
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evolution of branched peptides as novel biomaterials
Matthew J.
Little
,
Jody M.
Mason
and
Nazia
Mehrban
 *
*
University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: nm2051@bath.ac.uk
First published on 13th January 2025
Abstract
Branched peptide-based materials draw inspiration from dendritic structures to emulate the complex architecture of native tissues, aiming to enhance the performance of biomaterials in medical applications. These innovative materials benefit from several key features: they exhibit slower degradation rates, greater stiffness, and the ability to self-assemble. These properties are crucial for maintaining the structural integrity and functionality of the materials over time. By integrating bioactive peptides and natural polymers within their branched frameworks, these materials offer modularity and tunability and can accommodate a range of mechanical properties, degradation rates, and biological functions making them suitable for biomedical applications, including drug delivery systems, wound healing scaffolds, and tissue engineering constructs. In drug delivery, these materials can be engineered to release therapeutic agents in a controlled manner, enhancing the efficacy and safety of treatments. In wound healing, they provide a supportive environment which promotes rapid and efficient tissue repair. The combination of biomimetic design and functional adaptability makes branched peptide-based materials a promising candidate for the development of next-generation biomaterials, paving the way for significant advancements in healthcare.
1. Introduction
Biomaterials have progressed significantly over the last century, transitioning from simple systems replicating tissue mechanics, such as titanium alloys1 and calcium-phosphate ceramics,2 to biologically active materials that mimic native tissue chemistry and architecture, enhance repair, or prevent immunological rejection, such as collagen3 and gelatin methacryloyl (GelMA).4 Of note is the shift from inert materials to biologically functional systems capable of interacting with cells and promoting regeneration.5–9 Polymers are particularly attractive due to their ease of synthesis and flexibility for molecular modification.6 Polymers encompass a wide range of natural and synthetic materials, each with distinct physical and chemical characteristics.10,11 Synthetic polymers, such as polyethylene glycol (PEG), are hydrophilic and scalable12,13 but biologically inert without chemical modification.14 In contrast, natural proteins, like collagen, offer cellular compatibility by preserving amino acid sequences, such as RGDS, recognised by cell membrane integrin receptors like αVβ5.15 Natural proteins, however, suffer from limitations in structural integrity and batch-to-batch consistency.16 Peptides, particularly branched peptides, offer a compelling alternative combining the bioactivity of natural proteins with the tunability and reproducibility of synthetic systems.Peptides are short chains of amino acids that can be synthesised and modified to control their chemical and physical properties, including charge, isoelectric point, and solubility, as well as their three-dimensional (3D) assembly,17,18 stiffness,19 and porosity.20,21 When used in vitro, these alterations can dictate specific cellular responses, such as proliferation,22 differentiation8 and migration23 which in turn support advancements in biomaterial design, including tissue regeneration.24–29 Unlike linear peptides, branched peptides introduce branching points at amide or carboxyl side chains, enabling diverse structural architectures and functionalities such as self-assembly,30 bio-active functionalisation,7,31,32 and the ability to act as drug carriers.33 These branching points provide unique opportunities for tailoring mechanical stiffness,7,30 fibril diameter,32 and biodegradation rates34 to meet specific biomedical needs. Novel and updated methods of synthesis have further decreased the cost and expertise required for their development, which previously hindered progress.35–37
A key enabling technology for the development of peptide-based biomaterials is solid-phase peptide synthesis (SPPS), a method introduced by Merrifield in 1963 which revolutionised peptide chemistry.38 SPPS involves the sequential addition of amino acids to a growing peptide chain formed via a condensation reaction between amines and carboxylic acids anchored to an insoluble resin (Fig. 1).38 This approach enables precise control over peptide sequence and allows for the incorporation of non-natural amino acids, functional groups, and branching points with high efficiency and purity.39 SPPS chemistry relies on the activated ester from the incoming amino acid attacking the amine of the amino acid bound to the resin. Following the same SPPS principles, the process can be applied to the amine side chain on lysine, enabling the creation of branching points. To utilise the amine side chain, SPPS employs orthogonal protecting groups, including; 4-methyltrityl (MTT),40 allyloxycarbonyl (Alloc),41 and 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl (ivDdE),42 which can be selectively removed under specific conditions. Protecting groups such as these provide selective functionalisation and the incorporation of branching points. Alternatively, the carboxylic acid side chains on aspartic acid and glutamic acid can also be used as branching points although the orientation is reversed. The activated ester is bound to the resin, and the amine will be the incoming amino acid, typically using the Alloc protecting group41 or bis(2-sulfanylethyl)amido (SEA) via native peptide ligation.43 The use of orthogonal protecting groups allows the synthesis of complex architectures, such as dendrimers and multi-arm peptides, which are challenging to achieve through other methods such as liquid phase peptide synthesis.44 Furthermore, SPPS facilitates the incorporation of novel amino acid sequences, functional groups or bioactive motifs, enabling precise tuning of peptide properties for applications such as self-assembly, extracellular matrix (ECM) mimicry, or drug delivery.
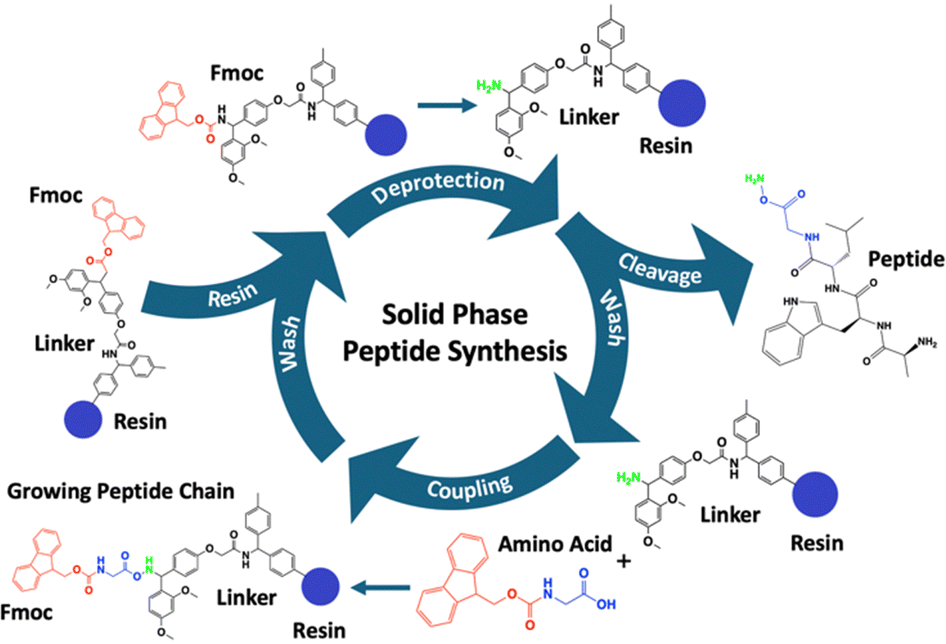 | ||
| Fig. 1 The key steps of solid-phase peptide synthesis (SPPS). The resin is first saturated in an organic polar solvent such as N,N-dimethylformamide (DMF) or dichloromethane (DCM). This is followed by deprotection of the Fmoc group to expose the amino group for subsequent coupling reactions. Washing steps to remove unbound reagents and by-products, ensure that the resin is free of contaminants. Amino acid coupling then follows, facilitated by activating agents, to attach the next amino acid in the sequence. This is followed by additional washing to maintain reaction fidelity. Finally, cleavage of the synthesised peptide from the resin is performed, along with sidechain deprotection, to release the final peptide product. | ||
The composition of amino acids in the peptide-based material can directly dictate cellular behaviour. Self-assembling peptides utilising combinations of polar and non-polar amino acids to direct folding, tangling, can be used to create biomaterials. By utilising the natural secondary structures, α-helices and β-sheets, peptides can support the propagation of self-assembling 3D materials such as hSAF (hydrogelating self-assembly fibres)8,17 and RADA16-I systems.45 α-Helices require specific polar and non-polar regions in the peptide sequence to create turns in the linear chain, whereas β-sheets utilise alternating polar and non-polar amino acids to create large folds, forming stacked sheets with a myriad of examples utilising them to create 3D materials.8,17,46,47 The creation of 3D secondary structures, whether α-helical or β-sheet based, allows these materials to more closely mimic fibril structures typically seen in the ECM. The dynamic fibrous environment of the ECM influences cell signalling transduction, as well as degradation and reconstruction of tissue matrices.48 Mimicking the ECM is an ideal way of mitigating a negative immunological response to biomaterials and influencing cell–cell paracrine signalling to promote acceptance by the body. This strategy has further been shown to improve damaged ECM reconstruction.49–51 Other functional moieties can also be appended to a peptide chain to increase cellular compatibility.
Functionalisation of peptides or polymers aims to chemically couple additional moieties to improve for example, the biological properties of materials, such as targeting the cell receptors/integrins which control cellular response to environmental stimuli; crucial for tissue repair, immune response, and development, such as cell survival and proliferation.52,53 A popularly used moiety for material functionalisation is the RGDS peptide. This sequence is repeated in many proteins, for example fibrinogen, and can interact directly with integrin αVβ5, promoting cell adhesion and proliferation, and contributing to biomaterial integration with cells both in vitro and in vivo.54–57 Functionalisation is achieved by chemical ligation to modified side chains through reactions such as Cu-catalysed cycloaddition (CuAAC),58 strain-promoted azide–alkyne cycloaddition (SPAAC),59 thiol–ene,60 Staudinger ligation,61 and others.62
Given their versatile properties, branched peptides are underrepresented in biomaterial research. This review introduces branched peptides and provides an update in this field, such as novel orthogonal deprotection methods, functionalisation with bioactive peptides, such as RGDS and IKVAV and other long-chain polymers, including lipids and carbohydrates. We also review the current state of supramolecular self-assembly using branched α-helices and β-sheets focussing on how these structures may be adapted to different solvent environments, particularly for modifying material stiffness and degradation rates and the limitations associated with current experimental designs. Finally, we outline how branched peptide research has influenced other fields including oncology and infectious diseases, and discuss the future of branched peptide-based biomaterials.
2. Branched peptide design, structure, and synthesis
2.1 Overview of branched peptides
Early iterations of branched peptides were synthesised using thermal homo- and co-polymerisation, primarily of lysine, glutamic acid and aspartic acid,63 creating arbitrary structures with limited applications.35,64 Current peptide syntheses rely heavily on SPPS, rather than liquid-phase peptide synthesis (LPPS) offering greater chemical stability by controlled fluorenylmethyloxycarbonyl (Fmoc) deprotection, efficient couplings, and a variety of side chain protecting groups, removable under conditions such as tert-butyloxycarbonyl (Boc or tBu) in 50% trifluoroacetic acid (TFA) and 2-acetyldimedone (Dde) in hydrazine.65Most branched peptides can be separated into three broad categories based largely on the AB2 branching method, where the starting amino acid (A) can form two branches (B2)37,66 (Fig. 2A). Hyperbranched peptides are highly branched, 3D structures with multiple functional end groups (Fig. 2B). They are synthesised through global deprotection and one-pot coupling reactions,67 where, due to the limits of SPPS and steric hindrance, some branches may be longer, others shorter, and some non-existent; a problem faced by most hyperbranched polymer systems.67,68 This creates the least organised of all three structures but does make them suitable for exploring extended molecular interactions and scalable syntheses.
