
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali triel chalcogenide nanocrystals: a molecular reactivity approach to ternary phase selectivity
Md Riad Sarkar
Pavel
a,
Anuluxan
Santhiran
 ab,
Seth
Dalberg
a,
Aaron J.
Rossini
ab and
Javier
Vela
*ab
ab,
Seth
Dalberg
a,
Aaron J.
Rossini
ab and
Javier
Vela
*ab
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: vela@iastate.edu
bAmes National Laboratory, Ames, Iowa 50011, USA
First published on 6th November 2025
Abstract
Alkali-metal-based materials are promising building blocks for energy conversion and storage technologies. Here, we use a molecular reactivity-based solution-phase approach to selectively synthesize multiple phases and specific polymorphs of lithium- and sodium-containing triel chalcogenide nanocrystals, LiTrCh2, NaTrCh2, and NaTr3Ch5, where Tr = Ga and In and Ch = S, Se, and Te. Analogous to the case of binary II–VI and III–V tetrahedral semiconductors, where the two commonly isolated zinc blende and wurtzite polymorphs are separated by only 1–50 meV f.u.−1, we find that LiTrCh2 nanocrystals easily adopt tetragonal (chalcopyrite) and orthorhombic polymorphs separated by only 2.7–6.2 meV f.u.−1 Because of this small energy difference, soft colloidal synthesis succeeds in accessing either one of these polymorphs, depending on the specific dichalcogenide precursor used. Highly reactive diethyl diselenide favors the thermodynamically more stable tetragonal I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d phase, whereas mildly reactive diphenyl diselenide favors the kinetic, metastable orthorhombic Pna21 phase. Density functional theory calculations confirm the relative energies among multiple LiTrCh2 polymorphs and also model the observed powder X-ray diffraction pattern of a new C2 NaIn3Te5 phase. 7Li, 69Ga, and 77Se solid-state NMR spectra are consistent with phase-pure ternary LiGaSe2 nanocrystals. A majority of the nanocrystal compositions are visible-light emitters. This work opens the door to new Li/Na-based ternary triel chalcogenide nanostructures for energy storage and conversion applications.
2d phase, whereas mildly reactive diphenyl diselenide favors the kinetic, metastable orthorhombic Pna21 phase. Density functional theory calculations confirm the relative energies among multiple LiTrCh2 polymorphs and also model the observed powder X-ray diffraction pattern of a new C2 NaIn3Te5 phase. 7Li, 69Ga, and 77Se solid-state NMR spectra are consistent with phase-pure ternary LiGaSe2 nanocrystals. A majority of the nanocrystal compositions are visible-light emitters. This work opens the door to new Li/Na-based ternary triel chalcogenide nanostructures for energy storage and conversion applications.
Introduction
From their discovery in 1807 (ref. 1) to the next generation of batteries2–4 and solar cells,5 alkali metals have become indispensable elements at the forefront of research and development in new energy conversion and storage materials and devices. Alkali ion batteries provide on-demand power to small portable electronics and electric vehicles.6 High energy density, lightweight Li batteries are most widespread.7,8 Similarly, Na and K batteries continue to gain momentum due to their wide elemental geodistribution, low potential, fast ion diffusion, and low toxicity compared to Pb acid batteries.9,10 Alkali metal chalcogenides have shown promise beyond battery anodes,11 for example, as earth-abundant, environmentally benign, and biocompatible materials for photovoltaic devices.12 By mitigating adverse interfacial recombination pathways, alkali ions help to effectively reduce photovoltaic losses and increase the open-circuit current and fill factor of perovskite13 and copper indium gallium diselenide (CIGS) solar cells.14In this context, alkali triel chalcogenides (Alk–IIIx–VIy; Alk = Li, Na, and K; III = Ga and In; VI = S, Se, Te, etc.) belong to an interesting family of ternary semiconductors with rich structural chemistry and useful optoelectronic properties. While related I–III–VI2 materials (i.e. CuInSe2, AgGaSe2, etc.) adopt the chalcopyrite structure (I2d), lithium triel chalcogenides (Li–III–VI2) crystallize in a wide range of space groups. Depending on their composition and specific crystallization conditions, they can adopt orthorhombic (Pna21/P21nb), cubic (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), and trigonal (RmH) phases.12 (Fig. 1). In turn, sodium triel chalcogenides (Na–IIIx–VIy, x = 1, 3, y = 2, 5) adopt mainly trigonal (RmH/P32) or monoclinic (C2) phases, with some tetragonal and orthorhombic variations. From a colloidal synthesis perspective, this polymorphism offers an opportunity to observe, study, and isolate metastable or kinetic phases with unprecedented chemical and physical properties.15,16 From the perspective of applications, replacement of Cu or Ag with Li widens the semiconducting band gap, increasing the threshold for laser-induced damage and overall photostability. Thus, in addition to batteries or solar cells,17 alkali triel chalcogenides may be useful in near- and mid-IR nonlinear optics,18–20 water splitting21 and CO2 photocatalysis,22 and also in neutron detectors,23 and even phase-change memory materials.24 Already, experimental examples underscoring the potential of this new family of nanostructured materials include the use of LiInS2 or LiInSe2 nanosheets in batteries,25,26 NaGaS2 in radiation detectors,27 and KGaS2 nanocrystals in the conversion of CO2 to CO.28
m), and trigonal (RmH) phases.12 (Fig. 1). In turn, sodium triel chalcogenides (Na–IIIx–VIy, x = 1, 3, y = 2, 5) adopt mainly trigonal (RmH/P32) or monoclinic (C2) phases, with some tetragonal and orthorhombic variations. From a colloidal synthesis perspective, this polymorphism offers an opportunity to observe, study, and isolate metastable or kinetic phases with unprecedented chemical and physical properties.15,16 From the perspective of applications, replacement of Cu or Ag with Li widens the semiconducting band gap, increasing the threshold for laser-induced damage and overall photostability. Thus, in addition to batteries or solar cells,17 alkali triel chalcogenides may be useful in near- and mid-IR nonlinear optics,18–20 water splitting21 and CO2 photocatalysis,22 and also in neutron detectors,23 and even phase-change memory materials.24 Already, experimental examples underscoring the potential of this new family of nanostructured materials include the use of LiInS2 or LiInSe2 nanosheets in batteries,25,26 NaGaS2 in radiation detectors,27 and KGaS2 nanocrystals in the conversion of CO2 to CO.28
 | ||
| Fig. 1 Common crystal structures of ternary lithium- and sodium-based triel chalcogenide semiconductors. | ||
Common approaches to achieve synthetic control during nanocrystal preparations include optimization of reaction conditions (reaction time, temperature), as well as utilization of different ligands and solvents.29,30 A powerful tool in nanocrystal synthesis is leveraging predictable trends in the reactivities of molecular precursors based on their molecular structure, in order to control the kinetics of nanocrystal nucleation and growth.31,32 Disubstituted (diorganyl) dichalcogenides (RChChR; R = ethyl, methyl, phenyl, etc.; Ch = S, Se, etc.) are prime examples of this approach because their reactivities strongly depend on the identity of the organic substituent (R).33–36 In this way, different dichalcogenide precursors can enable the preparation of metastable phases depending on their C–Ch bond dissociation energies.31
At present, high-temperature reactions between the elements result in bulk (polycrystalline) or single crystals of alkali triel chalcogenides. A few solvothermal and molten-salt syntheses,37 as well as high-temperature reactions in flux, also produce alkali triel chalcogenides with large particle size, although binary impurities are sometimes present.38–40 A few colloidal syntheses of (Alk)–Tr–Ch2 nanocrystals have appeared recently.41–43 Interestingly, only pressure-induced Alk–III–VI2 phase transformations are reported.44 Here, we describe a molecular reactivity approach to the phase-selective synthesis of lithium- and sodium-based alkali triel chalcogenide (Ch = S, Se, and Te) nanocrystals using readily available alkali metal, triel, and disubstituted dichalcogenide RChChR precursors.
Results and discussion
Colloidal alkali triel chalcogenide nanocrystals
Reaction of Li(acac) and Ga(acac)3 or In(acac)3 with disubstituted dichalcogenides (RChChR) in oleylamine (oleylNH2) and 1-octadecene (ODE) at 270–330 °C produces LiTrCh2 nanocrystals (Scheme 1). Because of its mild reactivity and moderate solubility, Li(acac) is more reliable and versatile than other Li precursors, such as nitrates, halides, or carbonates.45 Harsher precursors such as LiH, Li(NPr2), PhLi, or n-BuLi react too fast under these conditions, resulting in lower order, binary triel chalcogenides (Tr2Ch3) rather than the desired ternary nanocrystals. | ||
| Scheme 1 Solution-phase synthesis of colloidal alkali triel chalcogenide nanocrystals. | ||
The experimental powder X-ray diffraction (XRD) patterns of the nanocrystals match the reported Pna21 and I2d (LiGaSe2 and LiInSe2) or P21nb (LiInS2) phases of these materials (Fig. 2). The specific polymorph observed is highly dependent on the structure and reactivity of the specific disubstituted dichalcogenide precursor used—see further phase selectivity and computations sections below. The single crystalline domain (Scherrer) sizes calculated from the powder XRD peak widths for LiTrCh2 nanocrystals range from 7 to 20 nm, respectively (Table 1).
 | ||
| Fig. 2 Powder XRD of (a) Pna21 LiGaSe2 (ICSD-53093), I2d LiGaSe2 (ICSD-89176), P21nb LiInS2 (ICSD-53093), Pna21 LiInSe2 (ICSD-60838), and I2d LiInSe2 (ICSD-56532) nanocrystals; (b) RmH NaInS2 (ICSD-25557), P32 NaIn3Se5 (ICSD-243563), and C2 NaIn3Te5 nanocrystals (calculated using VASP, see Methods). *In2S3 impurity (ICSD-23844). | ||
| Precursors (mmol) | Conditionsa | Products | ||||
|---|---|---|---|---|---|---|
| Alkali metal | Triel | Chalcogen | T (°C) | t (min) | Phase | Scherrer, TEM/SEM (nm) |
| a 4 mL ODE + 4 mL oleylNH2. b Minor impurity. c Estimated from match. d Total span × arm width of hexapods. | ||||||
| Li(acac) (0.1) | Ga(acac)3 (0.1) | PhSeSePh (0.2) | 330 | 60 | Pna21 LiGaSe2 | 15 ± 4.9, 11 ± 4.2 |
| Li(acac) (0.1) | Ga(acac)3 (0.1) | EtSeSeEt (0.6) | 300 | 60 |
I2d LiGaSe2 |
20 ± 5.5, 22 ± 5.6 |
| Li(acac) (0.15) | In(acac)3 (0.2) | PhSSPh (0.6) | 330 | 60 | P21nb LiInS2 + In2S3b | 8.1 ± 1.3 |
| Li(acac) (0.1) | In(acac)3 (0.1) | PhSeSePh (0.2) | 270 | 60 |
Pna21 + I2d (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)c LiInSe2 :1)c LiInSe2 |
18 ± 8.3, 13 ± 6.7 |
| Li(acac) (0.1) | In(acac)3 (0.1) | EtSeSeEt (0.4) | 270 | 60 |
I2d LiInSe2 |
7.4 ± 1.6, 19 ± 5.5 |
| Na(oleate) (0.1) | In(acac)3 (0.1) | PhSSPh (0.2) | 280 | 30 |
RmH NaInS2 |
507 ± 83 |
| Na(oleate) (0.1) | In(acac)3 (0.1) | PhSeSePh (0.1) | 290 | 60 | P32 NaIn3Se5 | 957 ± 182 |
| Na(oleate) (0.1) | In(acac)3 (0.1) | PhTeTePh (0.05) | 260 | 30 | C2 NaIn3Te5 | 946 × 256d |
Using a similar approach, the reaction of Na(oleate) with In(acac)3 and PhChChPh (Ch = S, Se, Te) enables the preparation of nanocrystalline sodium triel chalcogenides. Powder XRD patterns match those reported for RmH NaInS2, P32 NaIn3Se5 and—for the first time—a previously unreported C2 NaIn3Te5 phase (Fig. 2). This new ternary compound has a relatively large monoclinic unit cell comprised of octahedral [NaTe6], tetrahedral [InTe4], and four-coordinate [Na2SeIn2] sites (see the SI). Rietveld refinement of the experimental powder XRD pattern matches well with the calculated C2 XRD (see calculations below). The higher reactivity of Na(oleate) compared to Li(acac) results in generally larger 510–960 nm particles for Na- vs. Li-based materials (Table 1).
Phase selectivity and morphology
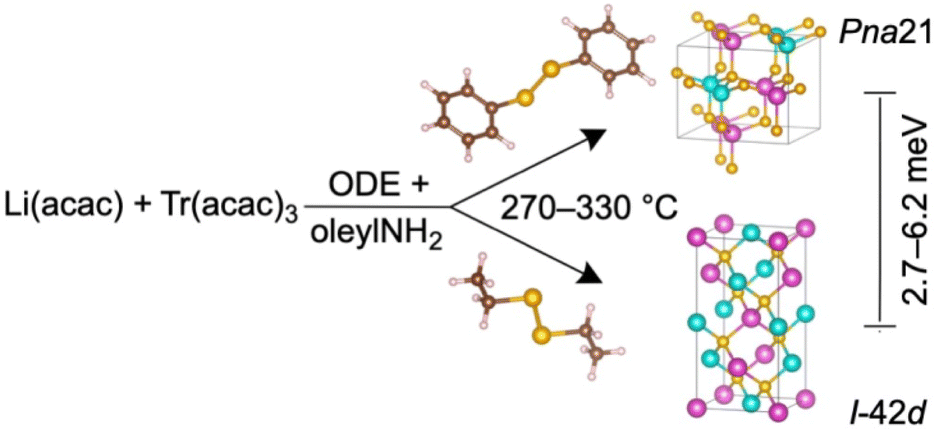
The colloidal LiTrCh2 nanocrystals made by our method typically adopt the tetragonal I2d chalcopyrite and/or orthorhombic Pna21 (LiTrSe2) and the P21nb (LiInS2) structures (Table 1). This agrees with our calculations showing that these two phases have very similar energies. In the case of LiGaSe2 and LiInSe2, the chalcopyrite (I2d) polymorph is the most thermodynamically stable, but only by about 2.7–6.2 meV compared to the orthorhombic (Pna21) phase (see computations below). For comparison, the two most isolated, zinc blende and wurtzite polymorphs of binary tetrahedral semiconductors (II–VI, III–V) are only 1–50 meV per formula unit (f.u.) apart.46,47
To effectively deliver chalcogen to the reaction medium, both of the C–Ch and Ch–Ch bonds in the dichalcogenide precursor must break. Aromatic disubstituted dichalcogenides such as PhChChPh have strong C(Aryl)–Ch bonds (64.4–69.8 kcal mol−1) and weak Ch–Ch bonds (43.6–45.7 kcal mol−1, as judged by their bond dissociation energies).31 As a result, PhChChPh precursors release [Ch] very slowly but quickly release [PhCh·]; these can act as surface passivating agents that stabilize small reactive clusters and seeds, slowing nanocrystal growth. Therefore, the use of aryl-substituted dichalcogenides, such as PhSeSePh, enables the preparation of metastable, kinetic orthorhombic (Pna21) polymorphs of LiGaSe2 and LiInSe2 (Scheme 2 and Table 1). These observations agree with the isolation of metastable polymorphs of CuInSe2—and, very recently, CsGaCh2—from reactions using PhSeH35 and PhSeSePh,43 respectively, both of which bear strong, hard-to-break C–Se bonds.
 | ||
| Scheme 2 Phase-selective synthesis of colloidal LiTrCh2 nanocrystals. | ||
In contrast, aliphatic substituted dichalcogenides have much weaker C–Ch bonds (by about 11–12 kcal mol−1),31 compared to those bearing aromatic substituents. Consequently, both C–Ch and Ch–Ch bonds in EtChChEt precursors break more easily, releasing [Ch] relatively fast and accelerating the rates of nucleation and growth. Thus, the use of the much more reactive EtSeSeEt precursor results in the isolation of the more stable, thermodynamic chalcopyrite (I2d) polymorphs of LiGaSe2 and LiInSe2 (Scheme 2). Using PhSSPh also results in the formation of the orthorhombic (P21nb) phase of LiInS2, the only experimentally reported polymorph for this material, albeit with a small binary In2S3 impurity (Table 1). To further increase the phase purity of LiInS2, future work will explore the use of precursors with weaker C–S bonds compared to that of PhSSPh. Alternatively, more reactive Li precursors could also decrease the amount of In2S3 byproduct by virtue of releasing Li much faster into the reaction mixture.
Bright-field transmission electron microscopy (TEM) shows that the morphology of LiTrCh2 nanocrystals mainly depends on their composition rather than on the specific polymorph present. For example, both Pna21 and I2d LiGaSe2 nanocrystals are spheroidal, even though high-resolution (HR) TEM and fast Fourier transform (FFT) of selected area electron diffraction (SAED) patterns confirm the presence of different sets of planes for the Pna21 [(110), (221)] and I2d [(112), (101), (204)] phases, as shown in Fig. 3a and b, respectively. P21nb LiInS2 nanocrystals are also spheroidal (see SI). In contrast, both Pna21 and I2d LiInSe2 nanocrystals adopt a triangular plate morphology, even though HR TEM and FFT SAED analyses confirm the presence of different sets of planes for the Pna21 [(002), (210), (113), and (231)] and I2d [(112), (103), and (204)] phases, as shown in Fig. 3c and d, respectively. Energy dispersive X-ray spectroscopy (EDS) under scanning electron microscopy (SEM) is consistent with the nanocrystals' compositions; for example, (Li)Ga1.0Se2.4 and (Li)In1.0Se1.9 for the Pna21 phases of LiGaSe2 and LiInSe2, respectively (the light Li atoms are not detected).
 | ||
| Fig. 3 Representative TEM images of (a) Pna21 LiGaSe2 (TEM particle size: 10 ± 4.2 nm), (b) I2d LiGaSe2 (22 ± 5.6 nm), (c) Pna21 LiInSe2 (13 ± 6.7 nm), and (d) I2d LiInSe2 (19 ± 5.5 nm) nanocrystals. | ||
Interestingly, the fact that LiGaSe2 and LiInS2 nanocrystals are spheroidal while LiInSe2 nanocrystals containing the heavier Se chalcogen are highly anisotropic (plate-like) has some precedence in the literature. For example, NaSbSe2 nanocrystals are quasispheroidal while NaBiSe2 nanocrystals are anisotropic (elongated, rods).48,49 Particle shapes with lower aspect ratios are normally preferred thermodynamically.49 In our case, the less reactive, lighter Ga and S precursors required significantly higher reaction temperatures (300–330 °C, Table 1) compared to the more reactive, Se precursors (270 °C), which likely explains why the former trended toward the lower aspect ratio, spheroidal shapes.
SEM images reveal that NaInS2, NaIn3Se5, and NaIn3Te5 nanocrystals adopt different morphologies (Fig. 4 and SI). Very much like the Li-ternary nanocrystals, Na-containing ternary nanocrystals undergo shape evolution from lower to higher anisotropy on going down the chalcogen group from S to Se to Te. Specifically, NaInS2 crystals are spheroidal, NaIn3Se5 crystals exhibit some anisotropy and are cuboidal, and NaIn3Te5 crystals are very anisotropic and adopt a hexapod-like shape. Thus, the idea that composition plays a key role in determining the shape of Li- and Na-containing ternary chalcogenide nanocrystals is strongly supported by the available data. Nanocrystals containing lighter atoms often adopt more thermodynamically favorable, isotropic shapes, while those containing heavier atoms tend to be much more anisotropic. Recent reports on AInSe2 (A = K, Rb, Cs) nanocrystals also showed a variety of morphologies depending on the different cations that were present and the chalcogen precursors used.41 Selected EDS area scans under the SEM show average compositions of Na1.2In1.0S2.0, Na1.4In3.0Se5.1, and Na4.5In2.5Te5.0 (see SI). An excess of Na and Ch atoms in the latter may be attributed to some remaining unreacted sodium precursor or amorphous impurity, as it is absent in the powder XRD.
 | ||
| Fig. 4 Representative SEM images of (a) NaInS2, (b) NaIn3Se5, and (c) NaIn3Te5 nanocrystals. | ||
Stability and phase transformations
The phase-selective synthesis and structural transformations of alkali metal-based ternary chalcogenides continue to be the subject of intense research activity. For example, CsGaCh2 (Ch = S, Se) adopts a TlGaSe2-type (mC64) layered (2D) structure at low temperature and a KFeS2-type (mC16) structure containing one-dimensional (1D) chains at high temperature. Density functional theory (DFT) calculations show these two polymorphs are only 0.01 eV per atom apart.42 In the bulk, mC64 CsGaCh2 reversibly phase transitions to the mC16 polymorph at ∼600 °C. At the nanoscale, mC64 CsGaCh2 irreversibly converts to the thermodynamic mC16 polymorph at 320 °C in solution43 and at 1000 °C in the solid state.42In this work, we find that LiTrCh2 nanocrystals possess significant air and thermal stability, as evidenced by continued monitoring of their powder XRD over time and by thermal analyses (see SI). Notably, LiInSe2 nanocrystals remain phase pure even after 5 months under ambient conditions of temperature and air, while LiGaSe2 nanocrystals only start showing signs of partial decomposition to lower order, binary impurities after 2 months. Both are remarkably thermally stable in N2, losing ≤3% mass to 1000 °C.
SSNMR spectroscopy
Solid-state NMR (SSNMR) spectroscopy experiments were performed on ternary LiGaSe2 and LiInSe2 samples to identify their chemical environments. 7Li is a spin 3/2 nucleus with 92.4% natural abundance.507Li has a chemical shift of 1 ppm and 3 ppm for the LiGaSe2 and LiInSe2, respectively, differing by 2 ppm (Fig. 5a). 69Ga is a spin 3/2 nucleus with 60.1% natural abundance.50 Quadrupolar nuclei, such as 69Ga, often have sizable first- and second-order quadrupolar interactions, which lead to broader central transition SSNMR spectra.51,52 The static 69Ga SSNMR spectrum was acquired with a wideband uniform rate smooth truncation QCPMG (WCPMG) pulse sequence.53,54 The WCPMG echoes were coadded using a C code to make the coadded echo SSNMR spectrum (Fig. 5b). The electric field gradients (EFG) tensor parameters were measured by fitting the coadded SSNMR spectrum, summarized in Table 2. 77Se is a spin ½ nucleus with 7% natural abundance.50 Most 77Se compounds have large spin–lattice relaxation rates (T1), which results in a longer experimental time.55,56 The MAS 77Se NMR spectrum of LiGaSe2 was acquired using a Carr–Purcell Meiboom–Gill (CPMG) pulse sequence. The 77Se SSNMR spectrum shows a single peak that has an isotropic chemical shift (diso) of 794 ppm with a span (W) of ∼475 ppm. | ||
| Fig. 5 (a) Comparison of 7Li spin-echo SSNMR spectra of LiGaSe2 and LiInSe2 nanocrystals. The MAS frequency was 10 kHz. Spinning side bands are marked with an asterisk. (b) Static 69Ga WCPMG SSNMR spectrum and MAS 77Se CPMG SSNMR spectrum of LiGaSe2 nanocrystals. The MAS frequency was 22 kHz. | ||
| Material | Nuclei | δ iso (ppm) | Ω (ppm) | κ | η Q | C Q (MHz) |
|---|---|---|---|---|---|---|
| LiGaSe2 | 69Ga | 233 | — | — | 1.0 | 4.9 |
| 77Se | 794 | 473 | 0.2 | — | — |
Optical properties
The band gaps of 1.8–3.4 eV in the synthesized LiTrCh2 nanocrystals closely match the values reported in the literature57,58 (Table 3). It is noteworthy that, despite their distinct structures, the tetragonal (I2d) and orthorhombic (Pna21) forms of LiTrCh2 have similar band gaps, which is consistent with previous reports on single crystals.59 All LiTrCh2 nanocrystals display UV or Vis photoluminescence, with maxima (PLmax) between 380 nm and 620 nm, and quantum yields (QY) ranging from 1.2–4.6% (Fig. 6).
| Nanocrystal phase | Band gap (eV) | |||
|---|---|---|---|---|
| Exp. | Theo. | |||
| (Abs.) | Lit. | (DFT) | Lit. | |
| Pna21 LiGaSe2 | 3.0 | 2.1–3.7 | 2.0–2.1 | 2.4–3.4 |
|
I2d LiGaSe2 |
2.7 | 1.7–2.0 | 2.0 | 2.8 |
| P21nb LiInS2 | 3.4 | 3.6 | 2.1 | 1.3–3.3 |
| Pna21 LiInSe2 | 1.8 | 1.6–2.8 | 1.5–1.6 | 2.6 |
|
I2d LiInSe2 |
2.0 | 2.9 | 1.5–1.6 | — |
|
RmH NaInS2 |
2.5 | 2.4 | — | 0.7–3.3 |
| P32 NaIn3Se5 | 2.1 | 2.2 | — | — |
| C2 NaIn3Te5 | 1.7 | — | 0.6 | — |
 | ||
| Fig. 6 (a) Optical absorption (dashed lines) and photoluminescence (solid lines) of LiTrCh2 nanocrystals. (b) Experimental (▲) vs. calculated (●) band gaps. | ||
Multiple literature reports indicate that the band gap and PLmax of LiTrCh2 semiconductors are strongly dependent on defects.60 For example, LiInSe2 has an intrinsic bandgap of around 2.8 eV,61 but this narrows with increasing defect density.62,63 Density functional theory (DFT) calculations suggest that InLi and LiIn antisites are the main defects responsible for this behavior.63 Defect types in yellow single crystals of Li1.01In1Se2 are VSe+ and LiIn2−, while those in red crystals are InLi2+ and VLi−.23 In this study, we observe that the Pna21 and I2d phases of LiInSe2 have band gaps of 1.8 and 2.0 eV. Thus, we assume that both synthesized nanocrystals possess defects to some degree, with a higher density in the Pna21 LiInSe2 nanocrystals. Further work will address ways to decrease the number of defects as a way to further increase the PL QYs of these materials.
Sodium-based ternary nanocrystals, on the other hand, have band gaps of 1.7–2.5 eV (see SI). Importantly, NaInS2 and NaIn3Se5 nanocrystals show visible-range PL, while NaIn3Te5 lacks PL in either Vis or NIR regions. Previously, NaInS2 nanoplates and NaIn3Se5 single crystals showed band gaps of 2.35 eV (ref. 64) and 2.17 eV,40 respectively.
Correlating theory and experiment
Seeking to obtain deeper physical insight into our results, we used the Vienna Ab initio Simulation Package (VASP)65 and the Tight Binding Linear Muffin-tin Orbital Atomic Sphere Approximation (TB-LMTO-ASA)66 to estimate the relative energies, bonding, and optical properties of all phases for each LiTrCh2 composition. We used coloring patterns16,67,68 for phases with mixed-cation sites (below).VASP and LMTO calculations show direct band gaps in the range of 1.5–2.1 eV for these LiTrCh2 nanocrystals and a small theoretical band gap of 0.6 eV for the hypothetical C2 NaIn3Te5 (Table 3). These band gaps are underestimated by 20–54% compared to the experimentally observed values (Fig. 6). Such underestimation is common in semiconductor chalcogenides because the excited states are not considered in the DFT calculations.69 Density of states (DOS) shows that the Ch-p orbital dominates the valence bands, with some contributions from the Tr-s and Tr-p orbitals in all the phases (see SI). Previous DFT calculations on LiTrCh2 and NaTrCh2 also indicated a substantive contribution of the covalent component from the Ch-p and Tr-p orbitals in the chemical bonding of these materials.70–72
A comparison of relative energies among the PBE-relaxed crystal structures for each composition yields useful insights into the outcome of our nanocrystal preparations. LiGaSe2 has two reported phases, I2d and Pna21, which are only 6.2 meV apart, with I2d being the lowest energy polymorph.73 As noted above, this explains our ability to prepare either phase selectively using molecular precursors with different reactivity. More specifically, the more reactive EtSeSeEt leads to the quick formation of nanocrystals adopting the thermodynamic I2d phase, while the relatively mildly reactive PhSeSePh enables the isolation of nanocrystals adopting the kinetic (metastable) Pna21 phase (Scheme 2). In contrast, LiInS2 has only one crystallographically reported phase, P21nb, as observed experimentally.
The inorganic crystal structure database (ICSD) contains four reported polymorphs for LiInSe2, including a disordered cubic Fmm phase with mixed cation (Li/In) sites. In this case, we first created two different supercells using different coloring patterns while keeping the stoichiometry intact,67 which we labeled (Fmm)S1 and (Fmm)S2 (Fig. 7). As in the case of the lighter, gallium-containing analogue, I2d LiInSe2 has the lowest energy, followed closely by Pna21 LiInSe2, which is only ca. 2.7 meV higher than the first. Interestingly, the even smaller energy difference compared to the previous gallium case explains why it is even more challenging in the indium case to isolate metastable Pna21 LiInSe2 nanocrystals in 100% pure form (Table 1).
 | ||
| Fig. 7 Calculated (a) band structure and (b) density of states for I2d LiInSe2, along with (c) relative energies for its different polymorphs. | ||
The other polymorphs of LiInSe2, including RmH (291 meV higher), and the colored supercells (Fmm)S1 and (Fmm)S2 (715–732 meV higher), contain octahedrally coordinated cation sites and shorter bonds. These high-pressure phases, which are metallic in character (see SI), are too high-energy to be relevant under the relatively low temperature and ambient pressure, colloidal synthesis conditions used here. Lastly, because the ICSD lacks a reported structure for NaIn3Te5, we computationally built a unit cell by replacing Ga with In in the reported R32H crystal structure of NaGa3Te5. The calculated XRD pattern matches well with the powder XRD pattern obtained experimentally (Fig. 2b).
Conclusions
In summary, we have synthesized eight different polymorphs of Li/Na-based ternary chalcogenide nanocrystals using a reliable colloidal synthesis method. A combination of powder XRD, high-resolution (HR) TEM, fast Fourier transform (FFT) of selected area electron diffraction (SAED) patterns, energy-dispersive X-ray spectroscopy (EDS), and, in the case of LiGaSe2 nanocrystals, 7Li, 69Ga, and 77Se solid state (SS) NMR analysis, confirms the identity and phase purity of the different crystalline materials. All nanocrystals except NaIn3Te5 are photoluminescent, with modest PL QYs between 1.2% and 4.6%.Critically, we can selectively isolate either of the tetragonal (chalcopyrite) (I2d) or orthorhombic (Pna21) phases of LiGaSe2 and LiInSe2 nanocrystals using different dichalcogenide (RSeSeR) precursors. Specifically, highly reactive EtSeSeEt yields the most stable, thermodynamic I2d phase while mildly reactive PhSeSePh yields the metastable (kinetic) Pna21 phase. Density functional theory (DFT) calculations on both compositions confirm the relative energy order between these polymorphs—separated by only 2.7–6.4 meV per formula units—as well as other, higher energy phases in the case of LiInSe2. Further, DFT predicts a powder XRD pattern of a previously unreported, C2 NaIn3Te5 phase that matches the powder XRD observed experimentally.
Experimental
Materials
Oleylamine (oleylNH2, 70%), 1-octadecene (ODE, 90%), diphenyl diselenide (98%), diphenyl ditelluride (98%), and indium(III) acetylacetonate (99.99+% In) were purchased from Sigma-Aldrich; gallium(III) acetylacetonate (99.99%+ Ga), indium(III) acetylacetonate (99.99+% In), lithium acetylacetonate (99.99+% Li), and sodium oleate (99%) from Strem; diphenyl disulfide (99%) from Acros. Caution: oleylamine is very corrosive and should be handled with great care.Synthesis of LiTrCh2 nanocrystals
Lithium acetylacetonate (0.10 mmol, 11 mg for LiInSe2 and LiGaSe2; 0.15 mmol, 16 mg for LiInS2) and gallium or indium acetylacetonate (0.10 mmol, 37 mg for Ga; 0.10–0.20 mmol, 41–82 mg for In) were dissolved and stirred in a mixture of oleylNH2 (2 mL) and ODE (4 mL) in a round-bottom (RB) flask at 100 °C for 1 h under dynamic vacuum. After refilling the RB flask with N2, diphenyl disulfide/diselenide (0.20–0.60 mmol, 44–131 mg for S; 0.20 mmol, 62 mg for Se) in oleylNH2 (2 mL) was swiftly injected. The temperature was raised to 270–330 °C, and stirring was continued for 30–60 min. The crude solution was treated with an equivalent amount of ethanol and centrifuged at 4500 rpm for 5 min. After discarding the supernatant, the precipitate containing nanocrystalline solids was redispersed in hexanes, treated with an equivalent amount of ethanol and a few drops of methanol, and centrifuged again at 4500 rpm for 5 min.NaTrCh2 or NaTr3Ch5 nanocrystals
These were synthesized in a similar procedure with sodium oleate (0.10 mmol, 37 mg), indium acetylacetonate (0.10 mmol, 37 mg), and diphenyl disulfide/diselenide/ditelluride (0.2 mmol, 44 mg for S; 0.10 mmol, 31 mg for Se; 0.05 mmol, 21 mg for Te) dissolved in oleylNH2 (2 mL). The reaction temperatures for specific compositions are listed in Table 1.Structural characterization
Powder X-ray diffraction (XRD) was collected on a Rigaku Ultima IV diffractometer (40 kV, 44 mA) with a zero-background quartz sample holder using Cu K radiation. A K value of 0.9 was used to determine average single-crystalline domain sizes using the Scherrer equation. A JOEL JSM-IT200 scanning electron microscope was used for area scans and elemental mapping. Samples were put on double-sided carbon tape and examined at a voltage of 10–15 keV. A JEOL 2100 transmission electron microscope was utilized to carry out transmission electron microscopy imaging. Size distributions contain information from at least 100–200 particles. The samples were prepared by drop-casting a dilute nanocrystal solution in hexane on a Cu grid with a 200-mesh carbon coating.Optical characterization
A photodiode array Agilent 8453 UV/vis spectrophotometer was used to measure the absorption spectra of the nanocrystals in solution. All spectra are reported after subtracting the absorption of the solvent. Diffuse reflectance spectra were recorded using an SL1 Tungsten Halogen lamp (vis-IR), an SL3 Deuterium Lamp (UV), and a BLACK-Comet C-SR-100 spectrometer (200–1080 nm). Band gap values were estimated by extrapolating the linear slope of Tauc plots of (Ahν)1/rversus hν (A = absorbance, hν = incident photon energy in eV, r = 1/2 for direct and r = 2 for indirect semiconductors).74 A Horiba-Jobin Yvon Nanolog scanning spectrofluorometer with a photomultiplier detector was used to measure steady-state PL spectra. Rhodamine 440 or rhodamine 460 dye was utilized as a standard for measuring the relative PL quantum yields (QYs).75 Replicates of three or more were used to measure the absorption and PL emission spectra, and the average QYs were recorded.Solid-state NMR
SSNMR spectroscopy experiments (except 69Ga) were performed on a Bruker wide-bore 9.4 T [ν0(1H) = 400 MHz] NMR spectrometer equipped with a Bruker Advance III HD console. 7Li SSNMR spectra were acquired with a Bruker 4 mm HX MAS probe configured in 1H–7Li mode. A rotor-synchronized spin-echo pulse sequence was used with 62.5 kHz radio frequency (RF), CT-selective pulses. 3072 scans (LiGaSe2) or 300 scans (LiInSe2) were acquired with a 2 s recycle delay between scans. The MAS frequency was 10 kHz. 77Se SSNMR spectra were acquired with a Bruker 2.5 mm HX MAS probe configured in 1H–77Se mode. A rotor-synchronized spin-echo pulse sequence was used with 83.3 kHz radio frequency (RF), CT-selective pulses. 560 scans were acquired with a 100 s recycle delay between scans and 23 CPMG loops. The MAS frequency was 22 kHz. 69Ga SSNMR experiments were performed on a Bruker wide-bore 14.1 T [ν0(1H) = 600 MHz] NMR spectrometer equipped with a Bruker Advance NEO console, WCPMG pulse sequence with a 1 s delay, 25 ms WURST pulse, 12 CPMG loops, 40960 with a Bruker 4 mm HX MAS probe configured in 1H–115In mode. Peak fitting of the SSNMR spectra was performed in the solid line-shape analysis module version 3.6.3 included in the Bruker Topspin version 3.6.5 software. 1H chemical shifts were referenced to tetramethylsilane by using adamantane [δiso(1H) = 1.82 ppm] as a secondary chemical shift standard. 7Li, 69Ga, and 77Se chemical shifts were referenced indirectly to the established chemical shift scale [LiCl, δiso(7Li) = 0 ppm], [Ga(NO3)2, δiso(69Ga) = 0 ppm], [SeMe2, δiso(77Se) = 0 ppm] using the previously published relative NMR frequencies.76
Electronic structure calculations
Calculations were carried out using the Vienna Ab initio Simulation Package (VASP)65 and the Tight Binding Linear Muffin-tin Orbital Atomic Sphere Approximation (TB-LMTO-ASA) package.66 PBE band structures and DOS were calculated using the tetrahedron method after converging the total energy on a k-mesh of 1/a × 1/b × 1/c in the irreducible wedge of the Brillouin zone. Unit cell representations were generated using VESTA.77Author contributions
Md Riad Sarkar Pavel: writing – original draft, conceptualization, methodology, synthesis and computational investigation. Anuluxan Santhiran: solid-state NMR investigation, methodology. Seth Dalberg: synthesis investigation, methodology. Aaron J. Rossini: solid state NMR and writing – review & editing. Javier Vela: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information: powder XRD, electron microscopy images, SS NMR measurements, and DFT calculations. See DOI: https://doi.org/10.1039/d5ta07992f.Acknowledgements
This invited manuscript is dedicated to celebrating Prof. D. D. Sarma's contributions to materials science. This work was supported by the U.S. National Science Foundation, Division of Chemistry, Macromolecular, Supramolecular, and Nanochemistry Program (2305062) by a grant awarded to J. V. SSNMR work by A. S. and A. J. R. was supported by the U.S. Department of Energy (DOE) Ames National Laboratory, Materials Science and Engineering Division. Ames National Laboratory is operated for the U.S. DOE by Iowa State University, under contract no. DE-AC02-07CH11358. We thank Gordie Miller and Federico Zahariev for discussions.References
- J. L. Dye, Philos. Trans. R. Soc. A, 2015, 373, 20140174 CrossRef.
- Y. Shen, Z. Zhu, Z. Xu and Y. Li, Energy Adv., 2024, 3, 1844–1868 RSC.
- H. Hao, T. Hutter, B. L. Boyce, J. Watt, P. Liu and D. Mitlin, Chem. Rev., 2022, 122, 8053–8125 CrossRef CAS.
- P. Xu, F. Huang, Y. Sun, Y. Lei, X. Cao, S. Liang and G. Fang, Adv. Funct. Mater., 2024, 34, 2406080 CrossRef CAS.
- A. Kausar, A. Sattar, C. Xu, S. Zhang, Z. Kang and Y. Zhang, Chem. Soc. Rev., 2021, 50, 2696–2736 RSC.
- A. Van der Ven, Z. Deng, S. Banerjee and S. P. Ong, Chem. Rev., 2020, 120, 6977–7019 CrossRef CAS PubMed.
- Y. Zhang, R. Cao, C. Ouyang, L. Jiang, Y. Wang, M. Yang and H. Xia, J. Mater. Chem. A, 2025, 13, 3973–3990 RSC.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- F. Zhang, B. He, Y. Xin, T. Zhu, Y. Zhang, S. Wang, W. Li, Y. Yang and H. Tian, Chem. Rev., 2024, 124, 4778–4821 CrossRef CAS.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS.
- Y. Zhang, B. Han, S. Tan, Q. Gao, Z. Cai, C. Zhou, J. Li, R. Sun and K. Amine, Adv. Energy Mater., 2025, 15, 2404796 CrossRef CAS.
- H. McKeever, N. N. Patil, M. Palabathuni and S. Singh, Chem. Mater., 2023, 35, 9833–9846 CrossRef CAS.
- C. A. Aranda, A. O. Alvarez, V. S. Chivrony, C. Das, M. Rai and M. Saliba, Joule, 2024, 8, 241–254 CrossRef CAS.
- A. Karami, M. Morawski, H. Kempa, R. Scheer and O. Cojocaru-Mirédin, Sol. RRL, 2024, 8, 2300544 CrossRef CAS.
- B. A. Tappan and R. L. Brutchey, ChemNanoMat, 2020, 6, 1567–1588 CrossRef CAS.
- M. A. White, A. M. Medina-Gonzalez and J. Vela, Chem.–Eur. J., 2018, 24, 3650–3658 CrossRef CAS.
- H. Dong, J. Zhao, H. Yang and Y. Zheng, Phys. Rev. Mater., 2022, 6, 104001 CrossRef CAS.
- L. I. Isaenko and A. P. Yelisseyev, Semicond. Sci. Technol., 2016, 31, 12300 CrossRef.
- N. Jia, S. Wang, P. Wang, C. Li, T. Yu, J. Qiao, C. Li, X. Xiong, J.-L. Sun and X. Tao, J. Mater. Chem. C, 2018, 6, 12615–12622 RSC.
- T. V. Vu, A. A. Lavrentyev, B. V. Gabrelian, D. D. Vo, P. D. Khang, L. I. Isaenko, S. I. Lobanov, A. F Kurus and O. Y. Khyzhun, RSC Adv., 2020, 10, 26843–26852 RSC.
- Y. Fan, X. Song, S. Qi, X. Ma and M. Zhao, J. Mater. Chem. A, 2019, 7, 26123–26130 RSC.
- T. Biswas and A. K. Singh, npj Comput. Mater., 2021, 7, 189 CrossRef CAS.
- Z. Zheng, H. Yu, M. Zhu, Z. Zhang, Z. Gao, M. Xu, R. Zhang and Y. Xu, Sci. Rep., 2024, 14, 24779 CrossRef CAS.
- Z. Liu, Y. Sun, D. J. Singh and L. Zhang, Adv. Electron. Mater., 2019, 5, 1900089 CrossRef.
- Y. Zhao, L. Huang, D. Zhao and J. Yang Lee, Angew. Chem., Int. Ed., 2023, 62, 202308976 CrossRef.
- W. Hua, H. Li, C. Pei, J. Xia, Y. Sun, C. Zhang, W. Lv, Y. Tao, Y. Jiao, B. Zhang and S. Z. Qiao, Adv. Mater., 2021, 33, 2101006 CrossRef CAS PubMed.
- J. Yang, X. Huang, X. Xu, H. Lu, S. Wang and S. Wu, ACS Appl. Mater. Interfaces, 2024, 16, 15050–15058 CrossRef CAS PubMed.
- V. Bikbaeva, O. Perez, N. Nesterenko and V. Valtchev, Inorg. Chem. Front., 2022, 9, 5181–5187 RSC.
- J. De Roo, Chem. Mater., 2022, 34, 5766–5779 CrossRef CAS.
- M. A. White, K. J. Baumler, Y. Chen, A. Venkatesh, A. M. Medina-Gonzalez, A. J. Rossini, J. V. Zaikina, E. M. Chan and J. Vela, Chem. Mater., 2018, 30, 6173–6182 CrossRef CAS.
- Y. Guo, S. R. Alvarado, J. D. Barclay and J. Vela, ACS Nano, 2013, 7, 3616–3626 CrossRef CAS PubMed.
- M. P. Hendricks, M. P. Campos, G. T. Cleveland, I. Jen-La Plante and J. S. Owen, Science, 2015, 348, 1226–1230 CrossRef CAS.
- M. R. S. Pavel, A. Santhiran, S. Dalberg, A. J. Rossini and J. Vela, ACS Nano, 2025, 19, 33413–33422 CrossRef CAS PubMed.
- M. R. S. Pavel, Y. Chen, A. Santhiran, E. Gi, K. Ochoa-Romero, G. J. Miller, G. Guirado, A. J. Rossini and J. Vela, ACS Energy Lett., 2024, 9, 5012–5018 CrossRef CAS PubMed.
- B. A. Tappan, G. Barim, J. C. Kwok and R. L. Brutchey, Chem. Mater., 2018, 30, 5704–5713 CrossRef CAS.
- E. J. Endres, J. R. Bairan Espano, A. Koziel, A. R. Peng, A. A. Shults and J. E. Macdonald, ACS Nanosci. Au, 2024, 4, 158–175 CrossRef CAS PubMed.
- Y. Fu, X. Duan, M. Xing, N. Zhang, X. Luo, H. Wang and Y. Ma, Mater. Lett., 2014, 124, 141–143 CrossRef CAS.
- A. Adhikary, H. Yaghoobnejad Asl, P. Sandineni, S. Balijapelly, S. Mohapatra, S. Khatua, S. Konar, N. Gerasimchuk, A. V. Chernatynskiy and A. Choudhury, Chem. Mater., 2020, 32, 5589–5603 CrossRef CAS.
- V. V. Klepov, A. A. Berseneva, K. A. Pace, V. Kocevski, M. Sun, P. Qiu, H. Wang, F. Chen, T. M. Besmann and H. C. Zur Loye, Angew. Chem., Int. Ed., 2020, 59, 10836–10841 CrossRef CAS PubMed.
- S. F. Li, X. M. Jiang, B. W. Liu, D. Yan, C. S. Lin, H. Y. Zeng and G. C. Guo, Chem. Mater., 2017, 29, 1796–1804 Search PubMed.
- Z. Sun, C. M. Perez, O. V. Prezhdo and R. L Brutchey, ACS Nanoscience Au, 2024, 4, 381–390 CrossRef CAS PubMed.
- Z. Sun, C. P. Pakhanyan, U. Saleem, E. I. Feldman, S. Mallikarjun Sharada and R. L. Brutchey, Inorg. Chem., 2025, 64, 21796–21800 CrossRef CAS PubMed.
- S. K. O’Boyle, A. Naeem and R. E. Schaak, Chem. Mater., 2025, 37, 8889–8900 CrossRef.
- H. Yan, L. Chen, L. Feng, Y. Chen, M. Zhang and Q. Wei, Vacuum, 2024, 225, 113256 CrossRef CAS.
- R. F. Ali and B. D. Gates, Nanoscale, 2021, 13, 3214–3226 RSC.
- C.-Y. Yeh, Z. Lu, S. Froyen and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 10086–10097 CrossRef CAS PubMed.
- Y. E. Maidebura, V. G. Mansurov, T. V. Malin, I. A. Aleksandrov, K. S. Zhuravlev and B. Pecz, CrystEngComm, 2025, 27, 2307–2316 RSC.
- N. Kapuria, B. Nan, T. E. Adegoke, U. Bangert, A. Cabot, S. Singh and K. M. Ryan, Chem. Mater., 2023, 35, 4810–4820 CrossRef CAS PubMed.
- M. Strach, V. Mantella, J. R. Pankhurst, P. Iyengar, A. Loiudice, S. Das, C. Corminboeuf, W. van Beek and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 16312–16322 CrossRef CAS PubMed.
- R. K Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef.
- S. E. Ashbrook, Phys. Chem. Chem. Phys., 2009, 11, 6892–6905 RSC.
- S. E. Ashbrook and S. Sneddon, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef CAS PubMed.
- R. W. Schurko, Acc. Chem. Res., 2013, 46, 1985–1995 Search PubMed.
- L. A. O'Dell, A. J. Rossini and R. W. Schurko, Chem. Phys. Lett., 2009, 468, 330–335 CrossRef.
- W. H. Dawson and J. D. Odom, J. Am. Chem. Soc., 1977, 99, 8352–8354 CrossRef CAS.
- J. D. Odom, W. H. Dawson and P. D. Ellis, J. Am. Chem. Soc., 1979, 101, 5815–5822 CrossRef CAS.
- A. Eifler, V. Riede, J. Brückner, S. Weise, V. Krämer, G. Lippold, W. Schmitz, K. Bente and W. Grill, Jpn. J. Appl. Phys., 2000, 39, 279 CrossRef CAS.
- L. Isaenko, I. Vasilyeva, A. Yelisseyev, S. Lobanov, V. Malakhov, L. Dovlitova, J. J. Zondy and I. Kavun, J. Cryst. Growth, 2000, 218, 313–322 CrossRef CAS.
- M. Jomaa, V. Mishra, D. Mumbaraddi, R. Sikdar, D. Sarkar, M. Sun, J. Yao, V. K. Michaelis and A. Mar, Inorg. Chem., 2023, 62, 7491–7502 CrossRef CAS PubMed.
- D. Clark, J. Zhang, A. Craig, A. Weiland, J. Brant, J. Cho, Y. Kim, J. Jang and J. Aitken, J. Alloys Compd., 2022, 917, 165381 CrossRef CAS.
- E. Tupitsyn, P. Bhattacharya, E. Rowe, L. Matei, Y. Cui, V. Buliga, M. Groza, B. Wiggins, A. Burger and A. Stowe, J. Cryst. Growth, 2014, 393, 23–27 CrossRef CAS.
- L. Isaenko, A. Yelisseyev, S. Lobanov, V. Petrov, F. Rotermund, G. Slekys and J.-J. Zondy, J. Appl. Phys., 2002, 91, 9475–9480 CrossRef CAS.
- L. Guo, Y. Xu, H. Zheng, W. Xue, J. Dong, B. Zhang, Y. He, G. Zha, D. Y. Chung, W. Jie and M. G. Kanatzidis, Cryst. Growth Des., 2018, 18, 2864–2870 CrossRef CAS.
- A. K. P. Mann, S. Wicker and S. E. Skrabalak, Adv. Mater., 2012, 24, 6186–6191 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- O. Jepsen, A. Burkhardt and O. K. Andersen, The Program TB-LMTO-ASA, version 4.7, Max-Planck-Institut fur Festkorperforschung, Stuttgart, Germany, 1999 Search PubMed.
- G. J. Miller, Eur. J. Inorg. Chem., 1998, 1998, 523–536 CrossRef.
- B. A. Rosales, M. A. White and J. Vela, J. Am. Chem. Soc., 2018, 140, 3736–3742 CrossRef CAS PubMed.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Phys. Rev. Lett., 2008, 100, 146401 CrossRef PubMed.
- A. A. Lavrentyev, B. V. Gabrelian, V. T. Vu, L. N. Ananchenko, L. I. Isaenko, A. P. Yelisseyev and O. Y. Khyzhun, Opt. Mater., 2017, 66, 149–159 CrossRef CAS.
- W. Chen, J. M. Zhang, Q. L. Xia, Y. Z. Nie and G. H. Guo, Phys. Chem. Chem. Phys., 2020, 22, 16007–16012 RSC.
- M. S. Khan, B. Gul, G. Khan, S. A. Khattak, M. Ajaz, T. Khan and S. Zulfiqar, J. Solid State Chem., 2022, 307, 122853 CrossRef.
- W. B. Cai, A. Abudurusuli, C. W. Xie, E. Tikhonov, J. J. Li, S. L. Pan and Z. H. Yang, Adv. Funct. Mater., 2022, 32, 2200231 CrossRef CAS.
- B. D. Viezbicke, S. Patel, B. E. Davis and D. P. Birnie, Phys. Status Solidi B, 2015, 252, 1700–1710 CrossRef CAS.
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |