophilic corona structure with highly concentrated cationic charges can strengthen the non-specific interactions between the peptide vesicle and the anionic membrane of bacteria, giving rise to synergistic antibacterial activity. Chen and colleagues reported a polypeptide–polymer conjugate [PCL34-b-pGlu30-b-p(Lys16-stat-Phe12)], which self-assembled into micelles with an anionic surface (ξ ≈ −26.7 mV) under alkaline conditions.82 The micelles exhibited a synergistic antibacterial effect due to the potent antimicrobial activity of the polypeptide chains together with the loaded drugs. In another report, a biodegradable and noncytotoxic tri-block copolymer polycaprolactone–polyethyleneglycol-3-poly-lysine (PCL–PEG–pLys) was also synthesized to improve the self-assembling ability and antimicrobial efficacy.83 In this case, the tri-block copolymer formed a water-dispersed vesicle with PCL as the vesicle membrane and PEG and pLys blocks as the vesicle coronas. The self-assembled vesicles exhibited good antimicrobial activity with a MIC of 62.5 μg mL−1. More importantly, the vesicles, functioning as carriers, can load antimicrobial drugs and exert controllable drug release via lipid enzyme-triggered degradation. Apart from PCL, poly-lactide (pLA),84 poly(vinyl alcohol)85 and amphiphilic ciprofloxacin peptide-based polymers (PACs)86 have been conjugated with polypeptides, respectively. All of the resulting micelles, nanofibers and nanoparticles with polypeptide crowns exhibited enhanced antimicrobial efficacy and biocompatibility.
 | ||
| Fig. 11 The molecular structure of PCL19-b-p[Phe12-stat-Lys9-stat-(Lys-FA)6] and the schematic diagram of the self-assembled vesicle and antimicrobial behaviour. Reproduced with permission from ref. 81. Copyright 2015 American Chemical Society. | ||
The conjugation of the polypeptide with polymers shows the following advantages: (1) hydrophobic polymers can greatly hasten the self-assembly of the polypeptides, which may simplify the molecular design of the peptide sequences; (2) the hydrophilic polymers have the ability to reduce the toxicity of the assemblies via shielding the highly concentrated cationic residues of polypeptides on the surface of the assemblies; (3) the resulting nanostructures can function as ideal carriers to elongate the circulation lifespan of the polypeptides and to effectively deliver the assemblies into the infection area before degradation. In addition, the polypeptide–polymer assemblies can load drug molecules to exert antimicrobial action in a cooperative manner.
4. Peptide/polymer complexes
Besides the self-assembly of polypeptides or polypeptide–polymer conjugates, co-assembly between peptides and other objects has recently become increasingly prevalent since the modular strategy opens up a simple and flexible way in the creation of antimicrobial systems. Wan et al. designed a class of antimicrobial hydrogels via ionic co-assembly of cationic polypeptide amphiphiles and anionic sodium alginate (SA).49 The incorporation of the pLys segment into the polypeptide amphiphiles endowed them with antimicrobial activity. Anionic SA was used to enhance the mechanical performance of the co-assembled hydrogels. The formed hydrogels showed potent antimicrobial activity. To balance the antimicrobial activity and the cytotoxicity of antimicrobial peptides, D. Pranantyo et al. designed a co-assembled system based on cationic peptide–Au particles and anionic polymers.87 As illustrated in Fig. 12(a), the cysteine-terminated oligopeptide or thiol-capped pLys have been grafted on the surface of Au particles to form antimicrobial peptide–Au nanoparticles. Anionic polymers (Fig. 12(b)), such as polyphenyleneethynylene ether with a sodium carboxylate side chain (PPE) or with sodium carboxylate and a PEG side chain (PPEG), were utilized to co-assemble with Au–peptide nanoparticles. As shown in Fig. 12(c), the ionic co-assembled systems were biologically inert under physiological conditions and showed low cytotoxicity because the cationic surface of the peptide–Au nanoparticles was shielded by the anionic counter polymers. Once the co-assemblies were exposed to bacterial envelopes, competitive substitution of counter polymers from the surface of peptide–Au nanoparticles was initiated by the negatively charged bacteria. As a result, the cationic peptide–Au nanoparticles can interact with the bacterial membrane and quickly disrupt and kill the bacteria. This work demonstrates that ion co-assembly is a powerful strategy for balancing the contradictory relationship between the antimicrobial activity and the cytotoxicity of nano-antimicrobial peptides. | ||
