DOI:
10.1039/D5TA06475A
(Paper)
J. Mater. Chem. A, 2025,
13, 42101-42117
Double transition metal MXenes as anode materials for high-capacity multivalent metal-ion batteries: a computational study
Received
10th August 2025
, Accepted 7th October 2025
First published on 30th October 2025
Abstract
The growing global energy crisis and increasing demand for sustainable energy storage solutions have intensified the search for efficient and high-capacity battery technologies. Conventional lithium-ion batteries, although widely used, face challenges of resource scarcity, limited energy density, and environmental concerns. As a promising alternative, multivalent metal-ion (e.g., Mg2+, Zn2+, and Al3+) batteries offer higher charge storage capabilities and improved cost-effectiveness compared to monovalent Li+, Na+, and K+-based metal-ion batteries. Here, we explore the potential of novel double-transition-metal (DTM) MXenes as electrode materials for multivalent metal-ion batteries using density functional theory (DFT). Geometrical stability, electronic properties, ion adsorption behavior, and electrolyte compatibility are systematically analyzed to evaluate their electrochemical performance. Our results reveal that these MXenes exhibit excellent specific capacities of 3096.64, 2064.43, and 688.14 mAh per g (VNbC); 1978.76, 1319.17, and 439.72 mAh per g (VTaC); and 844.14, 1125.52, and 375.17 mAh per g (NbTaC) for Al, Mg, and Zn, respectively. Additionally, we demonstrate low diffusion barriers of 0.26, 0.13, and 0.19 eV (on VNbC); 0.25, 0.10, and 0.16 eV (on VTaC); and 0.18, 0.06, and 0.16 eV (on NbTaC) for Al, Mg, and Zn, respectively. This study shows that the VNbC monolayer provides a much higher Al3+-ion storage capacity than that of the widely commercialized graphite in lithium-ion batteries. This investigation unearths key insights into the fundamental mechanisms governing ion intercalation in DTM MXene-based anodes, which is encouraging for their application in advanced rechargeable battery technologies.
1. Introduction
The growing emphasis on clean and renewable energy sources stems from increasing concerns about energy shortages and environmental degradation caused by fossil fuel consumption. In recent times, there has been significant advancement in eco-friendly renewable energy technologies, particularly solar and wind power.1–4 These sustainable alternatives are now viewed as promising substitutes for traditional fossil fuels. The surge in new energy sources necessitates the development of eco-friendly and efficient energy storage technologies and devices.5 Among the various energy storage options, rechargeable metal-ion batteries, particularly lithium-ion batteries (LIBs), stand out as a primary choice. LIBs offer several advantages over other secondary batteries, including a broad voltage range and high energy density.6–8 These features have made LIBs the dominant power source for electric vehicles and portable electronic devices in today's market.9–14 Nevertheless, the electrode materials currently employed in LIBs are nearing their capacity limitations, and concerns regarding lithium's scarcity and safety pose significant obstacles to the widespread adoption of LIBs in the coming years.15–17 In response to these LIB-related challenges, numerous next-generation secondary batteries utilizing alternative metal ions have emerged. Although sodium and potassium could be viable alternatives for the development of metal-ion batteries, bivalent metal ions such as Mg2+, Ca2+, and Zn2+ may potentially offer greater capacity compared to monovalent ions, provided that the transfer of two electrons per ion is achieved.18–23 However, the larger radius and significantly higher mass of Na, K, and Mg in comparison to Li present new obstacles in developing suitable battery electrode and electrolyte materials. For instance, graphite is not suitable for SIBs due to its insufficient interlayer spacing (0.340 nm) to accommodate Na ions (ionic radius 0.095 nm) that can provide its easy intercalation and deintercalation.5,24,25 Consequently, the identification of appropriate electrode materials capable of effectively hosting metal-ions is a pressing challenge in the advancement of next-generation metal-ion batteries.
The revolutionary discovery of graphene by Geim and Novoselov in 2004 ushered in a new age of research and development in two-dimensional (2D) materials. These atomically thin substances, known for their exceptional characteristics, have shown significant promise for use in energy storage systems. Their large surface area relative to volume, exceptional electrical conductivity, and structural durability make them particularly well-suited as electrode components in metal-ion batteries.26–29 Two-dimensional materials, particularly transition-metal-dichalcogenides (TMDs) such as MoS2, WSe2, ScTe2, Nb2S2, TiS2, and V2S2 have been theoretically investigated to examine their potential as electrodes in metal ion batteries.30–35 The TiS2 monolayer was found to demonstrate high adsorption capacities for alkali and alkaline earth metal ions, with Mg and Ca offering the highest capacities (1914 mAh g−1). It is important to note that Mg ions exhibit a very low open-circuit voltage (0.13 V) and favorable diffusion properties, making TiS2 suitable for replacing Li in high-energy-density rechargeable batteries.36
MXenes, a class of 2D materials derived from MAX phases (M = early transition metal, A = group 13–16 element, and X = C or N), exhibit high conductivity, mechanical strength, chemical stability, and tunable surface functionalities. Their hydrophilicity combined with electrical conductivity makes them attractive for energy storage.37 Recent efforts have focused on optimizing synthesis via selective etching and intercalation to improve yield, flake size control, and stability.38–42 Ti3C2 has been investigated as an anode, showing storage capacities of 447.8 (Li), 351.8 (Na), 191.8 (K), and 319.8 (Ca) mAh g−1,43 while carbides of Mo, Ti, V, and Nb also demonstrate promise for Li-ion batteries.44–47 Surface terminations significantly affect performance; asymmetric Ti2CFBr and Ti2CFOH deliver higher Na-ion storage capacity (518.9 and 559.5 mAh g−1) than symmetric Ti2CF2 (367.9 mAh g−1).48 Beyond LIBs and SIBs, multivalent ion batteries (MIBs) such as Mg2+-, Zn2+-, and Al3+-based systems provide higher charge storage per ion, lower cost, and improved safety.49 AIBs, in particular, benefit from the abundance, stability, and cycling durability of Al,49–51 with AlCl3-based ionic liquids serving as electrolytes and active materials.52 Mg-ion batteries also offer dendrite-free operation and enhanced safety. Overall, MIBs, ZIBs, and AIBs represent sustainable and scalable alternatives to conventional LIBs for next-generation energy storage.53,54
Ordered double-transition-metal (DTM) MXenes have recently attracted significant attention due to their tunable electronic and electrochemical properties. Various DTM MXenes, including TiNbC, TiVC, TiVN, Mo2VC2, Mo2NbC2, Mo2TiC2Tx, Mo2Ti2C3Tx, Cr2VC2, CrVN, Cr2NbC2, and Cr2TiC2Tx, have been synthesized or theoretically predicted.55,56 Structurally, they consist of two distinct transition-metal layers sandwiching carbon atoms in an octahedral arrangement, enabling promising applications in energy storage and magnetic devices. Naguib et al. first synthesized TiNbC MXene by etching Al from TiNbAlC, confirming its metallic character, while subsequent studies revealed that introducing a second transition metal enhances ion adsorption, multilayer adsorption mechanisms, and diffusion kinetics.57,58 For instance, WCrC- and WCrCO2-based MXenes exhibit lower migration barriers, superior ion transport, and higher stability, with WCrC delivering ∼40% higher capacity than W2C. Similarly, MoWC outperforms Mo2C and W2C, with a storage capacity of 670 mAh g−1 and a diffusion barrier of 0.029 eV, compared to 530 mAh g−1/0.045 eV and 259 mAh g−1/0.040 eV, respectively.59,60 The synthesis of VNbCTx multilayer MXene by selectively etching Al from its MAX phase, VNbAlC, was reported by Cheng et al. and they investigated its electrochemical performance.61 Their work demonstrated the practical feasibility of VNbCTx, multilayer MXene, achieving a specific capacity of 520.5 mAh g−1 in lithium-ion batteries (LIBs) with good charge/discharge reversibility, thereby positioning this material as a strong candidate for next-generation energy storage devices. Zhou et al. theoretically reported specific storage capacities of 601.85, 302.20 and 328.13 mAh g−1 for sodium ions on the VNbC, VTaC and NbTaC monolayers.62 These double transition metal carbides exhibit 75%, 37.5% and 75% higher Na-ion storage capacities compared to their single transition metal counterparts.62
The successful synthesis of multilayer double transition metal carbides and their demonstration as promising anode materials in LIBs and subsequent theoretical reports on their use in SIBs, motivated us perform a first-principles investigation of VNbC, VTaC, and NbTaC nanosheets for application as electrodes in the multivalent metal-ion batteries. We provide in-depth insights into their thermodynamic and mechanical stability, electronic properties, and electrochemical behavior (ion diffusion, voltage profiles, and theoretical capacities), which have not been reported before. This investigation reveals the performance of double transition metal carbides as high-capacity anode materials in multivalent rechargeable metal-ion batteries. The thermal stability, compatibility with conventional electrolytes—ethylene carbonate (EC), dimethyl carbonate (DMC), diethyl carbonate (DEC), and propylene carbonate (PC)—have also been explored. These research findings will motivate experimentalists to synthesize anodes for multi-valent metal-ion batteries using these DTM MXenes.
2. Computational details
All calculations were performed using first-principles “density functional theory” (DFT) with the “Vienna Ab initio Simulation Package” (VASP) code.63,64 The plane wave basis set for the wave functions were considered up to 500 eV. The “projector-augmented wave (PAW)” pseudopotential and “Perdew–Burke–Ernzerhof (PBE)” generalized gradient approximation (GGA) framework were employed to describe the core electrons and exchange–correlation interactions, respectively.65–67 The two-dimensional monolayers in the x–y plane were realized by considering a vacuum of ∼32.96 Å normal to the monolayer plane. Geometrical relaxations were carried out with a Monkhorst–Pack (MP) 4 ×4 ×1 k-point grid for the Brillouin zone (BZ).68 The PBE functional are used for band structure calculations.69 The energy threshold was set to 10−6 eV for the convergence criterion during the self-consistent field iteration in the structural relaxations. The monolayers were relaxed until the Hellmann–Feynman forces on each atom became less than 0.01 eV Å−1. The van der Waals (vdW) interactions between the adsorbate metals and the DTM (VNbC, VTaC, and NbTaC) monolayers, were accounted for using “Grimme's DFT-D3” dispersion correction in the calculations.65 We utilized a 3 ×3 ×1 supercell of the two-dimensional monolayers in the ab initio molecular dynamics (AIMD) simulation under the canonical ensemble (NVT) using the Nöse–Hoover thermostat at 300 K and 500 K.67 Mechanical stability analysis was carried out using the energy-strain method, confirming the stability of the DTM MXenes.70 The charge density difference (CDD) provides qualitative information regarding the charge transfer process between the adsorbates and the monolayers.71 The VTST tool was employed to generate intermediate images between the initial and final positions for the determination of the diffusion energy barrier using the “Climbing Image Nudged Elastic Band (CI-NEB)” method.72 VESTA was used to render all of the atomic models and charge density difference plots.73 Vaspkit was also used for the pre and post processing of the VASP simulation data.66
3. Results and discussion
3.1. Geometry optimized XYC (X = Y = V, Nb, and Ta) DTM MXene monolayers
We denote the double transition metal MXenes as XYC, where X (V or Nb) represents the top layer; Y (Nb or Ta) represents the bottom layer and C is sandwiched in between them. Three possible XYC MXene nanosheets namely, VNbC, VTaC, and NbTaC, are considered in this present study on the basis of higher formation energy. The most stable structures of these sheets obtained by complete relaxation are depicted in Fig. 1 and all the lattice parameters, bond lengths, bond angles, and thickness, are given in Table 1. The space group for these structures is P3m1, featuring a hexagonal arrangement. The lattice constants are 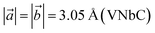 ,
, 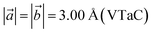 , and
, and 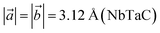 , which are close to the previously reported values
, which are close to the previously reported values 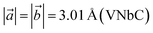 ,
, 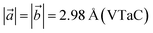 , and
, and 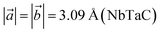 by Zhou et al. which benchmark our calculations.62 The thicknesses of the three sheets are different due to the different atomic radii of the metal atoms. The formation energy (Ef) is determined to assess the stability of the XYC two-dimensional sheets by using the following expression,
by Zhou et al. which benchmark our calculations.62 The thicknesses of the three sheets are different due to the different atomic radii of the metal atoms. The formation energy (Ef) is determined to assess the stability of the XYC two-dimensional sheets by using the following expression,| | 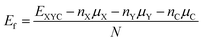 | (1) |
Here, EXYC denotes the total energy of the XYC monolayer; µ represents the chemical potential of each element in its bulk phase while n indicates the number of particular X, Y or C atoms in the sheet and N (= nX + nY + nC) refers to the total number of atoms within the sheet. The calculated formation energies are shown in Table 1. The negative values of the formation energies confirm the stability of the pristine sheets.
 |
| | Fig. 1 (a–c) The top view of relaxed 1 × 1 × 1-unit cells and (d–f) the top panel: top view; bottom panel: cross-sectional view of the optimized 3 × 3 × 1 supercell of VNbC, VTaC, and NbTaC DTM carbide monolayers. | |
Table 1 Lattice parameter, bond length, bond angle, formation energy, and thickness of the XYC MXene monolayers
| System, XYC |
Lattice parameter, 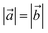
(Å) |
Bond length, dX–C (Å) |
Bond length, dY–C (Å) |
Angle, φ∠C–X–C (°) |
Angle, φ∠C–Y–C (°) |
Formation energy, Ef (eV) |
Thickness, t (Å) |
| VNbC |
3.05 |
1.99 |
2.17 |
88.43 |
99.15 |
−2.30 |
2.25 |
| VTaC |
3.00 |
2.00 |
2.16 |
98.71 |
88.44 |
−2.77 |
2.27 |
| NbTaC |
3.12 |
2.16 |
2.15 |
91.72 |
92.86 |
−2.39 |
2.40 |
3.2. Thermodynamic and mechanical stability of XYC nanosheets
To check the thermal stability of pristine XYC monolayers we conducted “ab initio molecular dynamics (AIMD)” simulations at 500 K for 10 ps. The variations in energies of the systems with respect to time along with their final structures are depicted in Fig. 2(a–c). We have also shown the bond length fluctuations with time in Fig. S1 (S represents the SI). We calculated the average energy, bond length and their percentage fluctuations over the simulation time as listed in Table S1. The average energy values of VNbC, VTaC and NbTaC are −242.67, −259.07, and −269.04 eV, respectively. The energy fluctuations diminish after 2 ps and the structure reaches equilibrium as shown in Fig. 2(a–c). The average energy fluctuations of the VNbC, VTaC, and NbTaC monolayers are 1.91%, 0.81%, and 2.30%. The geometrical structures remain intact after 10 ps AIMD simulation at 500 K. The thermal stability of the pristine nanosheets is thus confirmed. Zhou et al. carried out phonon dispersion calculations using density functional perturbation theory (DFPT) and observed the absence of imaginary frequencies in the phonon spectra of VNbC, VTaC and NbTaC, confirming that these three DTM MXenes are dynamically stable.62 To check the mechanical stability of XYC monolayers, we calculated the elastic constants of the elastic tensor which are presented in Table 2. It is clear that the elastic constants values satisfy the Born–Huang mechanical stability criteria, C11C22 − C122 > 0 and C66 > 0, confirming the mechanical stability of these sheets.70,74
 |
| | Fig. 2 (a–c) The energy vs. time plot calculated using the ab initio molecular dynamics (AIMD) simulations for 10 ps at 500 K on the DTM (VNbC, VTaC, and NbTaC) monolayers. | |
Table 2 Elastic constant values for XYC MXenes
| MXene structure |
C
11 (N m−1) |
C
22 (N m−1) |
C
12 (N m−1) |
C
66 (N m−1) |
| VNbC |
42.23 |
42.94 |
13.88 |
14.82 |
| VTaC |
46.69 |
44.85 |
18.23 |
12.4 |
| NbTaC |
51.74 |
51.96 |
24.59 |
13.33 |
The elastic constants which define a material's elasticity, indicate how a material responds to the application of external forces on it. The elastic constants C11 and C22 are not equal, indicating the anisotropic Young's modulus of these nanosheets.75 The angular dependence of Young's modulus, Y(θ), and Poisson's ratio, ν(θ), of the two-dimensional pristine VNbC, VTaC, and NbTaC monolayers are calculated and shown in Fig. S2(a–f) respectively. Young's modulus, Y(θ) of the VTaC and NbTaC monolayers is anisotropic with maximum (minimum) values of 39.28 and 40.10 N m−1 (35.70 and 39.53 N m−1) respectively in the directions of 0° and 180° (45°, 135°, 225°, and 315°) with respect to the x-axis. Young's modulus of the VNbC monolayer is anisotropic with maximum (minimum) values of 38.91 N m−1 (37.74 N m−1) in the directions of 45°, 135°, 225°, and 315° (0° and 180°) with respect to the x-axis. As compared to graphene (342 N m−1)76 and MoS2 (130 N m−1),77 Young's modulus of these XYC MXene nanosheets is much smaller, indicating that these double transition metal carbide nanosheets are quite stretchable and flexible. Also, the observed Poisson's ratio ν(θ) in DTM nanosheets lies in the range of 0.32–0.49 (VNbC), 0.41–0.57 (VTaC) and 0.47–0.61 (NbTaC) which is larger than that of graphene (ν(θ) = 0.15).76 This indicates that DTM nanosheets exhibit greater transverse deformation under strain, suggesting a higher degree of mechanical flexibility and ductility compared to graphene.
3.3. Electronic properties of double transition metal MXenes
The performance of rechargeable metal-ion batteries is significantly influenced by the electrical conductivity of the electrode material. To investigate this, we computed the electronic band structure and density of states (DOS) for two-dimensional XYC nanosheets using the PBE exchange–correlation functional. Fig. 3(a–c) present the electronic band structures and total density of states plots for VNaC, VTaC, and NbTaC nanosheets, respectively. The results indicate that all three pristine MXene nanosheets exhibit metallic behavior, characterized by a significant density of electronic states at the Fermi level. This metallic nature is crucial for efficient charge transport, making these materials suitable for battery applications.
 |
| | Fig. 3 (a–c) The electronic band structure and density of states (DOS) plots of pristine VNbC, VTaC, and NbTaC monolayers in the energy range of −3.0 to +3.0 eV, respectively. The Fermi energy level is set at zero eV and is indicated by a red dotted line. | |
3.4. Adsorption of multivalent metal atoms on the pristine DTM MXenes and their electronic properties
The spatial variation of electron distribution on the surface of MXene nanosheets gives an idea of the sites for the intercalation of metal atoms (Al, Ca, Mg, and Zn).30 The electron localization function (ELF) plots of the DTM MXenes are shown in Fig. S3. High ELF values indicate regions of strong electron localization, while low values suggest regions of weak electron localization. From Fig. S3(a–c), two less electron localized sites are identified on the DTM nanosheets which may act as adsorption sites: the c-site (on top of a carbon atom) and the h-site (at the center of a hexagonal ring).
To investigate the adsorption behavior, we placed Al, Ca, Mg, and Zn atom at these two sites on VNbC, VTaC, and NbTaC monolayers and calculated the adsorption energy using the standard formula as described in ref. 30. All the adsorption energies at two adsorption sites are given in Table S2. The results indicate that the c-site is energetically the most favorable adsorption site for all studied metal atoms. The Al adatom adsorption energies at the favorable site are −0.68 eV (VNbC), −0.79 eV (VTaC), and −0.76 eV (NbTaC), with corresponding bond lengths of 2.72 Å, 2.58 Å, and 2.72 Å, respectively. For Ca, the adsorption energies are −1.30 eV (VNbC), −1.11 eV (VTaC), and −1.27 eV (NbTaC), with bond lengths of 3.18 Å, 3.05 Å, and 3.15 Å respectively. Similarly, the adsorption energies are −0.91 eV (VNbC), −0.81 eV (VTaC), and −0.91 eV (NbTaC) for Mg, with the bond lengths of 2.79 Å, 2.79 Å, and 2.93 Å, respectively. In the case of Zn, the adsorption energies are −0.82 eV (VNbC), −0.71 eV (VTaC), and −0.80 eV (NbTaC), with corresponding bond lengths of 2.61 Å, 2.61 Å, and 2.73 Å respectively. In some cases, the adsorption energies at the two sites appear identical because the adsorbed atoms initially placed at the h-site migrated to the more stable c-site during structural relaxation. The most energetically favorable relaxed configurations (at the c-site) are shown in Fig. S4, while the alternative relaxed configurations (at the h-site) for the other adsorption site are provided in Fig. S5.
The band structures and density of states (DOS) for Al-, Ca-, Mg-, and Zn-adsorbed 3 ×3 ×1 XYC MXene supercells are shown in Fig. 4(a–l) respectively. The presence of energy bands crossing the Fermi level with a significant density of states confirms the metallic nature of the metal-adsorbed MXene systems. We further note that the metallicity of the metal-adsorbed MXenes is enhanced when compared with that of the corresponding pristine sheets. This metallic behavior facilitates easy transport of charge carriers.
 |
| | Fig. 4 Electronic band structures and total density of states of single Al-, Ca-, Mg-, and Zn-adsorbed 3 × 3 × 1 DTM MXene (VNbC, VTaC, and NbTaC) supercells, are shown in (a–c), (d–f), (g–i), and (j–l) respectively. All the metal-adsorbed nanosheets exhibit good metallicity. | |
3.5. Analysis of the charge density difference and work function
Charge density difference (CDD) calculations analyze how electron density redistributes upon metal adsorption on MXene monolayers and the visualization of CDD reveals regions of charge accumulation and depletion.78 We conducted a qualitative charge transfer analysis for a single metal-adsorbed system employing charge density difference calculations.78 We computed the difference in electrical charge density for the metal-adsorbed DTM MXenes using the following relation79,80 and the obtained CDD results are visualized in Fig. 5.where, Δρ is the charge density difference caused by the adsorption of alkali metals on DTM MXenes, ρML+M is the charge density of the metal-adsorbed DTM nanosheets, ρML is the charge density of the pristine DTM nanosheets, and ρM denotes the charge density of the isolated metal atom (Ca/Mg/Zn/Al). In Fig. 5, yellow indicates charge accumulation, while cyan represents charge depletion. Cyan iso-surfaces are observed surrounding the aluminum, calcium, magnesium, and zinc atoms, whereas yellow iso-surfaces are present on the DTM nanosheets near the metal adsorbed sites. This indicates the electronic charge transfer from the metal atom to the DTM nanosheets.
 |
| | Fig. 5 The charge density difference plots for Al, Ca, Mg and Zn adsorbed on VNbC, VTaC, and NbTaC nanosheets are shown in (a, e, i), (b, f, j), (c, g, k), and (d, h, l), respectively. The iso-surface values of all the CDD plots are set at 0.001 e Å−3 except for the Mg (0.0005 e Å−3) and Zn (0.0002 e Å−3) adsorbed VNbC monolayers as shown at the bottom of each plot. | |
We calculated the work function of the pristine and single metal-intercalated DTM MXenes. The values of the work function of pristine VNbC, VTaC and NbTaC sheets are 4.6 eV, 4.7 eV and 4.7 eV respectively (Table S4). After adsorption of Al, Ca, Mg, and Zn the work function values decrease to 4.3/4.6/4.6 eV, 3.9/4.3/4.3 eV, 4.4/4.5/4.5 eV, and 4.5/4.6/4.6 eV on the metal adsorbed VNbC/VTaC/NbTaC nanosheets. The decrease in work function confirms charge transfer from the metal atom to the nanosheets. The results obtained from the work function calculations re-affirm the results obtained from the CDD calculations. This charge transfer from the metal atom to the host nanosheets causes interaction between them resulting in easy ion-storage.
3.6. Diffusion kinematics
The mobility of metal atoms on DTM carbide (VNbC, VTaC, and NbTaC) nanosheets plays a crucial role in determining the charging/discharging rate of metal-ion batteries. To assess this, we calculated the diffusion energy barriers for the migration of Al, Ca, Mg, and Zn atoms on the DTM monolayers using the CI-NEB method.72Fig. 6 illustrates the diffusion energy profiles for Al, Mg, and Zn atoms migrating directly from one c-site to the nearest c-site (Path 1) and via the h-site (Path 2) on XYC nanosheets in (a, d, g), (b, e, h) and (c, f, i) respectively. The schematic diagrams of two migration paths are shown in Fig. S6. The diffusion energy barrier is much higher along Path 1 compared to Path 2 for the migration of all the considered metal atoms. Hence, the diffusion path from c-site to the nearest c-site and via the h-site (Path 2) turns out to be the minimum energy path (MEP). The diffusion barrier of the Ca ion is quite high (>3.0 eV) on all the three DTM MXene monolayers; hence Ca is not suitable for efficient multivalent metal-ion battery applications using these DTM nanosheets. For aluminum migration along the minimum energy pathway, following the c–h–c trajectory, the diffusion energy barriers are 0.26 eV, 0.25 eV, and 0.18 eV on VNbC, VTaC, and NbTaC nanosheets, respectively. Magnesium exhibits migration barriers of 0.13 eV, 0.10 eV, and 0.06 eV along the MEP on VNbC, VTaC, and NbTaC, respectively. Similarly, zinc shows migration barriers of 0.19 eV, 0.16 eV, and 0.16 eV along the MEP on VNbC, VTaC, and NbTaC, respectively as provided in Table S3. Such a small diffusion energy barrier on the XYC nanosheets will provide faster diffusion of Al, Mg and Zn ions. The diffusion coefficient (D) and mobility (µ) are also calculated using the following equations:| | 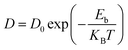 | (3) |
| | 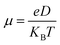 | (4) |
In this equation, D0 = l2v0, where l represents the diffusion length, and v0 is the vibration frequency, typically having a value of ∼1013 s−1 as obtained from transition state theory (TST) (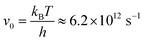 at 300 K), D is the diffusion coefficient and µ is the ionic mobility, Eb denotes the diffusion barrier, KB stands for the Boltzmann constant, and T represents the temperature.81–83 The computed diffusion coefficients (ionic mobility) along the favourable diffusion path are found to be 4.22 × 10−7, 6.00 × 10−7, and 9.59 × 10−6 cm2 s−1 (1.62 × 10−5, 2.31 × 10−5, and 3.69 × 10−4 cm2 s−1 V−1) for Al on VNbC, VTaC, and NbTaC nanosheets, respectively. Similarly, 6.27 × 10−5, 1.92 × 10−4, and 9.68 × 10−4 cm2 s−1 (2.41 × 10−3, 7.39 × 10−3, and 3.72 × 10−2 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Mg on VNbC, VTaC, and NbTaC nanosheets, respectively and 6.24 × 10−6, 1.91 × 10−5, and 2.07 × 10−5 cm2 s−1 (2.40 × 10−4, 7.36 × 10−4, 7.96 × 10−4 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Zn on VNbC, VTaC, and NbTaC nanosheets, respectively, as shown in Table 3. The diffusion coefficients follow the order VNbC < VTaC < NbTaC among the studied DTM MXenes. The Mg2+-ion diffusion on NbTaC is an order of magnitude faster than on VNbC, showing the highly favorable ionic transport characteristics in NbTaC. Li+ diffusion coefficient in graphite ranges from 1.0 × 10−11 to 4.0 × 10−10 cm2 s−1;84 the diffusion coefficients of Li+ in Si3N, SiC2 and SiC4 are ∼1.02 × 10−10, 4.26 × 10−5 and 5.10 × 10−12 cm2 s−1, respectively,85,86 while those in silicon and CNTs are ∼1.2 × 10−6 and 1.2 × 10−11 cm2 s−1.87,88 For Mg2+, AlN exhibits a diffusion coefficient of 1.51 × 10−3 cm2 s−1.89 Na+ diffusion coefficient in Si3N is ∼2.26 × 10−7 cm2 s−1
at 300 K), D is the diffusion coefficient and µ is the ionic mobility, Eb denotes the diffusion barrier, KB stands for the Boltzmann constant, and T represents the temperature.81–83 The computed diffusion coefficients (ionic mobility) along the favourable diffusion path are found to be 4.22 × 10−7, 6.00 × 10−7, and 9.59 × 10−6 cm2 s−1 (1.62 × 10−5, 2.31 × 10−5, and 3.69 × 10−4 cm2 s−1 V−1) for Al on VNbC, VTaC, and NbTaC nanosheets, respectively. Similarly, 6.27 × 10−5, 1.92 × 10−4, and 9.68 × 10−4 cm2 s−1 (2.41 × 10−3, 7.39 × 10−3, and 3.72 × 10−2 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Mg on VNbC, VTaC, and NbTaC nanosheets, respectively and 6.24 × 10−6, 1.91 × 10−5, and 2.07 × 10−5 cm2 s−1 (2.40 × 10−4, 7.36 × 10−4, 7.96 × 10−4 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Zn on VNbC, VTaC, and NbTaC nanosheets, respectively, as shown in Table 3. The diffusion coefficients follow the order VNbC < VTaC < NbTaC among the studied DTM MXenes. The Mg2+-ion diffusion on NbTaC is an order of magnitude faster than on VNbC, showing the highly favorable ionic transport characteristics in NbTaC. Li+ diffusion coefficient in graphite ranges from 1.0 × 10−11 to 4.0 × 10−10 cm2 s−1;84 the diffusion coefficients of Li+ in Si3N, SiC2 and SiC4 are ∼1.02 × 10−10, 4.26 × 10−5 and 5.10 × 10−12 cm2 s−1, respectively,85,86 while those in silicon and CNTs are ∼1.2 × 10−6 and 1.2 × 10−11 cm2 s−1.87,88 For Mg2+, AlN exhibits a diffusion coefficient of 1.51 × 10−3 cm2 s−1.89 Na+ diffusion coefficient in Si3N is ∼2.26 × 10−7 cm2 s−1 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 83 while Li+, Na+, and Mg2+ in Y2C(OH)2 exhibit diffusion coefficients of 1.52 × 10−18, 1.52 × 10−12 and 1.52 × 10−8 cm2 s−1,90 In comparison, Nb2Sc2C demonstrates significantly higher diffusion coefficients of ∼9.5 × 10−5 cm2 s−1 for Na and ∼2.89 × 10−4 cm2 s−1 for K.91 The diffusion coefficient of Na+ in hard carbon has been experimentally reported to be ∼1.0 × 10−9 cm2 s−1 during charge–discharge processes.92 Similarly, MoSe2 shows a Li diffusion coefficient of ∼1.31 × 10−13 cm2 s−1,93 while Ti2C0.5N0.5Tx exhibits ∼2.3 × 10−13 cm2 s−1 for Na-ions.94 Commercially used hard carbon shows a K-ion diffusion coefficient of ∼1.73 × 10−9 cm2 s−1,95 whereas graphite demonstrates ∼1.0 × 10−8 cm2 s−1 for K-ions.96 The values obtained for Al3+, Mg2+, and Zn2+ on the studied MXenes are comparable to or higher than those reported theoretically and/or experimentally for other electrode materials, indicating favorable ion transport and high ionic mobility.
83 while Li+, Na+, and Mg2+ in Y2C(OH)2 exhibit diffusion coefficients of 1.52 × 10−18, 1.52 × 10−12 and 1.52 × 10−8 cm2 s−1,90 In comparison, Nb2Sc2C demonstrates significantly higher diffusion coefficients of ∼9.5 × 10−5 cm2 s−1 for Na and ∼2.89 × 10−4 cm2 s−1 for K.91 The diffusion coefficient of Na+ in hard carbon has been experimentally reported to be ∼1.0 × 10−9 cm2 s−1 during charge–discharge processes.92 Similarly, MoSe2 shows a Li diffusion coefficient of ∼1.31 × 10−13 cm2 s−1,93 while Ti2C0.5N0.5Tx exhibits ∼2.3 × 10−13 cm2 s−1 for Na-ions.94 Commercially used hard carbon shows a K-ion diffusion coefficient of ∼1.73 × 10−9 cm2 s−1,95 whereas graphite demonstrates ∼1.0 × 10−8 cm2 s−1 for K-ions.96 The values obtained for Al3+, Mg2+, and Zn2+ on the studied MXenes are comparable to or higher than those reported theoretically and/or experimentally for other electrode materials, indicating favorable ion transport and high ionic mobility.
 |
| | Fig. 6 Energy profiles of Al (a, d, g), Mg (b, e, h), and Zn (c, f, i) along the two possible diffusion pathways, c–c (Path 1: red line) and c–h–c (Path 2: green line), on the DTM nanosheets. The diffusion path via the hollow site i.e., Path 2 is the minimum energy path (MEP) for all the multivalent metal ions. | |
Table 3 The diffusion co-efficient and ionic mobility values of Al, Mg, and Zn on XYC MXene monolayers determined using the barrier height obtained from the CI-NEB method
| System |
Metal-ion |
Diffusion coefficient (cm2 s−1) |
Ion mobility (cm2 s−1 V−1) |
| VNbC |
Al |
4.22 × 10−7 |
1.62 × 10−5 |
| Mg |
6.27 × 10−5 |
2.41 × 10−3 |
| Zn |
6.24 × 10−6 |
2.40 × 10−4 |
| VTaC |
Al |
6.00 × 10−7 |
2.31 × 10−5 |
| Mg |
1.92 × 10−4 |
7.39 × 10−3 |
| Zn |
1.91 × 10−5 |
7.36 × 10−4 |
| NbTaC |
Al |
9.59 × 10−6 |
3.69 × 10−4 |
| Mg |
9.68 × 10−4 |
3.72 × 10−2 |
| Zn |
2.07 × 10−5 |
7.96 × 10−4 |
3.7. Gravimetric storage capacity and open-circuit-voltage
To assess the maximum storage capacity of XYC (SCXYC) monolayers, we carried out the layer-by-layer adsorption of metal atoms on both surfaces of the XYC nanosheets until one of the following conditions was encountered: (a) Al, Ca, Mg, and Zn atoms started to migrate from the cell boundary leading to structural deformation, (b) the adsorption energy became positive, and (c) the structural stability of the monolayer began to degrade. To obtain the maximum storage capacity, first we adsorbed 9 metal atoms (Al, Mg, and Zn) and 3 Ca atoms in a layer on one surface of the XYC monolayer; then we proceeded to bilayer, tetralayer and up to hexalayer adsorption. The layer-by-layer adsorption energies were calculated and are given in Table S5 and the fully intercalated XYC DTM MXenes are shown in Fig. S7. A maximum of 54, 54, and 36 aluminum (Al); 54, 54, and 54 magnesium (Mg); 18, 18, and 18 zinc (Zn) and 12, 12, and 6 calcium (Ca) atoms are adsorbed on VNbC, VTaC, and NbTaC nanosheets respectively. Beyond these metal coverages, agglomeration of the metal atoms is observed. However, the calcium (Ca) populated system exhibits thermal instability due to dimer formation as shown in Fig. S8. For Al adsorption the storage capacities of VNbC, VTaC, and NbTaC nanosheets are 3096.64, 1978.76, and 844.14 mAh g−1, respectively; for Mg adsorption the storage capacities of VNbC, VTaC, and NbTaC nanosheets are 2064.43, 1319.17, and 1125.52 mAh g−1 respectively and the storage capacities upon Zn adsorption on VNbC, VTaC, and NbTaC nanosheets are 688.14, 439.72, and 375.17 mAh g−1 respectively, as determined using the following equation:| | 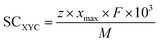 | (5) |
where, z is the valency of the decorated metal atoms (3 for Al and 2 for Ca, Mg and Zn ions), xmax is the number of alkali metal atoms in the highest populated geometries, and F represents the Faraday constant (=26.810 Ah mol−1), M is the molar mass of the XYC nanosheets. The calculated specific storage capacities are represented in the scatter plot as shown in Fig. 7(a–c) for the Al-, Mg-, and Zn-intercalated VNbC, VTaC and NbTaC DTM nanosheets at each intermediate state.
 |
| | Fig. 7 (a–c) The specific storage capacity of Al-, Mg-, and Zn-intercalated VNbC, VTaC and NbTaC nanosheets at each intermediate state. (d–f) The scatter plot of the open circuit voltages of these metal-decorated DTM nanosheets at each intermediate state. | |
The open-circuit voltage (OCV) plays a critical role in determining whether a system can serve as an anode or cathode. The average OCV for the multivalent metal-adsorbed DTM MXene nanosheets are estimated by considering the following charging/discharging half-reaction.30,97
| | | XYC + xMz+ + zxe ↔ XYCMx (X = Y = V, Nb and Ta; M = Al, Mg and Zn) | (6) |
Here,
z is the valency of the metal ions. The chemical reactions during the charge–discharge process of the fully discharged electrode configurations at their maximum theoretical capacities for these DTM nanosheets are provided in the SI. We calculated OCV values using the following equations for the alkali metal adsorbed systems:
| | 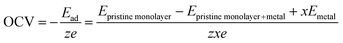 | (7) |
The estimated average open circuit voltage (OCV) values are 0.39, 0.37, and 0.44 V for Al; 0.48, 0.47, and 0.53 V for Mg; and 0.54, 0.54, and 0.58 V for Zn on VNbC, VTaC, and NbTaC nanosheets, respectively. The open circuit voltages of these metal-decorated DTM nanosheets at each intermediate state are shown in Fig. 7(d–f). Since the OCV values for Al, Mg and Zn are less than 1.5 V in all three MXenes, these materials are suitable for use as anode materials in rechargeable metal-ion batteries according to the standard convention for selecting anode materials based on OCV values.48,98 We also examined the volume change at each intermediate state during the loading of Al, Mg, and Zn on DTM MXenes as shown in Table S6. The volume changes for the intercalation of Al (3.52–9.39%), Mg (0.07–1.79%), and Zn (1.89–3.10%) are well within the acceptable limit when compared with that of commercial anode material graphite (13.2%).99,100
3.8. Molecular dynamics and convex hull curves of metal loaded nanosheets
We performed ab initio molecular dynamics (AIMD) simulations at 300 K on fully loaded nanosheets containing aluminum, calcium, magnesium, and zinc to evaluate their thermal stability after metal loading. As shown in Fig. 8, the structures remain stable with only small energy fluctuation, confirming their robustness and stability at this temperature.
 |
| | Fig. 8 (a–i) Variations in energy during AIMD simulations at 300 K for fully loaded Al (a, d, g), Mg (b, e, h), and Zn (c, f, i) on VNbC, VTaC, and NbTaC nanosheets, respectively. The final atomic structures at the end of the simulation of fully loaded systems are shown in the inset. | |
To further understand the interactions between the intercalated metal atoms and transition metals (X and Y) in XYC nanosheets, we calculated the radial distribution function (RDF) of Ca, Al, Mg, and Zn atoms relative to X, Y, and other metal atoms. The results, illustrated in Fig. S8(a–c) and S9(a–i), reveal how the density of metal atoms varies with distance from X, Y, and other metals. In RDF plots, the vertical axis represents the RDF value, which indicates the probability of finding an atom at a given distance (horizontal axis) from a reference atom. In Fig. S8(a–c), the red lines show the density variation of Ca with respect to other Ca atoms in XYC nanosheets. In Fig. S9(a–i), the green lines (Al–Al, Mg–Mg, and Zn–Zn) represent the density variation of Al, Mg, and Zn relative to other Al, Mg, and Zn atoms in VNbC, VTaC, and NbTaC nanosheets. The blue (X–Al, X–Mg, and X–Zn) and red (Y–Al, Y–Mg, and Y–Zn) lines depict the RDF values of metal atoms relative to X and Y in XYC MXene nanosheets, respectively. The first RDF peaks for Al–Al occur at 2.90 Å (VNbC), 2.87 Å (VTaC), and 2.95 Å (NbTaC). For Ca–Ca, the peaks appear at 3.74 Å (VNbC), 3.70 Å (VTaC), and 3.66 Å (NbTaC), while for Mg–Mg, they appear at 3.04 Å (VNbC and VTaC) and 3.12 Å (NbTaC). Similarly, the Zn–Zn peaks appear at 3.05 Å (VNbC), 3.00 Å (VTaC), and 3.11 Å (NbTaC). The peak positions for Al, Mg, and Zn are higher than their respective dimer bond lengths (Al–Al: 2.67 Å, Mg–Mg: 2.85 Å, and Zn–Zn: 2.63 Å), indicating no cluster formation and confirming a well-dispersed metal distribution. However, the Ca–Ca peaks appear at shorter distances than the Ca–Ca dimer distance (3.90 Å), suggesting that calcium tends to aggregate, which makes it unsuitable for electrode materials in metal-ion battery applications. Furthermore, the RDF curves for X–Al, X–Mg, and X–Zn and Y–Al, Y–Mg, and Y–Zn exhibit significant peaks at shorter distances compared to Al–Al, Mg–Mg, and Zn–Zn, implying a higher probability of metal atoms being located near X and Y rather than other metal atoms. This strong interaction prevents the aggregation of Al, Mg, and Zn, ensuring their stability and uniform dispersion within the monolayer. At greater distances, the RDF values approach 1, which suggests that the influence of X and Y weakens, allowing the metal atoms to diffuse freely throughout the nanosheets. This behavior is beneficial for multivalent metal-ion battery applications, as it facilitates efficient ion transport without strong binding to specific transition metal sites.
We then calculated the velocity autocorrelation function for Al, Mg, and Zn atoms, followed by a Fourier transform to obtain the vibrational density of states (VDOS), as shown in Fig. S10(a–i). The peak vibrational frequencies for Al are 3.38, 4.42, and 3.86 THz; those for Mg are 7.69, 7.55, and 6.32 THz; and those for Zn are 3.90, 1.63, and 3.86 THz. The VDOS analysis provides valuable insights into the structural stability of the system. The absence of imaginary frequencies confirms that the system remains stable at 300 K. Collectively, these findings suggest that these DTM MXenes are promising material for Al-, Mg-, and Zn-ion battery anodes, offering strong structural integrity and stability under operating conditions.
We also evaluated the thermodynamic stability of intermediate configurations formed during the intercalation of Al and Mg ions by calculating their formation energies (Ef). The formation energy of a partially intercalated structure was determined using the following expression:71,101
| | 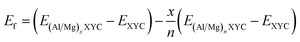 | (8) |
Here,
E's represent the total energies of the corresponding configurations indicated by the subscripts,
n denotes the maximum possible concentration of Al/Mg (which is 6/6 for VNbC and VTaC, and 3/6 for NbTaC), and
x is a particular ion concentration in the partially loaded structure. The computed convex hull diagrams in
Fig. 9(a–f) for both Al and Mg intercalation pathways show that most of the intermediate phases lie on the convex hull curve but a few intermediate phases lie very close to the convex hull curve. The proximity of these phases to the hull curve suggests that the ion intercalation process proceeds
via a series of stable or nearly stable intermediate configurations, allowing smooth ion insertion and extraction without significant energy barrier. This implies that these phases are energetically favorable and can feasibly form during electrochemical cycling.
 |
| | Fig. 9 Convex hull diagram plots in (a), (c), and (e) for Al and plots in (b), (d), and (f) for Mg intercalation on VNbC, VTaC, and NbTaC, respectively. Relaxed structures corresponding to few points lying on the tie-line have also been displayed with the ball-stick atomic model (color code: Al (blue), Mg (orange), and remaining color codes are the same as in Fig. 1). | |
3.9. Charge distribution and interactions in metal-intercalated DTM-MXenes
The electron distribution between the intercalated metal atoms and the MXene nanosheets has been investigated qualitatively using the ELF plots as shown in Fig. 10 with the color scale bar at the bottom. It is clear from the cross-sectional view that the charge is more localized around the carbon atoms than around the transition metals of the DTM nanosheets. The lower electron localization near the transition metal is attributed to their lower electronegativity that rises from the partially filled d-orbitals.102Fig. 10 indicates that a significant number of electrons move from the inner layer to the outer layer, forming a uniformly distributed negatively charged electron cloud that surrounds the metal atom layer. When compared with the charge localization around the metal atoms, after maximum intercalation, the charge becomes more localized near the metal (Al, Ca, Mg, and Zn) atoms that are far away from the nanosheet. This is unique to double transition metal (DTM) MXenes and is responsible for enhancing the theoretical capacity of multivalent metal-ion batteries. This type of electron redistribution behavior in metal adsorbed nanosheets known as the “Collaborative Activation” mechanism has also been observed by Zhou et al. for monovalent-ion intercalation.62
 |
| | Fig. 10 The ELF plot of the fully Al-, Ca-, Mg-, and Zn-intercalated VNbC, VTaC, and NbTaC MXenes are shown in (a–d), (e–h), and (i–l) respectively. | |
The non-covalent interactions between the metal atoms and the DTM nanosheets are analyzed using the promolecular density approach.103 Fig. S11(a–i) present the two-dimensional (2D) reduced density gradient (RDG) scatter plots, which map the RDG function against ρ(r)sign(λ2). Here, ρ(r) is the electron density and λ2 is the second eigenvalue of the Hessian matrix of the electron density.103 The position of peaks along the x-axis provides insight into the nature of the interactions: peaks toward the negative side (with high electron density) suggest covalent bonding, while peaks near zero indicate non-covalent interactions such as van der Waals forces. To further visualize these interactions in real space, three-dimensional (3D) RDG iso-surfaces are shown in Fig. S12(a–i). A color scheme is employed where red denotes strong steric repulsion, green and yellow-green highlight weak van der Waals or dispersive interactions, and blue indicates strong attractive interactions, typically associated with hydrogen bonding or electrostatic attraction. From the 3D RDG iso-surfaces, bluish-green lobes are observed at the metal layer near the DTM surface, suggesting significant attractive interactions between the metal atoms and the DTM nanosheets. However, as the distance from the DTM nanosheet increases (i.e., in the upper metal layers), these regions gradually shift to pure green, indicating a transition to weaker, more dispersive interactions with the DTM nanosheets. This implies that the metal atom layer near the DTM nanosheets experiences stronger interactions compared to the metal layers away from the nanosheets.
3.10. Interaction of electrolytes with the DTM-MXenes
To gain deeper insight into the electrode–electrolyte interface and assess the compatibility of common battery electrolytes, we extended our study by explicitly modeling the adsorption of typical solvent molecules used in multivalent metal-ion batteries, including ethylene carbonate (EC, C3H4O3), dimethyl carbonate (DMC, C3H6O3), diethyl carbonate (DEC, C5H10O3), and propylene carbonate (PC, C4H6O3), on the DTM MXene nanosheets.104–106 To understand the effect of the electrolyte on the anode material, one may investigate the interaction of the electrolyte molecule with the anode material (here, DTM MXenes) by calculating their adsorption energy. The adsorption energies (Ead) were calculated using the following equation:| | | Ead = EDTM MXenes+elctrolyte − (EDTM MXenes + Eelectrolyte) | (9) |
where EDTM MXenes+electrolyte is the total energy of the MXene–electrolyte system, EDTM MXenes is the energy of pristine MXene, and Eelectrolyte is the energy of the isolated electrolyte molecule. The obtained adsorption energies (Table 4) are all negative, indicating thermodynamically favorable interactions between the solvent molecules and the MXene nanosheets. The relaxed adsorption configurations are shown in Fig. S13 (SI). The adsorption energies of DEC, DMC, EC, and PC electrolyte molecules on these three DTM nanosheet range from −4.03 to −4.17 eV; from −1.04 to −1.16 eV; from −1.27 to −3.87 eV and from −6.25 to −8.07 eV respectively. Among these studied electrolyte molecules, DEC and PC interact strongly with these MXene nanosheets. However, DMC and EC interact moderately with the nanosheets. Recently Verma et al. reported that the adsorption energy of the EC, DMC, DEC, and PC molecules on pristine and Al and Si-doped BeSeCl monolayer ranges from −2.90 to −1.99 eV; from −2.68 to −0.74 eV; from −3.19 to −0.72 eV; and from −2.75 to −1.90 eV respectively. This study reveals that the DMC and EC electrolytes exhibit better compatibility with these DTM MXene nanosheets than DEC and PC electrolytes.
Table 4 The adsorption energy of the DEC, DMC, EC and PC electrolyte molecules on the VNbC, VTaC and NbTaC MXenes
| Electrolytes (solvent molecules) |
Adsorption energy (eV) |
| VNbC |
VTaC |
NbTaC |
| DEC |
−4.03 |
−4.17 |
−4.07 |
| DMC |
−1.04 |
−1.16 |
−1.15 |
| EC |
−3.87 |
−1.27 |
−2.25 |
| PC |
−6.25 |
−7.26 |
−8.05 |
The choice of double transition metal (DTM) carbides over traditional single transition metal (STM) carbides stems from their enhanced electrochemical properties, including excellent capacity, improved structural stability, and lower diffusion barriers. DTM carbides exhibit a synergistic effect due to the presence of two different transition metal atoms, leading to an optimized electronic structure and stronger metal–carbon bonding, which enhances ion storage and diffusion kinetics. Additionally, their ability to accommodate multivalent ions (Al3+, Mg2+, and Zn2+) with low diffusion barriers makes them superior for next-generation metal-ion batteries. To support this, we provide a comparison in Table 5 and Fig. S14, showcasing the capacity and diffusion barriers of graphite, single transition metal carbides, and the studied DTM carbides, demonstrating that our results surpass those reported in previous studies in both energy storage potential and ion transport efficiency. These advantages position DTM carbides as more efficient and versatile electrode materials compared to their single transition metal counterparts.
Table 5 A comparison of the capacity and diffusion barrier with previously reported results
| System |
Metal atom |
Capacity (mAh g−1) |
Diffusion barrier (eV) |
Reference |
| Graphite |
Li |
372 |
0.30 |
107
|
| Mo2C |
Na |
438 |
0.017 |
108
|
| Mo2C |
Mg |
526 |
0.070 |
108
|
| Mn2C |
Li |
887.6 |
0.024 |
109
|
| Mn2C |
Na |
443.6 |
0.022 |
109
|
| Ca2C |
Li |
582 |
0.027 |
110
|
| Ca2C |
Na |
582 |
0.059 |
110
|
| Ca2C |
K |
582 |
0.028 |
110
|
| W2C |
Li |
259 |
0.045 |
111
|
| Nb2C |
Li |
542 |
0.032 |
112
|
| Nb2C |
Na |
271 |
0.015 |
112
|
| Nb2C |
K |
136 |
0.004 |
112
|
| Nb2C |
Mg |
1084 |
0.096 |
112
|
| Nb2C |
Ca |
271 |
0.039 |
112
|
| Zr2C |
Li |
826.95 |
0.067 |
113
|
| Zr2C |
Na |
551.27 |
0.014 |
113
|
| Zr2C |
K |
68.90 |
0.033 |
113
|
| V2C |
Li |
940 |
0.045 |
44
|
| Sc2C |
Li |
462 |
0.018 |
41
|
| Sc2C |
Na |
362 |
0.012 |
41
|
| VNbC |
Al/Mg/Zn |
3096.64/2064.43/688.14 |
0.26/0.13/0.19 |
Our result |
| VTaC |
Al/Mg/Zn |
1978.76/1319.17/439.72 |
0.25/0.10/0.16 |
Our result |
| NbTaC |
Al/Mg/Zn |
844.14/1125.52/375.17 |
0.18/0.06/0.16 |
Our result |
4. Conclusions
This study presents an in-depth theoretical investigation of VNbC, VTaC, and NbTaC MXene nanosheets as potential electrode materials for post-lithium-ion batteries. Using first-principles DFT calculations, we examined their structural, electronic, and electrochemical properties. All three nanosheets exhibit thermal and mechanical stability, along with metallic conductivity—characteristics that are highly desirable for battery applications. The adsorption of Al, Mg, and Zn ions was found to be energetically favorable, with adsorption energies significantly lower than the cohesive energy of their bulk state. The theoretical storage capacities of VNbC, VTaC, and NbTaC are 3096.64, 1978.76, and 844.14 mAh g−1 for Al; 2064.43, 1319.17, and 1125.52 mAh g−1 for Mg; 688.14, 439.72, and 375.17 mAh g−1 for Zn, surpassing those of many existing two-dimensional carbon-based materials, including commercial graphite. The calculated diffusion barriers using the Cl-NEB method also confirm that these ions can migrate smoothly across the DTM surfaces, suggesting that the materials could support fast charge/discharge cycles. There is no formation of metal clusters even in the highest adsorption configuration. The open circuit voltage (OCV) values suggest that VNbC, VTaC, and NbTaC can act as efficient anode materials for Al, Mg, and Zn-ion batteries. These interactions also maintain the structural integrity of the nanosheets under ion loading, with minimal volume change. These nanosheets also exhibit compatibility with conventional electrolytes, dimethyl carbonate and ethylene carbonate. The unique combination of structural robustness, high electrical conductivity, favorable ion diffusion characteristics, compatibility with conventional electrolytes highlights the potential of these MXenes for next-generation rechargeable battery technologies. Further experimental validation and material optimization could pave the way for their practical application in high-performance energy storage systems.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data supporting the findings in the article have been included in the article itself and as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ta06475a.
Acknowledgements
T. Dey thanks the University Grant Commission for the UGC-JRF fellowship (Fellowship Ref. No. 231610133807). J. C. Mahato is thankful to UGC-DAE CSR Indore for partial support under the CRS project (Ref: CRS/2022-23/01/705) dated 15.05.2023. We are thankful to Dr Ashok Kumar, Central University of Punjab, Bathinda for providing structural information.
References
- B. Chen, D. Chao, E. Liu, M. Jaroniec, N. Zhao and S.-Z. Qiao, Energy Environ. Sci., 2020, 13, 1096–1131 RSC.
- A. Taranova, E. Moretti, K. Akbar, G. Dastgeer and A. Vomiero, Nano Energy, 2024, 128, 109872 CrossRef.
- B. Liang, X. Chen, X. Wang, H. Yuan, A. Sun, Z. Wang, L. Hu, G. Hou, Y. Zhao and X. Zhang, J. Mater. Chem. A, 2025, 13, 2441–2477 RSC.
- G. H. Gu, J. Noh, I. Kim and Y. Jung, J. Mater. Chem. A, 2019, 7, 17096–17117 RSC.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 RSC.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- X. Meng, J. Mater. Chem. A, 2017, 5, 10127–10149 RSC.
- M. L. Chaudhary, R. Patel, S. Princy, A. Bhatnagar and R. K. Gupta, J. Mater. Chem. A, 2025, 13, 28718–28748 RSC.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gómez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- W. Liu, M. Song, B. Kong and Y. Cui, Adv. Mater., 2017, 29, 1603436 CrossRef PubMed.
- G. Liang, V. K. Peterson, K. W. See, Z. Guo and W. K. Pang, J. Mater. Chem. A, 2020, 8, 15373–15398 RSC.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- L. Wang, Z. Su, R. Wang, H. Liang, B. Fang and R. Mo, J. Mater. Chem. A, 2025, 13, 21116–21171 RSC.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. T. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442–8447 CrossRef CAS.
- M. Du, Z. Miao, H. Li, Y. Sang, H. Liu and S. Wang, J. Mater. Chem. A, 2021, 9, 19245–19281 RSC.
- D. D. Vargas, G. L. Cardoso, P. C. Piquini, R. Ahuja and R. J. Baierle, ACS Appl. Mater. Interfaces, 2022, 14, 47262–47271 CrossRef CAS PubMed.
- E. Kohan, R. Khoshnavazi, M. G. Hosseini, A. Salimi and M. Salami-Kalajahi, J. Mater. Chem. A, 2024, 12, 30190–30248 RSC.
- X. Ye, X. Xiao, Z. Wu, Y. Zhan, X. Wu and S. Liu, J. Mater. Chem. A, 2024, 12, 23337–23363 RSC.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 RSC.
- Y. Liu, B. V. Merinov and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3735–3739 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS.
- K. S. Novoselov, V. I. Fal’ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- T. Dey, S. Chowdhury, S. G. Kang, P. Sen, B. C. Gupta and J. C. Mahato, Comput. Mater. Sci., 2025, 253, 113824 CrossRef CAS.
- S. Chowdhury, J. C. Mahato, J. S. Chung, S. G. Kang and B. C. Gupta, Surf. Interfaces, 2024, 51, 104750 CrossRef CAS.
- K. Chaoui, H. Zaari, Z. Mansouri, F. Caballero-Briones, A. Benyoussef, A. El Kenz and A. Sibari, J. Energy Storage, 2025, 109, 115110 CrossRef.
- L. Zhao, Y. Wang, C. Wei, X. Huang, X. Zhang and G. Wen, Particuology, 2024, 87, 240–270 CrossRef CAS.
- J. Hao, J. Zheng, F. Ling, Y. Chen, H. Jing, T. Zhou, L. Fang and M. Zhou, Sci. Rep., 2018, 8, 2079 CrossRef.
- S. Chowdhury, P. Sen and B. C. Gupta, Comput. Mater. Sci., 2023, 230, 112539 CrossRef CAS.
- A. Samad, A. Shafique and Y.-H. Shin, Nanotechnology, 2017, 28, 175401 CrossRef.
- T. S. Mathis, K. Maleski, A. Goad, A. Sarycheva, M. Anayee, A. C. Foucher, K. Hantanasirisakul, C. E. Shuck, E. A. Stach and Y. Gogotsi, ACS Nano, 2021, 15, 6420–6429 CrossRef CAS.
- J.-C. Lei, X. Zhang and Z. Zhou, Front. Phys., 2015, 10, 276–286 CrossRef.
- A. N. Enyashin and A. L. Ivanovskii, J. Solid State Chem., 2013, 207, 42–48 CrossRef CAS.
- M. Khazaei, M. Arai, T. Sasaki, M. Estili and Y. Sakka, Phys. Chem. Chem. Phys., 2014, 16, 7841–7849 RSC.
- Q. Hu, H. Wang, Q. Wu, X. Ye, A. Zhou, D. Sun, L. Wang, B. Liu and J. He, Int. J. Hydrogen Energy, 2014, 39, 10606–10612 CrossRef CAS.
- Z. Li, L. Wang, D. Sun, Y. Zhang, B. Liu, Q. Hu and A. Zhou, Mater. Sci. Eng., B, 2015, 191, 33–40 CrossRef CAS.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS.
- J. Hu, B. Xu, C. Ouyang, S. A. Yang and Y. Yao, J. Phys. Chem. C, 2014, 118, 24274–24281 CrossRef CAS.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- M.-Q. Zhao, M. Torelli, C. E. Ren, M. Ghidiu, Z. Ling, B. Anasori, M. W. Barsoum and Y. Gogotsi, Nano Energy, 2016, 30, 603–613 CrossRef CAS.
- D. Çakır, C. Sevik, O. Gülseren and F. M. Peeters, J. Mater. Chem. A, 2016, 4, 6029–6035 RSC.
- T. Dey, B. C. Gupta and J. C. Mahato, Int. J. Hydrogen Energy, 2025, 154, 150274 CrossRef CAS.
- S. K. Das, S. Mahapatra and H. Lahan, J. Mater. Chem. A, 2017, 5, 6347–6367 RSC.
- D. Pal, S. Chakraborty and S. Chattopadhyay, Energ. Tech., 2021, 9, 2100382 CrossRef CAS.
- A. Kumar, L. Sharma and A. Verma, J. Energy Storage, 2024, 98, 113039 CrossRef.
- K. L. Ng, Z. Lu, Y. Wang, C. V. Singh and G. Azimi, J. Phys. Chem. C, 2021, 125, 15145–15154 CrossRef CAS.
- V. Le, M. Wang and N. V. Medhekar, J. Power Sources, 2025, 630, 236043 CrossRef CAS.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- W. Hong, B. C. Wyatt, S. K. Nemani and B. Anasori, MRS Bull., 2020, 45, 850–861 CrossRef.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef.
- H. Liu, H. Wang, Z. Jing, K. Wu, Y. Cheng and B. Xiao, J. Phys. Chem. C, 2020, 124, 25769–25774 CrossRef.
- M. Zhou, Y. Shen, J. Liu, L. Lv, X. Gao, Y. Zhang, X. Meng, X. Yang, Y. Zheng and Z. Zhou, Vacuum, 2022, 200, 111054 CrossRef.
- V. Mehta, H. S. Saini, S. Srivastava, M. K. Kashyap and K. Tankeshwar, J. Mater. Sci., 2022, 57, 10702–10713 CrossRef.
- Y. Cheng, L. Yang and S. Yin, Compos. Commun., 2023, 40, 101588 CrossRef.
- M. Zhou, Y. Shen, J. Liu, L. Lv, Y. Zhang, X. Meng, X. Yang, B. Zhang and Z. Zhou, Vacuum, 2023, 213, 112150 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- J. Moellmann and S. Grimme, J. Phys. Chem. C, 2014, 118, 7615–7621 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- V. Shukla, N. K. Jena, S. R. Naqvi, W. Luo and R. Ahuja, Nano Energy, 2019, 58, 877–885 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Zhang, S.-X. Cui, W. Feng and J. Li, Phys. B, 2025, 715, 417558 CrossRef CAS.
- B. Roondhe, R. Ahuja and W. Luo, Appl. Surf. Sci., 2024, 667, 160404 CrossRef CAS.
- G. Cao, Polymers, 2014, 6, 2404–2432 CrossRef.
- R. C. Cooper, C. Lee, C. A. Marianetti, X. Wei, J. Hone and J. W. Kysar, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 035423 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- S. Chowdhury, P. Sarkar and B. C. Gupta, Phys. Chem. Chem. Phys., 2024, 26, 16240–16252 RSC.
- S. Chowdhury, M. Parvin, J. S. Chung, S. G. Kang and B. C. Gupta, J. Power Sources, 2025, 653, 237725 CrossRef CAS.
- O. R. Inderwildi and M. Kraft, ChemPhysChem, 2007, 8, 444–451 CrossRef CAS.
- M. Miletic, K. Palczynski and J. Dzubiella, J. Chem. Phys., 2020, 153, 164713 CrossRef CAS PubMed.
- H. Li, J. Hou and D. Jiang, J. Electron. Mater., 2020, 49, 4180–4185 CrossRef CAS.
- J. H. Park, H. Yoon, Y. Cho and C.-Y. Yoo, Materials, 2021, 14, 4683 CrossRef CAS PubMed.
- J. Liu, S. Wang, Y. Qie, J. Yu and Q. Sun, Carbon, 2018, 140, 680–687 CrossRef CAS.
- Y. Qie, S. Wang and Q. Sun, Nano Energy, 2019, 63, 103862 CrossRef.
- G. F. I. Toki, M. K. Hossain, W. U. Rehman, R. Z. A. Manj, L. Wang and J. Yang, Ind. Chem. Mater., 2024, 2, 226–269 RSC.
- G. Qi and T. Rabczuk, Carbon, 2019, 155, 727–733 CrossRef CAS.
- T. Mei, J. Wu, S. Lu, B. Wang, X. Zhao, L. Wang and Z. Yin, Comput. Theor. Chem., 2021, 1203, 113352 CrossRef CAS.
- D. Wang, H. Li, L. Zhang, Z. Sun, D. Han, L. Niu, X. Zhong, X. Qu and L. Yang, Appl. Surf. Sci., 2019, 478, 459–464 CrossRef CAS.
- Y. Jing, J. Liu, Z. Zhou, J. Zhang and Y. Li, J. Phys. Chem. C, 2019, 123, 26803–26811 CrossRef CAS.
- K. Wang, Y. Jin, S. Sun, Y. Huang, J. Peng, J. Luo, Q. Zhang, Y. Qiu, C. Fang and J. Han, ACS Omega, 2017, 2, 1687–1695 CrossRef CAS PubMed.
- H. Wang, X. Wang, L. Wang, J. Wang, D. Jiang, G. Li, Y. Zhang, H. Zhong and Y. Jiang, J. Phys. Chem. C, 2015, 119, 10197–10205 CrossRef CAS.
- K. Liang, A. Tabassum, A. Majed, C. Dun, F. Yang, J. Guo, K. Prenger, J. J. Urban and M. Naguib, InfoMat, 2021, 3, 1422–1430 CrossRef CAS.
- X. Lu, H. Peng, G. Liu, F. Qi, C. Shi, S. Wu, Y. Wu, H. Yang, J. Shan and Z. Sun, Energy Adv., 2023, 2, 1294–1308 RSC.
- L. Wang, J. Yang, J. Li, T. Chen, S. Chen, Z. Wu, J. Qiu, B. Wang, P. Gao, X. Niu and H. Li, J. Power Sources, 2019, 409, 24–30 CrossRef CAS.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- A. Eftekhari, Energy Storage Mater., 2017, 7, 157–180 CrossRef.
- S. Schweidler, L. de Biasi, A. Schiele, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2018, 122, 8829–8835 CrossRef CAS.
- Y.-X. Yu, J. Phys. Chem. C, 2016, 120, 5288–5296 CrossRef CAS.
- V. Shukla, R. B. Araujo, N. K. Jena and R. Ahuja, Nano Energy, 2017, 41, 251–260 CrossRef CAS.
- R. P. Jadav, D. Singh, R. Ahuja and Y. Sonvane, J. Energy Storage, 2024, 101, 113995 CrossRef.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
- N. Verma and A. Kumar, J. Mater. Chem. A, 2025, 13, 28559–28573 RSC.
- S. Lei, X. Chen, B. Xiao, W. Zhang and J. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 28830–28840 CrossRef CAS.
- S. Lin, M. Xu, F. Wang, J. Hao and Y. Li, Phys. Rev. Res., 2024, 6, 013028 CrossRef CAS.
- K. Toyoura, Y. Koyama, A. Kuwabara and I. Tanaka, J. Phys. Chem. C, 2010, 114, 2375–2379 CrossRef CAS.
- X. Li, Y. Fu, H. Wang, W. Yu, D. Li, Y. Liu, M. Wei, Q. Hu and A. Zhou, J. Energy Storage, 2024, 87, 111500 CrossRef.
- X. Zhang, W. Meng, T. He, L. Jin, X. Dai and G. Liu, Appl. Surf. Sci., 2020, 503, 144091 CrossRef CAS.
- K. Rajput, V. Kumar, S. Thomas, M. A. Zaeem and D. R. Roy, 2D Mater., 2021, 8, 035015 CrossRef CAS.
- Y. Zhang, Comp. Cond. Mat., 2017, 10, 35–38 Search PubMed.
- J. Hu, B. Xu, C. Ouyang, Y. Zhang and S. A. Yang, RSC Adv., 2016, 6, 27467–27474 RSC.
- Y. Wang, S. Wang, Y. Zhang, N. Song, S. Luo, B. Xu and F. Wang, Chem. Phys., 2025, 589, 112521 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Bikash
Chandra Gupta
*,
Bikash
Chandra Gupta
 ,
,  , and
, and  , which are close to the previously reported values
, which are close to the previously reported values  ,
,  , and
, and  by Zhou et al. which benchmark our calculations.62 The thicknesses of the three sheets are different due to the different atomic radii of the metal atoms. The formation energy (Ef) is determined to assess the stability of the XYC two-dimensional sheets by using the following expression,
by Zhou et al. which benchmark our calculations.62 The thicknesses of the three sheets are different due to the different atomic radii of the metal atoms. The formation energy (Ef) is determined to assess the stability of the XYC two-dimensional sheets by using the following expression,








 at 300 K), D is the diffusion coefficient and µ is the ionic mobility, Eb denotes the diffusion barrier, KB stands for the Boltzmann constant, and T represents the temperature.81–83 The computed diffusion coefficients (ionic mobility) along the favourable diffusion path are found to be 4.22 × 10−7, 6.00 × 10−7, and 9.59 × 10−6 cm2 s−1 (1.62 × 10−5, 2.31 × 10−5, and 3.69 × 10−4 cm2 s−1 V−1) for Al on VNbC, VTaC, and NbTaC nanosheets, respectively. Similarly, 6.27 × 10−5, 1.92 × 10−4, and 9.68 × 10−4 cm2 s−1 (2.41 × 10−3, 7.39 × 10−3, and 3.72 × 10−2 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Mg on VNbC, VTaC, and NbTaC nanosheets, respectively and 6.24 × 10−6, 1.91 × 10−5, and 2.07 × 10−5 cm2 s−1 (2.40 × 10−4, 7.36 × 10−4, 7.96 × 10−4 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Zn on VNbC, VTaC, and NbTaC nanosheets, respectively, as shown in Table 3. The diffusion coefficients follow the order VNbC < VTaC < NbTaC among the studied DTM MXenes. The Mg2+-ion diffusion on NbTaC is an order of magnitude faster than on VNbC, showing the highly favorable ionic transport characteristics in NbTaC. Li+ diffusion coefficient in graphite ranges from 1.0 × 10−11 to 4.0 × 10−10 cm2 s−1;84 the diffusion coefficients of Li+ in Si3N, SiC2 and SiC4 are ∼1.02 × 10−10, 4.26 × 10−5 and 5.10 × 10−12 cm2 s−1, respectively,85,86 while those in silicon and CNTs are ∼1.2 × 10−6 and 1.2 × 10−11 cm2 s−1.87,88 For Mg2+, AlN exhibits a diffusion coefficient of 1.51 × 10−3 cm2 s−1.89 Na+ diffusion coefficient in Si3N is ∼2.26 × 10−7 cm2 s−1
at 300 K), D is the diffusion coefficient and µ is the ionic mobility, Eb denotes the diffusion barrier, KB stands for the Boltzmann constant, and T represents the temperature.81–83 The computed diffusion coefficients (ionic mobility) along the favourable diffusion path are found to be 4.22 × 10−7, 6.00 × 10−7, and 9.59 × 10−6 cm2 s−1 (1.62 × 10−5, 2.31 × 10−5, and 3.69 × 10−4 cm2 s−1 V−1) for Al on VNbC, VTaC, and NbTaC nanosheets, respectively. Similarly, 6.27 × 10−5, 1.92 × 10−4, and 9.68 × 10−4 cm2 s−1 (2.41 × 10−3, 7.39 × 10−3, and 3.72 × 10−2 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Mg on VNbC, VTaC, and NbTaC nanosheets, respectively and 6.24 × 10−6, 1.91 × 10−5, and 2.07 × 10−5 cm2 s−1 (2.40 × 10−4, 7.36 × 10−4, 7.96 × 10−4 cm2 s−1 V−1) are the diffusion coefficients (ionic mobility) for Zn on VNbC, VTaC, and NbTaC nanosheets, respectively, as shown in Table 3. The diffusion coefficients follow the order VNbC < VTaC < NbTaC among the studied DTM MXenes. The Mg2+-ion diffusion on NbTaC is an order of magnitude faster than on VNbC, showing the highly favorable ionic transport characteristics in NbTaC. Li+ diffusion coefficient in graphite ranges from 1.0 × 10−11 to 4.0 × 10−10 cm2 s−1;84 the diffusion coefficients of Li+ in Si3N, SiC2 and SiC4 are ∼1.02 × 10−10, 4.26 × 10−5 and 5.10 × 10−12 cm2 s−1, respectively,85,86 while those in silicon and CNTs are ∼1.2 × 10−6 and 1.2 × 10−11 cm2 s−1.87,88 For Mg2+, AlN exhibits a diffusion coefficient of 1.51 × 10−3 cm2 s−1.89 Na+ diffusion coefficient in Si3N is ∼2.26 × 10−7 cm2 s−1 