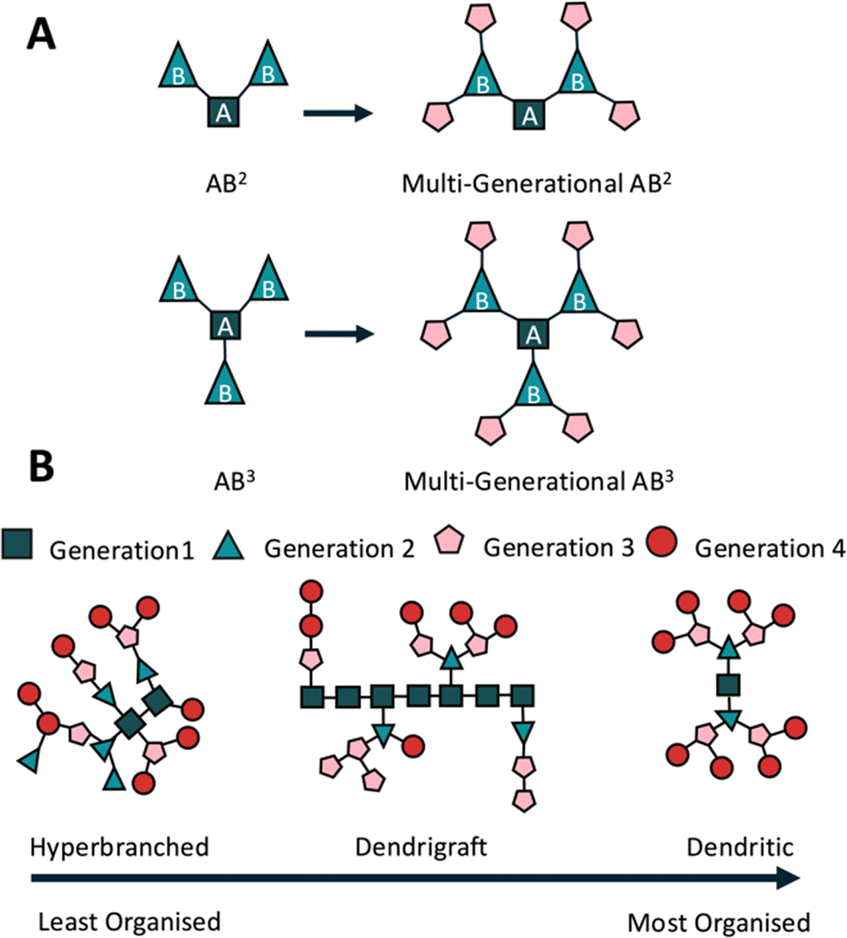 | ||
| Fig. 2 Polymer branching types. (A) AB2 & AB3 polymer branching types, dictating the formation of either two bonds (AB2) or three (AB3) from a starting amino acid. (B) Peptide branching conformations, hyperbranched peptides represent the least organised structure, synthesised through a one-pot synthesis originally using thermopolymerisation, forming complex 3D networks, but with an arbitrary structure. Dendrigraft peptides are typically formed through coupling of linear, or dendritic peptides, onto a linear chain, representing a relatively organised and most recently developed of all three branched peptide types. Dendritic peptides are synthesised through sequential couplings and deprotections to create stable structures (typically used as drug delivery vectors). | ||
Dendrimer peptides are tree-like macromolecules with a central core and multiple branched generations (Fig. 2B). They provide precise control over size, shape, and functionality and are ideal for gene delivery69 and bioimaging.70 Tam first described using the lysine core to synthesise a peptide dendrimer as an antigen presenter for vaccine design and multiple antigen peptides (MAPs).71 The polyvalency is generated by multiple outward-facing arms, allowing them to be used as drug carriers.72 The addition of natural peptide functionalities reduces targeted immunological events, thereby increasing peptide half-life in vivo.73 The typical radial shape also reduces enzymatic contact with molecules closer to the centre, reducing degradation and allowing them to be used as drug vectors.74
Dendrigraft peptides present a linear peptide in their centre, branching within the chain or at the termini66 (Fig. 2B). These branches may be other linear30 or dendritic structures,75 such as the dendrimer peptides described earlier. Dendrigraft synthesis relies on the coupling of peptide chains, as opposed to the single amino acid couplings used for dendrimer synthesis. This increases the ease of synthesis and promotes bulk multi-gram scale production.76 Using alternative side chain deprotection allows for multiple peptide couplings without compromising other side chains and branching points, enhancing adaptability and control over functionality. Multiple deprotection strategies, SPPS chemistry, and fewer branching points compared with the two previously described branched peptide systems improves dendrigraft synthesis, making them easier to manipulate and offering controllable functionality and degradation rates.30
2.2 Methods for branched peptide synthesis
Branched peptide synthesis follows two primary strategies: divergent and convergent.72 The divergent approach involves building the peptide directly on the resin, where the branches are added sequentially during synthesis; typically used with hyperbranched peptides and dendrimers such as MAPs.71 This method minimises purification steps and facilitates large-scale production but can lead to steric hindrance and reduced coupling efficiency as the branches grow. The convergent approach, in contrast, synthesises the branches independently before attaching them to the core peptide, and is often applied to the synthesis of dendrigraft peptides.77 This method allows for the optimisation of individual branches and reduces steric issues but requires additional purification and coupling steps. Advances in orthogonal ligation techniques, such as click chemistry, have made convergent synthesis increasingly accessible and efficient.58,78,79 Chemical ligation methods play a vital role in enhancing the complexity and functionality of branched peptides. Techniques such as native chemical ligation (NCL) enable the assembly of large, branched peptides by linking unprotected peptide segments through chemoselective reactions at cysteine residues.80,81 Similarly, Cu-catalysed azide–alkyne (CuAAC),82 strain-promoted azide–alkyne (SPAAC)83 and thiol–ene cycloaddition84 have facilitated the rapid and robust attachment of functional groups and peptide branches. Despite these advancements, synthesising branched peptides poses unique challenges. Steric hindrance during the addition of branches can lead to incomplete reactions and lower yields, necessitating the use of excess reagents or extended reaction times.31,85 The repeatability challenges associated with synthesising branched peptides and limited side chain chemistries have presented a significant factor in their slow development. Addressing early-stage synthesis issues would significantly advance the field.Innovations, such as automated SPPS protocols,86 microwave-assisted synthesis,87 and improved coupling agents like O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluor-ophosphate (HATU) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU),88 have mitigated these issues by enhancing reaction efficiency and reducing synthesis times. Additionally, the development of advanced resins, such as the 2-chlorotrityl chloride resin,89 offers improved swelling properties and stability, further optimising the synthesis of branched peptides (Table 1). Another area of research is the exploration of novel safety catch mechanisms to enable orthogonal deprotection for the implementation of on-resin branching points (Fig. 3). These safety catches can prevent unwanted deprotection, thereby preventing loss of yield.90 One notable study converted the side chain protecting groups of six amino acids, three of which can form stable branching points, into a new safety catch system by incubation with Me3SiCl-Ph3P in tetrahydrofuran. This created labile protecting groups enhancing the control over deprotection steps.91,92 Additionally, Zhou93 developed a reaction scheme to convert a Boc-protected lysine to a 2,4-dinitro-6-phenyl-benzene sulfenyl (DNPBS) protecting group, which can undergo orthogonal deprotection against Fmoc, contributing significantly to the construction of branched peptide systems with high precision and complexity. These advancements significantly enhance feasibility and scalability, making branched peptide synthesis more accessible (Table 1). By enabling precise control over deprotection steps, researchers can create more complex peptide structures such as cyclic branched peptides or orthogonal functionalisation, addressing some of the longstanding challenges associated with peptide synthesis such as low yield and purity issues.
| Name | Type | Sequence | Main properties | Activity | Applications | Ref. |
|---|---|---|---|---|---|---|
| Multiple antigen peptide (MAP) | Dendrimer | (Boc-peptide-Gly3)8-Lys4-Lys2-Lys-Ala-OCH2 | Large multi-arm dendritic structure | Capable of binding to different viral or bacterial antigens | Synthetic vaccine candidate | 71, 94 and 95 |
| P-alkyne and P-azide | Hyperbranched | Glu3-hexynoic acid or azidohexanoic acid | Dendritic structure functionalised with DNA binding domains | Functioning as a solid phase DNA hybridisation platform | Enhanced COVID-19 detection | 96 |
| D3K2 & D3G2 | Dendrigraft | Ala-Lys-Lys3-(Lys3)2-(Lys3)4-(Lys3)8-(Lys-NH3+)8 | Increase permeability through phospholipid bilayer | Transport of genes across the cell membrane | Induce targeted cytotoxicity to cancer cells. | 69 |
| HAIYPRH | Dendrimer | PAMAM-PEG-T7 | Multi-arm structure containing a gadolinium chelating moiety binding | Able to localise and bind to cancer cells through peptide recognition sequences | Increase magnetic resonance imaging resolution of cancer cells | 70 and 97 |
| FD2 | Dendrimer | (Fuc-Lys-Pro-Leu)4-(Lys-Phe-Lys-Ile)2-Lys-His-Ile | Localised binding to P. aeruginosa lectin receptors, LecB and LecA | Lectin binding to prevent the deposition of biofilm extra polymeric substance | Treatment the multi-drug-resistant bacteria P. aeruginosa | 98 and 99 |
| G3KL | Dendrimer | (Lys-Leu)8-(Lys-Lys-Leu)4-(Lys-Lys-Leu)2-Lys-Lys-Leu | Terminal positivity charged amino acids | Disrupt bacterial cell membranes, leading to cell death | Combating anti-microbial resistance | 100–102 |
| (LDLK)3 | Dendrigraft | (Leu-Asp-Leu-Lys)3 | Self-assembly branched peptide nanostructure | Utilising a lysine branching point as a cross-linking agent between three linear peptides | Enhanced hydrogel stiffness using branching points to create a more versatile SAP | 30 |
| Denpol | Dendrigraft | Cys-Cys-Lys(Lys(Lys2))-Cys-Cys-Lys-(Lys(Lys2)) | Combinatory pH responsive, hydrophilic/phobic branched structure | Permeability through cell membrane to deliver siRNA | Alternative non-viral siRNA delivery vector | 75 |
 | ||
| Fig. 3 Orthogonal and safety-catch protecting group strategies in solid-phase peptide synthesis. (A) Merrifield strategy using Boc for α-amino protection and Bzl for side chains, both removed by acidolysis but with different kinetics. (B) Bis-orthogonal strategy with Wang linker, Fmoc for α-amino protection, and tBu for side chains, enabling selective deprotection via acidolysis or base treatment. (C) Modified bis-orthogonal scheme with Cl-trityl chloride (CTC) resin, allowing peptide release with dilute TFA while retaining tBu-protected side chains. (D) Three-orthogonal approach using a photolabile linker with Fmoc and tBu groups, offering flexibility in deprotection and peptide cleavage. (E) and (F) Safety-catch linkers such as Mmsb, which resist TFA conditions but can be converted into labile forms via reductive treatment, providing compatibility with Boc and Fmoc chemistries. Reproduced (adapted) with permission from ref. 91 Copyright 2024, MDPI. | ||
3. Controlling the physical properties of branched peptides
3.1 Customisation of branched peptides for biomedical applications
Customisation of branched peptides is critical for developing functional materials tailored to applications such as drug delivery, tissue engineering, and ECM mimics. Customisation can include pH dependent degradation, modulation of material stiffness, or the incorporation of cyclic peptides or non-natural amino acids. Branched peptides offer unique advantages over linear peptides due to their multivalency, tuneable architecture, and enhanced stability. For instance, drug carriers leveraging branched peptide frameworks benefit from increased drug binding efficiency and controlled release kinetics.103 Tryptophan-rich peptide dendrimers (TRPDs) utilise lysine branching with tryptophan-terminated arms, improving drug stability and reducing degradation by enhancing interactions such as hydrophobic, π–π stacking, and hydrogen bonding, making them effective vehicles for gene therapies and DNA-targeting drugs (Fig. 4).104,105 Similar to TRPDs, dendrimers are also customisable, by changing the terminal amino acids and therefore changing the properties of the material. Mixson and colleagues added a histidine-rich tail to their branched dendrimer, discovering that the new design improved in vitro gene transfection efficiency.106 Similarly, poly(amidoamine) (PAMAM) dendrimers, using a lysine core, offer a high degree of customisability,107 having been functionalised with dodecapeptides to deliver interleukin-12 plasmids to mesenchymal stem cells, significantly improving the drug's cancer-targeting ability.33 Others have used un-natural amino acid derivatives to provide branching points, such as 2,3-diaminopropionic acid, which was used to develop a three-arm dative chelator for Ca2+ binding and transport.108 Branched peptides can also be designed to mimic the structures of other biological molecules, such as phospholipids, e.g. by using the terminal lysine as the polar head, and histidine/leucine as the non-polar body, to increase cell membrane transfection through the amphipathic structure.109 This versatility in designing branched peptides not only enables fine-tuning of their structural and functional properties but also opens avenues for combining them with other peptide systems, such as cyclic peptides, to further enhance their biomedical potential.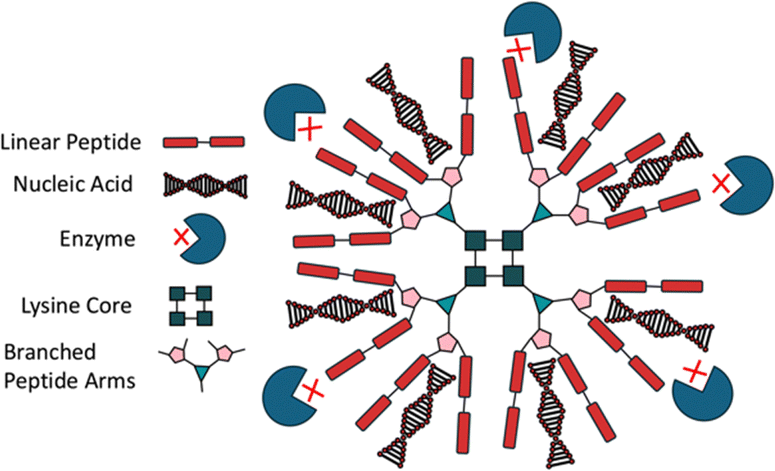 | ||
| Fig. 4 A dendrimer polylysine star. Of the three branched peptide types, dendrimers are typically used for drug delivery. Generating a lysine core can increase the production of available coupling points. In this case, a lysine core, depicted by the central squares, has created coupling points for red rectangle linear peptides. The densely packed structure and coupling of adhesive peptides (red rectangles) allows for the embedding of cargo such as nucleic acids (red & black triple helices) affording protection from enzymatic attack (outer blue circles). This allows for safe and practical transport of molecular mediators such as small-molecules and plasmids to the target cell. | ||
Cyclic peptides, characterised by the circular presentation of the amino acid sequence, either across the entire peptide or at the termini, offer distinct advantages in biomedical applications due to their conformational rigidity and enhanced proteolytic stability (Fig. 5).110,111 These features improve target binding affinity and selectivity while reducing cellular toxicity, making them promising candidates for therapeutic use.112,113 Their stability is underscored by the 18 cyclic peptides currently approved for clinical trials.113 When integrated with branched peptides, the resulting hybrid systems exhibit enhanced functionality, combining the stability and specificity of cyclic peptides with the multivalency and tunability of branched architectures.114 A practical demonstration of this synergy was provided by Lu et al., who coupled a cyclic peptide to the aspartic acid carboxyl group of a branching peptide, facilitating in situ ligation with an N-terminal cysteine.115 The resulting cyclic-branched peptide was synthesised with a yield of 50%, showcasing a novel approach to creating complex peptide structures. Such hybrid systems exemplify how customisation can generate multifunctional biomaterials, with potential applications in drug delivery, targeted therapeutics, and regenerative medicine. The iterative ‘denpol’ strategy developed by Zeng et al. further highlights this potential, demonstrating the construction of highly branched macromolecules through dendritic polypeptide coupling.75 This approach enables fine control over structural features, creating materials tailored for specific biomedical functions. Integrating cyclic motifs with branched systems also aligns with broader efforts to develop multi-receptor binding peptides, exemplified by polymer systems such as hyaluronic acid and N-(2-hydroxypropyl)methacrylamide, which are currently being trialled for similar applications.116,117 The ability to customise these hybrid structures further enhances their relevance, offering routes to creating tailored therapeutics with improved efficacy and specificity.
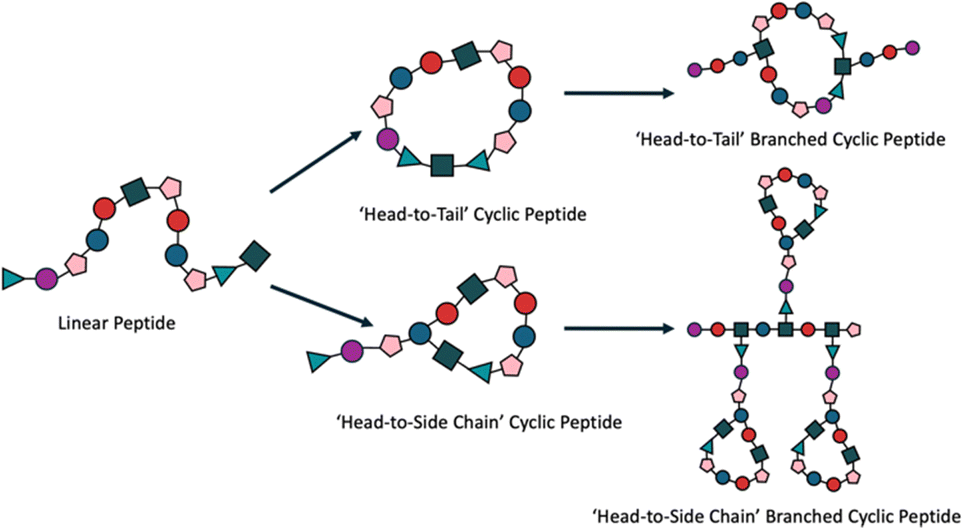 | ||
| Fig. 5 Generation and incorporation of cyclic peptides into branched peptide systems. The process begins with a linear peptide precursor, which can be cyclised into two distinct types of cyclic peptides: ‘head-to-side chain’ cyclic peptides, characterised by a cyclical end with a linear tail, and ‘head-to-tail’ cyclic peptides, featuring a simple closed-ring configuration. These cyclic peptides are subsequently incorporated as branching points into branched peptide systems, enhancing the versatility and stability of the resulting biomaterials. | ||
3.2 Control of degradation rates
Controlling the degradation rate of peptide-based structures has been highlighted throughout this manuscript. Various strategies have been employed to reduce peptide degradation and minimise premature loss of function while maintaining biological activity. Common strategies include the addition of motifs which cannot easily be cleaved by proteolytic enzymes, such as cyclisation,118 incorporation of D-amino acids,119 or the use of unnatural amino acids.120 These methods enhance resistance to degradation, increasing the material's versatility, which is crucial for in vivo applications such as cancer immunotherapies121 and biomaterial-based tissue engineering.122 Cross-linking is another method typically used to reduce degradation rates. However, in selecting crosslinking methods we must also consider the end application to avoid cytotoxicity. It is here that the introduction of branching points can create new opportunities to develop complex matrices without the need for potentially toxic crosslinking agents whilst benefiting from reduced degradation rates and an improved active half-life. This is demonstrated by Wang,123 who combined an α-helical peptide with a cystine lysine dendrimer, inducing cancer cell degradation (Fig. 6). They found that using a branched peptide was more effective than using the linear counterpart and reduced the degradation rate, proving a viable potential cancer therapy.123 The increased half-life of the peptide compared to the linear counterpart provides a suitable design template that could be applied to other peptide-based drug targets such as glucagon-like peptide-1 & 2 receptors in the treatment of type 2 diabetes,124–126 gonadotrophin-releasing hormone receptor as a novel drug candidate for prostate cancer,127 and parathyroid hormone-related protein receptor involved in osteoporosis therapies.128 By understanding and manipulating the degradation profile of peptide materials, we can better mimic the biological environment and improve clinical outcomes. | ||
| Fig. 6 Structural representations of linear and branched α-helical peptides. Schematic illustrations showcasing variations in peptide architecture. (A) The linear (LLKK)4 peptide with a single continuous sequence. (B) A 2-arm branched peptide, [(LLKK)2]2κC, with branching introduced via a lysine residue. (C) A 4-arm branched peptide, {[(LLKK)2]2κC}2, featuring a more complex branched structure derived from multiple lysine-mediated branching points. These designs highlight the increasing structural complexity and potential functional versatility of α-helical peptides with branching. Reproduced (adapted) with permission from ref. 122 Copyright 2024, Royal Society of Chemistry. | ||
As previously discussed, branched peptide systems are typically synthesised and assessed for their structural and biomimetic characteristics.32,46,47 However, research on the physical properties in vitro, such as degradation rates, is rarely assessed. One of the few examples where in vitro degradation studies have been employed is shown by Agazzi,129 where the branching structure was used to create pH- and enzyme-labile degradation profiles. This work highlights the potential of branched peptide structures to enhance biomaterial stability, thereby improving future material designs. Exploring this research area is crucial as it can reveal how branch points on peptides can enable greater stability and improve the longevity of biomaterials. It could also provide valuable data on material half-life when exposed to typical matrix proteases. Peptide degradation can be assessed from macro and molecular perspectives. At the macro level, rheological studies allow measurement of the viscoelastic properties of peptide-based solutions or soft solids, such as elastic (G′) and viscous (G′′) moduli. These measurements help in understanding the changes in the mechanical properties of peptides. Optical techniques, such as turbidity or light scattering, can also monitor bulk structural degradation.130,131 At the molecular level, the combination of high-performance liquid chromatography (HPLC)132 and mass spectrometry (MS)133 can separate, quantify, and identify degradation products or intact peptides. Spectroscopic methods such as circular dichroism (CD)134 track structural changes such as the formation or loss of secondary structures, α-helices and β-sheets. These methods together offer insights into the stability and breakdown of peptide-based materials. Such information is directly relevant to material usability and marketability since a biomaterial that degrades too quickly before achieving its therapeutic potential holds limited clinical value. Pharmacokinetic studies routinely assess degradation rates to determine the therapeutic effectiveness of drug candidates.135,136 These studies provide a deeper understanding of retention times and degradation rates, which are critical for predicting the in vivo behaviour of the therapeutic agents. Applying a similar rationale to branched peptides could help ascertain their in vitro degradation rates and ultimately lead to the development of more clinically relevant materials. This approach would ensure that branched peptide-based biomaterials maintain their functional integrity long enough to exert their intended therapeutic effects. This dual focus on structural integrity and material longevity is exemplified by innovations such as the lysine knot (discussed later), which enhances peptide stability and stiffness to better suit biological environments.
3.3 The impact of branching on the macromolecular structure
Utilising branched peptides to enhance secondary supramolecular structure is not a novel concept. A 2003 study on ‘T-SAFs’ introduced a branched helical coiled-coil peptide, exhibiting self-assembly branched nanofiber formations.137 Similar studies later followed, creating a branched peptide amphiphile which self-assembles into nanofibrils, for fibre-bonded poly(glycolic acid) scaffolds138 and RGDS epitope presentation.139 Building on the concept of using branching points to influence nanofiber formation, this work was continued in 2007, where branching extended the diameter of the nanofibers and reduced mechanical stiffness (Fig. 7).7 This was later adapted to guide neural cell network development, linked to the reduced stiffness of wider, branched, fibrils compared with linear fibres.32 The system developed by Sur and colleagues utilised two β-sheet peptide amphiphiles, one linear and one branched, with varying stiffnesses, between 7.3 kPa and 22.9 kPa, to guide fibroblast and neuron development. When assessed using 3T3 fibroblasts and mouse hippocampal neurons, they found different growth patterns between the materials, owing to the cellular preference of material stiffness. However, other branched peptide designs attempt to use branching points as a peptide-bond-based crosslinking strategy, enhancing mechanical strength.30 | ||
| Fig. 7 Morphological differences in peptide amphiphile (PA) nanofibers formed from branched and linear PAs. Cross-sectional representations of nanofibers highlighting structural variations based on the PA architecture. (A) Nanofibers derived from a branched PA featuring a single cyclic RGDS epitope. (B) Branched PA nanofibers incorporating two RGDS epitopes. (C) Nanofibers formed by a branched PA with a single RGDS epitope. (D) Linear PA nanofibers containing a single RGD epitope. These variations demonstrate how epitope arrangement and PA structure influences nanofiber morphology and organisation. Reproduced with permission from ref. 7 Copyright 2024, Elsevier. | ||
The lysine knot, developed by Pugliese, uses a Nε-di-Fmoc-lysine to bond three linear peptides simultaneously.30 Rheological studies showed that the linear peptides displayed weak associations, creating a loose network and a soft material. In contrast, when mixed with the lysine knot at a high molar ratio, the stiffness increased by approx. 100-Fold, with a final stress tolerance of 8.14 kPa.30 These studies collectively show how branch peptides can be customised to both reduce or enhance stiffness depending on the design. Such stiffness changes can have a significant impact on cell behaviour. For reference, fibroblasts exhibit a stiffness of ∼3 kPa, whereas lung tissue is ∼0.2 kPa, contrasted with the stiffness of bone at just under 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kPa.140 Several other studies detail how cells can transduce mechanical forces into biological signals, providing the basis for directing cell behaviour based on stiffness.141–143 Utilising cell receptors such as RHO-GTPase (guanosine triphosphatase)144 and transducers, including YAP (yes-associated protein) and TAZ (tafazin-associated protein),145 the ability to control a material's stiffness creates a more versatile 3D system that can be adapted to different tissue and cell types depending on the clinical need. By considering both the 3D environment as well as the stiffness, supramolecular designs could revolutionise other research fields such as organ-on-chip and organoid development. Many existing platforms in these areas do not offer modifiable stiffnesses that prevent natural gene expression146 with specific spatiotemporal mechanical properties.147,148 Therefore, innovations in peptide design, and the incorporation of branching points to fine-tune mechanical properties and degradation rates, are key to overcoming these challenges and advancing the development of realistic and functional organ-on-chip and organoid systems.