| Fig. 12 (a) The schematic drawing of the cationic peptide–Au nanoparticles; (b) anionic polymers PPE and PPEG; (c) ionic co-assembly of the cationic Au–peptide nanoparticles with anionic polymers (light blue) under physiological conditions, and the competitive displacement of the anionic polymers shield upon contact with bacteria (green). Reproduced with permission from ref. 87. Copyright 2021 American Chemical Society. | ||
In another work, Asmariah Ahmad et al. designed ionic complexes via the co-assembly of anionic colistimethate sodium (CMS) and a series of PEG-p(Lys/guanidinylated-Lys) cationic conjugates.88 The CMS is an anionic prodrug with poor activity, which can be rapidly hydrolyzed into cationic colistin exhibiting enhanced antimicrobial activity via cutting the sulfonic groups. However, the CMS is less stable and tends to be hydrolysed into colistin under physiological conditions before approaching the infection area. To reduce the nonspecific toxicity of colistin toward human cells and improve its bioavailability, the CMS/PEG-p(Lys/guanidinylated-Lys) ionic complex was explored as a carrier for on-site delivery of CMS. As shown in Fig. 13, the co-assembled complexes can form micelles or vesicles depending on the segment ratio of pLys to poly(guanidinylated-Lys). These self-assembled nanostructures tended to spontaneously dissociate in solution, enabling the sustained release of the cyclic CMS for maintaining long-term antimicrobial activity.
 | ||
| Fig. 13 Schematic diagram of the polyion complex resulting from colistimethate sodium (CMS) and a series of PEG-p(Lys/guanidinylated-Lys) conjugates. Reproduced with permission from ref. 88. Copyright 2022 Wiley-VCH GmbH. | ||
Similarly, a pH-responsive polyion nanocomplex (ε-pLys/DA@Magainin-I) was designed via electrostatic co-assembly of cationic Magainin-I and anionic DA modified ε-pLys. Under physiological conditions, the ε-pLys/DA@Magainin-I nanocomplex was stable, which can effectively reduce the cytotoxicity of Magainin-I. In the slightly acidic environment of the bacteria infected area, the nanocomplex will decompose into anionic ε-pLys and cationic Magainin-I, restoring the antimicrobial properties.89 Following the same way, an enzyme-responsive polyion complex has been generated by combining the cationic poly(ethylene imine) (PEI) with Cys-based anionic peptide targeted P. aeruginosa elastase (LasB).90 The introduction of Cys into the N- and C-termini of the anionic peptide can stabilize the polyion complex owing to the formation of covalent disulphide bonds. The polyion nanoparticles could be selectively degraded in the existence of P. aeruginosa elastase, leading to the release of the cationic PEI for inducing the apoptosis of P. aeruginosa. This type of antimicrobial system was selectively responsive to P. aeruginosa elastase. Thus, a suicide P. aeruginosa could be created. Recently, a dual responsive antimicrobial system has been designed via ionic co-assembly of the pLys dendrigraft (DGL) and tripolyphosphate anion (TPP) in aqueous solution (Fig. 14(a)).48 The resulting supraparticles (DGL/TPP) showed great ability to encapsulate diverse cargo molecules (Fig. 14(b)). Upon exposure to slightly acidic environments, these particles suffered a remarkable reduction in size and rapid disassembly to yield individual DGL blocks. Subsequently, model cargo molecules entrapped in DGL blocks were released via trypsin enzyme-mediated DGL degradation.
