
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous biocatalysis by magnetic nanoparticle immobilized biomass-degrading enzymes derived from microbial cultures†
Mani
Gupta
 a,
Arghya
Bandyopadhyay
ac,
Sushant Kumar
Sinha
a,
Suksham
a,
Abhisek
Samanta
a,
Surajit
Mondal
cd,
Sayan
Bhattacharyya
*cd and
Supratim
Datta
*abc
a,
Arghya
Bandyopadhyay
ac,
Sushant Kumar
Sinha
a,
Suksham
a,
Abhisek
Samanta
a,
Surajit
Mondal
cd,
Sayan
Bhattacharyya
*cd and
Supratim
Datta
*abc
aDepartment of Biological Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, India. E-mail: supratim@iiserkol.ac.in
bCenter for the Climate and Environmental Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, India
cCentre for Advanced Functional Materials, Indian Institute of Science Education and Research, Kolkata, Mohanpur 741246, India
dDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, India
First published on 3rd February 2025
Abstract
Recombinant enzymes have become increasingly popular and are frequently used as environmentally safe biocatalysts due to their wide range of applications and high specificity. Purifying these enzymes from the host cells, media, and other contaminants is essential for their characterization and applications. The widely utilized method for protein purification by nickel–nitrilotriacetic acid (Ni–NTA) resin-based affinity chromatography is a time-consuming, labor-intensive, and resource-demanding technique. In this study, we synthesized NTA–Ni@Fe3O4 nanoparticles (NPs) to capture enzymes from cell lysates and microbial culture media and developed a model system to show the efficacy of immobilizing and recycling biomass-degrading enzymes known as cellulases. Cellulases, which play an important role in biomass degradation and biofuel production, were baited with NTA–Ni@Fe3O4 NPs and purified in a single step. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis indicates efficient purification of the immobilized enzymes on the NPs from the cell lysate and extracellular media. Additionally, we successfully showed that cellulase-immobilized NTA–Ni@Fe3O4NPs can serve as a heterogeneous catalyst for the hydrolysis of p-nitrophenyl-β-D-glucopyranoside (pNPGlc) and carboxymethylcellulose (CMC). The NTA–Ni@Fe3O4 NPs immobilized with enzymes showed recyclability for up to five cycles. The applications of this methodology may be extended to various industries requiring efficient enzyme purification and recycling for promising advancements in biotechnology and sustainable biomanufacturing processes.
Introduction
Biocatalysis has emerged as a promising complementary approach to traditional chemical synthesis methods.1,2 Enzymes exhibiting high catalytic efficiency and selectivity are increasingly crucial in pharmaceutical and chemical manufacturing.3 However, enzyme purification methods are expensive, and the inability to reuse enzymes hinders their industrial applicability.4 The use of NPs to separate biomolecules, like nucleic acids or proteins, has proven superior to traditional methods due to its cost-effectiveness and efficiency.5 The primordial group of NPs used in separation methodologies are magnetic NPs. Iron-based NPs are a choice due to the availability and biocompatibility associated with the metal, apart from stable magnetic properties.6 All biomolecules do not interact with iron-based NPs spontaneously and with specificity and, therefore, may need to be functionalized. Nucleic acids spontaneously interact with iron oxide NPs and facilitate the separation from crude lysates.7 However, proteins need to be tagged for affinity-based separation techniques. Nickel–nitrilotriacetic acid (Ni–NTA) columns have been popular for several decades. However, these are not cost-effective, and the process takes time and effort.8 NTA functionalized magnetic nanoparticles are a good choice due to lower cost and reduced purification time compared to traditional Ni–NTA columns.9,10 The use of NPs also enhances the protein yield due to the intrinsic higher surface-to-volume ratio of the NP.11The enzymatic hydrolysis of cellulose, a naturally available biopolymer found in lignocellulose, into glucose by cellulases is crucial for generating fermentable sugars.12 This glucose can then be utilized in subsequent biochemical processes to produce biofuels.13–17 The three major components of cellulase are endoglucanase, cellobiohydrolase, and β-glucosidase.18,19 Many technical improvements are required for the efficient and economically feasible conversion of cellulose to biofuels. One critical limitation of this process is the high cost of the enzymes involved in converting cellulose into fermentable sugars.20 Any method to reuse enzymes across multiple biomass hydrolysis cycles will reduce enzyme production costs.
The His-tagged affinity-based method is the most popular and widely used protein purification technique.21,22 However, this study introduces a novel approach for heterologous protein purification from Escherichia coli (E. coli) using magnetic NPs. The nickel-based NPs introduced in this study exhibit a binding affinity comparable to His-tagged proteins, similar to commercially available Ni–NTA resin agarose beads. In the present study, we have demonstrated the application of magnetic NTA–Ni@Fe3O4 particles for immobilization of cellulases (β-glucosidase) (BGL), endoglucanase (EG), and cellobiohydrolase (CBH). These immobilized enzyme complexes exhibit effective cellulose hydrolysis into glucose. Furthermore, this enzyme complex can be recovered and reused for hydrolysis in subsequent reaction cycles. Additionally, by utilizing secretion tags, we facilitated the extracellular release of cellulases, which were then efficiently purified from the culture media. Our approach significantly reduces the time and effort required compared to the conventional protocols.
Results and discussion
The NTA-modified NPs were initially characterized through a zeta sizer to measure the surface charge, which is −11.2 mV along with the polydispersity index of 0.211 (Fig. S1, ESI†). These values correspond to a reasonable charge and size distribution.Furthermore, the NPs were characterized by transmission electron microscopy (TEM) for exact shape and size. Fig. 1a, b and d show that the synthesized NTA–Ni@Fe3O4 spherical NPs have an average length of 3.76 ± 0.302 nm (Fig. 1d). Fig. 1c is the selected area electron diffraction (SAED) pattern, and Fig. S2 shows the lattice spacing of 0.237 nm, corresponding to the (111) reflection of NiO.23,24 Also, the (220) plane of Fe3O4 and (012) plane of α-Fe2O3 are prominent (Fig. S2, ESI†). The high-angle annular dark-field scanning TEM (HAADF-STEM) map in Fig. 1e and f – shows the presence of Fe, Ni, and O over the surface, indicating the oxygenation of both Fe and Ni parts in the NPs.
 | ||
| Fig. 1 TEM and HR-TEM images (a) and (b) show near spherical nanoparticles. (c) SAED of the field from (a) shows the presence of both Fe and Ni oxides. The size of the nanoparticles is confirmed from the histogram (d) of the particle, showing 3.76 ± 0.3 nm size of the particle calculated from a population of 200 counts. (e) and (f) represent the HAADF-STEM analysis of the particle. The EDAX spectrum is represented in (e) and (f) corresponds to the mapping of individual elements overlayed on the dark-field HAADF-STEM image. (g) The magnetic moment vs. magnetic field hysteresis curve of the synthesized particle confirms the superparamagnetic nature of the particle at room temperature. | ||
The magnetization as a function of magnetic field (M–H) loops of the synthesized nanoparticles are represented in Fig. 1g. Due to slowing down of thermal fluctuations of electron spins and their progressive blocking with decreasing temperature, the magnetic anisotropy is enhanced at 3 K as compared to 300 K. Also, the presence of very negligible magnetic remanence and low coercivity at near room temperature (Fig. S3 and Table S1, ESI†), results in superparamagnetic ordering of the particles.25,26
The formation of an oxidized Ni layer is evident from the Fourier transform infrared spectroscopy (FTIR) spectral data, where the signature of the oxidized Ni layer is visible at 1764, 957, and 831 cm−1 corresponding to NO asymmetric stretch, the asymmetric stretch of NiO2, and a probable NiH(D)Ni stretch, respectively (Fig. S4a, ESI†). FTIR spectral analysis also validates the presence of NTA molecules over the NP surface. The NTA molecules can also interact with Fe, present in the NPs. Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis also confirmed the presence of iron and nickel on the particles. The results showed that the atomic percentage of iron and nickel is 95.8% ± 0.2% and 4.14% ± 0.006%, respectively. This result implies that the nickel is incorporated into the particles. A strong magnet was used to separate the NTA–Ni@Fe3O4NPs (Fig. S4b, ESI†). A good dispersity and easy separation of the NPs was observed upon application of an external magnetic field (Fig. S4c, ESI†).
The powder X-ray diffraction (PXRD) pattern of the Fe3O4 NPs reveals corresponding 2θ peaks at 30.2°, 35.5°, 43.2°, 53.7°, 57.1° and 62.8° related to the (220), (311), (400), (422), (511) and (440). All diffraction peaks align closely with the characteristic reflections of the Fe3O4 inverse spinel structure (PCPDFWIN v.2.02, PDF No. 89-0691).27,28 Similar PXRD peaks were observed after loading of Ni–NTA and enzymes (Fig. S5, ESI†).
The NTA–Ni@Fe3O4 magnetic NPs were proposed to be used to separate the desired tagged proteins from a mixture of proteins based on affinity to the NPs, and then protein-tagged NPs were separated by a magnet. To test our NPs for affinity to His-tagged proteins, we used pure His-tagged green fluorescent protein (GFP), the green colour of which can be visualized by the naked eye and under UV light. After washing with binding buffer (10 mM phosphate buffer, 40 mM imidazole, and 500 mM NaCl, pH 7.4), the protein could be separated from the NPs by elution with elution buffer (10 mM phosphate, 500 mM imidazole, and 500 mM NaCl, pH 7.4). Fig. 2a shows the washed 10 mg NTA–Ni@Fe3O4NPs mixed with 1 mg GFP (termed as 1 in Fig. 2b). After mixing, NTA–Ni@Fe3O4 NPs were separated by a magnet and the supernatant was collected (termed as 2 in Fig. 2b). After washing, elution buffer was used to elute GFP (termed as 3 in Fig. 2b). UV-vis spectroscopy was used to quantify the loading of GFP (Section S11, ESI†), confirming that 99% of the GFP from the stock solution was loaded onto the NTA–Ni@Fe3O4 NPs. This corresponded to approximately 10% (w/w) of the NTA–Ni@Fe3O4 NPs and was confirmed by thermogravimetric analysis (TGA) (Fig. S6, ESI†). After elution, about 85% of GFP was recovered from NTA–Ni@Fe3O4 NPs (Fig. 2c). The NPs were then heated and used for SDS-PAGE and western blot analysis to confirm the remaining amount of GFP. To compare the performance of the NPs prepared in this study, Promega Ni–NTA resin (Promega®, Madison, USA) was used to bind GFP. The analysis of the commercial resin showed a recovery of approximately 86% of the GFP, with the remaining GFP still bound to the resin, as confirmed by SDS-PAGE and western blot analysis (Fig. 2d–f). The combined results from UV-vis spectroscopy, SDS-PAGE, and western blot analysis showed that the synthesized NTA–Ni@Fe3O4NPs effectively purify His-tagged proteins. The surface immobilization of GFP on the NPs was confirmed by epifluorescence microscopy (Fig. S7, ESI†). For the binding adsorption experiment, His-tagged GFP was incubated with Ni–NTA@Fe3O4 NPs in a binding buffer under shaking conditions, using varying protein masses. The adsorption data were analyzed using the Langmuir isotherm equation,29 yielding a maximum binding capacity (qmax) of 124.36 μg of protein per mg of nanoparticles and a Langmuir constant (K) of 2.78 g L−1 (Section S11 and Fig. S11, S12, ESI†). As a control, non-His-tagged GFP was used under the same conditions, showing only about 13% of the binding observed with His-tagged GFP, confirming the high specificity of the NTA–Ni@Fe3O4NPs for His-tagged proteins (Fig. S13, ESI†).
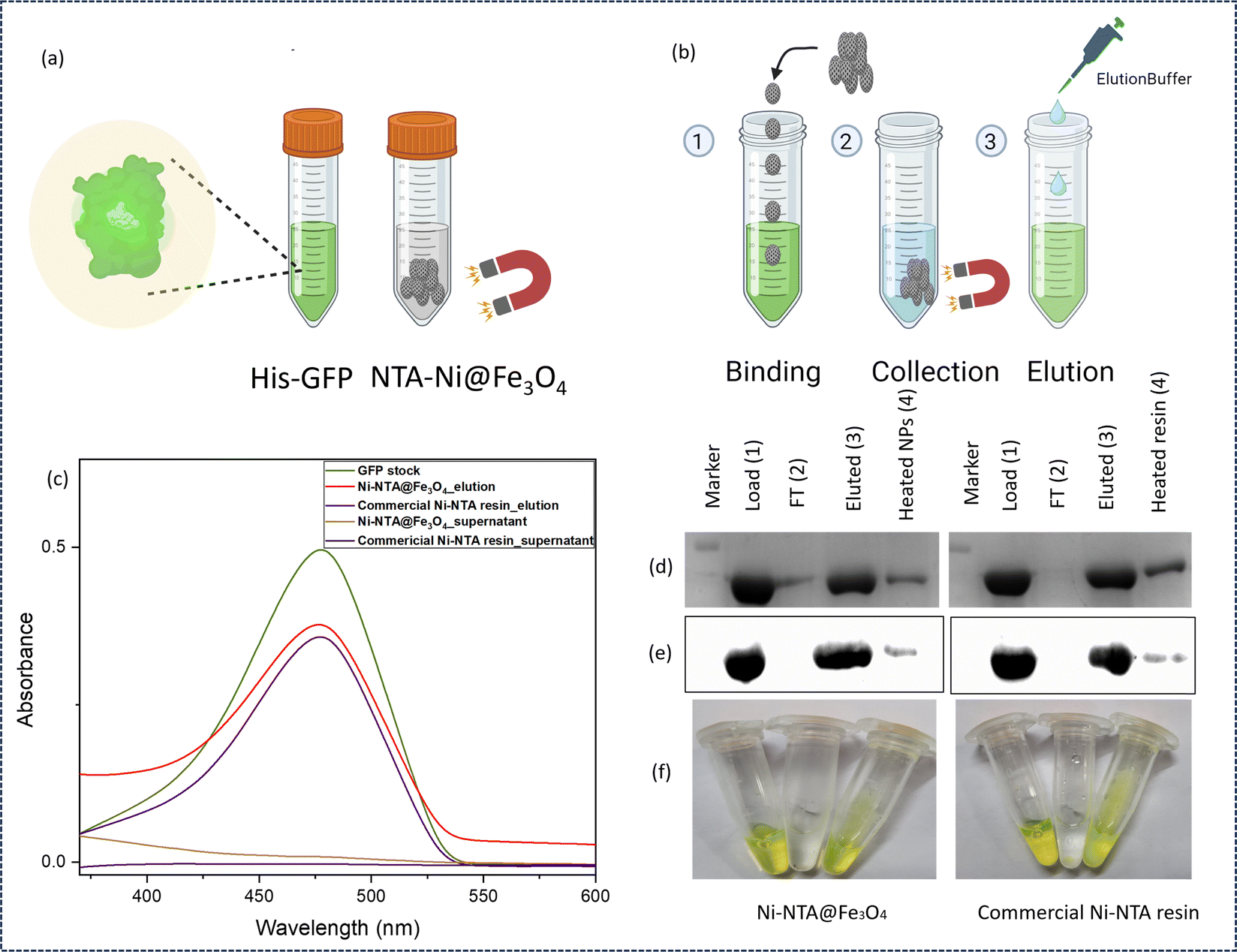 | ||
| Fig. 2 Preliminary binding test of the His6-GFP protein purified using NTA–Ni@Fe3O4 NPs. (a) Schematic diagram of protein separation using NTA–Ni@Fe3O4 NPs. A protein of interest, such as His6-GFP (green), was employed for protein-NTA–Ni@Fe3O4 NP capture, and the released GFP protein could be visualized during the whole process. (b) The study was performed by allowing the protein to bind (1) and to be collected by a magnet (2), followed by washing and elution (3). (c) UV-vis spectra of GFP stock, NTA–Ni@Fe3O4 NPs elution, commercial Ni–NTA resin elution, NTA–Ni@Fe3O4 NPs supernatant, and commercial Ni–NTA resin supernatant. (d) SDS-PAGE. (e) Western blot analysis of load-GFP (1), FT-flowthrough (2), eluted-elution samples (3), and heated NPs – the remaining bound protein on the NTA–Ni@Fe3O4 NPs or resin were denatured by heating (95 °C, 10 min) (4). (f) Left to right – image of GFP stock, flowthrough, NTA–Ni@Fe3O4 NPs, and commercial Ni–NTA resin eluted GFP in a microcentrifuge tube. | ||
After demonstrating the successful binding and elution of pure His-tagged protein by NTA–Ni@Fe3O4NPs, we evaluated the purification of His-tagged proteins by directly applying the cell lysate to the NTA–Ni@Fe3O4NPs. The genes encoding EG, CBH, and BGL with the His-tag were cloned, transformed, and overexpressed in E. coli, and then the cells were pelleted down and lysed to prepare the cell lysate containing a mixture of recombinant His-tagged protein and endogenous E. coli proteins (referred to as 1 in Fig. 3a). After washing the NTA–Ni@Fe3O4NPs with binding buffer, the NPs were mixed with the freshly prepared cell lysate. The leftover supernatant was labeled as 2 in Fig. 3a, and the NPs were separated with a magnet.
 | ||
| Fig. 3 (a) Schematic diagram of His-tagged protein purification using the NTA–Ni@Fe3O4 NPs from cell lysate. Washed NPs mixed with cell lysate (1), separation of NPs by a magnet (2), washing of enzyme immobilized NPs (3) and elution of his tagged enzyme (4). SDS-PAGE analysis of purified (b) BGL, (c) CBH, and (d) EG. Lane L represents the protein marker, lane 1 for cell lysate, lane 2 for supernatant (non-His-tagged proteins), lane 3 for washed eluent, and lane 4 for purified enzyme. | ||
The separated NTA–Ni@Fe3O4 NPs were washed with a binding buffer. The supernatant from these washes was collected and labeled as fraction 3 in Fig. 3a. Subsequently, proteins were eluted from the NPs using an elution buffer. The resulting eluted proteins are represented as fraction 4 in Fig. 3a. The effectiveness of the purification process was assessed through SDS-PAGE analysis, as depicted in Fig. 3b–d, which provided insights into the separation and purity of the proteins. The SDS-PAGE bands show that the cell lysate (lane 1) contains a variety of proteins. In lane 2, all of the unbound proteins are visible. Lane 3 displays the non-His-tagged proteins that emerged after washing the NPs with the binding buffer. Finally, upon washing with elution buffer, the pure protein was detected in lane 4 as a single band of corresponding molecular weight, confirming successful protein purification.
These results demonstrate that NTA–Ni@Fe3O4 NPs can achieve high specificity and purify proteins even from complex mixtures. This is consistent with previous reports on Ni–NTA functionalized materials, which have shown similar efficacy in capturing His-tagged proteins from crude lysates due to their high binding affinity.30,31
We then tagged our enzymes with secretion tag ansB at the N-terminal for extracellular secretion of the enzyme.32 Secreting recombinant proteins into the culture medium or the periplasm of E. coli offers several advantages over intracellular production. The benefits include simplified downstream processing, enhanced biological activity, increased product stability and solubility, and retention of the N-terminal of the expressed peptide. A secretion strategy significantly simplifies the recovery of recombinant gene products by minimizing contamination from host proteins. Moreover, if the product is secreted into the culture medium, cell disruption, and sophisticated equipment are unnecessary for recovery.33 The extracellular secretion of enzymes into media eliminates the need to pellet down and lyse cells through chemical or mechanical processes and is cost-effective.
The genes encoding the enzymes (EG, CBH, and BGL) with an ansB tag at the N-terminal and a His tag at the C-terminal were cloned, transformed, and overexpressed in E. coli. After expression, the media was collected. The NTA–Ni@Fe3O4 NPs were first washed with binding buffer, then added to the extracellular media containing the recombinant His-tagged proteins along with undesired native secretory proteins of E. coli (Fig. 4a). The proteins bound to NTA–Ni@Fe3O4 NPs were separated using a magnet and washed three times with binding buffer. Finally, the bound protein was eluted using an elution buffer. The purity of the secreted His-tagged BGL, CBH, and EG was assessed using SDS-PAGE (Fig. 4b). Prominent bands corresponding to BGL, CBH, and EG at their expected sizes confirmed the method's purity and efficiency.
 | ||
| Fig. 4 (a) Schematic representation of simultaneous purification and immobilization of recombinant His-tagged enzyme on NTA–Ni@Fe3O4 NPs from the extracellular medium. (b) SDS-PAGE analysis of purified lane 1 (EG), lane 2 (CBH), and lane 3 (BGL). | ||
This direct purification of recombinant proteins from the culture medium significantly simplifies the protein purification process. Unlike conventional methods requiring cell lysis and complex separation processes, our approach uses extracellular secretion to overcome these steps entirely. This reduces process complexity and cost and highlights the scalability and practicality of this methodology for industrial applications. Compared to prior reports focusing on protein purification from the cell lysate, our results establish a novel framework for directly purifying His-tagged proteins from extracellular media with high purity and efficiency.
We then investigated the maximum enzyme uptake capacity by loading different amounts of enzymes to saturate the loading of enzymes in the NPs. The maximum uptake capacity of BGL per milligram of NTA–Ni@Fe3O4 NPs is 196 μg, while for CBH, it is 215 μg mg−1 of the NTA–Ni@Fe3O4 NPs. Similarly, the extent of EG immobilization is 310 μg mg−1 of the NTA–Ni@Fe3O4 NPs (Fig. 5a). We also investigated the enzyme-loaded NTA–Ni@Fe3O4 NPs for heterogeneous biocatalysis and characterized these enzyme-loaded NTA–Ni@Fe3O4 through TEM, SEM, FTIR, and PXRD. SEM and TEM images reveal no morphology and structural changes in the enzyme-loaded NTA–Ni@Fe3O4 compared to pristine NTA–Ni@Fe3O4 NPs (Fig. S7 and S8, ESI†).
 | ||
| Fig. 5 (a) Maximum immobilization (μg) of BGL, CBH, and EG per milligram of the NTA–Ni@Fe3O4 NPs. The data are presented as mean ± SD; n = 3; the error bars represent standard deviations from three replicate measurements. Comparison of the FTIR spectra of the pristine NTA–Ni@Fe3O4 NPs and immobilized (b) EG, (c) CBH, and (d) BGL. Second derivate FT-IR spectra of (e) EG, EG@NTA–Ni@Fe3O4 NPs and NTA–Ni@Fe3O4 NPs. (f) CBH, CBH@NTA–Ni@Fe3O4NPs and NTA–Ni@Fe3O4 NPs and (g) BGL, BGL@NTA–Ni@Fe3O4 NPs and NTA–Ni@Fe3O4 NPs. | ||
PXRD analysis also reveals the (220), (311), (511), and (440) reflections, which are similar to pristine NTA–Ni@Fe3O4 NPs (Fig. S5, ESI†).
FTIR spectroscopy was performed to find the amide-I and amide-II bands of the enzymes@ NTA–Ni@Fe3O4 NPs. The two prominent characteristic bands on the enzyme's backbone are amide-I and amide-II. In particular, amide-I was detected in the 1600–1700 cm−1 range (Fig. 5b–d and Fig. S10, ESI†), whereas amide-II was identified in the 1500–1600 cm−1 range. With a modest contribution from the out-of-phase C–N stretching vibration, C–C–N deformation and N–H in-plane bending, amide-I predominantly corresponds to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.
O bond.
However, the out-of-phase combination of the N–H in-plane bend and the C–N stretching vibration, along with a lesser contribution from the C–O in-plane bend, C–C stretching vibration, and N–C stretching vibration, results in the amide-II vibration.34,35 The β-sheet, random coil, α-helix, and β-turn are all represented by the spectral components 1632, 1649, 1659, and 1680 cm−1 that make up the second derivative of amide-I.
The secondary structure of the enzyme backbone was examined using amide-I. The α-helix's assigned spectral component, amide-II, is visible at 1545 cm−1.34–37 The second derivative of the infrared spectra of the immobilized enzymes show all of the distinctive spectral components of amides I and II (Fig. 5e–g). The preservation of the amide I and II spectral features suggests that the secondary structure of the enzymes, such as alpha-helices and beta-sheets, remains intact upon immobilization. This structural integrity is essential for maintaining enzymatic activity, as the catalytic function of enzymes is closely linked to their three-dimensional structure. Thus, this analysis confirms that NTA–Ni@Fe3O4 NPs serve as a favorable platform for enzyme immobilization, providing a stable environment that preserves the structural and functional properties of the enzymes.
We investigated the catalytic efficiency and recyclability of enzyme-loaded NTA–Ni@Fe3O4 NPs using the cellulolytic enzymes EG, CBH, and BGL. Through the hydrolysis of CMC to reducing sugars, the activity of the two enzymes, EG-loaded NTA–Ni@Fe3O4 NPs and CBH-loaded NTA–Ni@Fe3O4 NPs, were examined (Fig. 6a). The dinitro salicylic acid (DNS) assay performed at 540 nm quantitated the amount of reducing sugar generated.38 The hydrolysis of pNPGlc to pNP was catalyzed by the BGL-loaded NTA–Ni@Fe3O4 NPs (Fig. 6d). By measuring the absorbance of the generated p-nitrophenol (pNP) at 405 nm, the catalytic activity of BGL@NTA–Ni@Fe3O4 was examined. Pristine NTA–Ni@Fe3O4 NPs exhibited no catalytic activity for these reactions.
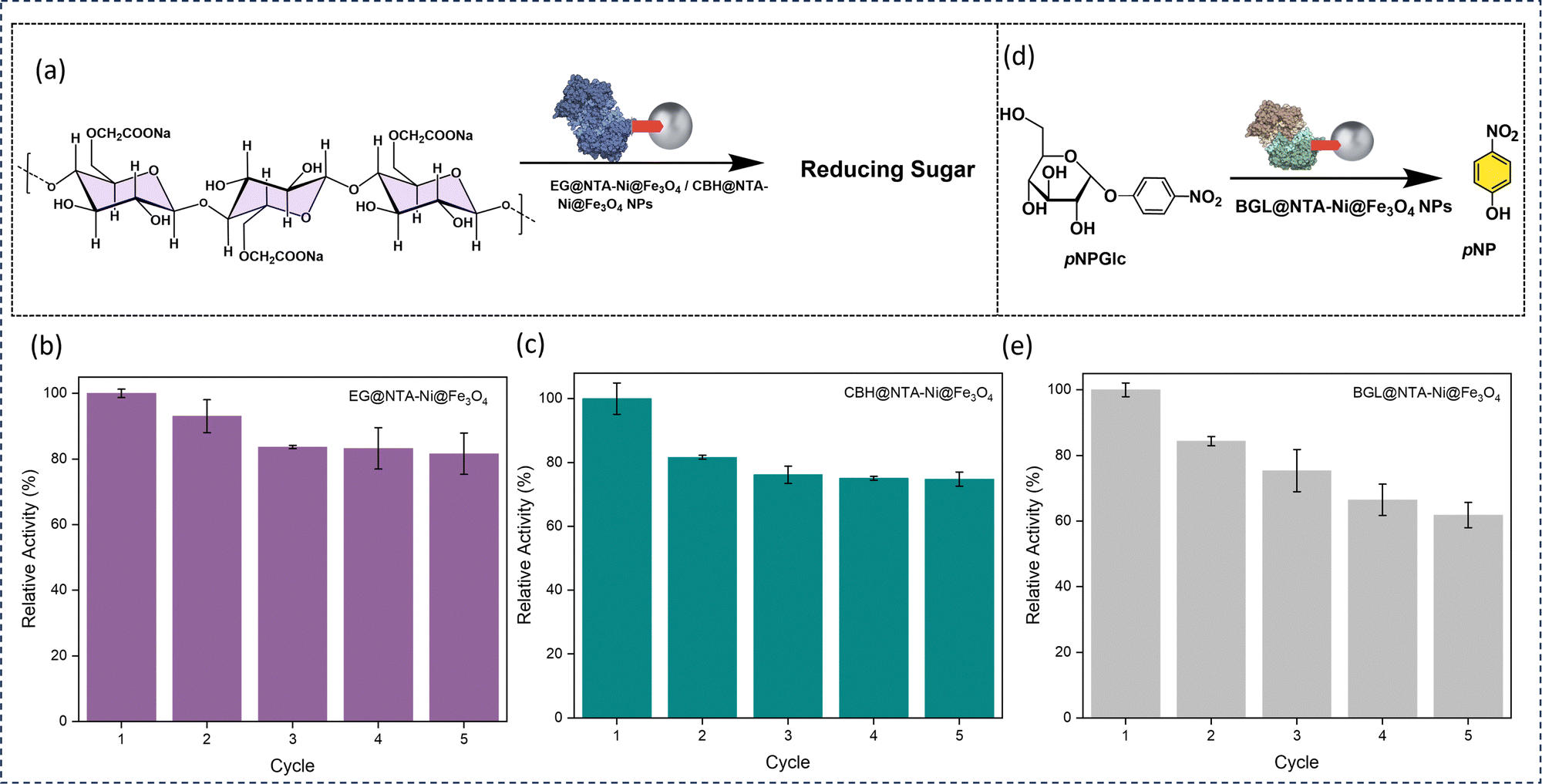 | ||
| Fig. 6 (a) Schematic of catalytic performance of enzyme@NTA–Ni@Fe3O4 NP hydrolysis of carboxymethylcellulose (CMC) to reducing sugar. (b) Recycling of EG@NTA–Ni@Fe3O4 NPs. (c) Recyclability of CBH@NTA–Ni@Fe3O4 NPs. (d) Schematic of catalytic performance of BGL@ NTA–Ni@Fe3O4 NPs towards hydrolysis of p-nitrophenyl-β-D-glucopyranoside (pNPGlc) to p-nitrophenol (pNP). (e) Recyclability of BGL@NTA–Ni@Fe3O4 NPs. The data are presented as mean ± SD; n = 3; the error bars represent standard deviations from three replicate measurements. | ||
Compared to free EG, CBH, and BGL, EG@NTA–Ni@Fe3O4, CBH@NTA–Ni@Fe3O4, and BGL@NTA–Ni@Fe3O4 NPs show 80%, 81%, and 88% relative activity, respectively (Fig. S15, ESI†). This slight reduction in activity upon immobilization may be due to reduced substrate accessibility or changes in enzyme conformation, which can influence enzyme performance. As previously reported in various studies, slight reductions in activity have been reported when enzymes are immobilized,39–43 which aligns with the findings in this study (Table S2, ESI†). The slight decline in activity over multiple cycles may be due to factors such as enzyme leaching or minor structural changes. Nevertheless, the immobilized enzyme offers significant advantages over the free enzyme, particularly in reusability, as it can be easily separated and recovered using a magnet, allowing for multiple cycles of use with consistent activity. This trade-off between minor activity loss and enhanced reusability highlights the practical benefits of immobilization on the NTA–Ni@Fe3O4 NPs. With 81 and 74% relative activity, respectively, the EG@NTA–Ni@Fe3O4 NPs and the CBH@NTA–Ni@Fe3O4 NPs are recyclable for up to five cycles (Fig. 6b and c). With 61% relative activity, the BGL@NTA–Ni@Fe3O4 NPs can be recycled for up to five cycles (Fig. 6e).
EG@NTA–Ni@Fe3O4 NPs hydrolyze CMC from the chain end by binding to the substrate until a minimum chain length is achieved. After that, CBH@NTA–Ni@Fe3O4 hydrolyzes one or a few accessible bonds in the chain from the surface. Finally, BGL@NTA–Ni@Fe3O4 NPs hydrolyze cellobiose and small soluble oligomers to glucose. We simultaneously utilized BGL@NTA–Ni@Fe3O4NPs, CBH@NTA–Ni@Fe3O4 NPs, and EG@NTA–Ni@Fe3O4NPs in one pot to determine the contribution of each enzyme@NTA–Ni@Fe3O4 NP to glucose conversion. The conversion of CMC to glucose was measured by GOD-POD assay (Fig. 7a). No products were observed when the reaction was performed with only NTA–Ni@Fe3O4 NPs or BGL@NTA–Ni@Fe3O4. The maximum conversion rate was observed when an equimolar mixture of all three nanoparticle complexes—EG@NTA–Ni@Fe3O4, CBH@NTA–Ni@Fe3O4 NPs and BGL@NTA–Ni@Fe3O4NPs—was used for catalysis, setting this condition as 100% relative activity for comparison (Fig. 7b). This shows a synergistic impact between the three enzyme-loaded nanoparticles. When only EG@NTA–Ni@Fe3O4 and BGL@NTA–Ni@Fe3O4 NPs were used in an equimolar mixture, the relative conversion rate dropped to 72%. This indicates that the absence of CBH@NTA–Ni@Fe3O4 NPs disrupts the synergistic interaction, leading to suboptimal substrate conversion. Similarly, the relative conversion was 64% for an equimolar mixture of BGL@NTA–Ni@Fe3O4 and CBH@NTA–Ni@Fe3O4 NPs, further indicating the critical role of EG@NTA–Ni@Fe3O4 in the catalytic efficiency of the system. For an equimolar combination of EG@NTA–Ni@Fe3O4 and CBH@NTA–Ni@Fe3O4 NPs, the relative conversion rate was 43%. This significant reduction shows the importance of BGL@NTA–Ni@ Fe3O4 in a cellulase cocktail, likely due to its role in breaking down intermediate products formed by EG and CBH enzymes, thereby enhancing the overall conversion efficiency. Individually, EG@NTA–Ni@Fe3O4 and CBH@NTA–Ni@Fe3O4 NPs showed relative conversions of 49% and 29%, respectively (Fig. 7b). The mixture of all three NP complexes—EG@NTA–Ni@Fe3O4, CBH@NTA–Ni@Fe3O4 NPs and BGL@NTA–Ni@Fe3O4 NPs demonstrated high recyclability over five cycles, maintaining excellent relative conversions of approximately 78% (Fig. 7c). This study demonstrates the practical advantages of enzyme immobilization on NTA–Ni@Fe3O4 NPs for heterogeneous biocatalysis, combining high catalytic activity with excellent recyclability. The ease of recovery and reusability positions NTA–Ni@Fe3O4 NPs as a promising platform for sustainable and cost-effective enzymatic processes.
 | ||
| Fig. 7 (a) Schematic diagram of the one-pot hydrolysis of carboxymethylcellulose (CMC) to glucose catalyzed by EG@NTA–Ni@Fe3O4 NPs, CBH@NTA–Ni@Fe3O4 NPs, and BGL@NTA–Ni@Fe3O4 NPs. (b) CMC conversions under different catalyst combinations. (c) Recycling of all three NP complexes—EG@NTA–Ni@Fe3O4, CBH@NTA–Ni@Fe3O4 NPs and BGL@NTA–Ni@Fe3O4 NPs required for one-pot glucose synthesis. The data are presented as mean ± SD; n = 3; the error bars represent standard deviations from three replicate measurements. | ||
Conclusions
For easier purification of histidine-tagged recombinant enzymes and proteins from cell lysate and extracellular medium, we synthesized NTA–Ni@Fe3O4. We successfully immobilized three enzymes, BGL, CBH, and EG, on the NTA–Ni@Fe3O4 NPs, purified each, and demonstrated their direct application in heterogeneous biocatalysis. The NTA–Ni@Fe3O4 NPs immobilized with enzymes showed considerable catalytic activity and were recyclable for up to five cycles. Additionally, we successfully demonstrated the one-pot tandem synthesis of glucose from CMC with good recyclability. We anticipate that our immobilization technology will open the way for developing low-cost, environmentally friendly biocatalysis-based solutions for various industrial applications.Author contributions
SD and SB obtained the funding, supervised the project, and finalized the manuscript. MG designed the project, performed assays, purified the enzymes, analyzed the data, and wrote the manuscript. AB synthesized the starting material and performed material characterization experiments. Suksham and AS performed the assays and analyzed the data. SKS helped with project design and verified the manuscript. SM synthesized the material and verified the manuscript.Data availability statement
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
MG acknowledges DST-INSPIRE for a Senior Research Fellowship. AB acknowledges SERB for the fellowship under sanction no. PDF/2018/001043/LS. SB is thankful for the financial support from SERB under sanction no. STR/2021/000001. SD acknowledges the SERB Core Research Grant, CRG/2023/002111, and BT/PR47801/BCE/8/1812/2023 (SD) from DBT. SM acknowledges CSIR–UGC for the senior research fellowship. We acknowledge Mr Shant Chettri for his help in PXRD and TGA data collection. We thank Grishma Kasle for her help in the fluorescence microscopy experiment. Fig. 2–4 were created with BioRender.com. We thank Mr Satyadip Paul for FTIR data collection. We thank Mr Anshuman Das for his assistance in this work.Notes and references
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC.
- G. K. Meghwanshi, N. Kaur, S. Verma, N. K. Dabi, A. Vashishtha, P. D. Charan, P. Purohit, H. S. Bhandari, N. Bhojak and R. Kumar, Biotechnol. Appl. Biochem., 2020, 67, 586–601 CrossRef CAS PubMed.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185–205 CrossRef PubMed.
- I. Yildiz, Nanotechnol. Rev., 2016, 5, 331–340 CAS.
- H. Nosrati, M. Salehiabar, M. Fridoni, M.-A. Abdollahifar, H. Kheiri Manjili, S. Davaran and H. Danafar, Sci. Rep., 2019, 9, 7173 CrossRef PubMed.
- L. Vanyorek, Á. M. Ilosvai, E. Szőri-Dorogházi, C. Váradi, F. Kristály, Á. Prekob, B. Fiser, T. Varga, Z. Kónya and B. Viskolcz, J. Dispersion Sci. Technol., 2021, 42, 693–700 CrossRef CAS.
- S. Paul, M. Gupta, A. Kumar Mahato, S. Karak, A. Basak, S. Datta and R. Banerjee, J. Am. Chem. Soc., 2024, 146, 858–867 CrossRef CAS PubMed.
- J. Xu, J. Sun, Y. Wang, J. Sheng, F. Wang and M. Sun, Molecules, 2014, 19, 11465–11486 CrossRef PubMed.
- W. Wang, F. Zhou, X. Cheng, Z. Su and H. Guo, IET Nanobiotechnol., 2020, 14, 67–72 CrossRef PubMed.
- L. Fei and S. Perrett, Int. J. Mol. Sci., 2009, 10, 646–655 Search PubMed.
- N. Ilić, M. Milić, S. Beluhan and S. Dimitrijević-Branković, Energies, 2023, 16, 3598 CrossRef.
- S. Datta, Curr. Metabolomics, 2016, 4, 14–22 CrossRef CAS.
- S. Aich and S. Datta, in Biorefineries: A Step Towards Renewable and Clean Energy, ed. P. Verma, Springer Singapore, Singapore, 2020, pp. 273–295 DOI:10.1007/978-981-15-9593-6_11.
- L. R. Lynd, M. S. Laser, D. Bransby, B. E. Dale, B. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan, J. Sheehan and C. E. Wyman, Nat. Biotechnol., 2008, 26, 169–172 CrossRef CAS PubMed.
- D. Klein-Marcuschamer, P. Oleskowicz-Popiel, B. A. Simmons and H. W. Blanch, Biotechnol. Bioeng., 2012, 109, 1083–1087 Search PubMed.
- S. Goswami, N. Gupta and S. Datta, Biotechnol. Biofuels, 2016, 9, 72 CrossRef PubMed.
- S. Paul, M. Gupta, K. Dey, A. K. Mahato, S. Bag, A. Torris, E. B. Gowd, H. Sajid, M. A. Addicoat and S. Datta, Chem. Sci., 2023, 14, 6643–6653 RSC.
- R. Datta, Heliyon, 2024, 10, e24022 CrossRef CAS PubMed.
- V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2011, 4, 3 CrossRef CAS PubMed.
- A. Spriestersbach, J. Kubicek, F. Schäfer, H. Block and B. Maertens, in Methods in Enzymology, ed. J. R. Lorsch, Academic Press, 2015, vol. 559, pp. 1–15 Search PubMed.
- M. Brownleader, Nat. Methods, 2007, 4, i–ii CrossRef.
- B. Liu, L. R. Liu, X. J. Liu, M. J. Liu and Y. S. Xiao, Mater. Sci. Technol., 2012, 28, 1345–1348 CrossRef CAS.
- B. Debnath, A. Bansal, H. G. Salunke, A. Sadhu and S. Bhattacharyya, J. Phys. Chem. C, 2016, 120, 5523–5533 CrossRef CAS.
- S. Patri, N. T. K. Thanh and N. Kamaly, Nanoscale, 2024, 16, 15446–15464 RSC.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- L. Zhuang, W. Zhang, Y. Zhao, H. Shen, H. Lin and J. Liang, Sci. Rep., 2015, 5, 9320 CrossRef CAS PubMed.
- S. Parvin, D. K. Chaudhary, A. Ghosh and S. Bhattacharyya, ACS Appl. Mater. Interfaces, 2019, 11, 30682–30693 CrossRef CAS PubMed.
- S. P. Schwaminger, P. Fraga-García, S. A. Blank-Shim, T. Straub, M. Haslbeck, F. Muraca, K. A. Dawson and S. Berensmeier, ACS Omega, 2019, 4, 3790–3799 CrossRef CAS PubMed.
- E. C. H. T. Lau, K. C. Dodds, C. McKenna, R. M. Cowan, A. Y. Ganin, D. J. Campopiano and H. H. P. Yiu, Sci. Rep., 2023, 13, 21549 CrossRef CAS PubMed.
- R. Minkner, J. Xu, K. Takemura, J. Boonyakida, H. Wätzig and E. Y. Park, J. Nanobiotechnol., 2020, 18, 159 CrossRef CAS PubMed.
- M. Hashemzaei, N. Nezafat, M. B. Ghoshoon and M. Negahdaripour, Inform. Med. Unlocked, 2023, 36, 101146 CrossRef.
- F. Mergulhão, D. K. Summers and G. A. Monteiro, Biotechnol. Adv., 2005, 23, 177–202 CrossRef PubMed.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- C. Xing, P. Mei, Z. Mu, B. Li, X. Feng, Y. Zhang and B. Wang, Angew. Chem., Int. Ed., 2022, 61, e202201378 CrossRef CAS PubMed.
- K. H. Sizeland, K. A. Hofman, I. C. Hallett, D. E. Martin, J. Potgieter, N. M. Kirby, A. Hawley, S. T. Mudie, T. M. Ryan, R. G. Haverkamp and M. H. Cumming, Materialia, 2018, 3, 90–96 CrossRef CAS.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- D. Yang, X. Wang, J. Shi, X. Wang, S. Zhang, P. Han and Z. Jiang, Biochem. Eng. J., 2016, 105, 273–280 CrossRef CAS.
- J. Lin, Q. Wen, S. Chen, X. Le, X. Zhou and L. Huang, Int. J. Biol. Macromol., 2017, 96, 377–383 CrossRef CAS PubMed.
- V. Mehnati-Najafabadi, A. Taheri-Kafrani and A.-K. Bordbar, Int. J. Biol. Macromol., 2018, 107, 418–425 CrossRef CAS PubMed.
- T. C. Coutinho, J. O. D. Malafatti, E. C. Paris, P. W. Tardioli and C. S. Farinas, ACS Appl. Nano Mater., 2020, 3, 12334–12345 CrossRef CAS.
- F. R. Paz-Cedeno, J. M. Carceller, S. Iborra, R. K. Donato, A. P. Godoy, A. Veloso de Paula, R. Monti, A. Corma and F. Masarin, Renewable Energy, 2021, 164, 491–501 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02011a |
| This journal is © The Royal Society of Chemistry 2025 |