000 kPa.140 Several other studies detail how cells can transduce mechanical forces into biological signals, providing the basis for directing cell behaviour based on stiffness.141–143 Utilising cell receptors such as RHO-GTPase (guanosine triphosphatase)144 and transducers, including YAP (yes-associated protein) and TAZ (tafazin-associated protein),145 the ability to control a material's stiffness creates a more versatile 3D system that can be adapted to different tissue and cell types depending on the clinical need. By considering both the 3D environment as well as the stiffness, supramolecular designs could revolutionise other research fields such as organ-on-chip and organoid development. Many existing platforms in these areas do not offer modifiable stiffnesses that prevent natural gene expression146 with specific spatiotemporal mechanical properties.147,148 Therefore, innovations in peptide design, and the incorporation of branching points to fine-tune mechanical properties and degradation rates, are key to overcoming these challenges and advancing the development of realistic and functional organ-on-chip and organoid systems.
4. Biomimetic functionalisation
4.1 Bioactive peptide functionalisation
By incorporating bioactive sequences, branched peptides can further enhance cell signalling leading to improved cell adhesion and differentiation, making them more effective than inert materials for drug delivery, tissue engineering, and regenerative medicine. From earlier discussions, it is unsurprising that several researchers have used RGDS to functionalise their initial branched peptide designs. Combining the biomimetic functionality of RGDS with multivalent branched peptides is demonstrated by Liu who synthesised a linear (RGD)3 peptide functionalised with two cyclic RGD peptides, creating two dendrigraft branched peptides.149 As discussed earlier the use of cyclic peptides has many physical advantages, but when used in conjunction with functional peptide moieties they can represent the in vivo environment more closely, resulting in an increased upregulation of cell proliferation through enhanced cellular attachment.150,151 Similarly, others have used an IKVAV-capped dendrimer embedded into a cross-linked collagen hydrogel which selectively upregulated rat Schwann cell proliferation and reduced human dermal fibroblast cell proliferation.152 These approaches demonstrate the versatility and efficacy of incorporating bioactive peptide sequences into biomaterials to achieve specific cellular responses.Functionalisation strategies have since developed from single epitope peptides to multiple epitopes, taking advantage of the availability of serval binding sites typically seen with branched peptide termini. The use of multiple peptide epitopes enhances cell signalling by simultaneously activating several integrin receptors, leading to a more robust cellular response.153 This approach better mimics the complex biochemical environment of native tissues, which often presents multiple binding sites to cells.154 Mas-Moruno utilised a lysine branching point to display RGDS and PHSRN simultaneously to promote integrin binding to metallic implants.31 Whilst bound to a titanium plate, the dual-functionalised system induced significantly increased cellular spreading and proliferation compared to non-coated surfaces or materials functionalised with a single peptide variant. Others have followed similar strategies to introduce more than one integrin-interacting peptide sequence to their own materials. For example, RGDS and IKVAV were presented simultaneously on another titanium implant, influencing cellular adhesion, proliferation, viability, and angiogenesis of endothelial cells.132 Dual-functionalised systems could reduce the risk of implant rejection and enhance integration with surrounding tissue, as seen with RGD and GFOGER.9 Furthermore, by selecting appropriate combinations of peptide epitopes to target desired cellular responses, this strategy allows for the customisation of biomaterials for specific applications.155
4.2 Hybrid-polymer functionalisation of branched peptides
Peptide-based functionalisation has been shown to increase bioactivity or provide sites of attachment for other cell-interacting chemistries, such as cell receptors or non-polar molecules. Although there are many examples of peptide functionalisation, less attention is focused on using other natural polymers to functionalise linear or branched peptide systems. In the body proteins readily undergo polymer functionalisation to enhance protein stability and secondary folding,156 reduce proteolytic degradation,157 and facilitate intra-/extra-cellular transport.158 Using a similar approach, bioactive polymers can be coupled to peptide-based materials to improve biological properties, which is not easily achieved through peptide chemistry, or the addition of small moieties, alone. Lipidated proteins, integral to native healthy tissues, matrices, and cells, facilitate metabolic transport,159 cell membrane anchoring,160 receptor binding,159 signal transduction,161 and intracellular trafficking162 by providing a means to non-polar interactions. Whilst this does include some non-polar amino acids such as alanine, valine, and leucine, they cannot achieve the same non-polar interactions as lipid molecules, owing to their natural amphipathic design. Combining the polar nature of peptides, multi-valency and the customisation of branched peptide designs, with the non-polarity of lipid molecules presents an opportunity to create dynamic biomaterials capable of interacting with multiple cell types, membranes and receptors. This was achieved in 2010, where researchers used a dendritic branching peptide with an aspartic acid core to create a self-assembling organogel which formed in various organic solvents at low concentrations163 (Fig. 8). Similarly, Haridas synthesised an asymmetrical branched peptide dendrimer using lysine, aspartic acid and glutamic acid branches, coupled to dodecylamine to create the lipidated compound.164 Due to the asymmetry of the design, the peptides self-assembled in both aqueous and organic solvents. Whilst being able to utilise organic solvents, organogels can sequester non-polar drugs and administer through direct cell membrane interaction, allowing for targeted drug delivery,165 and allowing access to cellular receptors otherwise inaccessible to polar peptides. This was also demonstrated by Sivades, who showed that lipidated glutamic acid dendrimers increased the chondrogenic differentiation of adipose-derived mesenchymal stem cells, with one lipopeptide regulating collagen type 1 production, suggesting potential for hyaline cartilage formation and prevention of hypertrophic development.166 While further research in in vitro models is necessary to fully understand the impact of these branched peptide hybrids, these findings indicate that providing interactions with non-polar molecules could aid drug delivery167 as well as an upregulation in ECM fibre formation which would otherwise have not been possible without lipidation. | ||
| Fig. 8 Self-assembling branched peptide organogels. Three different generations of branched peptides were compared, each undergoing the coupling of an additional generation of amino acids. (A)–(C) Field emission scanning electron micrographs of the resulting gels at the three different generations. (A) Shows the fewest number of generations and least dense network, whilst (B) is the median number of generations and density, and (C) is the largest number of generations forming the densest network. (D)–(F) High-resolution transmission electron micrographs (HR-TEM) of the branched gels obtained from the different generations of dendritic 1,2-dichlorobenzene. Reproduced with permission from ref. 162 Copyright 2024, American Chemical Society. | ||
Other natural polymers, such as carbohydrates also pose a significant benefit to branched peptides. Glycopeptides are used frequently by the body to promote cell–cell recognition,168 signalling,169 and to regulate the immune response.170 They facilitate interactions with lectins,171 and other carbohydrate-binding proteins,172 thereby influencing various physiological and pathological processes. When combined with biomaterials they can offer unique advantages as seen in next-generation antibiotics such as vancomycin173 and teicoplanin,174 anticancer drugs such as bleomycin175 and more recently supramolecular biomaterials.176,177 These developments have led to materials which react to pH,178–180 redox,181,182 and temperature.183,184 To this end, branched peptide systems have been further modified to adopt glycans, e.g., Li used a triazine core with three glycosylated phenylalanine residues bound in a ‘Y’ shaped conformation.185 After 24 hours of culturing with RAW264.7 macrophages, the studies showed that there was no evidence of cytotoxicity-driven cell death, though pro-inflammatory cytokines were observed. The presence of pro-inflammatory cytokines does suggest M1 macrophage polarisation, is not suited to ECM regeneration, although further studies would be needed to confirm. Other researchers have modified linear peptides for a variety of functions such as HIV inhibition186 and to cross the blood–brain barrier,187 as well as structural enhancements including in vivo peptide stability188 and half-life.189 Again, studies in this area remain limited and further research is needed to fully reap the benefits of branched peptide glycosylation as seen with linear peptides and to further understand, and exploit, their potential for biomedical applications. Of note is the need for comprehensive in vivo studies essential for the evaluation of their pharmacokinetic characteristics, biodistribution, and long-term effects. As these advances are realised, glycosylated branched peptides could revolutionise drug delivery, enhance therapeutic specificity, and contribute to innovations in immune regulation190 and interactions with glycan receptors including those involved in diabetes.191 With more than 90% of ECM proteins being glycosylated, this increases the mimetic qualities of ECM-inspired biomaterials.192
5. Medical & clinical application of branched peptides
The unique structural attributes of branched peptides, discussed throughout this review, including their multivalency103 and enhanced stability,30 have positioned them as promising candidates for medical and clinical applications. This is particularly evident within oncology and infectious diseases. Their modular nature allows for targeted interactions,104 precise delivery,33 with minimal immunogenicity.193 In oncology, branched peptides have shown considerable potential as targeted drug-delivery vehicles and anticancer agents. By leveraging multivalent interactions, branched peptides can selectively bind to overexpressed tumour receptors. Research in this area is primary focused on sulphated glycosaminoglycans (GAGs) and G protein-coupled receptors using the tetra-branched peptide human neurotensin (NT4).194–196 NT4 selectively binds to the NTS1 receptor overexpressed on certain malignant tumours,197 and was modified in 2011, creating a tetra-branched version. Comparatively, the branched version was found to increase the cytotoxicity of the drug doxorubicin, through increased binding affinity and tumour selectivity.198 This system has since been tested to compare linear and branched peptides,199 explore the role of heparin-sulphate proteoglycans receptors,194 and to create new derivatives with selective binding and in vitro cytotoxicity.200 The versatility and biocompatibility of branched peptides have also been used to increase the solubility of drugs, to preferentially accumulate in tumour tissues, and improve drug delivery efficiency. In one such example two branched amphiphilic peptides were self-assembled into nanofibrils, loaded with anti-cancer drugs, allowing for selective and prolonged drug release with increased cytotoxicity to cancer cells.201 Separately, branched peptides have been created with anti-cancer properties without requiring additional drug doping. Recncinai and colleagues developed two branched peptides, BOP7 and BOP9, which selectively bind to cancer cells, causing the release of damage-associated molecular patterns. This led to immunogenic cell death, equating to a 20% inhibition of tumour growth and grafting.202Other clinical applications of branched peptides include the treatment and reduction of microbial disease and infection. While antimicrobial resistance (AMR) continues to present considerable challenges, there is research on the potential use of peptides as therapeutic agents to act as front-line drugs to fight AMR.203 James Tam in 198871 first introduced this potential through the development of the earlier-mentioned MAPs as vaccine candidates. By 1999, the use of MAPs as antimicrobial agents was assessed, expressing eight copies of the antimicrobial peptide lactoferricin, and was noted to have had significant antimicrobial effects even on methicillin-resistant Staphylococcus aureus.95 More recently, antimicrobial peptide dendrimers (AMPDs) have been developed to interact with microbial membranes through multivalent binding, leading to membrane disruption and microbial death, with several reviews detailing their successes.73,204 Compared to linear peptides, branched AMPDs exhibit increased potency,100,205 reduced susceptibility to proteolytic degradation,206 and present scope for more complex designs.207 It has also been noted that bacteria are much slower to develop resistance against AMPDs once deployed in vitro.208–210 Biofilms present further complications, acting as a physical barrier against anti-microbial agents.211 In 2016 researchers developed a repeating Arg-Trp branched peptide (2D-24) which was shown to eradicate up to 94% of biofilm-producing multi-drug-resistant P. aeruginosa at concentrations that are not toxic to mammalian cells.193 Despite these early studies presenting clear potential for branched peptides to make significant impact on global health issues their progression from bench to bedside remains challenging.
Currently, only a handful of branched peptide-based therapies are in industrial development or have reached the market, such as the AMPD, VivaGel, which has been approved for clinical use in the United Kingdom and Australia for the treatment of bacterial vaginosis.212,213 AMPDs appear to show promise in moving from the laboratory to the clinic quicker than other branched peptide constructs with more work going into in vitro toxicity studies.214 A recent paper identified several tetra-branched AMPDs and tested these at 10 times the minimum inhibitory concentration and found that most showed no cytotoxicity against red blood cells.214 Although this is a limited view of cytotoxicity compared with in vivo studies, it does provide advancements in the field. Similarly, after their development in the 1980s, some MAP designs have been used for diagnostic tests215 or biosensors,216 but very few have reached clinical trials and those that did failed due to a low immunogenic response.217 Some more recent in vivo assessments have produced constructs that elicit promising immune responses, though these have yet to be approved for clinical trials.94,218 Thus, the popularity of MAPs appears to have decreased after clinical trial failure, with a decline in pharmaceutical interest leading to a lack of overall progression. Key to this lack of interest is addressing the limitations, including scalability of synthesis,219 high production costs,220 and concerns about long-term biocompatibility.195 Furthermore, many branched peptide therapeutics in the pipeline face regulatory hurdles related to their structural complexity and lack of precedent in clinical use.221 The few approved branched peptide systems highlight the need for further innovation to address these shortcomings. For instance, while their multivalency enhances drug delivery and target specificity, it may also increase the risk of off-target interactions or unexpected immunogenicity.195,222 These limitations underscore the importance of continued research into optimising the design and synthesis of branched peptides to balance efficacy, safety, and cost-effectiveness. By overcoming these challenges, branched peptides have the potential to redefine clinical therapeutics, offering novel strategies to address unmet clinical needs and paving the way for their adoption in clinical practice.
6. Other applications of branched peptides
In addition to their cell-promoting and tissue-integrating biomimetic qualities, branched peptides are being assessed for other applications including biosensors,223 3D printable bio-inks224 and antimicrobial drug candidates.225 However, similar to the aforementioned applications, the ability of branched peptides to imitate native chemical motifs remains key to creating these new platforms which interact with, and influence, molecular biology. An example of this is the novel photoelectrochemical biosensor developed for detecting cardiac troponin I in human serum. This system features a uniquely engineered branching peptide with enhanced antifouling properties223 (Fig. 9). Integrating a dual-photoelectrode system (C3N4/TiO2 photoanode and AuPt/PANI photocathode), the Y-shaped dendrimer branched peptide offers superior antifouling and recognition capabilities compared to linear peptides. The biosensor demonstrated high sensitivity, specificity, and anti-interference for detecting cardiac troponin I (cTnI). Similarly, Li combined a zwitterionic antifouling peptide and an antibacterial peptide with a recognition peptide aptamer226 to produce an electrochemical biosensor for detecting the receptor binding domain of the SARS-CoV-2 spike protein. Other uses for branched peptide-based biosensors include antibody mimics,227 clinical sample testing,228 antibody detection,229 cellular quantification230 and nucleic acid detection.96,231 | ||
| Fig. 9 Branched peptide photoelectrochemical biosensor design. The photoelectrochemical biosensor integrates a novel engineered branching peptide (EBP) into a dual-photoelectrode system. The EBP, has a Y-shaped configuration featuring a recognition backbone and two antifouling branches. The biosensor demonstrated a high photocurrent response and strong antioxidation properties with excellent sensitivity, specificity, and anti-interference in detecting the cardiac troponin I (cTnI) biomarker. Reproduced with permission from ref. 222 Copyright 2024, American Chemical Society. | ||
In recent years, companies such as CELLINK™ and 3DBio™ have developed the technology to create cell-embedded 3D biomaterials to produce tissues such as cardiovascular,232 bladder,233 and bone.234 A study on bioinks used a reinforced peptide-dendrimer224 combining a peptide-dendrimer branched PEG with end-grafted norbornene (PDN) and cysteamine-modified HA (HC), enhancing crosslinking. The combined HC-PDN showed improved mechanical and rheological properties, reduced reactive oxygen species accumulation, and enabled bioprinting of complex structures with high shape fidelity, such as a hepatic tissue model with HepG2-C3As, LX-2s, and EA.hy.926s. As described earlier, branched peptides are ideally suited to the generation of customisable 3D structures, with tailored degradation rates129 and stiffnesses,32 and for the incorporation of functional motifs such as RGDS.149 These features also allow branched peptides to be explored as potential candidates for complex 3D bioinks towards the generation of organoids and organ-on-chip models, to model disease progression,235 tumour growth,236 organ replacements,237 and others.238
By combining rapid production technological advancements with bottom-up peptide design strategies we are afforded limitless opportunities to explore healthcare applications that extend far beyond the tissue repair and regeneration need described at the beginning of this review. There are opportunities for growth in all the areas described and, as such, we are likely to see significant advances in the coming years, beyond those described here.
7. Conclusions
The exploration of branched peptide-based biomaterials represents a significant leap forward in biomedical research, inspired by nature's own designs. Enhanced by breakthroughs in SPPS, such as safety catches and novel amino acid side chain protecting groups, the precision, yield, and scalability of synthesis have all dramatically improved, presenting new opportunities for branched peptide research. Comprising three main categories—hyperbranched, dendrimer, and dendrigraft—branched peptides form a range of nanostructures and are easily tuneable for different applications including drug delivery, bioimaging, and tissue engineering. Advancements in the control of degradation and stiffness profiles of these designs have in particular led to materials that are capable of self-assembling into higher-ordered 3D structures. Additionally, the ability to functionalise branched peptides with short motifs such as RGDS, IKVAV, and PHSRN, as well as non-peptide polymers such as lipids and carbohydrates, facilitates physical, molecular, and cellular interactions that were previously thought to be unattainable via traditional synthetic material design.While challenges such as scalable syntheses, comprehensive degradation rates from in vivo studies, and the limited in vivo research in the area, still exist the progress made thus far, combined with technological advancements, indicates a bright future for branched peptide design, holding the promise of innovative therapeutic strategies and a transformative impact on biomedical applications in future.
Author contributions
Nazia Mehrban and Jody M. Mason were responsible for the conceptualisation and revision of the article. Matthew J. Little was responsible for the investigation, visualisation, drafting of the manuscript, and the production of all figures used in the article.Data availability
No primary or new data was collected for the purposes of this article, nor was any software, code or programs used to generate any of the information displayed in this article.Conflicts of interest
All Authors declare no conflict of interest.Acknowledgements
We thank the Academy of Medical Sciences (SBF008\1020) and the Engineering and Physical Sciences Research Council (EP/T020792/1) for supporting Nazia Mehrban and the Biotechnology and Biological Sciences Research Council for supporting Jody M. Mason (BB/X001849/1).References
- A. Bandyopadhyay, I. Mitra, S. B. Goodman, M. Kumar and S. Bose, Prog. Mater. Sci., 2023, 133, 101053 CrossRef PubMed.
- O. Mishchenko, A. Yanovska, O. Kosinov, D. Maksymov, R. Moskalenko, A. Ramanavicius and M. Pogorielov, Polymers, 2023, 15, 3822 CrossRef PubMed.
- Y. Wang, Z. Wang and Y. Dong, ACS Biomater. Sci. Eng., 2023, 9, 1132–1150 CrossRef PubMed.
- S. Bupphathong, C. Quiroz, W. Huang, P.-F. Chung, H.-Y. Tao and C.-H. Lin, Pharmaceuticals, 2022, 15, 171 CrossRef PubMed.
- D. Cao and J. Ding, Regener. Biomater., 2022, 9, rbac098 CrossRef PubMed.
- A. Revete, A. Aparicio, B. A. Cisterna, J. Revete, L. Luis, E. Ibarra, E. A. Segura, J. Molino and D. Reginensi, Int. J. Biomater., 2022, 2022, 1–16 CrossRef PubMed.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef PubMed.
- N. Mehrban, B. Zhu, F. Tamagnini, F. I. Young, A. Wasmuth, K. L. Hudson, A. R. Thomson, M. A. Birchall, A. D. Randall, B. Song and D. N. Woolfson, ACS Biomater. Sci. Eng., 2015, 1, 431–439 CrossRef PubMed.
- D. Fraser and D. Benoit, Biomater. Adv., 2022, 141, 213093 CrossRef PubMed.
- G. D. Mogoşanu and A. M. Grumezescu, Int. J. Pharm., 2014, 463, 127–136 CrossRef PubMed.
- F. Donnaloja, E. Jacchetti, M. Soncini and M. T. Raimondi, Polymers, 2020, 12, 905 CrossRef PubMed.
- S. Sun, Y. Cui, B. Yuan, M. Dou, G. Wang, H. Xu, J. Wang, W. Yin, D. Wu and C. Peng, Front. Bioeng. Biotechnol., 2023, 11, 1117647 CrossRef.
- Z. Wang, Q. Ye, S. Yu and B. Akhavan, Adv. Healthcare Mater., 2023, 12, 2300105 CrossRef CAS.
- S. P. Zustiak and J. B. Leach, Biomacromolecules, 2010, 11, 1348–1357 CrossRef CAS PubMed.
- B. S. Ludwig, H. Kessler, S. Kossatz and U. Reuning, Cancers, 2021, 13, 1711 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, Y. Li, Y. Yang, M. Jin, X. Lin, Z. Zhuang, K. Guo, T. Zhang and W. Tan, Gels, 2023, 9, 185 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- R. Wang, Z. Wang, Y. Guo, H. Li and Z. Chen, J. Biomater. Sci., Polym. Ed., 2019, 30, 713–736 CrossRef CAS.
- E. Thomas Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef.
- C.-Y. Lin, Y.-R. Wang, C.-W. Lin, S.-W. Wang, H.-W. Chien, N.-C. Cheng, W.-B. Tsai and J. Yu, BioRes. Open Access, 2014, 3, 297–310 CrossRef CAS PubMed.
- J. Liao, S. Wu, K. Li, Y. Fan, N. Dunne and X. Li, J. Biomed. Mater. Res., Part A, 2019, 107, 1491–1512 CrossRef CAS PubMed.
- M. Dzierżyńska, J. Sawicka, M. Deptuła, P. Sosnowski, P. Sass, B. Peplińska, Z. Pietralik-Molińska, M. Fularczyk, F. Kasprzykowski, J. Zieliński, M. Kozak, P. Sachadyn, M. Pikuła and S. Rodziewicz-Motowidło, Sci. Rep., 2023, 13, 6273 CrossRef PubMed.
- S. A. Khaliq, Z. Umair, M.-O. Baek, S. J. Chon and M.-S. Yoon, Front. Cell Dev. Biol., 2022, 10, 800181 CrossRef PubMed.
- K. Hosoyama, C. Lazurko, M. Muñoz, C. D. McTiernan and E. I. Alarcon, Front. Bioeng. Biotechnol., 2019, 7, 205 CrossRef.
- S. Chu, A. L. Wang, A. Bhattacharya and J. K. Montclare, Prog. Biomed. Eng., 2021, 4, 012003 CrossRef PubMed.
- S. B. Ebrahimi and D. Samanta, Nat. Commun., 2023, 14, 2411 CrossRef PubMed.
- R. Floriano, K. Edalati, K. D. Pereira and A. D. Luchessi, Sci. Rep., 2023, 13, 470 CrossRef PubMed.
- S. Abe and T. Ueno, RSC Adv., 2015, 5, 21366–21375 RSC.
- A. Solomonov, A. Kozell and U. Shimanovich, Angew. Chem., Int. Ed., 2024, 63, e202318365 CrossRef PubMed.
- R. Pugliese, F. Fontana, A. Marchini and F. Gelain, Acta Biomater., 2018, 66, 258–271 CrossRef PubMed.
- C. Mas-Moruno, R. Fraioli, F. Albericio, J. M. Manero and F. Javier Gil, ACS Appl. Mater. Interfaces, 2014, 6, 6525–6536 CrossRef PubMed.
- S. Sur, C. J. Newcomb, M. J. Webber and S. I. Stupp, Biomaterials, 2013, 34, 4749–4757 CrossRef PubMed.
- M. Amin Azimifar, Z. Salmasi, A. Doosti, N. Babaei and M. Hashemi, Biotechnol. Prog., 2021, 37, e3175 Search PubMed.
- A. A. Chis, C. Dobrea, C. Morgovan, A. M. Arseniu, L. L. Rus, A. Butuca, A. M. Juncan, M. Totan, A. L. Vonica-Tincu, G. Cormos, A. C. Muntean, M. L. Muresan, F. G. Gligor and A. Frum, Molecules, 2020, 25, 3982 CrossRef PubMed.
- M. R. Heinrich, D. L. Rohlfing and E. Bugna, Arch. Biochem. Biophys., 1969, 130, 441–448 CrossRef CAS PubMed.
- J.-A. Fehrentz, J. Martinez, G. Subra and M. Amblard, Mol. Biotechnol., 2006, 33, 239–254 CrossRef PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 Search PubMed.
- V. Molakaseema, A. Selvaraj, H.-T. Chen, Y.-W. Chen, Y.-C. Liu and C.-L. Kao, J. Org. Chem., 2022, 87, 1–9 Search PubMed.
- B. Sax, F. Dick, R. Tanner and J. Gosteli, Pept. Res., 1992, 5, 245–246 Search PubMed.
- N. Thieriet, J. Alsina, E. Giralt, F. Guibé and F. Albericio, Tetrahedron Lett., 1997, 38, 7275–7278 Search PubMed.
- Y. Xu, T. Wang, C.-J. Guan, Y.-M. Li, L. Liu, J. Shi and D. Bierer, Tetrahedron Lett., 2017, 58, 1677–1680 CrossRef CAS.
- E. Boll, J. Dheur, H. Drobecq and O. Melnyk, Org. Lett., 2012, 14, 2222–2225 CrossRef CAS PubMed.
- S. J. Kim and S. R. McAlpine, Molecules, 2013, 18, 1111–1121 CrossRef CAS PubMed.
- M. Ozeki, S. Kuroda, K. Kon and S. Kasugai, J. Biomater. Appl., 2010, 25, 663–684 CrossRef.
- C. Kumar Thota, D. J. Mikolajczak, C. Roth and B. Koksch, ACS Med. Chem. Lett., 2020, 12, 67–73 Search PubMed.
- G. Fichman, C. Andrews, N. L. Patel and J. P. Schneider, Adv. Mater., 2021, 33, 2103677 CrossRef CAS.
- B. Yue, J. Glaucoma, 2014, 23, S20–S23 CrossRef.
- A. Aazmi, D. Zhang, C. Mazzaglia, M. Yu, Z. Wang, H. Yang, Y. Yan and L. Ma, Bioact. Mater., 2024, 31, 475–496 CAS.
- R. Lemos, F. Raquel Maia, R. L. Reis and J. M. Oliveira, Adv. NanoBiomed Res., 2022, 2, 2100116 CrossRef CAS.
- A. Raspa and F. Gelain, Curr. Neuropharmacol., 2021, 19, 2110–2124 CrossRef CAS.
- O. J. Mezu-Ndubuisi and A. Maheshwari, Pediatr. Res., 2021, 89, 1619–1626 Search PubMed.
- L. R. Anderson, T. W. Owens and M. J. Naylor, Biophys. Rev., 2013, 6, 203–213 Search PubMed.
- L. V. Tapia-Lopez, M. A. Luna-Velasco, E. K. Beaven, A. S. Conejo-Dávila, M. Nurunnabi and J. S. Castro, ACS Biomater. Sci. Eng., 2023, 9, 5270–5278 CrossRef CAS PubMed.
- G. Gambino, T. Gambino, L. Connah, F. La Cava, H. Evrard and G. Angelovski, J. Med. Chem., 2021, 64, 7565–7574 CrossRef CAS.
- J. Carlos Forero, K. Carvajal, F. Guzmán, C. Acevedo, N. Osses and P. Santana, Biomimetics, 2023, 8, 323 CrossRef.
- J. Dvořáková, J. Trousil, B. Podhorská, Z. Mikšovská, O. Janoušková and V. Proks, Biomacromolecules, 2021, 22, 1417–1431 CrossRef.
- H. C. Kolb, M. G. Finn and K. Barry Sharpless, Angew. Chem., 2001, 40, 2004–2021 CrossRef CAS.
- E. Jain, S. Neal, H. Graf, X. Tan, R. Balasubramaniam and N. Huebsch, ACS Appl. Bio Mater., 2021, 4, 1229–1237 CrossRef CAS.
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 CrossRef CAS PubMed.
- C. Kitoun, S. Saidjalolov, D. Bouquet, F. Djago, Q. Blancart Remaury, B. Sargueil, P. Poinot, M. Etheve-Quelquejeu and L. Iannazzo, ACS Omega, 2023, 8, 3850–3860 Search PubMed.
- K. Johann, D. Svatunek, C. Seidl, S. Rizzelli, T. A. Bauer, L. Braun, K. Koynov, H. Mikula and M. Barz, Polym. Chem., 2020, 11, 4396–4407 RSC.
- K. Harada, Bull. Chem. Soc. Jpn., 1959, 32, 1007–1008 CrossRef CAS.
- K. Harada and S. W. Fox, Arch. Biochem. Biophys., 1965, 109, 49–56 Search PubMed.
- M. Conda-Sheridan and M. Krishnaiah, Methods Mol. Biol., 2019, 111–128 Search PubMed.
- M. Thompson and C. Scholz, Nanomaterials, 2021, 11, 1119 Search PubMed.
- M. Scholl, Z. Kadlecova and H.-A. Klok, Prog. Polym. Sci., 2009, 34, 24–61 CrossRef CAS.
- A. Kavand, N. Anton, T. Vandamme, C. Serra and D. Chan-Seng, J. Controlled Release, 2020, 321, 285–311 CrossRef CAS PubMed.
- M. Gorzkiewicz, M. Konopka, A. Janaszewska, I. I. Tarasenko, N. N. Sheveleva, A. Gajek, I. M. Neelov and B. Klajnert-Maculewicz, Bioorg. Chem., 2020, 95, 103504 CrossRef CAS PubMed.
- L. Han, J. Li, S. Huang, R. Huang, S. Liu, X. Hu, P. Yi, D. Shan, X. Wang, H. Lei and C. Jiang, Biomaterials, 2011, 32, 2989–2998 Search PubMed.
- J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5409–5413 CrossRef CAS PubMed.
- K. Sadler and J. P. Tam, Rev. Mol. Biotechnol., 2002, 90, 195–229 Search PubMed.
- S. Paul, S. Verma and Y.-C. Chen, ACS Infect. Dis., 2024, 10, 1034–1055 Search PubMed.
- C. Dannert, I. Mardal, R. Lale, B. T. Stokke and R. S. Dias, ACS Omega, 2023, 8, 44624–44636 CrossRef PubMed.
- H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef PubMed.
- H. Collet, E. Souaid, H. Cottet, A. Deratani, L. Boiteau, G. Dessalces, J.-C. Rossi, A. Commeyras and R. Pascal, Chemistry, 2010, 16, 2309–2316 CrossRef.
- A. J. Brouwer, S. J. E. Mulders and R. M. J. Liskamp, Eur. J. Org. Chem., 2001, 1903–1915 CrossRef.
- C. Dai, R. J. Stephenson, M. Skwarczynski and I. Toth, in Peptide Synthesis: Methods and Protocols, ed. W. M. Hussein, M. Skwarczynski and I. Toth, Springer US, New York, NY, 2020, pp. 13–27 Search PubMed.
- S. W. Millward, R. K. Henning, G. A. Kwong, S. Pitram, H. D. Agnew, K. M. Deyle, A. Nag, J. Hein, S. S. Lee, J. Lim, J. A. Pfeilsticker, K. B. Sharpless and J. R. Heath, J. Am. Chem. Soc., 2011, 133, 18280–18288 CrossRef PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef PubMed.
- I. Sánchez-Campillo and J. B. Blanco-Canosa, JACS Au, 2024, 4, 4374–4382 CrossRef PubMed.
- X. Li, Chem. – Asian J., 2011, 6, 2606–2616 CrossRef PubMed.
- C. Ornelas, J. Broichhagen and M. Weck, J. Am. Chem. Soc., 2010, 132, 3923–3931 CrossRef PubMed.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef PubMed.
- M. Bodanszky and R. J. Bath, J. Chem. Soc., Chem. Commun., 1969, 1259–1260 RSC.
- G. Sabatino, A. D’Ercole, L. Pacini, M. Zini, A. Ribecai, A. Paio, P. Rovero and A. M. Papini, Org. Process Res. Dev., 2021, 25, 552–563 CrossRef.
- G. S. Vanier, Methods Mol. Biol., 2013, 1047, 235–249 CrossRef CAS.
- L. A. Carpino, H. Imazumi, A. El-Faham, F. J. Ferrer, C. Zhang, Y. Lee, B. M. Foxman, P. Henklein, C. Hanay, C. Mügge, H. Wenschuh, J. Klose, M. Beyermann and M. Bienert, Angew. Chem., Int. Ed., 2002, 41, 441–445 CrossRef CAS.
- K. Barlos, D. Gatos, J. Kallitsis, G. Papaphotiu, P. Sotiriu, Y. Wenqing and W. Schäfer, Tetrahedron Lett., 1989, 30, 3943–3946 CrossRef CAS.
- G. W. Kenner, J. R. McDermott and R. C. Sheppard, J. Chem. Soc., Chem. Commun., 1971, 636–637 RSC.
- S. Noki, E. Brasil, H. Zhang, T. Bruckdorfer and F. Albericio, J. Org. Chem., 2022, 87, 9443–9453 CrossRef CAS.
- S. Noki and F. Albericio, Molecules, 2024, 29, 1429 CrossRef CAS PubMed.
- Y. Zhou, H. Li, Y. Huang, J. Li, G. Deng, G. Chen, Z. Xi and C. Zhou, Nat. Commun., 2023, 14, 5324 CrossRef CAS PubMed.
- Y. Gu, X. Sun, J. Huang, B. Zhan and X. Zhu, J. Immunol. Res., 2020, 2020, 2074803 Search PubMed.
- M. Azuma, T. Kojima, I. Yokoyama, H. Tajiri, K. Yoshikawa, S. Saga and C. A. Del Carpio, J. Pept. Res., 1999, 54, 237–241 CrossRef CAS PubMed.
- A. Kavand, P. Robin, L. Mayoraz, M. Mensi and S. Gerber-Lemaire, RSC Adv., 2023, 13, 34003–34011 RSC.
- L. Han, R. Huang, S. Liu, S. Huang and C. Jiang, Mol. Pharm., 2010, 7, 2156–2165 CrossRef CAS.
- J.-L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- B.-H. Gan, T. N. Siriwardena, S. Javor, T. Darbre and J.-L. Reymond, ACS Infect. Dis., 2019, 5, 2164–2173 CrossRef.
- J. Pires, T. N. Siriwardena, M. Stach, R. Tinguely, S. Kasraian, F. Luzzaro, S. L. Leib, T. Darbre, J.-L. Reymond and A. Endimiani, Antimicrob. Agents Chemother., 2015, 59, 7915–7918 CrossRef PubMed.
- V. S. Fluxà, N. Maillard, M. G. P. Page and J.-L. Reymond, Chem. Commun., 2011, 47, 1434–1436 RSC.
- M. Stach, N. Maillard, R. U. Kadam, D. Kalbermatter, M. Meury, M. G. P. Page, D. Fotiadis, T. Darbre and J.-L. Reymond, MedChemComm, 2012, 3, 86–89 RSC.
- K. C. Tjandra and P. Thordarson, Bioconjugate Chem., 2019, 30, 503–514 CrossRef.
- J. Shao, B. P. Kuiper, A.-M. W. H. Thunnissen, R. H. Cool, L. Zhou, C. Huang, B. W. Dijkstra and J. Broos, J. Am. Chem. Soc., 2022, 144, 13815–13822 CrossRef PubMed.
- X. Zhang, Z. Zhang, X. Xu, Y. Li, Y. Li, Y. Jian and Z. Gu, Angew. Chem., 2015, 54, 4289–4294 CrossRef PubMed.
- Q. Leng and A. J. Mixson, Nucleic Acids Res., 2005, 33, e40 CrossRef PubMed.
- V. K. Yellepeddi and H. Ghandehari, Tissue Barriers, 2016, 4, e1173773 CrossRef PubMed.
- Ł. Szyrwiel, Ł. Szczukowski, J. S. Pap, B. Setner, Z. Szewczuk and W. Malinka, Inorg. Chem., 2014, 53, 7951–7959 CrossRef.
- X. Yuan, S.-Z. Luo and L. Chen, Int. J. Pharm., 2022, 624, 121983 CrossRef PubMed.
- T. E. McAllister, T.-L. Yeh, M. I. Abboud, H. Leung, E. S. Hookway, B. Bhushan, S. T. Williams, R. J. Hopkinson, M. Münzel, N. D. Loik, R. Chowdhury, U. Oppermann, W. Claridge, Y. Goto, H. Suga, C. J. Schofield and A. Kawamura, Chem. Sci., 2018, 9, 4569–4578 RSC.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef PubMed.
- T. P. Smith, B. Bhushan, D. Granata, C. S. Kaas, B. Andersen, K. W. Decoene, Q. Ren, H. Liu, X. Qu, Y. Yang, J. Pan, Q. Chen, M. Münzel and A. Kawamura, RSC Chem. Biol., 2024, 5, 12–18 RSC.
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC.
- C. C. Lee, J. A. MacKay, J. M. J. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef PubMed.
- J. Lu, X.-B. Tian and W. Huang, Chin. Chem. Lett., 2015, 26, 946–950 CrossRef.
- D. Christopher Radford, J. Yang, M. C. Doan, L. Li, A. S. Dixon, S. C. Owen and J. Kopeček, J. Controlled Release, 2020, 319, 285–299 CrossRef.
- G. V. Dubacheva, T. Curk, B. M. Mognetti, R. Auzély-Velty, D. Frenkel and R. P. Richter, J. Am. Chem. Soc., 2014, 136, 1722–1725 CrossRef PubMed.
- Y. Zhang, H. Zhang, D. Ghosh and R. O. Williams, Int. J. Pharm., 2020, 587, 119491 CrossRef PubMed.
- L. Ageitos and C. de la Fuente-Nunez, Arch. Bronconeumol., 2022, 58, 383–385 CrossRef PubMed.
- A. E. Mattei, A. H. Gutierrez, W. D. Martin, F. E. Terry, B. J. Roberts, A. S. Rosenberg and A. S. De Groot, Front. Drug Discovery, 2022, 2, 952326 CrossRef.
- D. G. Leach, N. Dharmaraj, S. L. Piotrowski, T. L. Lopez-Silva, Y. L. Lei, A. G. Sikora, S. Young and J. D. Hartgerink, Biomaterials, 2018, 163, 67–75 CrossRef.
- T. T. Lee, J. R. García, J. I. Paez, A. Singh, E. A. Phelps, S. Weis, Z. Shafiq, A. Shekaran, A. del Campo and A. J. García, Nat. Mater., 2014, 14, 352–360 CrossRef PubMed.
- X. Wang, Y. Zheng, C. Bao, G. Zhong, S. Liu, N. Wiradharma, W. Fan, Y. Y. Yang, X. Wang and Y. Huang, Biomater. Sci., 2020, 8, 6387–6394 RSC.
- F. K. Knop, A. Brønden and T. Vilsbøll, Expert Opin. Pharmacother., 2017, 18, 555–571 CrossRef.
- A. B. Petersen, F. K. Knop and M. Christensen, Drugs Today, 2013, 49, 0537 CrossRef.
- E. Ladenheim, Drug Des., Dev. Ther., 2015, 1867 CrossRef.
- R. S. Kirby, J. M. Fitzpatrick and N. Clarke, BJU Int., 2009, 104, 1580–1584 CrossRef.
- S. Bhattacharyya, S. Pal and N. Chattopadhyay, Biochem. Pharmacol., 2019, 166, 185–191 CrossRef.
- M. L. Agazzi, S. E. Herrera, M. Lorena Cortez, W. A. Marmisollé and O. Azzaroni, Colloids Surf., B, 2020, 190, 110895 CrossRef CAS PubMed.
- K. Dauer, W. Kamm, K. G. Wagner and S. Pfeiffer-Marek, Mol. Pharm., 2021, 18, 1939–1955 CrossRef CAS.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- C. T. Mant, Y. Chen, Z. Yan, T. V. Popa, J. M. Kovacs, J. B. Mills, B. P. Tripet and R. S. Hodges, Pept. Charact. Appl. Protoc., 2007, 386, 3 CAS.
- D. R. Fuller, C. R. Conant, T. J. El-Baba, Z. Zhang, K. R. Molloy, C. S. Zhang, D. A. Hales and D. E. Clemmer, Eur. J. Mass Spectrom., 2019, 25, 73–81 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2527–2535 CrossRef CAS.
- B. Meibohm and H. Derendorf, J. Pharm. Sci., 2002, 91, 18–31 CrossRef PubMed.
- S. Kanji, M. Hayes, A. Ling, L. Shamseer, C. Chant, D. J. Edwards, S. Edwards, D. R. Foster, B. Hardy, T. H. Kiser, C. la Porte, J. A. Roberts, R. Shulman, S. Walker, S. Zelenitsky and D. Moher, Clin. Pharmacokinet., 2015, 54, 783–795 CrossRef PubMed.
- M. G. Ryadnov and D. N. Woolfson, Angew. Chem., Int. Ed., 2003, 42, 3021–3023 CrossRef PubMed.
- D. A. Harrington, E. Y. Cheng, M. O. Guler, L. K. Lee, J. L. Donovan, R. C. Claussen and S. I. Stupp, J. Biomed. Mater. Res., Part A, 2006, 78, 157–167 CrossRef PubMed.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef PubMed.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 CrossRef PubMed.
- S. Ghassemi, G. Meacci, S. Liu, A. A. Gondarenko, A. Mathur, P. Roca-Cusachs, M. P. Sheetz and J. Hone, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5328–5333 CrossRef.
- M. Caiazzo, Y. Okawa, A. Ranga, A. Piersigilli, Y. Tabata and M. P. Lutolf, Nat. Mater., 2016, 15, 344–352 CrossRef PubMed.
- R. Zaidel-Bar and B. Geiger, J. Cell Sci., 2010, 123, 1385–1388 CrossRef PubMed.
- B. Zhao, L. Li, L. Wang, C.-Y. Wang, J. Yu and K.-L. Guan, Genes Dev., 2012, 26, 54–68 CrossRef PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 CrossRef PubMed.
- M. G. Andrews and A. R. Kriegstein, Annu. Rev. Neurosci., 2022, 45, 23–39 CrossRef PubMed.
- H. N. Lee, Y. Y. Choi, J. W. Kim, Y. S. Lee, J. W. Choi, T. Kang, Y. K. Kim and B. G. Chung, Nano Convergence, 2021, 8, 35 CrossRef CAS PubMed.
- M. R. Blatchley and K. S. Anseth, Nat. Rev. Bioeng., 2023, 1, 329–345 CrossRef CAS PubMed.
- J. Liu, J. Li, X. Tian, F. Tang and W. Huang, Methods Mol. Biol., 2019, 189–198 Search PubMed.
- S. E. Ochsenhirt, E. Kokkoli, J. B. McCarthy and M. Tirrell, Biomaterials, 2006, 27, 3863–3874 CrossRef CAS.
- N. Li, S. Qiu, Y. Fang, J. Wu and Q. Li, Biology, 2021, 10, 688 CrossRef CAS.
- J. J. Kim, D. V. Bax, R. Murphy, S. M. Best and R. E. Cameron, Nanomaterials, 2021, 11, 1157 CrossRef CAS PubMed.
- J. Zhao, F. Santino, D. Giacomini and L. Gentilucci, Biomedicines, 2020, 8, 307 CrossRef CAS PubMed.
- L. Oliver-Cervelló, H. Martin-Gómez and C. Mas-Moruno, J. Pept. Sci., 2021, 28, e3335 CrossRef PubMed.
- Z. Fang, J. Chen, Y. Zhu, G. Hu, H. Xin, K. Guo, Q. Li, L. Xie, L. Wang, X. Shi, Y. Wang and C. Mao, Nat. Commun., 2021, 12, 3757 CrossRef CAS PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- J. M. Lee, H. M. Hammarén, M. M. Savitski and S. H. Baek, Nat. Commun., 2023, 14, 201 CrossRef.
- J. M. Carnino, K. Ni and Y. Jin, Front. Immunol., 2020, 11, 948 CrossRef PubMed.
- J. Glatz, J. Luiken and A. Bonen, Physiol. Rev., 2010, 90, 367–417 CrossRef.
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef PubMed.
- K. M. Eyster, Adv. Physiol. Educ., 2007, 31, 5–16 CrossRef.
- J. Bernd Helms and C. Zurzolo, Traffic, 2004, 5, 247–254 CrossRef.
- G. Palui, A. Garai, J. Nanda, A. K. Nandi and A. Banerjee, J. Phys. Chem. B, 2010, 114, 1249–1256 CrossRef PubMed.
- V. Haridas, P. Kumar, S. Dhawan and S. J. Devaki, ChemistrySelect, 2016, 1, 4582–4590 CrossRef.
- M. A. Kuzina, D. D. Kartsev, A. V. Stratonovich and P. A. Levkin, Adv. Funct. Mater., 2023, 33, 2301421 CrossRef.
- V. P. Sivadas, S. Dhawan, J. Babu, V. Haridas and P. D. Nair, Acta Biomater., 2019, 99, 196–210 CrossRef PubMed.
- S. K. Patel, A. Lavasanifar and P. Choi, Biomacromolecules, 2009, 10, 2584–2591 CrossRef PubMed.
- G. Ashwell and A. G. Morell, Trends Biochem. Sci., 1977, 2, 76–78 CrossRef.
- I. Canobbio, C. Balduini and M. Torti, Cell. Signalling, 2004, 16, 1329–1344 CrossRef CAS.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef CAS.
- J. T. Dulaney, Mol. Cell. Biochem., 1978, 21, 43–63 CrossRef CAS PubMed.
- J. Labat-Robert and L. Robert, Pathol. Biol., 2012, 60, 66–75 CrossRef CAS PubMed.
- R. Nagarajan, Antimicrob. Agents Chemother., 1991, 35, 605–609 CrossRef CAS PubMed.
- H.-K. Kang and Y. Park, J. Bacteriol. Virol., 2015, 45, 67 CrossRef.
- V. Murray, J. Chen and L. Chung, Int. J. Mol. Sci., 2018, 19, 1372 CrossRef PubMed.
- C. He, S. Wu, D. Liu, C. Chi, W. Zhang, M. Ma, L. Lai and S. Dong, J. Am. Chem. Soc., 2020, 142, 17015–17023 CrossRef CAS PubMed.
- V. I. B. Castro, A. R. Araújo, F. Duarte, A. Sousa-Franco, R. L. Reis, I. Pashkuleva and R. A. Pires, ACS Appl. Mater. Interfaces, 2023, 15, 29998–30007 CrossRef CAS.
- B. Mondal, B. Pandey, N. Parekh, S. Panda, T. Dutta, A. Padhy and S. S. Gupta, Biomater. Sci., 2020, 8, 6322–6336 RSC.
- E. S. Lee, H. J. Shin, K. Na and Y. H. Bae, J. Controlled Release, 2003, 90, 363–374 CrossRef CAS.
- L. Qiu, Z. Li, M. Qiao, M. Long, M. Wang, X. Zhang, C. Tian and D. Chen, Acta Biomater., 2014, 10, 2024–2035 CrossRef CAS PubMed.
- Z. Wang, R. Sheng, T. Luo, J. Sun and A. Cao, Polym. Chem., 2017, 8, 472–484 RSC.
- S. S.-S. Wang, S.-C. How, Y.-D. Chen, Y.-H. Tsai and J.-S. Jan, J. Mater. Chem. B, 2015, 3, 5220–5231 RSC.
- M. Mokrus and H. Menzel, Macromol. Biosci., 2022, 22, 2100518 CrossRef CAS.
- L. Monica, R. Petitdemange, M. Rosselin, E. Ibarboure, B. Garbay, E. Garanger, T. Deming and S. Lecommandoux, Biomacromolecules, 2020, 22, 76–85 Search PubMed.
- L. Li, L. Wu, M. Urschbach, D. Straßburger, X. Liu, P. Besenius and G. Chen, ACS Polym. Au, 2022, 2, 478–485 CrossRef CAS PubMed.
- S. Cheng, M. Li, Y. Feng, T. Liu, L. He, M. Xu, L. Ma and X. Li, J. Med. Chem., 2024, 67, 4225–4233 CrossRef CAS.
- E. J. Bilsky, R. D. Egleton, S. A. Mitchell, M. M. Palian, P. Davis, J. D. Huber, H. Jones, H. I. Yamamura, J. Janders, T. P. Davis, F. Porreca, V. J. Hruby and R. Polt, J. Med. Chem., 2000, 43, 2586–2590 CrossRef CAS.
- Q. Wei, J. Zhang, Y. Dao, M. Ye, D. Liu, W. Dong, N. Yuan, H. Li, C. Song, M. Li, X. Shi and S. Dong, CCS Chem., 2023, 5, 1623–1634 CrossRef CAS.
- M. Akhter Hossain, R. Okamoto, J. A. Karas, P. Praveen, M. Liu, B. E. Forbes, J. D. Wade and Y. Kajihara, J. Am. Chem. Soc., 2019, 142, 1164–1169 CrossRef.
- S. S. Pinho, I. Alves, J. Gaifem and G. A. Rabinovich, Cell. Mol. Immunol., 2023, 20, 1101–1113 CrossRef CAS PubMed.
- R. Naseri, S. J. Navabi, Z. Samimi, A. P. Mishra, M. Nigam, H. Chandra, A. Olatunde, H. Tijjani, R. P. Morais-Urano and M. H. Farzaei, Daru, J. Pharm. Sci., 2020, 28, 333–358 CrossRef.
- M. Lynch, J. Barallobre-Barreiro, M. Jahangiri and M. Mayr, J. Intern. Med., 2016, 280, 325–338 CrossRef CAS.
- A. A. Bahar, Z. Liu, F. Totsingan, C. Buitrago, N. Kallenbach and D. Ren, Appl. Microbiol. Biotechnol., 2015, 99, 8125–8135 CrossRef CAS.
- J. Brunetti, L. Depau, C. Falciani, M. Gentile, E. Mandarini, G. Riolo, P. Lupetti, A. Pini and L. Bracci, Sci. Rep., 2016, 6, 27174 CrossRef CAS.
- J. Brunetti, C. Falciani, L. Bracci and A. Pini, Pept. Sci., 2018, 110(5), e24089 CrossRef.
- L. Depau, J. Brunetti, C. Falciani, S. Scali, G. Riolo, E. Mandarini, A. Pini and L. Bracci, Oncotarget, 2017, 8, 76141–76152 CrossRef PubMed.
- I. U. Khan and A. G. Beck-Sickinger, Anticancer Agents Med. Chem., 2008, 8, 186–199 CrossRef CAS.
- C. Falciani, A. Accardo, J. Brunetti, D. Tesauro, B. Lelli, A. Pini, L. Bracci and G. Morelli, ChemMedChem, 2011, 6, 678–685 CrossRef CAS PubMed.
- C. Falciani, J. Brunetti, B. Lelli, A. Accardo, D. Tesauro, G. Morelli and L. Bracci, J. Pept. Sci., 2013, 19, 198–204 CrossRef.
- J. Brunetti, S. Piantini, M. Fragai, S. Scali, G. Cipriani, L. Depau, A. Pini, C. Falciani, S. Menichetti and L. Bracci, Molecules, 2020, 25, 1088 CrossRef PubMed.
- J. Chen, W. Wang, Y. Wang, X. Yuan, C. He, P. Pei, S. Su, W. Zhao, S.-Z. Luo and L. Chen, Eur. J. Pharm. Biopharm., 2022, 179, 137–146 CrossRef PubMed.
- A. Rencinai, E. Tollapi, G. Marianantoni, J. Brunetti, T. Henriquez, A. Pini, L. Bracci and C. Falciani, Front. Mol. Biosci., 2024, 11, 1429163 CrossRef PubMed.
- Antimicrobial Resistance Collaborators, Lancet, 2022, 399, 629–655 CrossRef PubMed.
- C. Galanakou, D. Dhumal and L. Peng, Biomater. Sci., 2023, 11, 3379–3393 RSC.
- X. Cai, M. Orsi, A. Capecchi, T. Köhler, C. van Delden, S. Javor and J.-L. Reymond, Cell Rep. Phys. Sci., 2022, 3, 101161 CrossRef PubMed.
- Z. Lai, X. Yuan, G. Li, H. Chen, B. Cheng and A. Shan, Chem. Eng. J., 2024, 496, 154171 Search PubMed.
- Y. Zhang, K. Kang, N. Zhu, G. Li, X. Zhou, A. Zhang, Q. Yi and Y. Wu, J. Mater. Chem. B, 2020, 8, 7428–7437 RSC.
- A. W. Young, Z. Liu, C. Zhou, F. Totsingan, N. Jiwrajka, Z. Shi and N. R. Kallenbach, MedChemComm, 2011, 2, 308–314 RSC.
- Y. Gou, X. Yang, L. He, X. Xu, Y. Liu, Y. Liu, Y. Gao, Q. Huang, K. Liang, C. Ding, J. Li, C. Zhao and J. Li, Polym. Chem., 2017, 8, 4264–4279 RSC.
- A. M. Schito and S. Alfei, Polymers, 2020, 12, 1818 CrossRef.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef PubMed.
- R. Rupp, S. L. Rosenthal and L. R. Stanberry, Int. J. Nanomed., 2007, 2, 561–566 Search PubMed.
- J. O’Loughlin, I. Y. Millwood, H. M. McDonald, C. F. Price, J. M. Kaldor and J. R. A. Paull, Sex. Transm. Dis., 2010, 37, 100–104 CrossRef PubMed.
- G. Meogrossi, E. Tollapi, A. Rencinai, J. Brunetti, S. Scali, E. Paccagnini, M. Gentile, P. Lupetti, S. Pollini, G. M. Rossolini, A. Bernini, A. Pini, L. Bracci and C. Falciani, J. Med. Chem., 2024, 67, 16145–16156 CrossRef PubMed.
- P. Saravanan, S. Kumar and J. M. Kataria, J. Immunol. Methods, 2004, 293, 61–70 CrossRef.
- Y. Xu, X. Qiao, Z.-L. Song, G.-C. Fan and X. Luo, Anal. Chem., 2023, 95, 14119–14126 CrossRef PubMed.
- R. Edelman, S. S. Wasserman, J. G. Kublin, S. A. Bodison, E. H. Nardin, G. A. Oliveira, S. Ansari, C. L. Diggs, O. L. Kashala, B. J. Schmeckpeper and R. G. Hamilton, Vaccine, 2002, 21, 269–280 CrossRef PubMed.
- Y. Tanaka, H. Wada, R. Goto, T. Osada, K. Yamamura, S. Fukaya, A. Shimizu, M. Okubo, K. Minamiguchi, K. Ikizawa, E. Sasaki and T. Utsugi, Sci. Rep., 2020, 10, 17284 CrossRef PubMed.
- J. Pei, X. Gao, D. Pan, Y. Hua, J. He, Z. Liu and Y. Dang, Curr. Res. Food Sci., 2022, 5, 2162–2170 CrossRef PubMed.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef PubMed.
- S. D. Jois, in Peptide Therapeutics: Fundamentals of Design, Development, and Delivery, ed. S. D. Jois, Springer International Publishing, Cham, 2022, pp. 287–305 Search PubMed.
- D. Zane, P. L. Feldman, T. Sawyer, Z. Sobol and J. Hawes, Internet J. Toxicol., 2021, 40, 108–124 CrossRef.
- Y. Xu, X. Qiao, Z.-L. Song, G.-C. Fan and X. Luo, Anal. Chem., 2023, 95, 14119–14126 CrossRef.
- K. Zhou, R. Ding, X. Tao, Y. Cui, J. Yang, H. Mao and Z. Gu, Acta Biomater., 2023, 169, 243–255 CrossRef.
- S. Shabani, S. Hadjigol, W. Li, Z. Si, D. Pranantyo, M. B. Chan-Park, N. M. O’Brien-Simpson and G. G. Qiao, Nat. Rev. Bioeng., 2024, 2, 343–361 CrossRef.
- Y. Li, R. Han, X. Yu, L. Jiang and X. Luo, Sens. Actuators, B, 2024, 405, 135322 Search PubMed.
- M. Chen, Z. Song, R. Han, Y. Li and X. Luo, Biosens. Bioelectron., 2021, 178, 113016 Search PubMed.
- N. Liu, Y. Ma, R. Han, S. Lv, P. Wang and X. Luo, Chem. Commun., 2021, 57, 777–780 RSC.
- N. Islam, P. V. Gurgel, O. J. Rojas and R. G. Carbonell, Bioconjugate Chem., 2019, 30, 815–825 CrossRef CAS PubMed.
- N. Liu, J. Song, Y. Lu, J. J. Davis, F. Gao and X. Luo, Anal. Chem., 2019, 91, 8334–8340 CrossRef CAS PubMed.
- S. Campuzano, M. Pedrero, R. Barderas and J. M. Pingarrón, Adv. Mater. Technol., 2022, 7, 2200310 CrossRef CAS.
- N. Noor, A. Shapira, R. Edri, I. Gal, L. Wertheim and T. Dvir, Adv. Sci., 2019, 6, 1900344 CrossRef PubMed.
- S. Chae, J. Kim, H.-G. Yi and D.-W. Cho, Micromachines, 2022, 13, 277 CrossRef PubMed.
- X. Qi, P. Pei, M. Zhu, X. Du, C. Xin, S. Zhao, X. Li and Y. Zhu, Sci. Rep., 2017, 7, 42556 CrossRef CAS.
- Z. Li, J. Hui, P. Yang and H. Mao, Biosensors, 2022, 12, 370 CrossRef CAS PubMed.
- A. A. Qureshi, C. J. Wehrle, S. Ferreira-Gonzalez, C. Jiao, H. Hong, N. Dadgar, J. Arpi-Palacios, Y. P. Phong, J. Kim, K. Sun, K. Hashimoto, D. C. H. Kwon, C. Miller, N. Leipzig, W. W. Ma, J. Melenhorst, F. Aucejo and A. Schlegel, JHEP Rep., 2024, 101164 CrossRef PubMed.
- J. A. Davies, I. Holland and H. Gül, Biochem. Soc. Trans., 2024, BST20231554 Search PubMed.
- A. Żuchowska, P. Baranowska, M. Flont, Z. Brzózka and E. Jastrzębska, Anal. Chim. Acta, 2024, 1301, 342413 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |