
Cellulose-based bioactive material and turmeric-impregnated flexible and biocompatible scaffold for bone tissue engineering applications†
Shital S.
Shendage
a,
Kranti
Kachare
a,
Kajal
Gaikwad
b,
Shivaji
Kashte
b,
Fu-Der
Mai
 c and
Anil Vithal
Ghule
*a
c and
Anil Vithal
Ghule
*a
aGreen Nanotechnology Laboratory, Department of Chemistry, Shivaji University, Kolhapur 416004, India. E-mail: avg_chem@unishivaji.ac.in
bDepartment of Stem Cell and Regenerative Medicine, Centre for Interdisciplinary Research, D. Y. Patil Education Society (Institution Deemed to Be University), Kolhapur, India
cDepartment of Biochemistry and Molecular Cell Biology, School of Medicine, College of Medicine, Taipei Medical University, No. 250, Wuxing St., Taipei 11031, Taiwan
First published on 11th February 2025
Abstract
Metal transplants, autografts, and allografts are currently used for the treatment of bone-related problems, but each comes with inherent limitations. However, advances in science and technology have underscored the need for the development of cost-effective, eco-friendly, and customized architectural scaffolds with desirable porosity and mechanical strength. Additionally, the synthesis of sustainable scaffolds using biowaste is being studied to decrease environmental pollution. Green fabrication of scaffolds has an inestimable influence on decreasing production costs and toxicity while increasing biological compatibility. With this motivation, in the present study, a 70S30C calcium silicate bioactive material (BM) was synthesized through a simple precipitation method, using recycled rice husk (as a silica source) and eggshells (as a calcium source). Further, the BM and turmeric powder (Tm) were impregnated onto cellulose-based cotton fabric (CF), considering its easy availability, flexibility, mechanical strength, and cost-effective nature. The prepared scaffolds were characterized using UV-visible spectroscopy, XRD, FTIR spectroscopy, SEM, and EDS mapping. Further, in vitro bioactivity and degradation studies were performed in simulated body fluid (SBF). The in vitro haemolysis study revealed less than 5% haemolysis. Ex ovo CAM results showed good neovascularization. Both in vitro and in vivo biocompatibility studies demonstrated non-toxic nature. Furthermore, in vivo osteogenesis results showed bone regeneration capacity, as confirmed by X-ray and histological analysis. Thus, the CF template impregnated with BM and Tm acts as a porous, flexible, bioactive, degradable, haemocompatible, osteogenic, antibacterial, cost-effective, and eco-friendly scaffold for bone tissue engineering applications.
1. Introduction
The demand for bone defect treatment resulting from traffic accidents, bone infections, sports injuries, and the surgical resection of bone tumors is progressively increasing. Although bone can heal itself, it fails when the bone defects exceed the critical size. As a treatment, a diverse array of transplant techniques, such as metal transplants, autografts, and allografts, have been used.1 Currently, autograft treatment remains the unique standard because of its biocompatibility, osteoconductive, and osteoinductive properties. However, autografts suffer from an inadequate supply and donor site morbidity at the site where the implant is harvested, while the use of allografts is relatively less due to the risk of disease transmission and graft rejection.2,3 Considering these challenges and the demand for bone defect treatments, the orthopedic field has focused on developing osteoconductive, osteoinductive, bioactive, biodegradable, and porous scaffolds. In addition, the development of multifunctional and tailorable flexible scaffolds is of great significance, as flexible scaffolds directly influence the healing process by mimicking the bone properties. Specifically, bone consists of both rigid and elastic components. If the scaffold is rigid, it might fail to replicate the natural environment. In contrast, a flexible scaffold can adapt to the dynamic mechanical forces of the body, such as movement and weight-bearing, which helps in promoting bone growth and remodelling. A scaffold with flexibility can effectively distribute mechanical stress, minimizing the risk of secondary fractures. In addition, bone defects often have complex shapes, and flexible scaffolds can fit with these irregularities, providing better integration with the surrounding tissue.4Further, considering the properties for a scaffold in bone defect treatment, it should give structural strength and create an environment favorable to cell proliferation, adhesion, infiltration, and migration until new tissue forms around the injured area.5,6 The scaffold should possess flexibility, biocompatibility, degradability, porosity, and antibacterial activity.7 In addition, one of the most significant properties of the scaffold is its design, which must balance porosity and mechanical strength.8–10 In search for these properties, to make scaffolds, biomaterials, such as calcium silicate bioactive material (BM), wollastonite, and polymers, have received more attention in the biomedical field because they are cost-effective, easy to store, carry, and prepare.11–14
Among them, the BM is widely used for bone treatment because of its ability to bond with bone tissue compared to other biomaterials. In addition, BM possesses bioactivity, osteogenesis, angiogenesis, biocompatibility, and osteointegration properties, and thus, BM acts as a foundation base for future breakthroughs in bone regeneration application.15 Apart from this, the BM has low mechanical strength. Based on the literature, researchers have synthesized composites of BM with polymers because polymers help enhance mechanical strength. Polymers, such as chitosan, gelatin, cellulose, collagen, polycaprolactone, and polyvinyl alcohol, were used owing to their biodegradability and mechanical properties.2,16,17 Generally, electrospinning, thermally induced phase separation, foaming, and 3D printing techniques are used for the preparation of BM/polymer composite scaffolds.18 In particular, 3D printing is the most recommended technique for obtaining composite scaffolds.19 However, it requires high manufacturing budgets (materials price, post-processing costs, and preservation costs), and its availability is limited.20 As an alternative, research has focused on the synthesis of cost-effective scaffolds for bone regeneration applications. In the search for synthesis of a cost-effective scaffold, it was found that researchers used cotton fabric as a template to create a porous scaffold.20 The cotton fabric consists of 90% cellulose, and the cellulose is a natural and biocompatible polymer with D-glucose residues (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) linked with β-(1 → 4)-glycosidic bonds.21 In addition, cellulose is a semi-flexible, stiff, and rigid molecule because it consists of intramolecular hydrogen bonding. Thus, cellulose-based cotton fabric (CF) was used in the present study to make the scaffold porous and sufficiently strong to withstand bone tissue. The porous nature helps with cell proliferation and cell adhesion, while the CF scaffold gives strength, which acts as a key factor in bone regeneration.11
000) linked with β-(1 → 4)-glycosidic bonds.21 In addition, cellulose is a semi-flexible, stiff, and rigid molecule because it consists of intramolecular hydrogen bonding. Thus, cellulose-based cotton fabric (CF) was used in the present study to make the scaffold porous and sufficiently strong to withstand bone tissue. The porous nature helps with cell proliferation and cell adhesion, while the CF scaffold gives strength, which acts as a key factor in bone regeneration.11
Furthermore, infections create serious and devastating complications in millions of patients, and the risk of infection due to open fractures is estimated to be up to 30%. Infections can occur during surgery, hematogenous bacteraemia, nearby infection site, biofilm formation, etc., which lead to significant morbidity and even death and or require complete implant removal, long-term antibiotic therapy, and surgical debridement,22 and thus, to circumvent these issues, the development of an antibacterial scaffold is urgently needed. Generally, metal ions (silver, copper, iron, etc.) are used as dopants to enhance the antibacterial activity of the BM and composites. However, high metal doping can cause side effects on the body; thus, a low metal doping percentage is required to optimize the desired properties.23–26 In addition to this and to address the challenge, in the present study, a naturally derived antibacterial turmeric powder (Tm) is also used. Curcuma longa (Turmeric) derived from Tm rhizome is a biologically active, natural polyphenolic phytochemical that belongs to the Zingiberaceae family and has been widely used in the treatment of bacterial infection in India since ancient times.27,28 Furthermore, curcumin has antioxidant, antitumor, and anti-inflammatory properties and has proven to be useful as a therapeutic material for bone regeneration and is also considered for wound healing, cardiovascular ailments, and diabetes treatment.22,28–30
Overviewing all the problems, in the present study, a scaffold with suitable flexibility, porosity, mechanical strength, and antibacterial activity is prepared using a cost-effective and eco-friendly approach. Primarily, the 70S30C BM was synthesized by applying a simple precipitation method using recycled rice husk (silica source) and eggshell (calcium source) biowaste materials without the use of an acid or binder. Furthermore, the synthesis of 70S30C from recycled biowaste offers sustainability, cost-effectiveness, and eco-friendly aspects. In addition, the 70S30C offers a balanced composition (70% silica (SiO2) and 30% calcium oxide (CaO)) that ultimately helps with bioactivity, degradation, bone regeneration, etc. In addition, recycled biowaste acts as a source of sustainable, economical, and scalable biomaterials to synthesize potentially therapeutic materials. Further, the Tm powder was synthesized by cleaning, chopping, drying, and grinding Tm rhizomes, which act as antibacterial and cost-effective materials. Further, the BM and Tm were coated on the CF by stirring and sonication, which further acts as a cost-effective approach. The synthesized samples were examined using UV-visible, XRD, FTIR, SEM, and EDS mapping analyses. The samples were further investigated owing to their mechanical properties, such as antibacterial, angiogenesis, haemolysis, in vitro bioactivity, degradation, in vitro and in vivo biocompatibility, and in vivo bone regeneration. It is important to note that the prepared scaffold is flexible and can bend or compress to fit into irregular wounds. Thus, the present work aims to synthesize biomaterials and make them more sustainable not only in the biomedical field but also in society by reducing their environmental impact.
2. Experimental
2.1 Synthesis of silica and calcium oxide
The rice husk was used as a precursor of silica taken from a rice mill in Gokul Shirgaon, Kolhapur, India. First, it was cleaned using 0.5 N hydrochloric acid (HCl-Sigma Aldrich), neutralized, and washed with distilled water (D.W.). After that, the cleaned rice husk was dried in an oven and calcined at 600 °C for 4 h. In the meantime, the collected eggshells (as a source of Ca, collected from the local market) were cleaned with D.W., dried, ground into fine powder, and finally calcined at 900 °C for 2 h to obtain calcium oxide.2.2 Synthesis of BM
The 70S30C BM (SiO2:CaO = 1:0.4) was synthesized by applying a simple precipitation method. In brief, 0.7 g of silica obtained from rice husk was added in 2 N sodium hydroxide (NaOH, Sigma-Aldrich) with stirring and heating for the formation of sodium silicate solution. Meanwhile, 0.3 g of calcium oxide obtained from the eggshell was dispersed in ammonium hydroxide (NH4OH, Sigma-Aldrich) to form calcium hydroxide. Then, the sodium silicate was added to calcium hydroxide under stirring and heating for 30 min, followed by 30 min of sonication. The solution was then kept overnight to allow homogenization. After that, the resultant solution was washed, filtered, dried, and calcined at 700 °C for 2 h in an air atmosphere.
2.3 Synthesis of Tm powder
The Tm was collected from a farm in Sangli, India. The Tm was then cleaned with D.W. to remove mud particles. The outer surface of the Tm was removed and chopped into small pieces, which were then boiled in water for 1 h and dried in an oven. Finally, dried Tm was ground in a fine powder form, and it was denoted as boiled turmeric powder (B-Tm). The same procedure was followed to prepare un-boiled turmeric powder (UB-Tm) excluding the boiling step.2.4 Impregnation of BM and Tm on CF
The CF was procured from Ichalkaranji, Kolhapur, India (textile hub), which was used as a template to produce the desired scaffold. First, the CF was cleaned with D.W. and used for further experiments. The BM powder was dispersed in an NH4OH solution and stirred for 30 min. After that, the CF was submerged in the solution and stirred continuously for 1 h. Then, Tm powder was added to it and further stirred for 1 h. For complete impregnation of BM and Tm on the CF surface, the solution was sonicated for another 1 h. Then, the CF-impregnated with BM and Tm (BM:Tm = 1:1) were kept for drying at room temperature and used for further experiments.
2.5 Characterization of cellulose-based scaffolds
The crystal structure and phase present in the synthesized material were analyzed by X-ray diffraction (XRD) using a Bruker D8-Phaser X-ray diffractometer (CuKα1 radiation-λ = 1.54 Å, 40 kV, and 40 mA). The crystallite size was calculated using the Scherrer equation: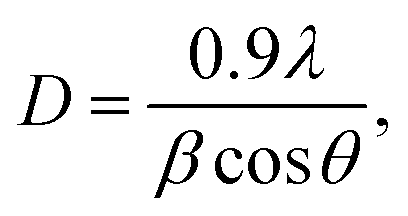 | (1) |
2.6 In vitro bioactivity
The in vitro bioactivity of the synthesized samples was performed in a simulated body fluid (SBF) solution for 3, 7, and 14 days. The SBF solution was prepared at 37 °C by maintaining the solution pH at 7.43 according to the Kokubo method.31 All the samples were immersed in SBF solution and incubated at 37 °C for the aforementioned days. After that, the samples were removed from SBF, washed with D.W., and dried. The HAp growth on the sample surface was analyzed by XRD, FTIR, SEM, and EDS mapping. Further, the degradation behavior of the synthesized samples was studied.2.7 Degradation study
To check the degradability of the samples, the study was performed in SBF (pH 7.4) solution, and samples were incubated at 37 °C for 3, 7, and 14 days. After that, the samples were removed from SBF, washed, dried, and weighed. The weight loss percentage was calculated using the following equation: | (2) |
2.8 Antibacterial activity
The antibacterial activity of UB-Tm and B-Tm powder was examined against Escherichia coli (E. coli-ATCC 9027-Gram-negative) and Staphylococcus aureus (S. aureus-ATCC 25923-Gram-positive). The well diffusion method was used to investigate the antibacterial activity of the UB-Tm and B-Tm powders. Before that, all samples and Petri plates used during the experiment were disinfected. Then, the bacterial culture was homogenously dispersed on the nutrient agar plates. After that, the wells were created and the samples were placed and incubated at 37 °C for 24 h. The antibacterial activity was checked after 24 h by observing the zone of inhibition, and the results were compared with the control (Penstrep is a combination of penicillin and streptomycin). To evaluate the haemocompatibility of the samples, a haemolysis study was performed.2.9 Haemolysis study
The in vitro haemolysis study was performed using blood from a healthy human donor. The collected blood was centrifuged at 3000 rpm for 15 min, and a settled blood pellet was used for further experiments. Then, the blood was mixed with the 2 mL phosphate-buffered saline (PBS). Meanwhile, 10 mL normal saline was added to a piece of a sample in the falcon tubes and then incubated at 37 °C for 30 min. in a water bath. After that, the tubes were removed from the water bath, and 200 μL of blood was added to it and again incubated in a water bath at 37 °C for 2 h. Finally, the samples were centrifuged at 3000 rpm for 15 min, and the absorbance of the supernatant of the experiments was checked in triplicate using colorimetry at 540 nm. The haemolysis percentage of the synthesized samples was calculated using the following equation: | (3) |
2.10 CAM assay
The eggs (fertilized-present day) were procured from the egg hatchery Kolhapur, Maharashtra, India. Three sets of eggs were used for each sample during the experiment. Further, the eggs were disinfected using alcohol (70%) and placed in an incubator for 3 days. After that, the eggs were removed, and further experiments were carried out in a laminar flow hood. To make a free space in the egg, 4–5 mL albumen was removed from the narrow side, and the hole was closed with cello tape. From the blunt side of the egg, a 2–3 cm area was broken, and rectangular-shaped samples were kept near the embryo and the control (without the addition of a sample). The opened eggshell was covered with cello tape and parafilm to avoid bacterial attack, and then the eggs were again placed in an incubator (humidified condition) at 37 °C. The growth of the blood vessels in the presence of the sample was examined after 24, 48, and 96 h and compared with the control.32 Further, an in vitro biocompatibility study was carried out to evaluate the biocompatibility of the samples.2.11 In vitro biocompatibility
The MTT assay was performed using MG-63 bone cells to study the biocompatibility of CF and BM–Tm–CF. The MG-63 cells (1 × 104 mL−1) were incubated in Dulbecco's modified Eagle's medium (DMEM) in a CO2 atmosphere for 24 h at 37 °C. Further, the 70 μL cells, culture media, and 100 μL sample solution (2.5, 5, and 10 mg mL−1) were transferred to 96-well plates. A solution of DMSO and MG-63 cells was used as a control. The triplicate sample plates were incubated at 37 °C for 24 h in a CO2 atmosphere. Then, 20 μL MTT was used to analyze the cell viability by measuring the optical density at 570 nm wavelength. The percentage inhibition and cell viability were calculated using eqn (4) and (5). The in vitro biocompatibility study was further supported by in vivo biocompatibility. | (4) |
| Percentage cell viability = 100 − percent inhibition. | (5) |
2.12 In vivo biocompatibility
The animal experiments were executed with prior approval from the Animal Ethics Committee, D. Y. Patil Medical College Kolhapur (Approval No. 6/IAEC/2017). Three sets of rats (n = 3) were used for this study. The Wistar rats (7–8 weeks old, ∼250 g weight) were cared for by following the principles of the Committee for Control and Supervision of Experiments on Animals (CCSEA) and provided with a food diet and water. The rats were anesthetized by applying intraperitoneal injection (ketamine-100 mg kg−1 and xylazine-5 mg kg−1). The subcutaneous part was cleaned by removing hairs and wiped using betadine. A skin excision (1 cm) was created using sterile forceps, and the skin pockets were designed. The disinfected CF-impregnated BM and Tm scaffolds were kept in the skin pockets of the rat, and the skin was sutured. The surgery was reopened after 14 days, images were taken, and skin tissue from the implanted area was harvested for the histology.The harvested skin tissue was fixed in formalin for 24 h. Further, the tissue was dehydrated, cleared in xylene, and embedded in paraffin wax using a rotary microtome to obtain the tissue sections (2–4 microns). Then, the tissue sections were rehydrated, dehydrated, cleared (in xylene), and stained with H&E staining. The interaction between tissue and implant material was analyzed by capturing the images using a compound microscope (Leica DM 750) equipped with a camera.
2.13 In vivo bone study
With prior approval from the Institutional Animal Ethics Committee (IAEC), D. Y. Patil Medical College Kolhapur (approval no. 6/IAEC/2017), the animal experiments were performed. For the in vivo bone experiments, the rats (7–8 weeks old, ∼250 g weight) in six sets (n = 6) were used, and caring was performed according to the principles of Guide Care with food and water. Ketamine-100 mg kg−1 and xylazine-5 mg kg−1 were used intraperitoneally for anesthesia. The thigh was shaved under anesthesia, and antiseptic was applied to avoid infection. Then, a 3 mm femur bone defect was formed for all the rats, and the synthesized scaffold was placed in the femur defect. The control was considered a 3 mm femur bone defect without the addition of the sample. X-ray images were taken 28 and 56 days after surgery to observe the healing of the bone defect area and to compare it with the control. Further, surgery was reopened after 56 days, photographic images were captured, and the femur bone was harvested from the rat for histological study.The harvested femur bones were kept in a neutral-buffering formalin solution and then in a formic acid solution to soften the bone. Then, the bones were decalcified in 0.1 M ethylene diamine tetraacetic acid (EDTA), and further, bone tissues were processed and embedded in paraffin wax for sectioning using a rotary microtome. The sections were stained with the H&E staining, and bone regeneration was analyzed by capturing the images using a compound microscope (Leica DM 750) equipped with a camera.
2.14 Statistical analysis
The experiments were conducted in three and six sets. For all experiments, the data were shown by calculating the mean ± standard deviation. Statistical significance was determined by one-way ANOVA. In all cases, the p-value is considered as p < 0.05 (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001) with the Dunnet comparison test.3. Results and discussions
The schematic of a simple, cost-effective, and green approach used for the synthesis of the BM–Tm–CF scaffold is shown in Fig. S1 (ESI†). The rice husk and eggshell were used as precursors of silica and calcium for the synthesis of 70S30C BM. The Tm powder was synthesized by cleaning, chopping, boiling (for B-Tm), drying, and grinding Tm rhizomes, as shown in Fig. S1a (ESI†). Further, Tm–CF, BM–Tm, BM–CF, and BM–Tm–CF scaffolds were prepared by impregnating BM and Tm on the CF, and changes observed after deposition were compared with CF. The structural and physicochemical properties of CF, Tm, Tm–CF, BM–Tm, BM–CF, and BM–Tm–CF were studied using analytical tools. The antibacterial activity, bioactivity, degradation rate, angiogenesis, and haemolysis properties were also examined considering bone regeneration applications.Fig. S1b (ESI†) shows the comparative UV-visible spectra for commercial Tm (C-Tm), UB-Tm, and B-Tm. The UB-Tm and B-Tm showed a broad peak from 300–500 nm with a main absorption peak at ∼425 nm along with a secondary peak at ∼242 nm due to the presence of curcumin.33 The absorption is because of the electronic dipole, which permitted π–π* excitation originating from the extended conjugation.33 It was observed that the B-Tm showed absorbance at the same wavelength, which was further compared with absorbance recorded with the commercial Tm. Interestingly, UB-Tm showed higher absorbance because the rhizomes were not boiled during synthesis. Further, this was supported by the antibacterial study.
It is important to note that for decades, Tm has been used in the medical field because of its antibacterial activity. The Tm rhizome consists of moisture, volatile oil, extraneous matter, turmerone, and coloring agent curcuminoids.34 Generally, during the synthesis of Tm powder, Tm rhizomes are boiled because boiling helps in the uniform distribution of curcuminoids, gelatinizes the starch, and removes the raw odor. However, more heating results in curcuminoid degradation, which is supported by the comparison of the antibacterial activity of B-Tm and UB-Tm against E. coli and S. aureus bacteria compared with C-Tm, as shown in Fig. S1c and d (ESI†). It was observed that B-Tm showed 26 mm and 26 mm zones of inhibition against E. coli and S. aureus, respectively, while UB-Tm showed 15 mm and 13 mm inhibition zones for E. coli and S. aureus, respectively. Thus, based on the results of UV-visible and antibacterial activity, B-Tm (Tm) was chosen for further study. The BM, CF, Tm, Tm–CF, BM-Tm, BM–CF, and BM–Tm–CF were further characterized with XRD and FTIR techniques to evaluate the impregnation of BM and Tm on the CF surface.
Fig. 1 illustrates the XRD patterns of the 70S30C BM, CF, Tm, Tm–CF, BM-Tm, BM–CF, and BM–Tm–CF samples. The 70S30C BM showed a crystalline nature, and the peaks were in good agreement with JCPDS-027-0088. The CF shows a crystalline nature with characteristic peaks at 2θ = 14.7°, 16.4°, and 22.8° for cellulose I at the (111), (002), and (004) planes, respectively (JCPDS-03-0289).35 A sharp peak at 22.8° reveals higher perfection of the crystal lattice of cellulose. The Tm demonstrates characteristic peaks for curcumin by matching well with JCPDS-09-0816. The Tm–CF showed the major peaks for cellulose along with small peaks for curcumin because the peaks for curcumin were observed between 2θ = 10–30° and those overlapped with the cellulose peaks. Further, the BM-Tm spectrum shows peaks for BM along with peaks for Tm, indicating curcumin at 2θ = 14.4°, 17.3°, and 18.1°. In the BM–CF, the BM was impregnated on the CF, and thus, it consists of the peaks for cellulose and BM as observed in the BM–CF spectrum. The BM–Tm–CF comprises characteristic peaks for cellulose with higher peak intensity along with the peaks for BM and Tm, as shown in Fig. 1a. The BM–Tm–CF shows a small shift in peak from 29.9° to 30.8°, which reveals the impregnation of the BM on the CF surface. Further, to support the XRD data and to determine the functional groups present in the synthesized materials, an FTIR study was performed. The FTIR spectra for 70S30C BM, CF, Tm, Tm–CF, BM-Tm, BM–CF, and BM–Tm–CF are shown in Fig. 1b. The Si–O–Si (asymmetric and symmetric) stretching, bending, and non-bridging oxygen vibrations were detected at ∼1012, ∼1084, ∼643, ∼452, ∼569, and ∼889 cm−1, which supports the formation of BM.32 The CF consists of cellulose as a major component with peaks at ∼2922 and ∼1327 cm−1 assigned for C–H stretching and deformation vibrations from glucose units, respectively.36 The absorption band at ∼3426 cm−1 is attributed to the stretching frequency of the hydroxy group.36 The Tm has curcumin, and the curcumin exhibits the C–H olefinic vibrations, C–C stretching vibrations, and C–O carbonyl frequency at ∼1156, ∼1519, and ∼1633 cm−1, respectively.37,38 Moreover, the peak at ∼2922 cm−1 could be attributed to the C–H vibration from the aromatic compound.37 The Tm–CF shows peaks for curcumin and cellulose, while BM-Tm reveals peaks for BM and curcumin. The impregnation of BM on the CF demonstrates the peaks for BM and cellulose. The BM–Tm–CF possesses peaks for BM, Tm, and CF (Fig. 1b). Thus, the XRD and FTIR data reveal the impregnation of BM and Tm on the CF, which is further supported by SEM and elemental mapping.
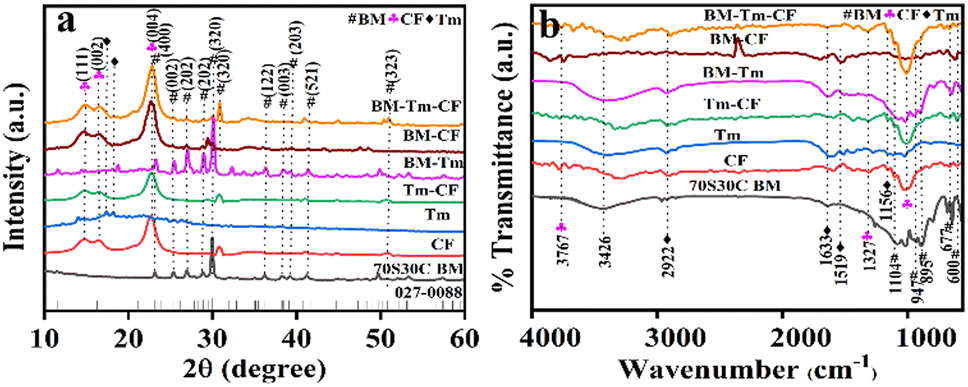 | ||
| Fig. 1 Representative (a) XRD and (b) FTIR spectra of CF, Tm, Tm–CF, BM-Tm, BM–CF, and BM–Tm–CF. | ||
Fig. 2a and b show the SEM images and elemental mapping for CF and BM–Tm–CF, respectively. It was observed that CF shows a plane surface of cotton fiber (inset of Fig. 2a), while the BM–Tm–CF shows a rough cotton surface. Further, the magnified SEM image of BM–Tm–CF reveals deposition of BM and Tm particles (inset of Fig. 2b), which is supported by the EDS elemental mapping study. The CF shows the presence of carbon and oxygen elements with 51.08 and 48.92 wt% ratios, respectively. Moreover, the BM–Tm–CF exhibits the presence of silicon, calcium, sodium, carbon, and oxygen elements (Fig. 2b), revealing 0.29, 0.99, 0.21, 64.33, and 34.18 wt% ratios, respectively (Table S1, ESI†). The increase in carbon content in BM–Tm–CF is justified by the presence of Tm on the CF surface, while BM has no carbon in the network. Further, it was observed that the silicon and calcium show lower percentages compared with carbon and oxygen because of the impregnation of a small amount of BM particles on the CF surface. The loading of BM and Tm on the CF can be further justified by TGA analysis.
 | ||
| Fig. 2 SEM and elemental mapping images of (a) CF and (b) BM–Tm–CF (inset shows magnified image). CF-scale-50 μm and BM–Tm–CF-5 μm. | ||
The TGA thermograms of CF, BM–CF, and BM–Tm–CF are presented in Fig. S2 (ESI†). The TGA thermogram of CF shows weight loss in the first step below 100 °C owing to moisture desorption. Further, the subsequent weight loss was observed at 370 °C and extending to 500 °C with no further weight loss beyond 500 °C, indicating 100% weight loss ascribed to the complete decomposition of cellulose. However, the BM–CF and BM–Tm–CF showed total weight loss of 92.5% and 91%, respectively. The difference in weight loss of 7.5% and 9%, respectively, was observed owing to the impregnation of BM and Tm particles on the CF compared to uncoated CF. The BM–Tm–CF showed a higher residual percentage compared with the BM–CF owing to the presence of BM and oxide formation of mineral content and carbonization of organics present in the Tm.39 In addition, it was observed that all the samples showed almost similar weight loss in the first step, while there was a difference during the second and third weight loss points, as shown in Fig. S2 (ESI†). The CF, BM9CF, and BM–Tm–CF showed weight losses of 71%, 60%, and 56%, respectively, in the second step. Interestingly, CF, BM–CF, and BM–Tm–CF showed similar starting at the third weight loss point in terms of temperature (370 °C) with a difference in weight percentage. The difference in weight loss during the third step for CF, BM–CF, and BM–Tm–CF is 20%, 23%, and 26%, which confirms the impregnation of BM and Tm.
Further, if the graft material is used as an implant, it should possess suitable strength. Thus, the tensile strength of the BM–Tm–CF was studied and compared with the strength of the CF. Fig. S3a (ESI†) shows the comparative tensile strengths for CF, Tm–CF, BM–CF, and BM–Tm–CF. The CF, Tm–CF, BM–CF, and BM–Tm–CF reveal 5.29 ± 1.25, 3.90 ± 0.64, 4.97 ± 1.40, and 6.68 ± 1.36 MPa tensile strength, respectively. It was observed that BM–Tm–CF achieved the highest tensile strength due to the impregnation of BM and Tm particles. The CF-impregnated silver-doped BM showed 28.77, 28.84, and 13.60 MPa tensile strength for BG1/Ag0.5%, BG2/Ag1%, and BG3 samples, respectively. The difference in tensile strength was observed due to the difference in CF used during the impregnation of silver-doped BG.20 Similarly, the Ag-SiO2 particles impregnated with CF reported an increase in mechanical properties owing to the incorporation of the nanoparticles.40 The Tm has antibacterial activity, as shown in Fig. S1 (ESI†). Further, the antibacterial activity of the BM–Tm–CF and Tm–CF was checked and compared with that of the CF, BM–CF, and control.
Fig. 3 shows the antibacterial activity of the control, CF, BM–CF, Tm–CF, and BM–Tm–CF against E. coli and S. aureus bacteria after 24 h using the disc diffusion method. The Tm–CF shows 15 mm and 15 mm zones of inhibition against E. coli and S. aureus, while BM–Tm–CF reveals 15 mm and 14 mm zones of inhibition against E. coli and S. aureus owing to the deposition of the Tm (Fig. 3). Interestingly, CF and BM–CF (the absence of Tm) did not show antibacterial activity. This suggests the importance of using Tm as an antibacterial agent. Further, an in vitro haemolysis study was performed using human blood collected from a healthy human donor.
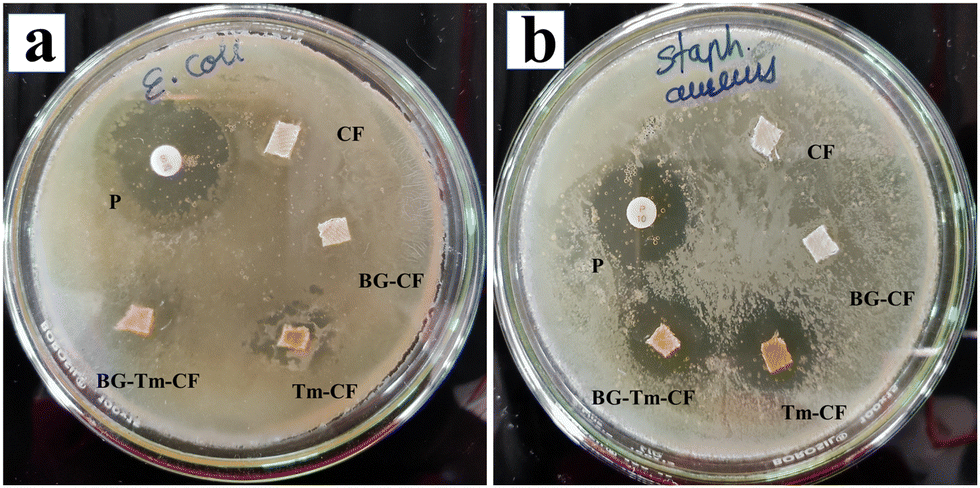 | ||
| Fig. 3 Antibacterial activity of CF, BM–CF, Tm–CF, and BM–Tm–CF against (a) E. coli and (b) S. aureus bacteria. | ||
The implant material comes in direct contact with blood according to ASTM 756-00; thus, in the present study, the haemocompatibility of the synthesized samples was studied.41 If the materials show less than 5% haemolysis, then the materials are considered haemocompatible. The CF, Tm–CF, BM–CF, and BM–Tm–CF samples show less than 5% haemolysis, revealing its haemocompatibility (Fig. 4a). After the completion of the experiment, the images of the CF, Tm–CF, BM–CF, and BM–Tm–CF were taken and compared with the image of unruptured blood cells, which means a negative control, as shown in Fig. 4b. Thus, the haemolysis images reveal that the CF, Tm–CF, BM–CF, and BM–Tm–CF are haemocompatible. A study is available on the use of cotton microfibres with proteins and apatite, and the results show the haemocompatibility of the samples.7 Further, the scaffold should have the ability to form new blood vessels from pre-existing vessels; thus, the angiogenesis study was performed to support the haemolysis.
 | ||
| Fig. 4 (a) Haemocompatibility study (in vitro study using human blood). (b) Suspension solution of (i) positive control, (ii) negative control, (iii) CF, (iv) Tm–CF, (v) BM–CF, and (vi) BM–Tm–CF. (c) Angiogenesis study of control, 70S30C BM, CF, Tm–CF, BM–CF, and BM–Tm–CF as a function of time. n = 3, p < 0.05 at *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 by one-way analysis of variance (ANOVA) with Dunnet comparison test. | ||
Angiogenesis plays a vital role in building the vascular network that helps to transport nutrients and waste during tissue regeneration.42 The ex ovo CAM study was performed to analyze the growth of the vascular network. Fig. 4c demonstrates the angiogenesis study for 70S30C, CF, BM–CF, Tm–CF, and BM–Tm–CF with respect to time. All samples show good neovascularization by showing the growth of blood vessels after 24, 48, and 96 h without any adverse effects. In addition, between Tm and BM, Tm is more directly associated with promoting angiogenesis owing to its active compounds, such as curcumin. Curcumin helps to increase angiogenic growth factors, such as Vascular Endothelial Growth Factor (VEGF), and promotes endothelial cell proliferation and migration. In addition, BM helps to create a conducive environment for angiogenesis, which helps to grow more blood vessels. Our observations reveal their synergistic effect in promoting angiogenesis. Further, for clarity of the image and to observe the blood vessels, the same image was inserted by processing with ImageJ software (Mexican Hat Filter), as shown in Fig. S4 (ESI†). Previous studies on the synthesis of cellulose microfibers/gelatin/egg white/nanohydroxyapatite composite scaffolds have shown improved angiogenesis through the formation of the blood vessels.7 Further, the bone consists of HAp as a major inorganic component; thus, HAp formation was studied. Before studying the HAp formation, a pH study was carried out to understand the ion exchange mechanism during HAp formation.
The pH study was performed in SBF solution, and changes in pH values were noted and plotted against time in days (Fig. 5a). It is observed that, when the 70S30C BM sample was placed in SBF solution, the pH was noted to be 7.43 and showed an incremental trend with time, with pH values of 7.70, 7.77, 7.93, 8.02, and 8.03 after 1, 2, 3, 4, and 5 days, respectively. After that, it remained constant. Similarly, with an initial pH value of 7.43, the change in pH values of the SBF solution was noted for CF (7.71, 7.73, 7.75, 7.77, and 7.8), Tm–CF (7.61, 7.73, 7.75, 7.84, and 7.85), and BM–CF (7.73, 7.8, 7.91, 7.97, 8.06, and 8.11) after 1, 2, 3, 4, and 5 days and remained constant thereafter. However, interestingly, the BM–Tm–CF sample showed a pH change (7.7, 7.9, 7.95, 7.99, 8.04, 8.14, and 8.15) extending to 7 days, after which it remained constant. It was observed that the CF and Tm–CF showed small changes in pH values because only OH functionalities were present in the CF and Tm–CF. BM–Tm–CF showed the highest change in the pH values owing to the presence of more functional groups compared with other samples. Consequently, ion exchange between the sample surface and the SBF solution occurs more efficiently. Particularly, the Si–O–Si network present on the BM surface breaks by OH− attack, which leads to silanol (Si–OH) formation, and then, the calcium-phosphate groups become adsorbed on it. The BM–Tm–CF has a higher pH compared with the BM because the BM consists of Na+ and Ca2+ ions on the surface, which are exchanged with the ions present in the simulated body fluid (SBF) solution. However, the BM–Tm–CF consists of the Na+ and Ca2+ ions along with OH functionalities present on the turmeric and cotton fabric. Thus, BM–Tm–CF has more functional groups than BM, leading to more exchange of the ions with SBF, which helps to enhance the pH value of the solution. In addition, a slightly higher pH is required for bone regeneration because alkaline phosphatase (ALP) occurs at a basic pH than the body pH (7.4); thus, it helps in HAp formation.43 Further, the changes in pH values were directly correlated with the degradation; thus, the degradation behavior of the synthesized samples was also performed in the SBF solution.
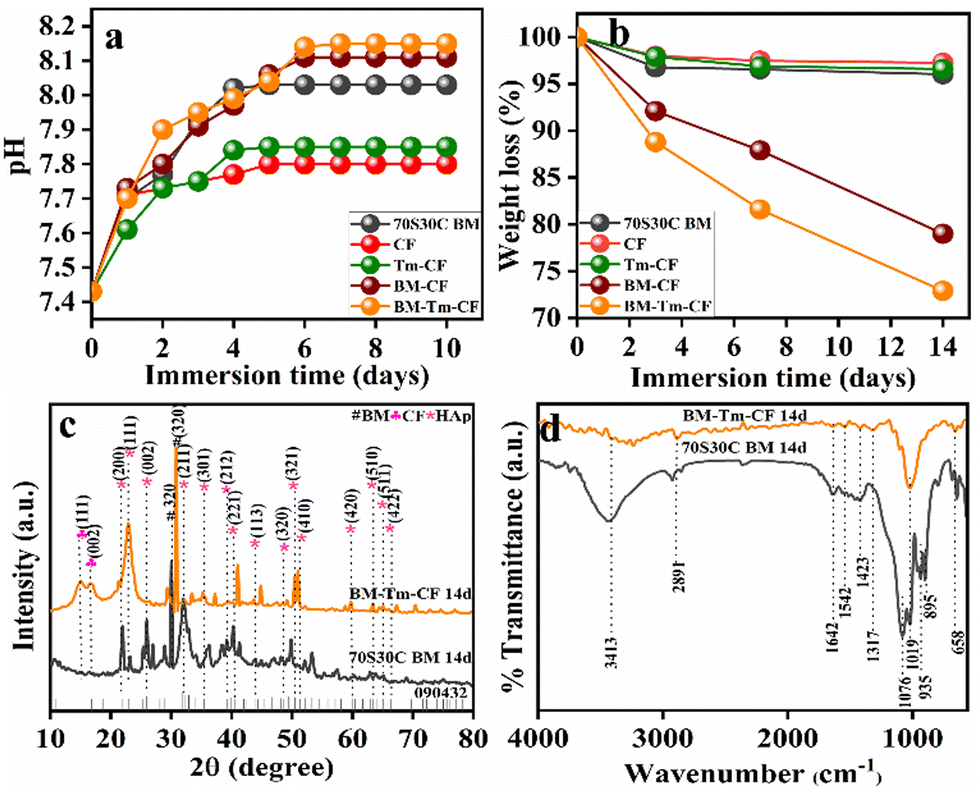 | ||
| Fig. 5 (a) Changes observed in pH values after 10 days of immersion of 70S30C BM in SBF, CF, Tm–CF, BM–CF, and BM–Tm–CF, measured over time. (b) Weight loss percentage of 70S30C BM, CF, Tm–CF, BM–CF, and BM–Tm–CF after 3, 7, and 14 days of immersion. (c) Representative XRD and (d) FTIR spectra of 70S30C BM and BM–Tm–CF after 14 days of in vitro bioactivity. | ||
In bone tissue engineering, the controlled degradation rate of the scaffold is considered a crucial parameter because it provides space for tissue growth and matrix deposition.44 Thus, the weight loss percentage of the sample was noted as a function of time (days) in the SBF solution. Fig. 5b illustrates the average percentage weight loss for 70S30C BM (3.96%), CF (2.74%), Tm–CF (3.41%), BM–CF (21%), and BM–Tm–CF (27.1%) as a function of time (after 14 days of immersion in SBF). Interestingly, the BM–Tm–CF showed a greater weight loss percentage, followed by BM–CF, 70S30C BM, BM–CF, Tm–CF, and CF. It was found that the degradation study showed a similar trend to the pH study. Thus, it can be inferred that more ion exchange facilitates more HAp formation. Based on the pH study and degradation study along with tensile strength, haemolysis, and angiogenesis results, the BM–Tm–CF was continued and characterized for further in vitro study to check the growth of HAp on the scaffold.
Fig. 5c demonstrates the XRD pattern of the BM–Tm–CF after 14 days of the SBF study, and it was compared with the 70S30C BM. It was observed that new peaks occurred at 2θ = 21.8°, 22.7°, 25.8°, 32.1°, 35.4°, 39.2°, 40.4°, 43.9°, 50.3°, 51.2°, 59.8°, 63.3°, 64.9°, and 66.3° were detected for HAp formation (JCPDS-09-0432)45 along with peaks for BM and cellulose. It was observed that 14 days after the SBF study, the intense peak for BM shifted from 29.9 to 30.7. In addition, it was observed that the main peak for HAp has a low intensity (32.1°) owing to minimum HAp deposition, which is further supported by FTIR analysis. Fig. 5d demonstrates the FTIR spectra of BM–Tm–CF after 14 days of SBF immersion. The newly noticed peaks for carbonate (CO32−) at ∼1423 cm−1 and phosphate (PO43−) at ∼1019 and ∼1076 cm−1 demonstrated the formation of HAp.32 Further, SEM and EDS mapping were studied to evaluate the actual formation of HAp on the sample surface.
Fig. 6 shows the surface structure and elemental mapping of the BM–Tm–CF after 14 days of SBF immersion. The BM–Tm–CF reveals the HAp formation all through the CF surface, as shown in Fig. 6a, while the HAp crystals were observed in the magnified SEM (Fig. 6b). In addition, the difference in surface morphology observed before and after the immersion of the BM–Tm–CF scaffold in the SBF solution is compared. Fig. S5 (ESI†) shows the growth of HAp on the BM–Tm–CF surface after 14 days of immersion. Further, SEM was supported by EDS mapping. Fig. 6c demonstrates the EDS layered image and elemental mapping for BM–Tm–CF after 14 days of SBF immersion. The presence of calcium and phosphate reveals the HAp formation. In addition, it was observed that after 14 days of the SBF study, the calcium and phosphate percentage (EDS data) increased to 10.08 and 5.24 wt% (Table S1, ESI†). Thus, the results demonstrate the HAp formation on the CF surface. Further, the synthesized material should be non-toxic; thus, the nontoxicity of the synthesized material is confirmed by the in vitro biocompatibility assay.
 | ||
| Fig. 6 (a) Low and (b) high-resolution SEM images of BM–Tm–CF after 14 days of bioactivity study. (c) EDS-layered image and elemental mapping of BM–Tm–CF after 14 days of bioactivity study. Scale-25 μm. | ||
Considering the bone regeneration application, if the material shows a toxic nature, then it is not applicable for use as an implant. Thus, the biocompatibility of CF and BM–Tm–CF was analyzed by MTT assay in the presence of MG-63 bone cells. The cell viability percentage is stated by calculating mean ± SD (p < 0.05) after 24 h. Fig. 7 illustrates the percentage cell viabilities of 98.41% ± 0.470%, 96.28% ± 1.141%, and 85.02% ± 0.537% for CF and 90.60% ± 1.23%, 91.00% ± 1.44%, and 85.01% ± 0.536% for BM–Tm–CF scaffold at 2.5, 5, and 10 mg mL−1 concentrations, respectively. The results demonstrate greater than 70% cell viability, which confirms that the sample is biocompatible. The in vitro biocompatibility data were further supported by the in vivo biocompatibility results.
 | ||
| Fig. 7 Percentage cell viability of CF and BM–Tm–CF using MG-63 bone cells. p < 0.05 at *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 by one-way analysis of variance (ANOVA) with the Dunnet comparison test. | ||
Depending on the bioactivity, angiogenesis, degradation, and biocompatibility results, the BM–Tm–CF scaffold was chosen for further study. Fig. 8 shows the in vivo biocompatibility results for the control and BM–Tm–CF scaffolds. The photographic images were taken during the surgery, as shown in Fig. 8a and b. Further, the surgery was reopened after 14 days, and it was observed that the BM–Tm–CF scaffold was completely degraded without any adverse or inflammatory effects (Fig. 8b′).
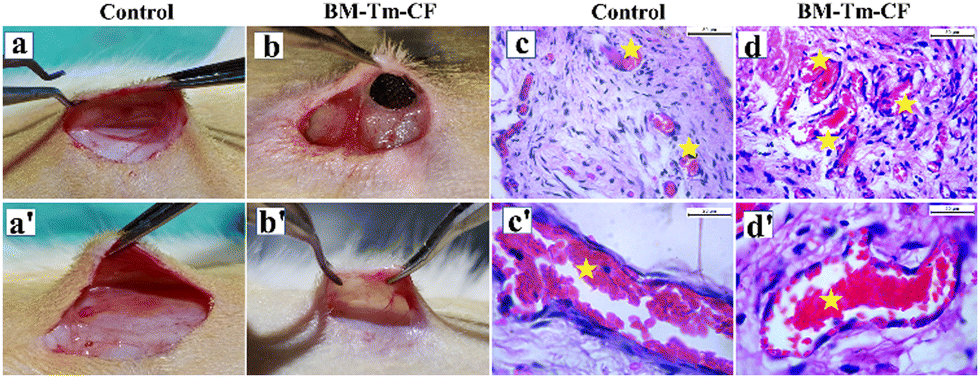 | ||
| Fig. 8 In vivo biocompatibility study. Photographs of (a) control and (b) BM–Tm–CF during the implantation and (a′) and (b′) after 14 days of implantation, respectively. Histological images of (c) and (c′) control (normal skin tissue); (d) and (d′) BM–Tm–CF. The figure shows blood vessels with endothelial cells (highlighted by a star mark). (n = 3). | ||
Further, the interaction between the scaffold and the implanted area of the rat was studied by recording the histology images. Fig. 8c and d′ shows the histological images for the control and BM–Tm–CF scaffold. The control and BM–Tm–CF scaffold shows the presence of mesenchymal cells, blood vessels having endothelial cells, and fibroblast cells. It was observed that the BM–Tm–CF scaffold showed more blood vessels than the control. Further, there were no signs of necrotic or fibrotic tissue formation at the site of implantation. This supports the biocompatibility of the scaffolds. Thus, the BM–Tm–CF scaffold was further used for bone regeneration applications.
To analyze the bone regeneration ability of the BM–Tm–CF scaffold, we investigated the osteogenic ability by implanting the scaffold in femur bone defect for 56 days. Initially, a 3 mm bone defect was formed and a scaffold was implanted in the defective area, as shown in Fig. 9a and b. Next, the bone was harvested from the control and BM–Tm–CF scaffold implanted rats after 56 days, and photographic images were taken, as shown in Fig. 9c and d. The control image is for a broken part of the femur bone, as shown in Fig. 9c. It was observed that the BM–Tm–CF implanted bone showed bone regeneration capacity with healing of the bone defect area compared with the control. Further, bone regeneration was monitored and supported with the X-ray images. Fig. 9e shows the X-ray images of the control and BM–Tm–CF implanted scaffold after 28 and 56 days of surgery. The X-ray image for control shows the formation of bone cracks or non-union bone defects after surgery because of a lack of support for the bone. The BM–Tm–CF implanted scaffold in bone defect demonstrates the bone regeneration and healing of the defected area, as depicted in Fig. 9e. Thus, the photographic and X-ray images confirm the bone regeneration ability of the BM–Tm–CF scaffold without any adverse or inflammatory effects on the rats. Further, new bone formation was determined by histological analysis.
 | ||
| Fig. 9 In vivo bone surgery process. Photographic images of (a) control and (b) BM–Tm–CF scaffold during implantation and (c) and (d) after 56 days of implantation. The 3 mm bone defect was formed in the bone. X-ray images of (e) and (g) control; and (f) and (h) BM–Tm–CF after 28 and 56 days of implantation. | ||
Fig. 10 shows the H&E-stained histological images of the control and BM–Tm–CF implanted scaffold after 56 days of surgery. The bone consists of outer cortical bone and inner cancellous bone, as illustrated in Fig. 10. Further, the histological images for the control show free space between the middle area of the bone in the presence of fat tissue. However, the implantation of the BM–Tm–CF composite reveals the presence of connective tissue, matrix mineralization, and osteoblast, confirming the formation of the new bone.46 Thus, the obtained histological results show new bone growth compared with the control and are analogous to the X-ray images.
 | ||
| Fig. 10 Histological images of (a) and (a′) control bone; (b) and (b′) bone defects implanted with BM–Tm–CF scaffold after 56 days of implantation. (red circle: matrix mineralization, red arrow: osteoblasts). | ||
4. Conclusion
The 70S30C BM was synthesized by applying a simple precipitation method using recycled biowaste materials, rice husk, and eggshell as a source of silica and calcium. The Tm powder was simply synthesized by cleaning, boiling, drying, and grinding Tm rhizomes. Further, the BM and Tm were impregnated on the CF surface. UV-visible, XRD, FTIR, and SEM-EDS studies were performed to check the impregnation of the BM and Tm. The in vitro bioactivity study showed the formation of HAp, while the degradation study revealed the desired degradation required for bone regeneration. The synthesized samples possess antibacterial activity against E. coli and S. aureus. Further, the haemocompatibility results revealed less than 5% haemolysis, which means that all the samples were not harmful to RBCs, WBCs, and platelets. To support the haemolysis results, an ex ovo angiogenesis study was carried out, which showed neovascularization. The in vitro biocompatibility results demonstrated more than 70% cell viability, while the in vivo biocompatibility also demonstrated its non-toxic nature. The in vivo bone study demonstrated almost complete bone repair in the bone defect rat model. Thus, the cost-effective cellulosic template acts as a flexible, cost-effective, eco-friendly scaffold, which makes the present work promising for bone regeneration applications.Author contributions
Shital S. Shendage: conceptualization, methodology, design experiment, investigation, data collection, and curation, writing – original draft. Kranti Kachare: synthesis, data curation, writing, and review. Kajal Gaikwad: angiogenesis, antibacterial, hemolysis, and in vivo study. Shivaji Kashte: in vivo experimental (Rat), data curation, writing, review, and editing. Fu-Der Mai: data curation, interpretation, supervision, review and editing the draft. Anil Vithal Ghule: conceptualization of the project, planning of the experiments, guidelines for execution of the project, following the investigation, methodology, data curation, interpretation, supervision, validation, drafting of the manuscript, review and editing the draft.Ethical approval statement
The animal experiments were executed with prior approval from the Institutional Animal Ethics Committee, D. Y. Patil Medical College, Kolhapur (Approval No. 6/IAEC/2017).Data availability
The data will be made available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author SSS is thankful to Mahatma Jyotiba Phule Research Fellowship (MJPRF), Nagpur, Government of Maharashtra for the fellowship (MJPRF-2021). We are thankful to Shivaji University, Kolhapur for providing analytical characterization facilities (CFC-SAIF). The authors are thankful to UGC-SAP and DST-FIST, DST-PURSE for instrument facilities at the Department of Chemistry, Shivaji University, Kolhapur. We are thankful to Shivaji University Group for Advanced Research “SUGAR” for the valuable discussion.References
- A. Chlanda, P. Oberbek, M. Heljak, E. Kijeńska-Gawrońska, T. Bolek, M. Gloc, Ł. John, M. Janeta and M. J. Woźniak, Mater. Sci. Eng., C, 2019, 94, 516–523 CrossRef CAS PubMed.
- S. Singh, S. Bhushan, A. Das, A. Barui and D. Dutt, Ceram. Int., 2023, 49, 1114–1127 CrossRef CAS.
- S. Kashte, A. K. Jaiswal and S. Kadam, Tissue Eng. Regener. Med., 2017, 14, 1–14 CrossRef CAS PubMed.
- Q. Chen, C. Zhu and G. A. Thouas, Prog. Biomater., 2012, 1, 2 CrossRef PubMed.
- M. Araújo, R. Viveiros, A. Philippart, M. Miola, S. Doumett, G. Baldi, J. Perez, A. R. Boccaccini and E. Verné, Mater. Sci. Eng., C, 2017, 77, 342–351 CrossRef PubMed.
- A. L. Torres, V. M. Gaspar, I. R. Serra, G. S. Diogo, R. Fradique, A. P. Silva and I. J. Correia, Mater. Sci. Eng., C, 2013, 33, 4460–4469 CrossRef CAS PubMed.
- S. Singh, S. Bhushan, H. Khan, L. R. Chaudhari, A. Ali, A. Das, A. Barui, Y. S. Negi, M. G. Joshi and D. Dutt, Int. J. Biol. Macromol., 2023, 236, 123812 CrossRef CAS PubMed.
- A. Kirillova, T. R. Yeazel, D. Asheghali, S. R. Petersen, S. Dort, K. Gall and M. L. Becker, Chem. Rev., 2021, 121, 11238–11304 CrossRef CAS PubMed.
- B. Sarker, W. Li, K. Zheng, R. Detsch and A. R. Boccaccini, ACS Biomater. Sci. Eng., 2016, 2, 2240–2254 CrossRef CAS PubMed.
- P. Feng, P. Wu, C. Gao, Y. Yang, W. Guo, W. Yang and C. Shuai, Adv. Sci., 2018, 5, 1–15 Search PubMed.
- E. Babaie and S. B. Bhaduri, ACS Biomater. Sci. Eng., 2018, 4, 1–39 CrossRef CAS PubMed.
- G. Tang, Z. Liu, Y. Liu, J. Yu, X. Wang and Z. Tan, Front. Cell Dev. Biol, 2021, 9, 1–18 Search PubMed.
- V. H. Ingole, K. Hany Hussein, A. A. Kashale, K. Ghule, T. Vuherer, V. Kokol, J. Y. Chang, Y. C. Ling, A. Vinchurkar, H. N. Dhakal and A. V. Ghule, J. Biomed. Mater. Res., Part A, 2017, 105, 2935–2947 CrossRef CAS PubMed.
- S. S. Shendage, V. H. Ingole, A. Rasal, J. Y. Chang, N. B. Birajdar, A. P. Tiwari, S. Kashte, T. Vuherer, G. Schmitt, M. Cucchiarini and A. V. Ghule, ACS Sustainable Chem. Eng., 2024, 12, 2598–2610 CrossRef CAS.
- Z. Yang, F. Zhao, W. Zhang, Z. Yang, M. Luo, L. Liu, X. Cao, D. Chen and X. Chen, Chem. Eng. J., 2021, 419, 129520 CrossRef CAS.
- G. Vergnol, N. Ginsac, P. Rivory, S. Meille, J. M. Chenal, S. Balvay, J. Chevalier and D. J. Hartmann, J. Biomed. Mater. Res., Part B., 2016, 104, 180–191 CrossRef CAS PubMed.
- F. V. Ferreira, L. P. Souza, T. M. M. Martins, J. H. Lopes, B. D. Mattos, M. Mariano, I. F. Pinheiro, T. M. Valverde, S. Livi, J. A. Camilli, A. M. Goes, R. F. Gouveia, L. M. F. Lona and O. J. Rojas, Nanoscale, 2019, 11, 19842–19849 RSC.
- A. K. Badekila and S. Kini, J. Cell. Physiol., 2020, 1–22 Search PubMed.
- F. Zhao, W. Zhang, X. Fu, W. Xie and X. Chen, RSC Adv., 2016, 6, 91201–91208 RSC.
- M. A. Ali, N. M. Aly, M. Mabrouk, S. A. M. El-sayed and H. H. Beherei, Int. J. Biol. Macromol., 2021, 181, 905–918 CrossRef CAS PubMed.
- T. L. B. Ha, T. M. Quan, D. N. Vu and D. M. S. Additional, Regener. Med. Tissue Eng., 2013, 248–274 Search PubMed.
- S. Wang, Z. Gu, Z. Wang, X. Chen, L. Cao, L. Cai, Q. Li, J. Wei, J. W. Shin and J. Su, Sci. Rep., 2018, 8, 1–12 Search PubMed.
- A. A. El-Rashidy, G. Waly, A. Gad, A. A. Hashem, P. Balasubramanian, S. Kaya, A. R. Boccaccini and I. Sami, J. Non-Cryst. Solids, 2018, 483, 26–36 CrossRef CAS.
- N. Gupta, D. Santhiya, S. Murugavel, A. Kumar, A. Aditya, M. Ganguli and S. Gupta, Colloids Surf., A, 2018, 538, 393–403 CrossRef CAS.
- C. Shuai, W. Guo, P. Wu, W. Yang, S. Hu, Y. Xia and P. Feng, Chem. Eng. J., 2018, 347, 322–333 CrossRef CAS.
- C. Shuai, X. Shi, F. Yang, H. Tian and P. Feng, Int. J. Extreme Manuf., 2024, 6, 015101 CrossRef CAS.
- G. S. Kumar, D. Muthu, G. Karunakaran, S. Karthi, E. K. Girija and D. Kuznetsov, J. Sol-Gel Sci. Technol., 2018, 86, 610–616 CrossRef CAS.
- A. Konwar, R. Kandimalla, S. Kalita and D. Chowdhury, ACS Sustainable Chem. Eng., 2018, 6, 5806–5817 CrossRef CAS.
- M. Faiz, M. Ahmad, R. Bahru, R. Amin, M. Umar, A. Khan, S. Izwan, A. Razak, S. Abu, M. Ra, A. Kadir, N. Hasraf and M. Nayan, J. Cleaner Prod., 2021, 290, 125792 CrossRef.
- V. D. Hiremani, S. Khanapure, T. Gasti, N. Goudar, S. Kumar, S. P. Masti, R. B. Malabadi, B. S. Mudigoudra and R. B. Chougale, Int. J. Biol. Macromol., 2021, 193, 2192–2201 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- S. S. Shendage, K. Gaikwad, K. Kachare, S. Kashte and A. V. Ghule, Mater. Chem. Phys., 2024, 313, 128789 CrossRef CAS.
- M. A. Subhan, K. Alam, M. S. Rahaman, M. A. Rahman and R. Awal, J. Sci. Res., 2013, 6, 97–109 CrossRef.
- A. J. Ruby, G. Kuttan, K. Dinesh Babu, K. N. Rajasekharan and R. Kuttan, Cancer Lett., 1995, 94, 79–83 CrossRef CAS PubMed.
- S. Thambiraj and D. Ravi Shankaran, Appl. Surf. Sci., 2017, 412, 405–416 CrossRef CAS.
- B. Abderrahim, E. Abderrahman, A. Mohamed, T. Fatima and T. Abdesselam, World J. Environ. Eng., 2015, 3, 95–110 Search PubMed.
- Z. Khodabandeh, N. Tanideh, F. S. Aslani, I. Jamhiri, S. Zare, N. Alizadeh, A. Safari, N. Farshidfar, M. Dara and M. Zarei, Mater. Today Commun., 2022, 31, 103339 CrossRef CAS.
- S. S. Shendage, K. Gaikwad, K. Kachare, S. Kashte, J. Y. Chang and A. V. Ghule, J. Mater. Sci., 2024, 59, 10744–10762 CrossRef CAS.
- S. Mishra and B. Goel, Adv. Zool. Bot., 2020, 8, 83–86 CAS.
- K. Panwar, M. Jassal and A. K. Agrawal, Carbohydr. Polym., 2018, 187, 43–50 CrossRef CAS PubMed.
- S. K. S. Hossain, S. Yadav, S. Majumdar, S. Krishnamurthy, R. Pyare and P. K. Roy, Ceram. Int., 2020, 46, 833–843 CrossRef CAS.
- S. Kargozar, F. Baino, S. Hamzehlou, R. G. Hill and M. Mozafari, Trends Biotechnol., 2018, 36, 430–444 CrossRef CAS PubMed.
- G. Luo, Y. Ma, X. Cui, L. Jiang, M. Wu, Y. Hu, Y. Luo, H. Pan and C. Ruan, RSC Adv., 2017, 7, 11880–11889 RSC.
- J. A. Sowjanya, J. Singh, T. Mohita, S. Sarvanan, A. Moorthi, N. Srinivasan and N. Selvamurugan, Colloids Surf., B, 2013, 109, 294–300 CrossRef CAS PubMed.
- L. Ji, W. Wang, D. Jin, S. Zhou and X. Song, Mater. Sci. Eng., C, 2015, 46, 1–9 CrossRef CAS PubMed.
- S. Kashte, R. Dhumal, P. Chaudhary, R. K. Sharma, V. Dighe and S. Kadam, Mater. Today Commun., 2021, 26, 102049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 (a) Synthesis of Tm powder (cleaning, chopping, boiling (for B-Tm), drying, and grinding process) (b) UV-visible spectra of the UB-Tm, B-Tm, and commercial Tm. Antibacterial activity of UB-Tm and B-Tm against (c) E. coli and (d) S. aureus bacteria. Fig. S2 TGA thermograms of CF, BM-CF, and BM–Tm–CF. Fig. S3 (a) Tensile strength study of CF, Tm–CF, BM–CF, and BM–Tm–CF. Images taken during tensile testing (b) CF (c) Tm–CF (d) BM–CF (e) BM–Tm–CF. n = 3, p < 0.05 at *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 by one-way analysis of variance (ANOVA) with Dunnet comparison test. Fig. S4 Corresponding angiogenesis images for control, 70S30C BM, CF, Tm–CF, BM–CF, and BM–Tm–CF as a function of time obtained after processing the images using ImageJ software (Mexican Hat Filter). Fig. S5 SEM images of BM–Tm–CF (a) and (a′) before and (b) and (b′) after 14 days of immersion in SBF solution. Table S1 EDS elemental composition of CF, BM–Tm–CF, and BM–Tm–CF after 14 days of bioactivity study in SBF solution. See DOI: https://doi.org/10.1039/d4tb02028f |
| This journal is © The Royal Society of Chemistry 2025 |