 | ||
| Fig. 14 (a) Schematic illustration of the co-assembly of the pLys dendrigraft (DGL) and tripolyphosphate (TPP); (b) responsive drug release of the co-assembled supraparticles triggered by pH and enzyme. Reproduced with permission from ref. 48. Copyright 2020, Elsevier. | ||
Apart from electrostatic interactions, the hydrophobic effect has also been explored to create co-assembled antimicrobial systems. As shown in Fig. 15, an antimicrobial peptide FKRLKKLISWIKRKRQQC-CONH2 (HNMC) was coupled with amphiphile 1,2-distearoyl-sn-glycero-3-phospho-ethanolamine (DSPE) through a PEG linker to form DSPE-PEG-HNMC.91 DSPE-PEG-HNMC can self-assemble into micelles under aqueous conditions, but the stability of the micelles is poor due to insufficient amphiphilic nature. After co-assembling with block polymer poly(lactic-co-glycolic acid)–poly(ethylene glycol) (PLGA–PEG), the DSPE-PEG-HNMC exhibited both structural stability and enzymatic hydrolysis stability. The hydrophilic PEG located on the surface of the micelles can effectively prevent the specific interactions between HNMC and enzymes, resulting in enhanced stability and elongated circulatory lifetime in the blood. However, the PEG chains cannot prevent the non-specific interaction between the cationic HNMC and the anionic membrane of drug-resistant bacteria, giving rise to the apoptosis of the bacteria via destroying the outer membrane.
 | ||
| Fig. 15 Schematic drawing of the biodegradable block polymer (PLGA–PEG), the antimicrobial DSPE-PEG-HNMC and the co-assembled micelles. Reproduced with permission from ref. 91. Copyright 2020 American Chemical Society. | ||
Harnessing the hydrophobic effect, a thermo-responsive hydrogel, consisting of poly(N-isopropylacrylamide) (PNIPAM) and a short peptide Ac-I3K-NH2 (I3K), could be conveniently constructed via one-step co-assembly (Fig. 16(a)).92 PNIPAM had a lower critical solution temperature (LCST) of 32–34 °C. Below the LCST, the PNIPAM chain was well-hydrated and adopted a fully expanded coil conformation. The interactions between the peptide nanofibrils and PNIPAM were weak, and the solution was in a free-flowing state. Above the LCST, the polymer chains collapsed into compact globules, which function as physical crosslinks to connect the peptide nanofibers into a networked hydrogel (Fig. 16(b)). This co-assembled hydrogel exhibited reversible sol–gel transition, which enabled the effective load and continuous release of an antimicrobial peptide G(IIKK)3I-NH2 in a controllable manner. The aforementioned results demonstrate that the non-covalent co-assembly shows significant potential in regulating the antimicrobial activity, stability and cytotoxicity without time-consuming synthesis because the complementary counterparts could be flexibly chosen from a broad range of natural and artificial building blocks. Apart from the pH-responsive system, the host–guest controlled antimicrobial polypeptide was designed by Huang and co-workers. Water-soluble cucurbit[7]uril (CB[7]) was mixed with ε-poly-lysine with the positive charge and hydrophobic components in an aqueous solution to form a polypseudorotaxane via host–guest interactions. The antibacterial efficacy of the co-assembled system can be well regulated by adjusting the ratio of CB[7] to the cationic polypeptide.93 Since the formation of the polypeptides/polymer complexes depends on the modular co-assembly via non-covalent interactions, the main charm of these antimicrobial systems is the simplicity and availability in synthesis by neat choice of the starting components and the flexible regulation of the bioactivity of the assemblies by controlling the stoichiometry.
 | ||
| Fig. 16 (a) The fabrication of reversible thermoresponsive peptide–PNIPAM hydrogels; (b) schematic diagrams of the proposed states of the I3K/PNIPAM networks at a temperature either below or above the PNIPAM LCST. Reproduced with permission from ref. 92. Copyright 2019 American Chemical Society. | ||
5. Summary and outlook
The peptide/polymer assembly has shown astonishing growth in the creation of nano-antimicrobial systems. In this review, we describe the assembly of block-polypeptides, peptide–polymer conjugates and peptide/polymer complexes in aqueous solution. Either hydrophobic or hydrophilic polymer (or polypeptide) blocks have been employed to combine with cationic polypeptides. The hydrophobic blocks enabled the cationic polypeptides to assemble into stable assemblies, such as micelles, vesicles, and nano-sheets, while the concentrated cationic polypeptides located on the surface of the assemblies enhanced the electrostatic binding affinity. Meanwhile, the assemblies with tight stacking of polypeptides tended to prevent the contact between protease and the hydrolysis sequences of polypeptides. Beneficial from the interfacial and bulk effects of the assemblies, the antimicrobial efficacy, the hydrolysis stability, in vivo circulation lifespan and bioavailability of polypeptides could be largely improved. Overall, the conjugation or complexation between polypeptides and polymers offers a straightforward protocol for the development of effective AMPs. It is thus expected that the principles of AMP design gleaned by studying sequence–property relationships might be unshackled from lots of trial and error constraints. Despite the advantages, the crucial conflict observed between the antimicrobial efficacy and toxicity of the assemblies needed to be confronted seriously. In this vein, the conjugation of hydrophilic polymers with polypeptides provided an alternative way of balancing the conflict via shielding the concentrated positive charges of peptide/polymer assemblies. However, ongoing efforts should be invested to understand the action mechanism behind the apparent phenomenon. Dynamic assembly of peptide–polymer conjugates or peptide/polymer complexes may afford a promising solution, where the antimicrobial capability of the peptide/polymer assemblies can only be on-demand activated by environmental stimuli. Although dynamic and responsive assemblies have been reported, the research remains in its infancy. There is still significant room for further development. With extensive validation, it becomes clear that in vitro investigation of polypeptide–polymer antimicrobial assemblies cannot directly reflect the complicated in vivo environments. In the future, translating the design principles into clinically applicable materials needs a shift in antimicrobial studies from in vitro solutions to in vivo environments by collaboration with multidisciplinary researchers from chemistry, biology, and medicine.Author contributions
Conceptualization: W. L.; software: J. S. and Y. C.; validation: W. L.; formal analysis: H.Z., J. S., S.N., and W. L.; data curation: H. Z., J. S., Y. C., S. N. and W. L.; writing – original draft preparation: H. Z.; writing – review and editing: W. L.; supervision: W. L.; project administration: W. L.; funding acquisition: W. L. All authors have read and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China under grant number 22372069 and the Natural Science Foundation of Jilin Province under grant number 20220101049JC.Notes and references
- D. E. Morris, D. W. Cleary and S. C. Clarke, Front. Microbiol., 2017, 8, 1041 CrossRef PubMed.
- M. Kolpen, K. N. Kragh, J. B. Enciso, D. Faurholt-Jepsen, B. Lindegaard, G. B. Egelund, A. V. Jensen, P. Ravn, I. H. M. Mathiesen, A. G. Gheorge, F. B. Hertz, T. Qvist, M. Whiteley, P. Ø. Jensen and T. Bjarnsholt, Thorax, 2022, 77, 1015–1022 CrossRef.
- J. Y. Quek, E. Uroro, N. Goswami and K. Vasilev, Mater. Today Chem., 2022, 23, 100606 CrossRef CAS.
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 Search PubMed.
- E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520–526 CrossRef CAS.
- E. M. Halawa, M. Fadel, M. W. Al-Rabia, A. Behairy, N. A. Nouh, M. Abdo, R. Olga, L. Fericean, A. M. Atwa, M. El-Nablaway and A. Abdeen, Front. Pharmacol., 2024, 14, 1305294 CrossRef PubMed.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- D. Hu, L. Zou, Y. Gao, Q. Jin and J. Ji, View, 2020, 1, 20200014 CrossRef.
- G. Xiao, J. Li and Z. Sun, Int. J. Mol. Sci., 2023, 24, 15493 CrossRef CAS PubMed.
- R. T. Khan, V. Sharma, S. S. Khan and S. Rasool, Front. Microbiol., 2024, 15, 1455759 CrossRef.
- Y. Yang, P. He, Y. Wang, H. Bai, S. Wang, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16239–16242 CrossRef CAS PubMed.
- N. E. Eleraky, A. Allam, S. B. Hassan and M. M. Omar, Pharmaceutics, 2020, 12, 142 CrossRef CAS.
- X. Li, H. Bai, Y. Yang, J. Yoon, S. Wang and X. Zhang, Adv. Mater., 2018, 31, 1805092 CrossRef.
- H. Moulahoum, F. G. Zamani, S. Timur and F. Zihnioglu, Probiotics Antimicrob. Proteins, 2019, 12, 48–63 CrossRef PubMed.
- R. Spohn, L. Daruka, V. Lázár, A. Martins, F. Vidovics, G. Grézal, O. Méhi, B. Kintses, M. Számel, P. K. Jangir, B. Csörgő, Á. Györkei, Z. Bódi, A. Faragó, L. Bodai, I. Földesi, D. Kata, G. Maróti, B. Pap, R. Wirth, B. Papp and C. Pál, Nat. Commun., 2019, 10, 4538 CrossRef PubMed.
- G. Li, Z. Lai and A. Shan, Adv. Sci., 2023, 10, 2206602 CrossRef CAS.
- J. Xuan, W. Feng, J. Wang, R. Wang, B. Zhang, L. Bo, Z.-S. Chen, H. Yang and L. Sun, Drug Resist. Updates, 2023, 68, 100954 CrossRef CAS PubMed.
- M. Mohamed-Benkada, Y. F. Pouchus, P. Vérité, F. Pagniez, N. Caroff and N. Ruiz, Chem. Biodiversity, 2016, 13, 521–530 CrossRef CAS.
- P. Zou, W.-T. Chen, T. Sun, Y. Gao, L.-L. Li and H. Wang, Biomater. Sci., 2020, 8, 4975–4996 RSC.
- H. Kim, J. H. Jang, S. C. Kim and J. H. Cho, Eur. J. Med. Chem., 2020, 185, 111814 CrossRef CAS.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- W. F. Porto, L. N. Irazazabal, V. Humblot, E. F. Haney, S. M. Ribeiro, R. E. W. Hancock, A. Ladram and O. L. Franco, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129633 CrossRef CAS PubMed.
- Z. Qiao, W. Zhang, Y. Wu, W. Jiang, N. Shao, J. Xie, G. Xia, Q. Chen, Z. Liu, J. Zou, J. Gu, S. Luan, H. Lin and R. Liu, Sci. China: Chem., 2023, 66, 1824–1833 CrossRef CAS.
- Q. Chen, Y. Xu, J. Feng, X. Lv, X. Fu, S. Yuan and Z. Li, Macromol. Biosci., 2023, 24, 2300388 CrossRef.
- Y. Yang, Z. Yu, Z. Ba, X. Ouyang, B. Li, P. Yang, J. Zhang, Y. Wang, Y. Liu, T. Yang, Y. Zhao, X. Wu, C. Zhong, H. Liu, Y. Zhang, S. Gou and J. Ni, Eur. J. Med. Chem., 2024, 271, 116451 CrossRef CAS.
- A. Doherty, R. Murphy, A. Heise, F. Fitzpatrick and D. Fitzgerald-Hughes, J. Med. Microbiol., 2024, 73, 001886 CrossRef CAS.
- L. Chen, N. Patrone and J. F. Liang, Biomacromolecules, 2012, 13, 3327–3333 CrossRef CAS.
- X. Zhu, W. Tang, X. Cheng, H. Wang, T. Sang and Z. Ye, Coatings, 2022, 12, 1456 CrossRef CAS.
- R. Lei, J. Hou, Q. Chen, W. Yuan, B. Cheng, Y. Sun, Y. Jin, L. Ge, S. A. Ben-Sasson, J. Chen, H. Wang, W. Lu and X. Fang, ACS Nano, 2018, 12, 5284–5296 CrossRef CAS.
- L. Sun, A. Li, Y. Hu, Y. Li, L. Shang and L. Zhang, Part. Part. Syst. Charact., 2019, 36, 1800420 CrossRef.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Tan, W. Fan, S. S. Venkatraman, L. Li and Y. Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS.
- C. X. Chen, F. Pan, S. Z. Zhang, J. Hu, M. W. Cao, J. Wang, H. Xu, X. B. Zhao and J. R. Lu, Biomacromolecules, 2010, 11, 402–411 CrossRef CAS.
- Z. Ma, X. Liu, J. Nie, H. Zhao and W. Li, Biomacromolecules, 2022, 23, 1302–1313 CrossRef CAS.
- R. Teng, Y. Yang, Z. Zhang, K. Yang, M. Sun, C. Li, Z. Fan and J. Du, Adv. Funct. Mater., 2023, 2214454 CrossRef CAS.
- K. Li, X. Ju, X. Li, G. Lu, J. Ou, D. Xu, C. Wan, M. Zhu, C. Du, Y. Tian and Z. Niu, Chem. Eng. J., 2024, 489, 151475 CrossRef CAS.
- H. Sun, X. Fu, C. Yang, C. Yuan and X. Yan, Curr. Opin. Colloid Interface Sci., 2024, 73, 101828 CrossRef CAS.
- Y. Wang, Y. Zhang, R. Su, Y. Wang and W. Qi, J. Mater. Chem. B, 2024, 12, 5061–5075 RSC.
- X.-Y. Guo, L. Yi, J. Yang, H.-W. An, Z.-X. Yang and H. Wang, Chem. Commun., 2024, 60, 2009–2021 RSC.
- R. Mu, D. Zhu, S. Abdulmalik, S. Wijekoon, G. Wei and S. G. Kumbar, Bioact. Mater., 2024, 35, 181–207 CAS.
- J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2592–2595 CrossRef CAS.
- X. Xie, B. Gao, Z. Ma, J. Liu, J. Zhang, J. Liang, Z. Chen, L. Wu and W. Li, CCS Chem., 2021, 3, 1949–1962 CrossRef CAS.
- J. Wang, D. L. Cooper, W. Zhan, D. Wu, H. He, S. Sun, S. T. Lovett and B. Xu, Angew. Chem., Int. Ed., 2019, 58, 10631–10634 CrossRef CAS PubMed.
- S. Gao, X. Yan, G. Xie, M. Zhu, X. Ju, P. J. Stang, Y. Tian and Z. Niu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23437–23443 CrossRef CAS.
- F. Tian, R. C. Guo, C. Wu, X. Liu, Z. Zhang, Y. Wang, H. Wang, G. Li and Z. Yu, Angew. Chem., Int. Ed., 2024, 63, e202404703 CrossRef CAS.
- P. Tan, Q. Tang, S. Xu, Y. Zhang, H. Fu and X. Ma, Adv. Sci., 2022, 9, 2105955 CrossRef CAS.
- X. Ju, J. Chen, M. Zhou, M. Zhu, Z. Li, S. Gao, J. Ou, D. Xu, M. Wu, S. Jiang, Y. Hu, Y. Tian and Z. Niu, ACS Appl. Mater. Interfaces, 2020, 12, 13731–13738 CrossRef CAS PubMed.
- M. L. Agazzi, S. E. Herrera, M. L. Cortez, W. A. Marmisolle and O. Azzaroni, Colloids Surf., B, 2020, 190, 110895 CrossRef CAS.
- Y. Wan, L. Liu, S. Yuan, J. Sun and Z. Li, Langmuir, 2017, 33, 3234–3240 CrossRef CAS.
- J. Peng, D. Cao, Z. He, J. Guo, P. Hapala, R. Ma, B. Cheng, J. Chen, W. J. Xie, X.-Z. Li, P. Jelínek, L.-M. Xu, Y. Q. Gao, E.-G. Wang and Y. Jiang, Nature, 2018, 557, 701–705 CrossRef CAS PubMed.
- Y. Tian, Y. Song, Y. Xia, J. Hong, Y. Huang, R. Ma, S. You, D. Guan, D. Cao, M. Zhao, J. Chen, C. Song, K. Liu, L.-M. Xu, Y. Q. Gao, E.-G. Wang and Y. Jiang, Nat. Nanotechnol., 2023, 19, 479–484 CrossRef PubMed.
- A. Song, A. A. Rane and K. L. Christman, Acta Biomater., 2012, 8, 41–50 CrossRef CAS PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. H. Wong, Y.-Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 16162 CrossRef CAS.
- S. J. Shirbin, I. Insua, J. A. Holden, J. C. Lenzo, E. C. Reynolds, N. M. O'Brien-Simpson and G. G. Qiao, Adv. Healthcare Mater., 2018, 7, 1800627 CrossRef.
- S. J. Lam, E. H. H. Wong, N. M. O’Brien-Simpson, N. Pantarat, A. Blencowe, E. C. Reynolds and G. G. Qiao, ACS Appl. Mater. Interfaces, 2016, 8, 33446–33456 CrossRef CAS.
- J. Zhao, Z. Dong, H. Cui, H. Jin and C. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 42058–42067 CrossRef CAS PubMed.
- Z. Zhang, X. Wang, J. Liu, H. Yang, H. Tang, J. Li, S. Luan, J. Yin, L. Wang and H. Shi, Angew. Chem., Int. Ed., 2024, 63, e202318011 CrossRef CAS PubMed.
- B. Cheng, H. Cui, N. Zhang, H. Feng, D. Chu, B. Cui, Z. Li, J. Zhang, S. Cao and J. Li, ACS Appl. Polym. Mater., 2022, 4, 7250–7257 CrossRef CAS.
- J. Gao, M. Wang, F. Wang and J. Du, Biomacromolecules, 2016, 17, 2080–2086 CrossRef CAS.
- J. Zhu, J. Tian, C. Yang, J. Chen, L. Wu, M. Fan and X. Cai, Small, 2021, 17, 2101495 CrossRef CAS.
- M. Xiong, Y. Bao, X. Xu, H. Wang, Z. Han, Z. Wang, Y. Liu, S. Huang, Z. Song, J. Chen, R. M. Peek, Jr., L. Yin, L. F. Chen and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12675–12680 CrossRef CAS.
- C. Gong, J. Sun, Y. Xiao, X. Qu and M. Lang, Adv. Healthcare Mater., 2021, 10, 2101244 CrossRef CAS.
- K. Yang, D. Liu, R. Teng, C. Li, Z. Fan and J. Du, ACS Biomater. Sci. Eng., 2023, 9, 1900–1908 CrossRef CAS.
- J. Wang, J. Song, Z. Yang, S. He, Y. Yang, X. Feng, X. Dou and A. Shan, J. Med. Chem., 2019, 62, 2286–2304 CrossRef CAS PubMed.
- P. Tan, C. Wu, Q. Tang, T. Wang, C. Zhou, Y. Ding, H. Fu, S. Xu, Y. Feng, Y. Zhang, Q. Dai and X. Ma, Adv. Mater., 2023, 2210766 CrossRef CAS.
- X. Zhu, A. Shan, Z. Ma, W. Xu, J. Wang, S. Chou and B. Cheng, Antimicrob. Agents Chemother., 2015, 59, 3008–3017 CrossRef CAS.
- W. Yu, Y. Sun, W. Li, X. Guo, X. Liu, W. Wu, W. Yu, J. Wang and A. Shan, ACS Appl. Mater. Interfaces, 2023, 15, 494–510 CrossRef CAS PubMed.
- D. Xu, Q. Ran, Y. Xiang, J. Linhai, B. M. Smith, F. Bou-Abdallah, R. Lund, Z. Li and H. Dong, RSC Adv., 2016, 6, 15911–15919 RSC.
- Z. Cui, M. A. Crawford, B. A. Rumble, M. M. Krogh, M. A. Hughes and R. A. Letteri, ACS Polym. Au, 2023, 4, 45–55 CrossRef PubMed.
- J. Song, M. G. J. Schmitz, M. Riool, S. Guo, S. A. J. Zaat and P. Y. W. Dankers, J. Polym. Sci., 2023, 61, 2866–2877 CrossRef CAS.
- D. Hu, Y. Deng, F. Jia, Q. Jin and J. Ji, ACS Nano, 2020, 14, 347–359 CrossRef CAS.
- Q. Xin, Y. Zhang, P. Yu, Y. Zhao, F. Sun, H. Zhang, Z. Ma, S. Sun, X. Yang, S. Tao, X. Xu, C. Ding and J. Li, Chem. Mater., 2024, 36, 1691–1706 CrossRef CAS.
- H. Asokan-Sheeja, K. Awad, J. Xu, M. Le, J. N. Nguyen, N. Nguyen, T. P. Nguyen, K. T. Nguyen, Y. Hong, V. G. Varanasi, X. Liu and H. Dong, Biomacromolecules, 2024, 25, 2814–2822 CrossRef CAS.
- C. Zhou, M. Wang, K. Zou, J. Chen, Y. Zhu and J. Du, ACS Macro Lett., 2013, 2, 1021–1025 CrossRef CAS PubMed.
- G. B. Qi, D. Zhang, F. H. Liu, Z. Y. Qiao and H. Wang, Adv. Mater., 2017, 29, 1703461 CrossRef.
- H. Yu, Z. Ma, S. Meng, S. Qiao, X. Zeng, Z. Tong and K. C. Jeong, Carbohydr. Polym., 2021, 253, 117309 CrossRef CAS PubMed.
- Z. Shi, X. Zhang, Z. Yu, F. Yang, H. Liu, R. Xue, S. Luan and H. Tang, Biomacromolecules, 2021, 22, 2373–2381 CrossRef CAS PubMed.
- S. Wang, Y. Sun, S. Xu and H. Liu, Langmuir, 2021, 37, 8840–8846 CrossRef CAS PubMed.
- M. L. Becker, J. Liu and K. L. Wooley, Biomacromolecules, 2005, 6, 220–228 CrossRef CAS PubMed.
- J.-B. Zhen, M.-H. Zhao, Y. Ge, Y. Liu, L.-W. Xu, C. Chen, Y.-K. Gong and K.-W. Yang, Biomater. Sci., 2019, 7, 4142–4152 RSC.
- M. Wang, C. Zhou, J. Chen, Y. Xiao and J. Du, Bioconjugate Chem., 2015, 26, 725–734 CrossRef CAS.
- L. Chen, Y. Hong, S. He, Z. Fan and J. Du, Acta Phys.-Chim. Sin., 2021, 37, 1910059 Search PubMed.
- C. Zhou, X. Zhou and X. Su, RSC Adv., 2017, 7, 39718–39725 RSC.
- Y. Xi, T. Song, S. Tang, N. Wang and J. Du, Biomacromolecules, 2016, 17, 3922–3930 CrossRef CAS.
- N. Keikha, M. H. Yadegari, M. Rajabibazl, J. Amani and S. Hosseinzadeh, Infect., Genet. Evol., 2019, 70, 36–41 CrossRef CAS.
- J.-B. Zhen, J.-J. Yi, B.-X. Liu, Y.-J. Liu, X.-Y. Bu, X.-J. Wu and D. Tang, New J. Chem., 2023, 47, 22377–22387 RSC.
- D. Pranantyo, C. Raju, Z. Si, X. Xu, K. Pethe, E. T. Kang and M. B. Chan-Park, Nano Lett., 2021, 21, 899–906 CrossRef CAS PubMed.
- A. Ahmad, T. Nii, T. Mori, Y. Katayama, M. Toyofuku and A. Kishimura, Macromol. Rapid Commun., 2022, 2200316 CrossRef CAS.
- S. Wang, Y. Yu, H. Li, Y. Huang, J. Wang, Q. Jin and J. Ji, J. Polym. Sci., 2022, 60, 2289–2297 CrossRef CAS.
- I. Insua, E. Liamas, Z. Zhang, A. F. Peacock, A. M. Krachler and F. Fernandez-Trillo, Polym. Chem., 2016, 7, 2684–2690 RSC.
- S. C. Park, C. Ko, H. Hyeon, M. K. Jang and D. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 54306–54315 CrossRef CAS PubMed.
- M. Cao, Y. Wang, X. Hu, H. Gong, R. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- Z. Huang, H. Zhang, H. Bai, Y. Bai, S. Wang and X. Zhang, ACS Macro Lett., 2016, 5, 1109–1113 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |