
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-concentration aqueous synthesis of salicylate-based metal–organic frameworks
Zayim M.
Jamil
a,
Tristan A.
Pitt
a,
Clarisse A.
Doligon
a,
Ronald T.
Jerozal
ab and
Phillip J.
Milner
 *a
*a
aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850, USA. E-mail: pjm347@cornell.edu
bDepartment of Chemistry, Hamilton College, Clinton, NY 13323, USA
First published on 3rd October 2025
Abstract
A current barrier to the practical applications of metal–organic frameworks (MOFs) is the vast quantity of organic solvents required for their preparation under dilute solvothermal conditions. Herein, we report the rapid, ambient-temperature, and high-concentration (up to 1.0 M) aqueous syntheses of three families of salicylate-based MOFs: M2(dobdc) (M = Mg, Co, Ni, Zn; dobdc4− = 2,5-dioxido-1,4-terephthalate), M2(dobpdc) (M = Mg, Co, Ni, Zn; dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate), and M2(m-dobdc) (M = Mg, Co, Ni; m-dobdc4− = 4,6-dioxido-1,3-benzenedicarboxylate). High-concentration MOF formation is accomplished by incorporating NaOH to deprotonate the linker molecules in situ, generally avoiding the crystallization of phases with partially protonated linkers favored under high-concentration solvothermal conditions. 77 K N2 surface area measurements confirm that the MOFs (especially Zn-based frameworks) demonstrate comparable or enhanced surface areas relative to traditionally prepared materials. Furthermore, this method enables the first synthesis of Zn2(m-dobdc), which does not form under standard solvothermal conditions. This material exhibits higher CO2 uptake and ideal adsorbed solution theory (IAST) CO2/N2 selectivities compared to the canonical framework Zn2(dobdc), highlighting the utility of aqueous high-concentration methods to facilitate the discovery of porous materials with improved gas sorption properties. Overall, our findings offer a practical and general alternative to dilute solvothermal syntheses of salicylate-based MOFs, paving the way for their production and implementation in industrial settings.
1 Introduction
Metal–organic frameworks (MOFs) are porous, crystalline coordination polymers with myriad potential applications in catalysis, chemical separations, gas storage, and beyond.1–4 Among these, frameworks bearing coordinatively unsaturated metal sites are particularly valued due to their strong interactions with guest molecules through Lewis acid–base interactions.5 The M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn, Cd; dobdc4− = 2,5-dioxidobenzene-1,4-dicarboxylate), MOF-74, or CPO-27 series, and related M2(dobpdc) (M = Mg, Mn, Fe, Co, Ni, Zn; dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate)6,7 and M2(m-dobdc) (M = Mg, Mn, Fe, Co, Ni; m-dobdc4− = 4,6-dioxido-1,3-benzenedicarboxylate) families,8 represent a privileged group of frameworks due to their high gravimetric and volumetric densities of five-coordinate M2+ centers, which contribute to their exceptional gas sorption properties.9,10 For example, M2(m-dobdc) possesses a high density of exposed cationic sites, leading to record-setting adsorption capacities for gases such as H2 and CO2.11,12 However, a barrier to the large-scale deployment of these salicylate-based frameworks is their synthesis; they are typically prepared under ultra-dilute (<0.01 M) solvothermal conditions, necessitating the use of liters of organic solvent(s) to produce only grams of material.13 Alternatives to traditional dilute solvothermal MOF synthesis methods have been developed,14 including mechanochemical,15,16 ionothermal,17 and hydrothermal18 approaches. Although mechanochemical methods can be employed to produce salicylate MOFs with minimal organic solvent use,11,19–22 they require specialized equipment such as ball mills or twin-screw extruders, which limits their broad applicability for non-experts. Ionothermal methods (i.e., using a metal salt as both the MOF precursor and solvent) to produce salicylate MOFs have also been reported but remain limited to specific frameworks.17 There are scattered reports of the aqueous syntheses of certain M2(dobdc) variants,23–30 but their generality towards other salicylate MOFs remains unclear. Moreover, these syntheses are generally conducted at dilute-to-intermediate concentrations (0.01–0.3 M), leading to significant generation of aqueous waste.High-concentration methods represent an emerging, operationally simple, and green alternative to dilute MOF syntheses.31,32 However, we33,34 and others35 have shown that higher reaction concentrations with N,N-dimethylformamide (DMF) as the solvent lead to the competing formation of kinetically-favored MOFs with partially protonated linkers when applied to the synthesis of M2(dobdc) materials. In particular, the M2(HCO2)2(H2dobdc) or CORN-MOF-1 (M) (CORN = Cornell University; M = Mg, Mn, Fe, Co, Ni, Zn) series of frameworks are persistent phases containing H2dobdc2− linkers that preclude the preparation of M2(dobdc) materials at all but the lowest concentrations (<0.05 M). Although we have shown that CORN-MOF-1 can be converted to M2(dobdc) by heating in DMF, this process requires long times (>5 days) and dilute conditions (∼0.05 M), leading to significant organic waste.33,34 As such, there remains an urgent need to develop simple and scalable methods to access M2(dobdc) and related salicylate-based MOFs with reduced organic solvent use.
Recently, we reported the high-concentration (1.00 M) aqueous synthesis of high-quality Mg2(dobdc) using NaOH as an exogenous base, which bypasses the formation of phases with partially protonated H2dobdc2− linkers.23 Herein, we demonstrate that this method can be generalized to enable the rapid, ambient-temperature synthesis of other M2(dobdc) variants (M = Mg, Co, Ni, Zn) as well as pore-expanded M2(dobpdc) (M = Mg, Co, Ni, Zn) and isomeric M2(m-dobdc) (M = Mg, Co, Ni, Zn) frameworks, for which aqueous syntheses have not been reported to date. Notably, this includes the previously unreported framework Zn2(m-dobdc), which cannot be prepared under traditional solvothermal conditions. Our findings offer a promising solution to replace wasteful and dilute syntheses of MOFs with green and scalable methods while retaining or even enhancing their gas sorption properties.
2 Results & discussion
2.1 Aqueous Synthesis of Mg2(dobdc)
Traditional solvothermal methods to prepare M2(dobdc) materials employ DMF/alcohol mixtures under ultra-dilute (∼0.01 M) solvothermal conditions.36 In contrast, we recently reported the 1.0 M aqueous synthesis of Mg2(dobdc) on >100 g scale.23 This method is performed without applied heating and only takes 1 h to complete. Such favorable conditions are enabled by the use of NaOH as a base to deprotonate H4dobdc and form dobdc4−in situ, rather than relying on the hydrolysis of DMF to slowly generate the required base. The dissolution of NaOH causes the reaction mixture to heat up naturally (SI Table S1), meaning that no applied heating is necessary. The promise of this method motivated further optimization and investigation of its generality.Because the aqueous synthesis of Mg2(dobdc) was only conducted on >100 g scale previously,23 we first confirmed that the method is similarly effective on a smaller scale. Additionally, concentration can have a significant impact on the success of MOF crystallization.32 For example, higher concentration conditions should lead to a faster rate of self-assembly and thus reduced crystallinity and/or increased defect content.37 Therefore, multiple concentrations were evaluated (0.1, 0.5, and 1.0 M) to determine which aqueous conditions are optimal for the preparation of Mg2(dobdc). For direct comparison, we also synthesized samples of all frameworks under traditional dilute solvothermal conditions in DMF/alcohol mixtures at a reaction concentration of 0.01 M (see SI Section 8 for details). Additionally, we attempted the syntheses of M2(dobdc) and M2(m-dobdc) at a higher reaction concentration of 0.1 M; this led to the wrong phase or low-surface-area materials in most cases, consistent with literature findings,33–35 although it was largely successful for Ni2(dobdc), Co2(m-dobdc), and Ni2(m-dobdc) (SI Table S17). This finding highlights the advantage of the aqueous conditions studied herein for high-concentration MOF synthesis.
To test the aqueous syntheses of Mg2(dobdc) on a smaller scale (SI Section 3.1), Mg(NO3)2·6H2O (2.5 equiv.) was dissolved in 5.0 mL, 1.0 mL, or 0.5 mL of H2O depending on the desired concentration (0.1 M, 0.5 M, or 1.0 M, respectively) in a 20 mL scintillation vial. In a separate 20 mL scintillation vial equipped with a stir bar, NaOH (4.0 equiv.), H4dobdc (1.00 equiv.), and 5.0 mL, 1.0 mL, or 0.5 mL of H2O were combined depending on the desired concentration (0.1 M, 0.5 M, or 1.0 M, respectively). The Mg(NO3)2·6H2O solutions were added to the H4dobdc solutions all at once, and the mixtures were stirred at ambient temperature for 1 h. The reaction mixtures were then filtered, and the obtained solids were soaked in methanol (MeOH) at 60 °C for six 12–24 h washes to remove soluble impurities prior to characterization by powder X-ray diffraction (PXRD) (Fig. 2b) and 77 K N2 adsorption/desorption measurements (Table 1, SI Fig. S9).
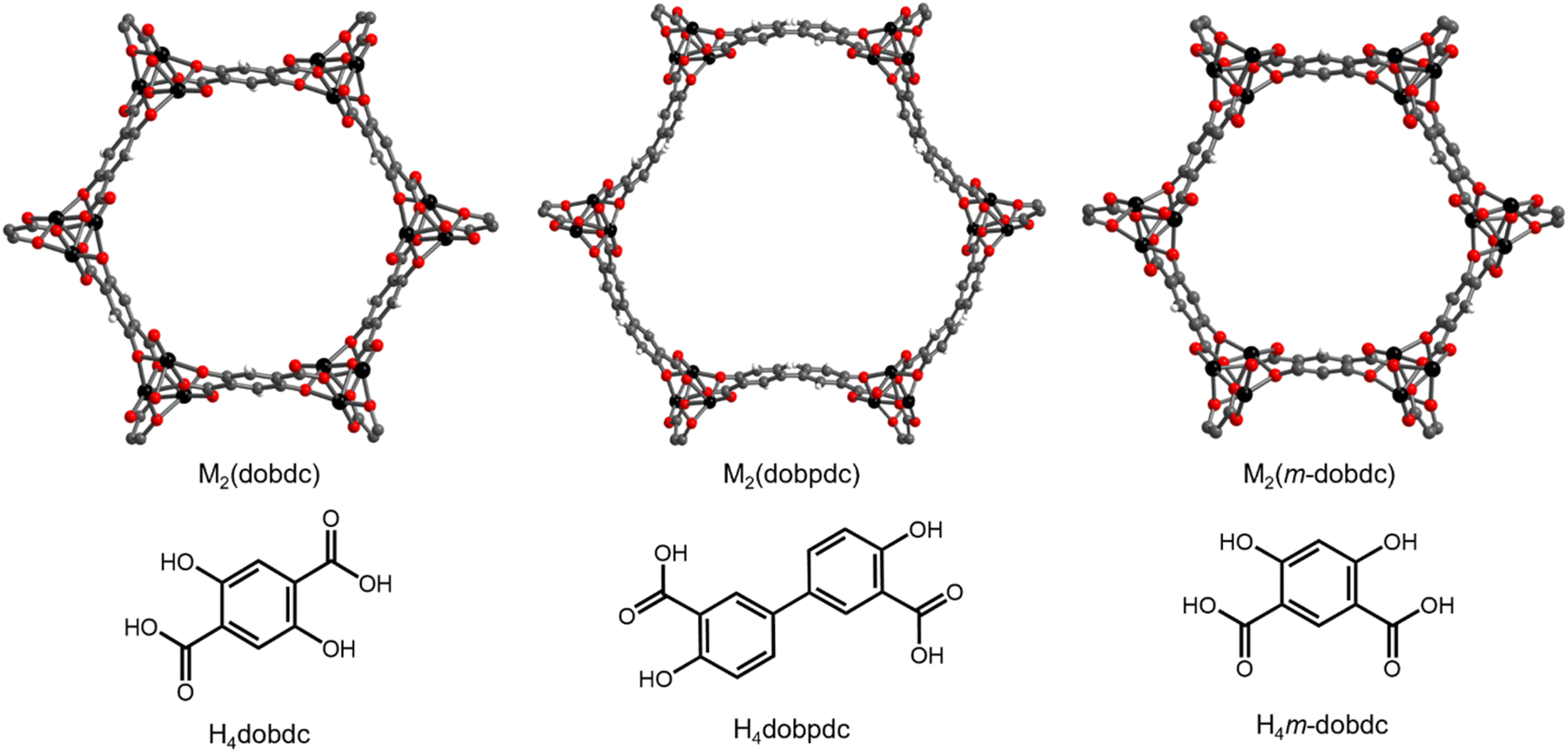 | ||
| Fig. 1 Structures and linkers of M2(dobdc), M2(dobpdc), and M2(m-dobdc). Black, grey, red, and white spheres represent metal, carbon, oxygen, and hydrogen atoms, respectively. | ||
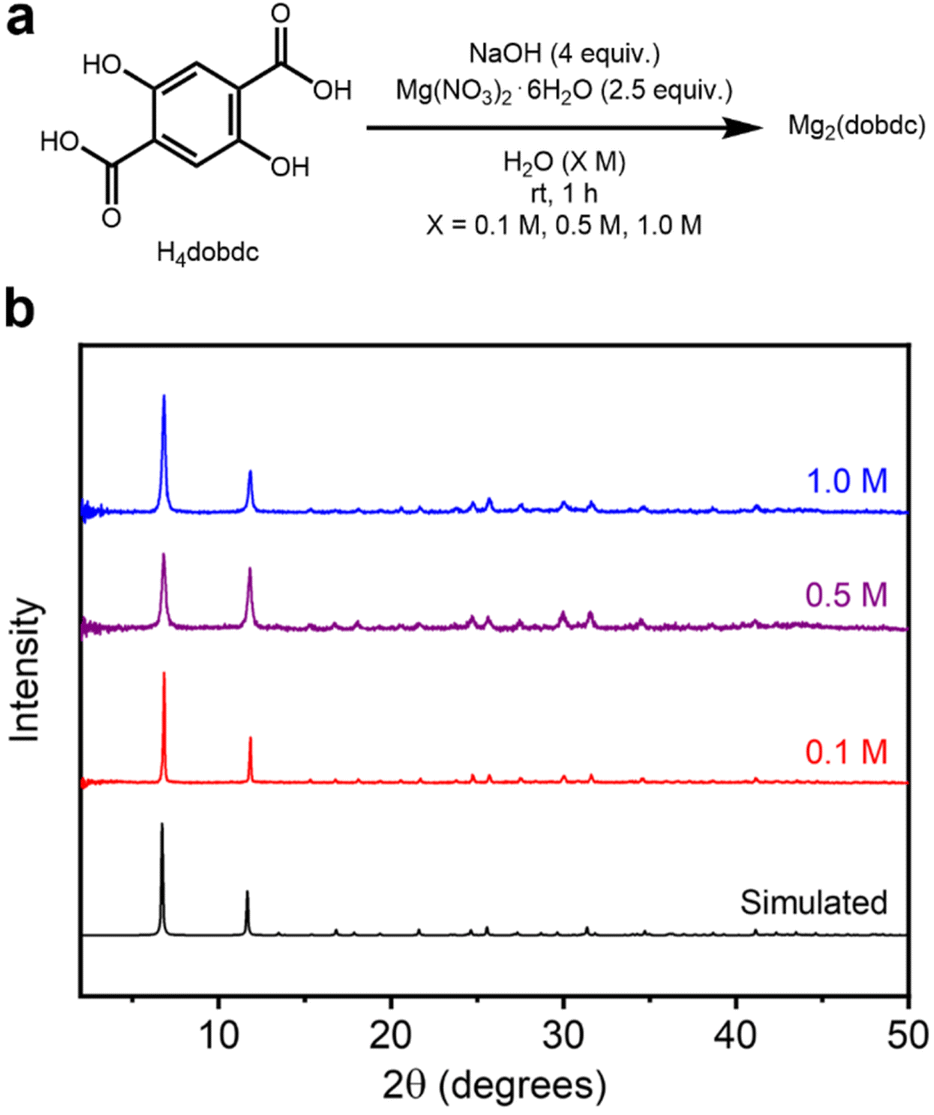 | ||
| Fig. 2 (a) Reaction conditions for the high-concentration aqueous synthesis of Mg2(dobdc). (b) Baseline-corrected PXRD patterns (λ = 1.5406 Å) of Mg2(dobdc) synthesized at different reaction concentrations. The simulated pattern based on the previously reported single-crystal X-ray diffraction (SCXRD) structure of the isostructural Zn2(dobdc) is included (ref. 38). | ||
The PXRD patterns of Mg2(dobdc) prepared at all concentrations agree well with the simulated pattern corresponding to the previously reported single-crystal X-ray diffraction (SCXRD) structure of Zn2(dobdc) (Fig. 2b).38 Nonetheless, differences in the broadness of reflections between samples indicate variations in crystallite sizes related to reaction concentration (see further analysis below). The formation of Mg2(dobdc) was also confirmed by infrared (IR) spectroscopy (SI Fig. S16). PXRD analysis of the crude solids obtained before washing in MeOH confirmed Mg2(dobdc) to be present in the 0.5 M and 1.0 M samples, indicating that it does not form during the soaking procedure (SI Fig. S8). However, the 0.1 M sample initially crystallized as an unknown phase that converts to Mg2(dobdc) after soaking in MeOH (SI Fig. S8). The PXRD pattern of this unknown phase was reliably indexed, but its unit cell does not match any reported H4dobdc-based materials (SI Fig. S140). Although a different phase is formed initially at 0.1 M, the properties of the obtained Mg2(dobdc) resemble those of the materials synthesized at higher concentrations.
The porosities of Mg2(dobdc) samples synthesized at different reaction concentrations were next evaluated. The samples were activated using supercritical CO2, slowly heated (250 °C, 0.1 °C min−1) under high vacuum (<10 μbar), and then held for a minimum of 12 h at 250 °C under high vacuum. Ramp rates faster than 0.1 °C min−1 often led to lower surface areas, likely due to partial framework collapse.20 The 77 K N2 BET surface areas of Mg2(dobdc) samples are all high and fall within a reasonably similar range: 1532 ± 1 m2 g−1 (0.1 M), 1597 ± 1 m2 g−1 (0.5 M), and 1569 ± 1 m2 g−1 (1.0 M) (Table 1, SI Fig. S9 and Table S2). These BET surface areas are also comparable to the value of 1506 ± 1 m2 g−1 obtained for a sample of Mg2(dobdc) prepared under traditional solvothermal conditions (Table 1 and SI Fig. S148). Together, these findings confirm that our previously developed high-concentration aqueous method23 can be scaled down and conducted at a range of reaction concentrations, which permits a broad evaluation of its applicably to other salicylate-based MOF syntheses. This is in contrast to solvothermal methods in DMF/alcohol mixtures, for which different (mixtures of) phases have been reported at different concentrations when attempting to synthesize Mg2(dobdc) (SI Fig. S141).33–35
2.2 Aqueous Synthesis of M2(dobdc) Variants (M = Co, Ni, Zn)2(dobdc)
Next, the generality of this method to other M2(dobdc) variants (M = Co, Ni, Zn) was investigated. Although these MOFs have all been previously synthesized under aqueous conditions,24 they have not been prepared at concentrations higher than ∼0.3 M. We applied the high-concentration aqueous method to access other variants of M2(dobdc) (M = Co, Ni, Zn) under similar conditions as Mg2(dobdc), changing only the metal salt precursor and activation conditions (180 °C instead of 250 °C) accordingly (Fig. 3a, SI Sections 3.2–3.4). This approach was successful in crystallizing all isostructural variants at reaction concentrations of 0.1 M, 0.5 M, and 1.0 M, as confirmed by PXRD (Fig. 3b). The PXRD patterns of the MOF samples with the highest BET surface areas (Table 1) are shown in Fig. 3b. Notably, the highest BET surface areas of Co, Ni, and Zn analogs were obtained at the highest reaction concentration of 1.0 M, potentially due to the formation of more defects under these conditions. These values match or exceed those of materials prepared under dilute solvothermal conditions (Table 1 and SI Fig. S148). In particular, the BET surface area of Zn2(dobdc) reported herein (1274 ± 1 m2 g−1 at 1.0 M) surpasses values previously reported for this MOF (702–1039 m2 g−1) and obtained herein under solvothermal conditions (1200 ± 1 m2 g−1), representing an advantage of mild aqueous synthesis.9,25,39,40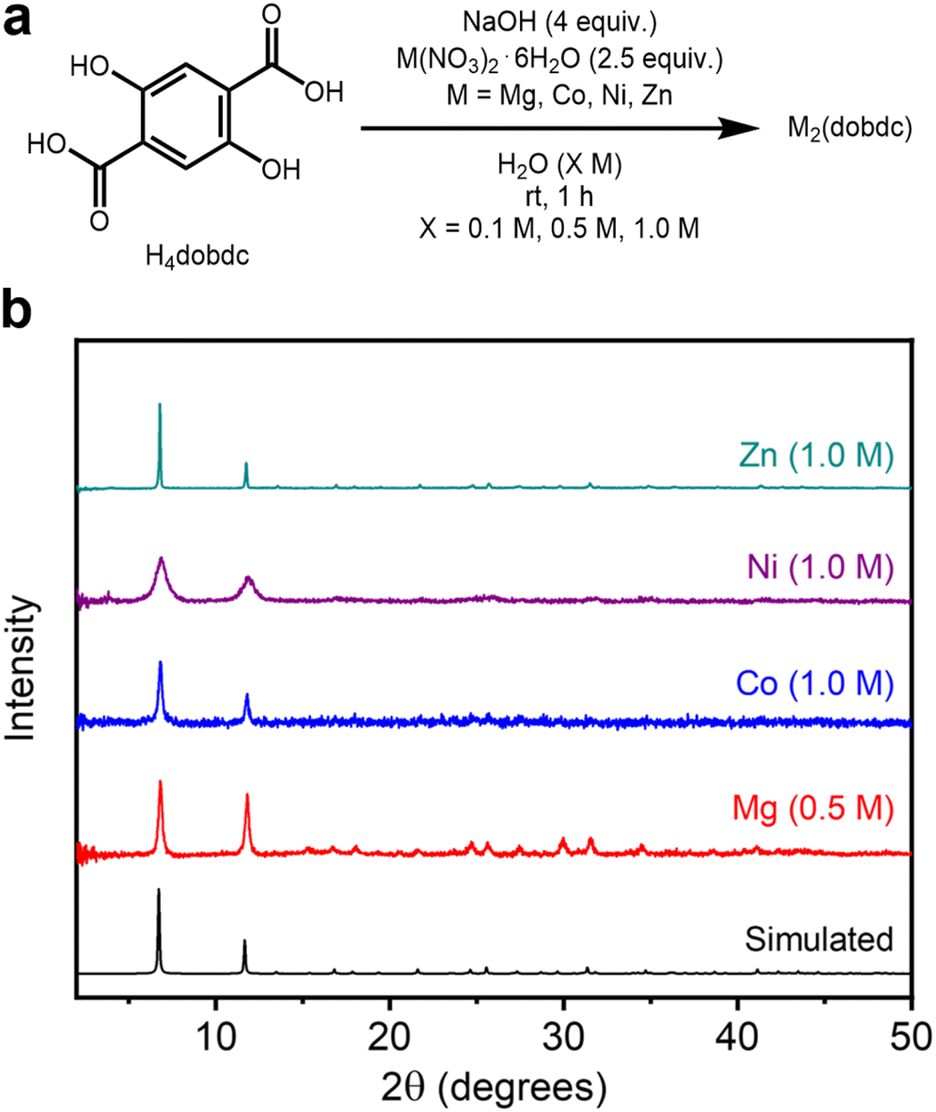 | ||
| Fig. 3 (a) Reaction conditions for the high-concentration aqueous synthesis of M2(dobdc) (M = Mg, Co, Ni, Zn). (b) Baseline-corrected PXRD patterns (λ = 1.5406 Å) of M2(dobdc). The patterns corresponding to samples exhibiting the highest BET surface areas are shown. The simulated pattern based on the previously reported SCXRD structure of Zn2(dobdc) is included (ref. 38). | ||
During the self-assembly of M2(dobdc), the relative bond strengths between different transition metal ions and the dobdc4− linkers are expected to follow the empirical Irving–Williams series and contribute to divergent reversibility during MOF crystallization, which is reflected in differences in relative crystalline domain sizes.41 This possibility was evaluated using whole powder pattern decomposition of experimental PXRD patterns to determine volume-weighted average crystalline domain sizes (LVol-IB) of each framework prepared at all concentrations (SI Section 6). Indeed, the crystallite sizes of the obtained MOFs reflect the expected trend in stability among metal variants (SI Fig. S137 and Table S13). At all concentrations, the largest crystalline domains were formed in Zn2(dobdc) (0.1 M: 773 ± 151 nm, 0.5 M: 145 ± 4 nm, 1.0 M: 289 nm ± 19 nm), and the smallest in Ni2(dobdc) (0.1 M: 11.3 ± 0.6 nm, 0.5 M: 10.4 ± 0.9 nm, 1.0 M: 12.9 nm ± 0.8 nm), exceeded by Co2(dobdc) (0.1 M: 56.3 ± 16.0 nm, 0.5 M: 45.8 ± 12.8 nm, 1.0 M: 63.2 nm ± 9.5 nm) by only a small amount. Mg2(dobdc) (0.1 M: 195 ± 11 nm, 0.5 M: 54.0 ± 3.4 nm, 1.0 M: 42.0 nm ± 3.1 nm) forms the second largest crystalline domains at 0.1 M and is close to Co2(dobdc) at higher concentrations. As the reaction concentrations increase from 0.1 to 0.5/1.0 M, the crystalline domain sizes of all samples generally decrease, consistent with faster self-assembly at the cost of inhibited crystallization at higher concentrations.37 The desired M2(dobdc) phase was identified as the major product of the initial aqueous syntheses of the Co, Ni, and Zn variants (SI Fig. S19, S30 and S41) prior to MeOH treatment, similar to the results obtained for Mg2(dobdc) at higher reaction concentrations (SI Fig. S8). Overall, these findings confirm that porous and crystalline M2(dobdc) variants can be readily prepared under high-concentration aqueous conditions.
2.3 Aqueous Synthesis of Mg2(dobdc)
To determine whether this method can be extended to the synthesis of MOFs bearing other salicylate linkers, it was applied to the isoreticularly expanded M2(dobpdc) (M = Mg, Co, Ni, Zn) series of frameworks, which are distinguished by larger pore diameters (18–22 Å) compared to M2(dobdc) (13–15 Å) (Fig. 1).43 These frameworks have been employed in specialized applications such as cooperative CO2 adsorption in amine-appended variants.6 The high-concentration, ambient-temperature aqueous method outlined above was modified simply by employing H4dobpdc in place of H4dobdc with no other changes (Fig. 4a, SI Section 4). As confirmed by PXRD (Fig. 4b, SI Fig. S51, S62, S73 and S84), all four M2(dobpdc) frameworks can be synthesized under these conditions, representing the first reported aqueous preparations thereof.36 The formation of each M2(dobpdc) variant was also confirmed via IR spectroscopy (SI Fig. S60, S71, S82 and S93).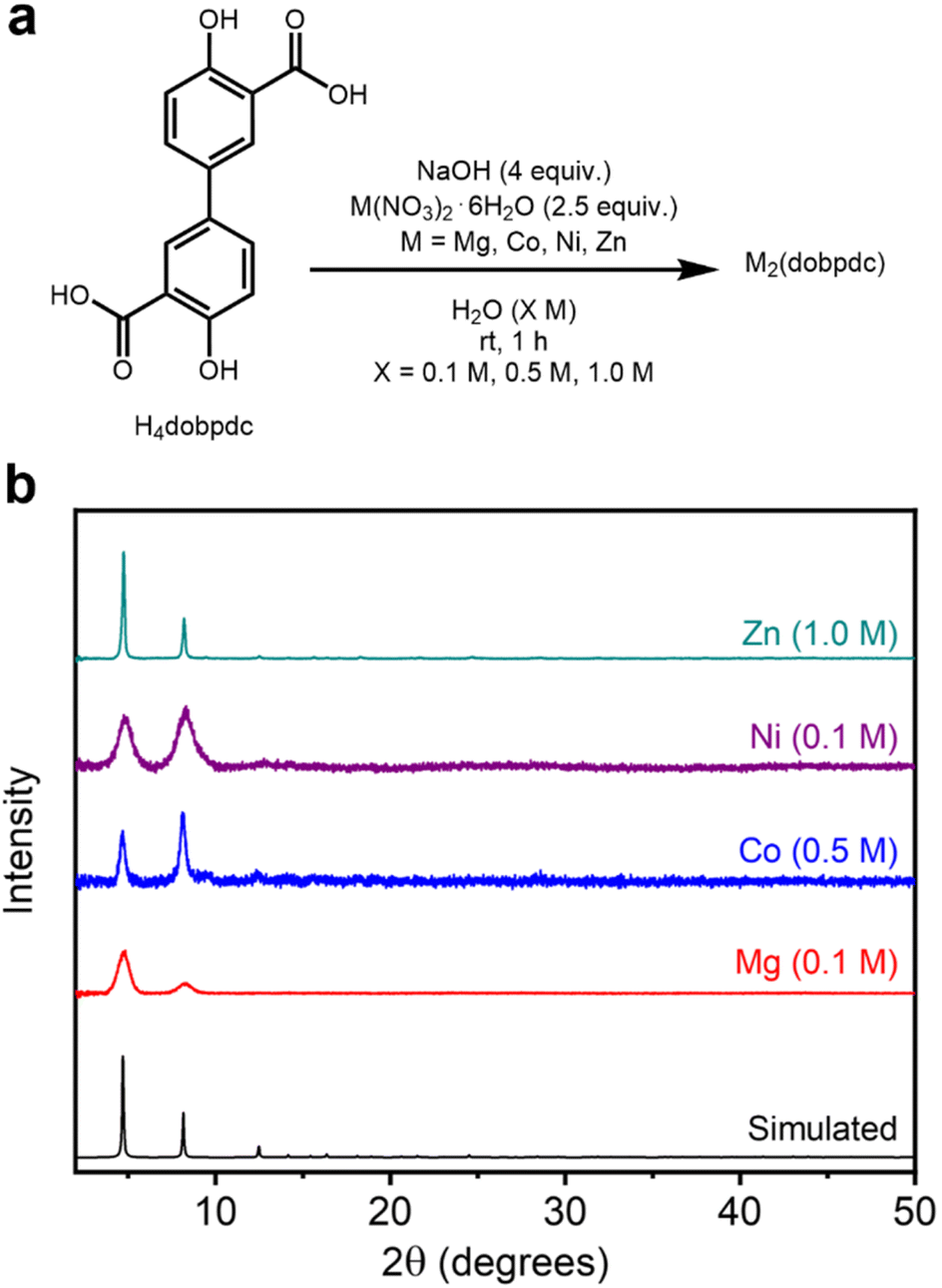 | ||
| Fig. 4 (a) Reaction conditions for the high-concentration aqueous synthesis of M2(dobpdc) (M = Mg, Co, Ni, Zn). (b) Baseline-corrected PXRD patterns (λ = 1.5406 Å) of M2(dobpdc). The patterns corresponding to samples exhibiting the highest BET surface areas are shown. The simulated pattern based on the previously reported SCXRD structure of Zn2(dobpdc) is included (ref. 44). | ||
The PXRD patterns of samples with the highest BET surface areas are presented in Fig. 4b. Although the PXRD patterns of all samples match the simulated pattern of Zn2(dobpdc),44 they show varied line broadening, indicating a range of crystallite sizes (SI Table S13 and SI Fig. S138). Reflecting a similar trend as the M2(dobdc) series, Zn2(dobpdc) (0.1 M: 523 ± 68 nm, 0.5 M: 157 ± 7 nm, 1.0 M: 153 nm ± 10 nm) contains the largest crystalline domains at all concentrations, followed by Co2(dobpdc) (0.1 M: 34.1 ± 4.6 nm, 0.5 M: 43.3 ± 4.9 nm, 1.0 M: 31.6 nm ± 9.6 nm), Mg2(dobpdc) (0.1 M: 10.5 ± 1.1 nm, 0.5 M: 30.0 ± 3.6 nm, 1.0 M: 15.7 nm ± 2.7 nm), and Ni2(dobpdc) (0.1 M: 9.0 ± 0.2 nm, 0.5 M: 9.7 ± 0.3 nm, 1.0 M: 10.4 nm ± 0.8 nm). The domain sizes of all variants other than Zn2(dobpdc) are relatively small and similar regardless of the concentration. Relatedly, whereas the Mg, Ni, and Co variants were all detected by PXRD after their initial aqueous syntheses (SI Fig. S52, S63 and S74), the Zn variant initially forms as an unknown crystalline phase or mixture of phases at all concentrations that subsequently transform(s) into Zn2(dobpdc) upon soaking in MeOH (SI Fig. S85). This distinct formation mechanism may account for its exceptional crystallinity among M2(dobpdc) variants. Notably, our attempt to prepare Zn2(dobpdc) under dilute solvothermal conditions was unsuccessful, highlighting the unique suitability of aqueous conditions for the preparation of this material (SI Fig. S143). Together with the good porosity and crystallinity of Zn2(dobdc) synthesized under aqueous conditions (Fig. 3a and Table 1), these results indicate that aqueous synthesis followed by soaking in MeOH may be particularly effective for the preparation of Zn-based salicylate MOFs.
To analyze the porosity of each M2(dobpdc) variant, supercritical CO2 activation and slow heating was employed. After activation, 77 K N2 adsorption/desorption isotherms for each MOF were collected (SI Tables S6–S9); the M2(dobpdc) samples with the highest BET surface areas are included in Table 2. The reaction concentrations that produced the highest surface areas among M2(dobpdc) frameworks were more variable than for M2(dobdc) (Table 2). The highest surface area is obtained at 1.0 M (2574 ± 77 m2 g−1) for Zn2(dobpdc), 0.5 M for Co2(dobpdc) (2371 ± 42 m2 g−1), 0.1 M for Mg2(dobpdc) (2369 ± 40 m2 g−1), and 0.1 M for Ni2(dobpdc) (1794 ± 17 m2 g−1), compared to the optimal 1.0 M synthesis for most M2(dobdc) frameworks. Notably, this is the highest BET surface area of Zn2(dobpdc) reported to date.43 The BET surface areas of other analogs of M2(dobpdc)—those formed directly and containing stronger metal-linker bonds—were generally less competitive with values obtained under dilute solvothermal conditions (Table 2). Metal-linker bond strengths may exert a more controlling influence on the crystallization of M2(dobpdc) frameworks than M2(dobdc). Regardless, these findings confirm that highly porous M2(dobpdc) variants can be readily prepared under high-concentration aqueous conditions.
2.4 Aqueous synthesis of M2(m-dobdc) variants (M = Mg, Co, Ni)
The developed mild synthetic method was applied to a third series of known salicylate-based MOFs: M2(m-dobdc) (M = Mg, Co, Ni) (SI Section 5). While these frameworks share the same overall pore size and hexagonal topology as the corresponding M2(dobdc) variants, the alteration of the linker makes their M2+ sites slightly more exposed and thus more Lewis acidic.11,12 Under the optimized aqueous conditions at ambient temperature, the known M2(m-dobdc) (M = Mg, Co, Ni) frameworks were successfully synthesized at all tested concentrations (Fig. 5a). The desired M2(m-dobdc) materials were obtained after MeOH washes, as determined through PXRD analysis and comparison to the simulated pattern of Co2(m-dobdc) (Fig. 5b, SI Fig. S95, S106 and S117).45 The formation of these MOFs was also confirmed using IR spectroscopy (SI Fig. S104, S115 and S126). These findings represent the first reported syntheses of all three MOFs under aqueous conditions.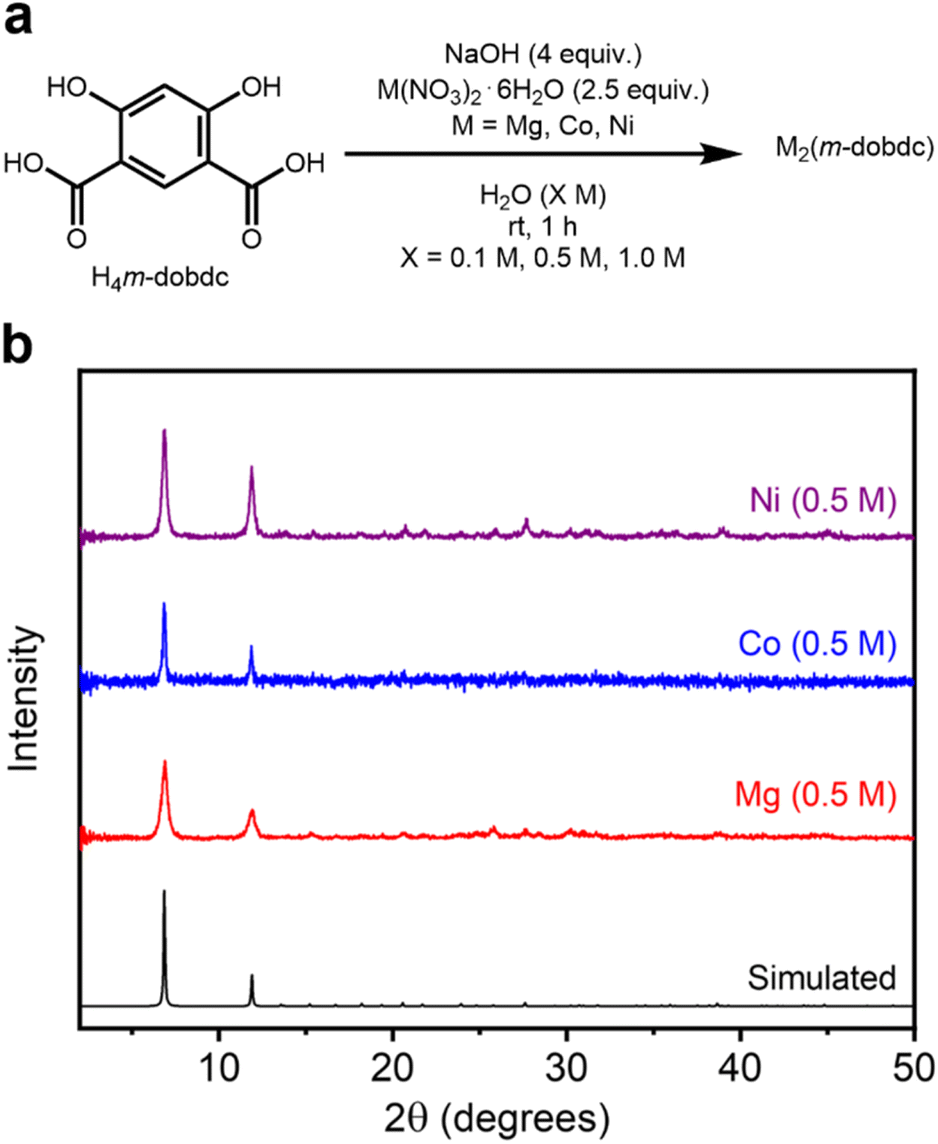 | ||
| Fig. 5 (a) Reaction conditions for the high-concentration aqueous synthesis of known M2(m-dobdc) (M = Mg, Co, Ni) variants. (b) Baseline-corrected PXRD patterns (λ = 1.5406 Å) of M2(m-dobdc). The patterns corresponding to samples exhibiting the highest BET surface areas are shown. The simulated pattern based on the previously reported SCXRD structure of Co2(m-dobdc) is included (ref. 45). | ||
The crystalline domain sizes of M2(m-dobdc) MOFs do not follow a uniform trend in reaction concentration but are generally largest at a reaction concentration of 0.1 M (SI Table S13 and SI Fig. S139). The crystallite sizes of Co2(m-dobdc) samples (0.1 M: 87.3 ± 11.0 nm, 0.5 M: 63.4 ± 7.7 nm, 1.0 M: 116 nm ± 3 nm) were slightly larger than Mg2(m-dobdc) (0.1 M: 67.8 ± 3.1 nm, 0.5 M: 37.0 ± 2.5 nm, 1.0 M: 59 nm ± 3 nm) and Ni2(m-dobdc) (0.1 M: 22.7 ± 2.3 nm, 0.5 M: 46.5 ± 2.4 nm, 1.0 M: 38.7 nm ± 2.0 nm) samples overall, which were similar. Much like the syntheses of Mg2(dobdc) at 0.1 M (SI Fig. S8) and Zn2(dobpdc) at all concentrations (SI Fig. S85), Mg2(m-dobdc) initially forms as an unidentified crystalline phase or mixture of phases at all concentrations that convert(s) to Mg2(m-dobdc) upon soaking in MeOH (SI Fig. S96). It is likely that mechanochemical synthesis of this material without added solvent proceeds through a similar step-wise mechanism.11 In contrast, the Co (SI Fig. S107) and Ni (SI Fig. S118) variants of this material crystallize directly from the aqueous reaction mixture.
The porosities of M2(m-dobdc) variants were analyzed through 77 K N2 adsorption/desorption measurements (SI Tables S10–S12). The reaction concentration that produced samples with the highest surface areas among M2(m-dobdc) frameworks was consistently 0.5 M, possibly reflecting an ideal balance between crystallinity and defects formed at higher concentrations (Table 3). Overall, these values are somewhat lower than optimized dilute solvothermal (Table 3) or mechanochemical syntheses of these frameworks. Nonetheless, our findings illustrate that M2(m-dobdc) variants can be readily prepared under high-concentration aqueous conditions, further supporting that this approach can be generalized across salicylate-based MOFs.
2.5 Aqueous synthesis of Zn2(m-dobdc)
Following the successful aqueous syntheses of salicylate MOFs, particularly Zn-based materials, we hypothesized that this method could facilitate the synthesis of a currently elusive framework: Zn2(m-dobdc). Despite the breadth of studies evaluating salicylate-based MOFs for various applications in gas storage and separations.5,9,47 Zn2(m-dobdc) has never been reported, implying that it does not form efficiently under traditional solvothermal conditions. Indeed, in six separate trials using representative combinations of organic solvents and common Zn salt precursors used to synthesize related MOFs,12,36 Zn2(m-dobdc) was not obtained, as determined by PXRD analysis (Fig. 6a, SI Section 5.4).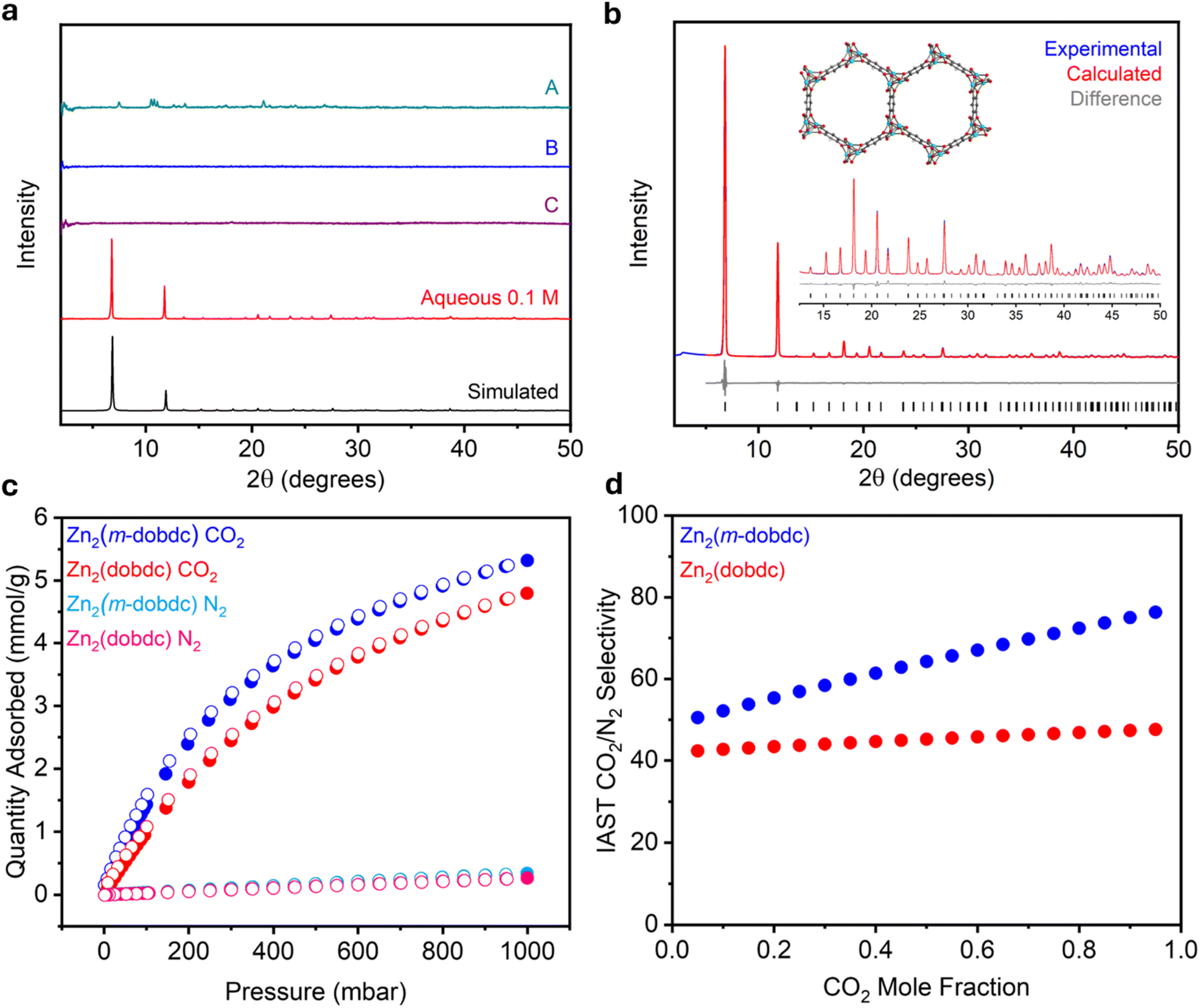 | ||
Fig. 6 (a) Baseline-corrected PXRD patterns (λ = 1.5406 Å) of Zn2(m-dobdc) prepared using different synthesis conditions: (A) 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 DMF:MeOH with Zn(NO3)2·6H2O as the metal salt at 0.15 M for 24 h at 120 °C, 13:7 DMF:MeOH at 0.015 M for 24 h at 120 °C using (B) ZnCl2 and (C) Zn(NO3)2·6H2O as the metal salts; and high-concentration aqueous synthesis at 0.1 M. The simulated pattern based on the previously reported SCXRD structure of the isostructural Co2(m-dobdc) is included (ref. 35). (b) Rietveld refinement of the PXRD (λ = 1.5417 Å) pattern of activated Zn2(m-dobdc) in an N2-filled capillary. Refined unit cell parameters: space group = R3m, a = 25.8808(5) Å, c = 6.78977(19). Black ticks correspond to calculated Bragg positions. Goodness of fit parameters: Rwp = 6.13%, Rp = 4.66%, Rexp = 0.32%, GoF = 19.3, Rwp (Rietveld)/Rwp (Pawley) = 1.09. GoF(Rietveld)/GoF(Pawley) = 1.08. (c) 30 °C CO2 and N2 adsorption (filled circles) and desorption (open circles) isotherms of activated Zn2(m-dobdc) and Zn2(dobdc). (d) IAST CO2/N2 selectivities for Zn2(m-dobdc) and Zn2(dobdc) at 25 °C and 1 bar total pressure. :7 DMF:MeOH with Zn(NO3)2·6H2O as the metal salt at 0.15 M for 24 h at 120 °C, 13:7 DMF:MeOH at 0.015 M for 24 h at 120 °C using (B) ZnCl2 and (C) Zn(NO3)2·6H2O as the metal salts; and high-concentration aqueous synthesis at 0.1 M. The simulated pattern based on the previously reported SCXRD structure of the isostructural Co2(m-dobdc) is included (ref. 35). (b) Rietveld refinement of the PXRD (λ = 1.5417 Å) pattern of activated Zn2(m-dobdc) in an N2-filled capillary. Refined unit cell parameters: space group = R3m, a = 25.8808(5) Å, c = 6.78977(19). Black ticks correspond to calculated Bragg positions. Goodness of fit parameters: Rwp = 6.13%, Rp = 4.66%, Rexp = 0.32%, GoF = 19.3, Rwp (Rietveld)/Rwp (Pawley) = 1.09. GoF(Rietveld)/GoF(Pawley) = 1.08. (c) 30 °C CO2 and N2 adsorption (filled circles) and desorption (open circles) isotherms of activated Zn2(m-dobdc) and Zn2(dobdc). (d) IAST CO2/N2 selectivities for Zn2(m-dobdc) and Zn2(dobdc) at 25 °C and 1 bar total pressure. | ||
As the presented aqueous procedure was effective for preparing other members of this series, we next applied it to the synthesis of Zn2(m-dobdc). At a concentration of 0.1 M, an off-white solid quickly formed; the PXRD pattern of this solid matched the simulated pattern of Co2(m-dobdc),45 indicating the direct preparation of Zn2(m-dobdc) for the first time (Fig. 6a and SI Fig. S128). As confirmation, Rietveld refinement of the powder pattern of Zn2(m-dobdc) using an initial model derived from the SCXRD structure of Co2(m-dobdc)45 produced an excellent match to the experimental data (Rwp = 6.13%, Fig. 6b). Upon activation, Zn2(m-dobdc) crystallizes in the R3m space group with unit cell parameters a = 25.8808(5) Å and c = 6.78977(19) Å.45 The formation of Zn2(m-dobdc) is also supported by IR spectroscopy (SI Fig. S133). Characterization of Zn2(m-dobdc) by scanning electron microscopy (SEM) revealed that it is composed of clusters of well-defined hexagonal rods approximately 5 μm in length (SI Fig. S135). This morphology is precedented in other variants of this series.48 Phase-pure Zn2(m-dobdc) only forms at 0.1 M out of the three evaluated concentrations, as amorphous products or a mixture of phases were obtained at higher concentrations (Fig. S128). PXRD analysis confirms that, after washing in MeOH, Zn2(m-dobdc) contains larger crystallites (330 ± 31 nm) compared to other M2(m-dobdc) samples (SI Table S13 and Fig. S139), matching the findings for Zn-based M2(dobdc) and M2(dobpdc) frameworks. After activation, 77 K N2 adsorption/desorption isotherms produced BET (1216 ± 1 m2 g−1) and Langmuir (1340 m2 g−1) surface areas for Zn2(m-dobdc), that are comparable to those of the structurally related Zn2(dobdc) (1274 ± 1 m2 g−1 and 1395 m2 g−1, respectively). Notably, the synthesis of Zn2(m-dobdc) could be scaled up ten-fold to produce material with comparable crystallinity and BET surface area (1228 ± 1 m2 g−1) to that prepared on a small scale (SI Section 9). This high surface area could be achieved without supercritical CO2 activation, indicating that this step may be unnecessary. Together, these data support the unique crystallization of Zn2(m-dobdc) under aqueous, ambient-temperature conditions.
Enhanced gas binding at unsaturated metal ions in M2(m-dobdc) materials relative to M2(dobdc) has been reported for some adsorbates,12 indicating that Zn2(m-dobdc) should show improved adsorption of gases such as CO2 compared to Zn2(dobdc). To evaluate this possibility, 30 °C CO2 and N2 adsorption isotherms of Zn2(m-dobdc) and Zn2(dobdc) synthesized under aqueous conditions were measured (Fig. 6c). Both materials exhibit steep uptake at low CO2 pressures, characteristic of the strong interaction of CO2 with coordinatively unsaturated metal sites. The maximum CO2 uptakes at 1 bar and 30 °C were 5.32 mmol g−1 and 4.80 mmol g−1 in Zn2(m-dobdc) and Zn2(dobdc), respectively, a marked increase of 0.52 mmol g−1 in the CO2 capacity of Zn2(m-dobdc). The isotherms were fit to dual-site Langmuir models, and competitive ideal adsorbed solution theory (IAST) CO2/N2 selectivities were calculated for the two MOFs over a range of simulated CO2:N2 mixtures (SI Section 7). Relative to Zn2(dobdc), Zn2(m-dobdc) was found to adsorb CO2 more selectively at all mole fractions of CO2 (Fig. 6d). This is likely due to Zn2(m-dobdc) exhibiting increased CO2 uptake and similar N2 uptake compared to Zn2(dobdc). These results emphasize the utility of aqueous methods not only as a facile and sustainable alternative to dilute solvothermal syntheses but also as a route to isolate new materials with enhanced gas sorption properties.
3 Conclusion
High-concentration, ambient-temperature aqueous synthesis enables the facile batch production of high-quality salicylate MOFs with reduced solvent consumption compared to traditional dilute syntheses. Beginning with the high-concentration aqueous synthesis of Mg2(dobdc), we extend this method to other M2(dobdc) variants (M = Mg, Co, Ni, Zn) and investigate concentration-based changes in MOF crystallization and porosity. This method was further employed to synthesize the salicylate-based MOFs M2(dobpdc) (M = Mg, Co, Ni, Zn) and M2(m-dobdc) (M = Mg, Co, Ni, Zn). High concentrations (0.5–1.0 M) produce high-quality M2(dobdc) and M2(m-dobdc) samples exhibiting competitive surface areas with materials derived from traditional methods, whereas careful control of the reaction concentration is needed to maximize porosity in M2(dobpdc) variants. Notably, this method produces record-setting porosity in Zn salicylate-based frameworks and facilitates the first synthesis of Zn2(m-dobdc), which does not crystallize under traditional solvothermal conditions. CO2 and N2 adsorption isotherms support that Zn2(m-dobdc) exhibits greater CO2 uptake and IAST CO2/N2 selectivity compared to the canonical framework Zn2(dobdc) synthesized under the same conditions, motivating the application of aqueous methods to the discovery of new MOFs. Overall, we attribute the success of this aqueous method for the synthesis of Zn salicylate-based MOFs to two factors: (i) the in situ generation of tetraanionic linkers by NaOH, effectively directing the growth of the desired phases over those incorporating partially protonated linkers, and (ii) labile metal-linker bonding relative to other metal analogs.33,34Overall, our findings have important implications for the production and implementation of MOFs on scale, as the reported procedure entirely avoids the use of DMF and specialized equipment in favor of cheaper and more sustainable solvents like H2O and MeOH. Moreover, this procedure produces MOFs at concentrations as high as 1.0 M at ambient temperature. Future research will focus on evaluating the generality of aqueous, high-concentration synthetic methods towards other families of MOFs.
Author contributions
P. J. M. and R. T. J. conceived the project. Z. M. J. carried out the synthesis, characterization, and gas sorption measurements of MOF samples under the supervision of R. T. J. and P. J. M. T. A. P. carried out characterization and PXRD analysis. C. A. D. conducted all solvothermal and large-scale MOF syntheses. Z. M. J. prepared the first draft of the manuscript, which was edited and approved by all co-authors.Conflicts of interest
P. J. M. and R. T. J. are listed as co-inventors on several patents related to metal–organic frameworks.Data availability
CCDC 2461999 contains the supplementary crystallographic data for this paper.49The data supporting this article have been included as part of the supplementary information (SI) and are available from the authors upon reasonable request. Supplementary information: synthesis procedures and characterization data for all MOFs. See DOI: https://doi.org/10.1039/d5ta04800a.
Acknowledgements
The development of methods for the scalable synthesis of MOFs was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138165. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The characterization of the CO2 adsorption properties of MOFs was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021000. We acknowledge the support of a Camille Dreyfus Teacher-Scholar Award to P. J. M. (TC-23-048). This work made use of the Cornell Center for Materials Research Shared Facilities. 1H NMR data were collected on a Bruker INOVA 500 MHz spectrometer that was purchased with support from the NSF (CHE-1531632). We thank Dr Ruth Mandel (Princeton University) for synthesizing some of the H4m-dobdc used in this work. We thank Dr Daewon Kim (Cornell University) and Alexandra Lim (Cornell University) for editorial assistance during the preparation of this manuscript.References
- B. I. Z. Ahmad, K. T. Keasler, E. E. Stacy, S. Meng, T. J. Hicks and P. J. Milner, MOFganic Chemistry: Challenges and Opportunities for Metal–Organic Frameworks in Synthetic Organic Chemistry, Chem. Mater., 2023, 35(13), 4883–4896, DOI:10.1021/acs.chemmater.3c00741.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives, Chem. Rev., 2020, 120(16), 8468–8535, DOI:10.1021/acs.chemrev.9b00685.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, Porous Metal-Organic Frameworks for Gas Storage and Separation: Status and Challenges, EnergyChem, 2019, 1(1), 100006, DOI:10.1016/j.enchem.2019.100006.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341(6149), 1230444, DOI:10.1126/science.1230444.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Coordinatively Unsaturated Metal Sites (Open Metal Sites) in Metal–Organic Frameworks: Design and Applications, Chem. Soc. Rev., 2020, 49(9), 2751–2798, 10.1039/c9cs00609e.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Cooperative Insertion of CO2 in Diamine-Appended Metal-Organic Frameworks, Nature, 2015, 519(7543), 303–308, DOI:10.1038/nature14327.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, Capture of Carbon Dioxide from Air and Flue Gas in the Alkylamine-Appended Metal–Organic Framework mmen-Mg2(dobpdc), J. Am. Chem. Soc., 2012, 134(16), 7056–7065, DOI:10.1021/ja300034j.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, M2(m-dobdc) (M = Mg, Mn, Fe, Co, Ni) Metal–Organic Frameworks Exhibiting Increased Charge Density and Enhanced H2 Binding at the Open Metal Sites, J. Am. Chem. Soc., 2014, 136(34), 12119–12129, DOI:10.1021/ja506230r.
- H. Kim and C. S. Hong, MOF-74-Type Frameworks: Tunable Pore Environment and Functionality through Metal and Ligand Modification, CrystEngComm, 2021, 23(6), 1377–1387, 10.1039/d0ce01870h.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, Rod Packings and Metal–Organic Frameworks Constructed from Rod-Shaped Secondary Building Units, J. Am. Chem. Soc., 2005, 127(5), 1504–1518, DOI:10.1021/ja045123o.
- E. Y. Chen, R. M. Mandel and P. J. Milner, Evaluating Solvothermal and Mechanochemical Routes towards the Metal–Organic Framework Mg2(m-dobdc), CrystEngComm, 2022, 24(41), 7292–7297, 10.1039/d2ce00739h.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, M2(m-dobdc) (M = Mg, Mn, Fe, Co, Ni) Metal–Organic Frameworks Exhibiting Increased Charge Density and Enhanced H2 Binding at the Open Metal Sites, J. Am. Chem. Soc., 2014, 136(34), 12119–12129, DOI:10.1021/ja506230r.
- T. J. Azbell, T. A. Pitt, R. T. Jerozal, R. M. Mandel and P. J. Milner, Simplifying the Synthesis of Metal–Organic Frameworks, Acc. Mater. Res., 2023, 4(10), 867–878, DOI:10.1021/accountsmr.3c00121.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, New Synthetic Routes towards MOF Production at Scale, Chem. Soc. Rev., 2017, 46(11), 3453–3480, 10.1039/c7cs00109f.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Mechanochemistry: Toward Green Synthesis of Metal–Organic Frameworks, Mater. Today, 2021, 46, 109–124, DOI:10.1016/j.mattod.2021.01.008.
- D. Chen, J. Zhao, P. Zhang and S. Dai, Mechanochemical Synthesis of Metal–Organic Frameworks, Polyhedron, 2019, 162, 59–64, DOI:10.1016/j.poly.2019.01.024.
- T. J. Azbell, T. A. Pitt, M. M. Bollmeyer, C. Cong, K. M. Lancaster and P. Milner, Ionothermal Synthesis of Metal-Organic Frameworks Using Low-Melting Metal Salt Precursors, Angew. Chem., Int. Ed., 2023, 62(17), e202218252, DOI:10.1002/anie.202218252.
- Z. Hu, Y. Wang and D. Zhao, Modulated Hydrothermal Chemistry of Metal–Organic Frameworks, Acc. Mater. Res., 2022, 3(11), 1106–1114, DOI:10.1021/accountsmr.2c00104.
- J. Beamish-Cook, K. Shankland, C. A. Murray and P. Vaqueiro, Insights into the Mechanochemical Synthesis of MOF-74, Cryst. Growth Des., 2021, 21(5), 3047–3055, DOI:10.1021/acs.cgd.1c00213.
- Z. Wang, Z. Li, M. Ng and P. J. Milner, Rapid Mechanochemical Synthesis of Metal–Organic Frameworks Using Exogenous Organic Base, Dalton Trans., 2020, 49(45), 16238–16244, 10.1039/d0dt01240h.
- G. Ayoub, B. Karadeniz, A. J. Howarth, O. K. Farha, I. Đilović, L. S. Germann, R. E. Dinnebier, K. Užarević and T. Friščić, Rational Synthesis of Mixed-Metal Microporous Metal–Organic Frameworks with Controlled Composition Using Mechanochemistry, Chem. Mater., 2019, 31(15), 5494–5501, DOI:10.1021/acs.chemmater.9b01068.
- P. A. Julien, K. Užarević, A. D. Katsenis, S. A. J. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann, M. Etter, R. E. Dinnebier, S. L. James, I. Halasz and T. Friščić, In Situ Monitoring and Mechanism of the Mechanochemical Formation of a Microporous MOF-74 Framework, J. Am. Chem. Soc., 2016, 138(9), 2929–2932, DOI:10.1021/jacs.5b13038.
- K. T. Keasler, M. E. Zick, E. E. Stacy, J. Kim, J.-H. Lee, L. Aeindartehran, T. Runčevski and P. J. Milner, Handling Fluorinated Gases as Solid Reagents Using Metal-Organic Frameworks, Science, 2023, 381(6665), 1455–1461, DOI:10.1126/science.adg8835.
- L. Garzón-Tovar, A. Carné-Sánchez, C. Carbonell, I. Imaz and D. Maspoch, Optimised Room Temperature, Water-Based Synthesis of CPO-27-M Metal–Organic Frameworks with High Space-Time Yields, J. Mater. Chem. A, 2015, 3(41), 20819–20826, 10.1039/c5ta04923g.
- M. Sánchez-Sánchez, N. Getachew, K. Díaz, M. Díaz-García, Y. Chebude and I. Díaz, Synthesis of Metal–Organic Frameworks in Water at Room Temperature: Salts as Linker Sources, Green Chem., 2015, 17(3), 1500–1509, 10.1039/c4gc01861c.
- S. Cadot, L. Veyre, D. Luneau, D. Farrusseng and E. Alessandra Quadrelli, A Water-Based and High Space-Time Yield Synthetic Route to MOF Ni2(dhtp) and Its Linker 2,5-Dihydroxyterephthalic Acid, J. Mater. Chem. A, 2014, 2(42), 17757–17763, 10.1039/c4ta03066d.
- X.-R. Shi, M. Qiao, Y. Wei, L.-X. Yun, J.-X. Wang and J.-F. Chen, Green, Efficient and Controllable Synthesis of High-Quality MOF-74 with High Gravity Technology, Green Chem., 2024, 26(10), 6209–6218, 10.1039/d4gc01350f.
- C. Yu, Y. Wang, J. Cui, D. Yu, X. Zhang, X. Shu, J. Zhang, Y. Zhang, R. Vajtai, P. M. Ajayan and Y. Wu, MOF-74 Derived Porous Hybrid Metal Oxide Hollow Nanowires for High-Performance Electrochemical Energy Storage, J. Mater. Chem. A, 2018, 6(18), 8396–8404, 10.1039/c8ta01426d.
- R. Sharma, D. Sürmeli, T. R. C. Van Assche, S. Tiriana, M.-P. Delplancke, G. V. Baron and J. F. M. Denayer, An Ultra-Permeable Hybrid Mg-MOF-74-Melamine Sponge Composite for Fast Dynamic Gas Separation, Microporous Mesoporous Mater., 2022, 343, 112146, DOI:10.1016/j.micromeso.2022.112146.
- T. Didriksen, A. I. Spjelkavik and R. Blom, Continuous Synthesis of the Metal-Organic Framework CPO-27-Ni from Aqueous Solutions, J. Flow Chem., 2017, 7(1), 13–17, DOI:10.1556/1846.2016.00040.
- T. J. Azbell and P. J. Milner, Cobalt(III) Halide Metal–Organic Frameworks Drive Catalytic Halogen Exchange, J. Am. Chem. Soc., 2024, 146(16), 11164–11172, DOI:10.1021/jacs.3c13872.
- R. T. Jerozal, T. A. Pitt, S. N. MacMillan and P. J. Milner, High-Concentration Self-Assembly of Zirconium- and Hafnium-Based Metal–Organic Materials, J. Am. Chem. Soc., 2023, 145(24), 13273–13283, DOI:10.1021/jacs.3c02787.
- A. Halder, D. C. Bain, T. A. Pitt, Z. Shi, J. Oktawiec, J.-H. Lee, S. Tsangari, M. Ng, J. J. Fuentes-Rivera, A. C. Forse, T. Runčevski, D. A. Muller, A. J. Musser and P. J. Milner, Kinetic Trapping of Photoluminescent Frameworks During High-Concentration Synthesis of Nonemissive Metal–Organic Frameworks, Chem. Mater., 2023, 35(23), 10086–10098, DOI:10.1021/acs.chemmater.3c02121.
- T. A. Pitt, D. C. Bain, M. Del Campo, A. J. Musser and P. J. Milner, Controlled Growth and Interconversion of Photoluminescent Metal–Organic Frameworks under High-Concentration Conditions, Chem. Mater., 2025, 37(8), 2964–2975, DOI:10.1021/acs.chemmater.5c00355.
- D. R. Du Bois, K. R. Wright, M. K. Bellas, N. Wiesner and A. J. Matzger, Linker Deprotonation and Structural Evolution on the Pathway to MOF-74, Inorg. Chem., 2022, 61(11), 4550–4554, DOI:10.1021/acs.inorgchem.1c03988.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Dramatic Tuning of Carbon Dioxide Uptake via Metal Substitution in a Coordination Polymer with Cylindrical Pores, J. Am. Chem. Soc., 2008, 130(33), 10870–10871, DOI:10.1021/ja8036096.
- D. R. Du Bois and A. J. Matzger, Metal–Organic Framework Seeding to Drive Phase Selection and Overcome Synthesis Limitations, Cryst. Growth Des., 2022, 22(11), 6379–6383, DOI:10.1021/acs.cgd.2c00762.
- P. D. C. Dietzel, R. E. Johnsen, R. Blom and H. Fjellvåg, Structural Changes and Coordinatively Unsaturated Metal Atoms on Dehydration of Honeycomb Analogous Microporous Metal–Organic Frameworks, Chem.–Eur. J., 2008, 14(8), 2389–2397, DOI:10.1002/chem.200701370.
- J. G. Flores, M. Díaz-García, I. A. Ibarra, J. Aguilar-Pliego and M. Sánchez-Sánchez, Sustainable M-MOF-74 (M = Cu, Co, Zn) Prepared in Methanol as Heterogeneous Catalysts in the Synthesis of Benzaldehyde from Styrene Oxidation, J. Solid State Chem., 2021, 298, 122151, DOI:10.1016/j.jssc.2021.122151.
- J. B. Lefton, K. B. Pekar, U. Haris, M. E. Zick, P. J. Milner, A. R. Lippert, L. Pejov and T. Runčevski, Defect Formation and Amorphization of Zn-MOF-74 Crystals by Post-Synthetic Interactions with Bidentate Adsorbates, J. Mater. Chem. A, 2021, 9(35), 19698–19704, 10.1039/d0ta10613e.
- H. Irving and P. Williams, Order of Stability of Metal Complexes, Nature, 1948, 162, 746–747, DOI:10.1038/162746a0.
- K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordiga and J. R. Long, Hydrogen Storage Properties and Neutron Scattering Studies of Mg2(dobdc)—a Metal–Organic Framework with Open Mg2+ Adsorption Sites, Chem. Commun., 2011, 47(4), 1157–1159, 10.1039/c0cc03453c.
- D. Gygi, E. D. Bloch, J. A. Mason, M. R. Hudson, M. I. Gonzalez, R. L. Siegelman, T. A. Darwish, W. L. Queen, C. M. Brown and J. R. Long, Hydrogen Storage in the Expanded Pore Metal–Organic Frameworks M2(dobpdc) (M = Mg, Mn, Fe, Co, Ni, Zn), Chem. Mater., 2016, 28(4), 1128–1138, DOI:10.1021/acs.chemmater.5b04538.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, Controlling Cooperative CO2 Adsorption in Diamine-Appended Mg2(dobpdc) Metal–Organic Frameworks, J. Am. Chem. Soc., 2017, 139(30), 10526–10538, DOI:10.1021/jacs.7b05858.
- J. E. Bachman, M. T. Kapelewski, D. A. Reed, M. I. Gonzalez and J. R. Long, M2(m-Dobdc) (M = Mn, Fe, Co, Ni) Metal–Organic Frameworks as Highly Selective, High-Capacity Adsorbents for Olefin/Paraffin Separations, J. Am. Chem. Soc., 2017, 139(43), 15363–15370, DOI:10.1021/jacs.7b06397.
- A. C. Forse, K. A. Colwell, M. I. Gonzalez, S. Benders, R. M. Torres-Gavosto, B. Blümich, J. A. Reimer and J. R. Long, Influence of Pore Size on Carbon Dioxide Diffusion in Two Isoreticular Metal–Organic Frameworks, Chem. Mater., 2020, 32(8), 3570–3576, DOI:10.1021/acs.chemmater.0c00745.
- J. H. Choe, H. Kim and C. S. Hong, MOF-74 Type Variants for CO2 Capture, Mater. Chem. Front., 2021, 5(14), 5172–5185, 10.1039/d1qm00205h.
- F. Wu, L. Li, Y. Tan, E.-S. M. El-Sayed and D. Yuan, The Competitive and Synergistic Effect between Adsorption Enthalpy and Capacity in D2/H2 Separation of M2(m-dobdc) Frameworks, Chin. Chem. Lett., 2021, 32(11), 3562–3565, DOI:10.1016/j.cclet.2021.02.063.
- CCDC 2461999: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nmxbk.
| This journal is © The Royal Society of Chemistry 2025 |