
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural transition in crystalline SiO2 during mechanical amorphization under frictional shear†
Jin Jung
Kweon
 a,
Hoon
Khim
a,
Yong-Hyun
Kim
a and
Sung Keun
Lee
*ab
a,
Hoon
Khim
a,
Yong-Hyun
Kim
a and
Sung Keun
Lee
*ab
aLaboratory of Physics and Chemistry of Earth and Planetary Materials, School of Earth & Environmental Sciences, Seoul National University, Seoul 08826, Korea. E-mail: sungklee@snu.ac.kr; Web: https://g2mat.snu.ac.kr
bInstitute of Applied Physics, College of Natural Sciences, Seoul National University, Seoul 08826, Korea
First published on 13th June 2025
Abstract
Mechanical milling increases structural disorder in crystalline materials, often resulting in a milling-induced amorphous phase. Prototypical SiO2 crystals under mechanical shear during milling produce an amorphous phase that has potential for a wide range of applications, from battery anodes to catalyst substrates. Despite its importance, the extent of mechanical amorphization, the mechanochemical reactions, and atomic structure of amorphous materials formed via mechanical deformation remain elusive, because of the lack of experimental probes. Here, multinuclear solid-state nuclear magnetic resonance (NMR) allowed us to uncover the nature of the amorphization of crystalline SiO2 produced by mechanical milling. The extensive NMR results reveal the milling-induced network depolymerization, hydroxylation, and topological contraction in SiO2 networks during amorphization. In particular, extreme friction activates chemical interactions among the materials, forming extrinsic chemical bonds via mechanical chemical reactions during deformation under amorphization of SiO2, indicating that the amorphous phase is not compositionally pure. The current findings with the extrinsic bonds may account for the anomalous increase in the storage capacity of amorphous materials formed by intense milling. The results establish the first quantitative kinetic model of a friction-induced increase in an amorphous SiO2 complex by a dynamic milling process, identifying the threshold milling rate and duration for the formation of amorphous products. The current results provide predictive and practical guidelines for controlling amorphization through mechanical shear, shedding light on novel synthetic routes to diverse amorphous materials.
1. Introduction
SiO2 is a prototypical oxide with critical importance in condensed matter physics,1 materials chemistry,2–4 and geosciences,5–7 and it has also been used as a potential Li-ion battery anode,8 catalyst substrate,9,10 and drug delivery system.11,12 Its amorphous analog (a-SiO2) can be produced by quenching molten SiO2, or via chemical routes (e.g., sol–gel synthesis or vapor deposition) and electron beam irradiation.13 The high melting temperature of SiO2 poses a challenge for the efficient production of a-SiO2 by melt quenching, and conventional synthesis methods may not be feasible for the production of water-free a-SiO2 nanoparticles with desirable physical properties.The mechanical deformation of SiO2 crystals (c-SiO2) can be an effective a-SiO2 synthesis technique,14,15 overcoming the challenges associated with the synthesis of a-SiO2 because amorphization via mechanical deformation can produce amorphous structures at room temperature (i.e., high temperatures above the melting point for glass synthesis); the deformation of crystalline materials by milling can lead to the destruction of crystalline periodicity and formation of disordered atomic configurations. Mechanical energy can be applied by rapidly shaking or rotating the materials in the presence of hard balls in a closed bowl (i.e., ball milling). The ball-milling process reduces the particle size by grinding the powders, thereby increasing the surface area.16 Extensive mechanical milling can amorphize condensed matters (i.e., mechanical amorphization), enabling the synthesis of amorphous materials that are difficult to prepare via conventional methods (e.g., melt quenching). Hazardous or expensive solvents that are typically required for the chemical synthesis of amorphous materials (e.g., sol–gel synthesis) may not be necessary for mechanical amorphization via milling, leading to the eco-friendly synthesis of novel amorphous materials. Additionally, mechanosynthesis has recently garnered attention as a viable method for material synthesis,17 including techniques that involve mechanochemistry,18,19 nanocomposites,20–22 isotopic labelling,23,24 multicomponent oxides,25 mechanocatalysis,26 and room-temperature thermochemical water splitting.27 Specifically, ball-milled SiO2 phases and composite systems have recently been used in Li-ion battery anode materials,28–30 where the amorphous phase of this SiO2-based material demonstrated superior electrode material performance; however, the detailed atomistic origin remains unknown.
The mechanical energy from ball milling may also activate chemical reactions by forming defects. During ball milling, the milling media (e.g., grinding balls and containers) or atmospheric components (e.g., ambient moisture, oxygen, and nitrogen) can potentially interact with materials, affecting their structures and properties.31,32 Thus, understanding the nature of such interactions is the key to designing mechanosynthetic routes. In particular, the structural characteristics of milling products and formation of milling-induced chemical bonds influence their macroscopic properties. Therefore, the desired functional properties can be optimized by controlling these structural parameters.33,34 Furthermore, information on the deformation and amorphization of c-SiO2 during mechanical milling can provide insight into earthquake rupture and slip weakening.5,6,35
Despite its importance and prospects in materials chemistry, condensed matter physics, and geosciences, the detailed nature of mechanical amorphization (e.g., the degree of mechanical amorphization under varying deformation conditions and/or structures of amorphous materials synthesized via mechanical milling) remains unknown. This is because of the limitations of conventional experimental techniques such as X-ray scattering in the quantification of the fractions of amorphous phase and crystalline components, as well as in the probing of amorphous structures. Challenges arise from the complex chemical interactions of the materials with the surrounding moisture and/or milling media, which form extrinsic and intrinsic chemical bonds and contribute to the structural complexity of amorphous materials produced via mechanical milling. Quantitative information on the extent of mechanical amorphization and structural evolution, and proper identification of chemical bonds in ball-milled non-crystalline oxides, including a-SiO2, remain to be explored.29,32
Solid-state nuclear magnetic resonance (NMR) is an ideal element-specific method for analyzing structural changes in mechanically deformed crystalline SiO2 and its amorphization (see, e.g., ref. 36–39). Solid-state 1H magic-angle spinning (MAS) NMR is useful for the analysis of hydrogen speciation and quantification of hydrogen content (e.g., ref. 40–42 and the references therein) and can thus effectively enable identification of the extent of potential hydration and hydroxylation during the mechanical milling of SiO2. 29Si MAS NMR spectroscopy can quantify the structural changes in the SiO4 networks in a-SiO2, which are often characterized by Qn speciation (where n is the number of bridging oxygen atoms in the SiO4 tetrahedra),37 thereby revealing the evolution of amorphous network structures in SiO2 during mechanical milling.431H–29Si heteronuclear correlation (HetCor) NMR enables the identification of potential water incorporation into SiO2 under extreme friction.37,44 Lastly, 17O NMR spectroscopy directly probes the oxygen environment in amorphous oxides (e.g., ref. 38 and 45–48 and the references therein). This technique can be used to explore the formation of novel oxygen environments during mechanical milling49 and evaluate the chemical interactions between SiO2 and the milling media.
Here, we note that ‘amorphization’ during intense ball-milling refers to a transition process that occurs along with the loss of crystallinity in c-SiO2. This process can be probed from the emergence of a broad diffraction peak and spectral broadening often observed unambiguously via high-resolution solid-state NMR. During amorphization under intense milling, the particle size could decrease, which may activate surface reactions. This could alter the chemical composition of the starting crystals. High-resolution spectroscopic techniques are necessary to understand the detailed nature of these interactions and for the identification of the species in the amorphous phases formed by milling of c-SiO2 (hereafter, we refer to the ‘a-SiO2 product formed via intense ball-milling’ as ‘milled amorphous silica product’. The phase should consist of a chemical component (such as hydrogen) added during milling-induced frictional amorphization). In the current study, we employed multinuclear 1H, 17O, and 29Si MAS NMR spectroscopy to quantitatively evaluate the structural changes in crystalline SiO2 and the milled amorphous silica product via rapid frictional shear. Although the quantification of mechanical amorphization has been challenging, our study constitutes the first study to yield quantitative information on the degree of mechanical amorphization (i.e., fractions of the milled amorphous silica product) at varying milling speeds and durations. Notably, we also discuss how experimental breakthroughs and conceptual advancements in mechanical amorphization in SiO2 can lead to more effective optimization of the material synthesis routes for diverse non-crystalline oxides during milling, as well as other relevant processes during frictional slip.
2. Experimental
2.1. Sample preparation
Quartz (SiO2) powder (≥99.995% trace metal basis, a few hundred μm in size on average) purchased from Sigma-Aldrich was deformed via milling at 200, 400, 600, and 800 rpm (revolutions per minute) under a milling duration of 5 h using a planetary ball mill machine (FRITSCH Pulverisette 7 premium line, Seoul National University). The crystalline SiO2 powder (∼600 mg) was loaded into a 20 mL ZrO2 bowl with 24 ZrO2 balls (diameter: 5 mm; total: ∼9 g). Ball-milled SiO2 with varying milling durations at 800 rpm was prepared by using a 20 mL ZrO2 bowl with five ZrO2 balls (8 mm in diameter). The SiO2 powder was dried in a furnace at 300 °C for more than 12 h. The dried SiO2 powder (of 200 mg) was ball-milled. The ball-to-powder weight ratio (BPR) was approximately 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Ball milling was performed for 10 min and then rested for 10 min to minimize heating. Ball milling was performed at milling durations of 10, 20, 30, 60, 120, and 180 min at 800 rpm. To observe the oxygen bonding configurations via17O MAS NMR, ∼100 mg of 17O-enriched (40%) amorphous SiO2 (ref. 50) was ball-milled by applying a 20 mL ZrO2 bowl, five ZrO2 balls (8 mm in diameter; BPR of ∼80:1), and a milling speed of 800 rpm for 15 and 60 min. We also synthesized 40% 17O-enriched crystalline SiO2 from melting at 1500 °C for 2 h using 40% 17O-enriched amorphous SiO2, which resulted in forming the crystalline SiO2 (cristobalite) phase. Then, ∼90 mg of 17O-enriched (40%) c-SiO2 (cristobalite) was ball-milled by applying a 20 mL ZrO2 bowl, five ZrO2 balls (8 mm in diameter; BPR of ∼90:1), and a milling speed of 800 rpm for 15 and 60 min. We note that 17O-enriched a-SiO2 of ∼100 mg and 17O-enriched c-SiO2 (cristobalite) of ∼90 mg were milled with milling durations of 15 and 60 min, respectively.
:1. Ball milling was performed for 10 min and then rested for 10 min to minimize heating. Ball milling was performed at milling durations of 10, 20, 30, 60, 120, and 180 min at 800 rpm. To observe the oxygen bonding configurations via17O MAS NMR, ∼100 mg of 17O-enriched (40%) amorphous SiO2 (ref. 50) was ball-milled by applying a 20 mL ZrO2 bowl, five ZrO2 balls (8 mm in diameter; BPR of ∼80:1), and a milling speed of 800 rpm for 15 and 60 min. We also synthesized 40% 17O-enriched crystalline SiO2 from melting at 1500 °C for 2 h using 40% 17O-enriched amorphous SiO2, which resulted in forming the crystalline SiO2 (cristobalite) phase. Then, ∼90 mg of 17O-enriched (40%) c-SiO2 (cristobalite) was ball-milled by applying a 20 mL ZrO2 bowl, five ZrO2 balls (8 mm in diameter; BPR of ∼90:1), and a milling speed of 800 rpm for 15 and 60 min. We note that 17O-enriched a-SiO2 of ∼100 mg and 17O-enriched c-SiO2 (cristobalite) of ∼90 mg were milled with milling durations of 15 and 60 min, respectively.
2.2. XRD & high-resolution TEM
X-ray diffraction (XRD) experiments were performed by using a Rigaku MiniFlex 600 with Cu Kα emission with a wavelength of 1.5406 Å (Seoul National University). In continuous mode with a scan speed of 2.5° min−1, we acquired the XRD pattern at 2θ intervals of 0.02° from 5 to 90° at 40 kV and 15 mA. High-resolution transmission electron microscopy (HRTEM) with energy-dispersive X-ray spectroscopy (EDS) was used to examine the morphologies and chemical composition of the SiO2 milled in our planetary ball-milling system.2.3. FT-IR spectroscopy
We collected the Fourier transform infrared (FT-IR) spectra for the milled oxides using a TENSOR27 spectrometer (Seoul National University) with a spectral resolution of 4 cm−1. The spectral range spans from 400 to 4000 cm−1. The scans were collected 32 times in attenuated total reflectance mode by using a mid-IR source. A diamond crystal plate was used to place a few milligrams of each sample in the optical window.2.4. NMR spectroscopy
A Varian 9.4 T (400 MHz) NMR spectrometer was used for acquiring the 29Si MAS NMR spectra with a 29Si Larmor frequency of 79.47 MHz (Seoul National University). A Doty 4 mm probe was used with a spinning speed of 11–12 kHz (12 kHz for the milled SiO2 from 200 to 800 rpm for 5 h, and 11 kHz for those deformed at a milling speed of 800 rpm with varying milling durations ranging from 10 to 180 min). The 29Si MAS NMR spectra were acquired using a ∼30° pulse (1.6 μs) with varying recycle delays. 1H MAS NMR spectra at a spinning speed of 20 kHz, 29Si MAS NMR spectra of 7 nm amorphous silica, 40% 17O-enriched a-SiO2 and 17O-enriched c-SiO2 (before and after milling at 800 rpm) at a spinning speed of 15 kHz (∼30° pulse of 1 μs with a recycle delay of 10 s), and 1H–29Si Lee-Goldburg cross-polarization HetCor NMR spectra with a contact time of 3 ms and a rotor spinning rate of 15 kHz were recorded on a Bruker 14.1 T (600 MHz) NMR spectrometer and a Bruker 3.2 mm probe (Seoul National University). Tetramethylsilane (TMS) was the external standard for 29Si and 1H NMR chemical shifts. To ensure complete relaxation, 1H MAS NMR spectra were collected with recycle delays exceeding 5 times spin-lattice relaxation time (T1)51 (see ESI-1†). Long recycle delays of ≥30 s with a 90° pulse (2.5 μs) were used for SiO2 deformed at 800 rpm with varying milling durations. To obtain the 1H MAS NMR spectra, the NMR signals of ball-milled SiO2 were subtracted from the background spectrum of the empty rotor. Adamantane (C10H16) was used as the standard to estimate the hydrogen contents.5217O MAS NMR spectra were recorded on a Bruker 14.1 T (600 MHz) NMR spectrometer with a 17O Larmor frequency of 81.39 MHz and a Bruker 3.2 mm probe with a rotor spinning speed of 20 kHz. 17O MAS NMR spectra were obtained with a recycle delay of 1 s and a pulse length of ∼0.2 μs. Tap water was used as the external standard. Here, we reported the 17O MAS NMR spectrum of 40% 17O-enriched amorphous SiO2 and also 40% 17O-enriched c-SiO2 (cristobalite). Because of the low natural abundance of 17O (0.037%), currently, it is challenging to obtain the 17O NMR spectrum for natural abundance a-SiO2. Note that ESI-1† describes the detailed spectroscopic and synthesis conditions.3. Results and discussion
3.1. Mechanical amorphization of SiO2via mechanical milling: XRD, TEM-EDS, and FT-IR analysis
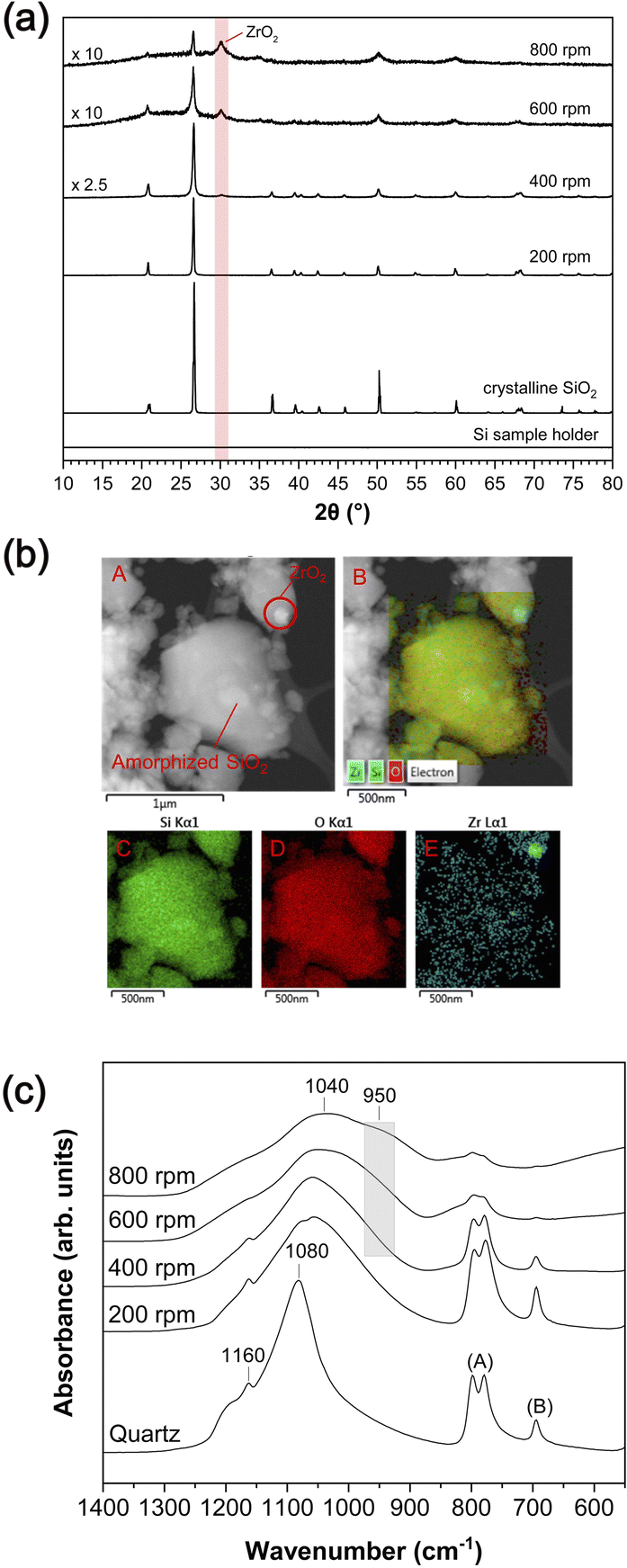 | ||
| Fig. 1 (a) X-ray diffraction patterns for milled SiO2 with varying milling speeds for 5 h. The XRD pattern of the sample holder is also shown. (b) TEM images for milled SiO2 with a milling speed of 600 rpm for 5 h (red A and B) and EDS mapping for Si (red C), O (red D) and Zr elements (red E). (c) FT-IR spectra for milled SiO2 with varying milling speeds for 5 h. | ||
Above ∼400 rpm, we observe an additional feature at ∼950 cm−1, and its intensity increases with increasing milling speed up to ∼800 rpm. This feature may be primarily attributable to the Si–O–Zr bond,55,58,59 indicating a chemical reaction between SiO2 and ZrO2 under the condition of milling above 400 rpm (see ESI-2 and Fig. S1†). Although this feature could also be attributable to Si–OH groups that can be expected at ∼980 cm−1,60 the intensity of this feature did not decrease upon heating the milled SiO2 up to 700 °C (see ESI-2†), which may have removed all potential hydroxyl groups.37 These results demonstrate that the amorphized SiO2 and worn ZrO2 particles from the milling media interact to form new chemical bonds during mechanical amorphization.
3.2. Deformation-induced change in network connectivity in the milled amorphous silica product under frictional shear and the interaction with milling media: 17O MAS NMR spectroscopy results
Fig. 2 shows the 17O MAS NMR spectra of 17O-enriched a-SiO2 before and after milling at 800 rpm. The dominant peak at ∼75 to ∼−25 ppm has been attributed to the Si–O–Si bonds, with an additional contribution from the Si–OH groups, whereas the features associated with Si–OH groups may not have fully resolved from the Si–O–Si bonds24,49,61 (see also 1H MAS NMR results below for further discussion on the effects of hydration). The spectral width of the Si–O–Si peak increases with prolonged milling duration (Fig. 2c), revealing an increase in the structural disorder and the extent of deformation.48,62 The 17O NMR spectrum of a-SiO2 subjected to mechanical shearing reveals additional broad features at ∼100–230 ppm, and they are attributable to Si–O–Zr bonds.49 Therefore, the 17O NMR results provide direct evidence that the enhanced interactions between ZrO2 and SiO2 during the intense milling-induced amorphization process resulted in Si–O–Zr bond formation. Additionally, the intensity of the Si–O–Zr peak increases with prolonged milling duration, and the bond fraction of Si–O–Zr [i.e., (Si–O–Zr)/(Si–O–Si + Si–O–Zr)] following 15 and 60 min of milling at 800 rpm are ∼4% and ∼9%, respectively (see Fig. S3†), confirming unequivocally that the degree of chemical interaction increases with increasing milling duration and amorphization (see ESI-4† for XRD, 1H and 29Si MAS NMR spectra). | ||
| Fig. 2 (a) 17O MAS NMR spectra with 17O-enriched a-SiO2 before and after milling at 800 rpm. The asterisks (*) indicate the spinning sidebands. (b) 17O MAS NMR spectra of 17O-enriched a-SiO2 before and after milling of 60 min at 800 rpm. (c) Enlarged 17O MAS NMR spectra between 75 ppm and −75 ppm for 17O-enriched a-SiO2 before and after milling at 800 rpm. (d) 17O MAS NMR spectra with 17O-enriched c-SiO2 (cristobalite) before and after milling at 800 rpm. The asterisks (*) indicate the spinning sidebands. (e) 17O MAS NMR spectra of 17O-enriched c-SiO2 (cristobalite) before and after milling of 60 min at 800 rpm. (f) 17O MAS NMR spectra of 17O-enriched c-SiO2 (cristobalite) and 17O-enriched a-SiO2 after milling for 60 min at 800 rpm. | ||
The amorphous phase formed via an intense milling of crystalline SiO2 during mechanical amorphization also consists of Si–OH; as shown from the 1H and 29Si spectra (Fig. S5 and S6†), OH is barely present in the 17O-enriched a-SiO2 before milling but is produced during milling. The amount of OH is comparable to the non-enriched SiO2. An earlier experimental study of 17O-labeled silica24 revealed the formation of Si–OH under shorter milling durations of 5 and 15 min using a mixer mill with a frequency of 25 Hz. The current study is consistent with the earlier study. In addition to Si–OH, the current results reveal the formation of Si–O–Zr in SiO2 under intense ball milling.
Fig. 2d shows the 17O MAS NMR spectra for 17O-enriched c-SiO2 (cristobalite) with milling durations of 15 and 60 min at 800 rpm (see ESI-4† for 29Si MAS NMR spectra, XRD and 1H MAS NMR results). The dominant peak with a typical quadrupolar line shape at ∼50 to ∼−25 ppm of 17O-enriched c-SiO2 (cristobalite) before milling has been assigned to the single bridging oxygen (BO) site from the Si–O–Si bonds.63,64 The crystalline quadrupolar pattern for the Si–O–Si peak disappears and the spectral width increases after milling durations of 15 and 60 min. This confirms increases in the structural disorder and amorphization of SiO2 cristobalite. The 17O NMR spectrum of milled 17O-enriched c-SiO2 (cristobalite) also shows broad features at ∼100–230 ppm, which are due to Si–O–Zr bonds. The intensity of the Si–O–Zr peak also increases with increased milling duration, consistent with those observed from the milling-induced transitions in amorphous SiO2 (Fig. 2a–c). The fractions of Si–O–Zr bonds in milled 17O-enriched c-SiO2 (cristobalite) with milling durations of 15 and 60 min at 800 rpm were ∼4% and ∼9% (Fig. 2d), respectively, which shows remarkable similarity to those of milled 17O-enriched a-SiO2 (Fig. 2a). After milling for 60 min at 800 rpm, the 17O NMR spectra of 17O-enriched c-SiO2 (cristobalite) and 17O-enriched a-SiO2 are similar (Fig. 2f). The 17O NMR results confirm that milling-induced amorphization is prevalent under a milling speed of 800 rpm and duration of 15 min with the formation of an extrinsic Si–O–Zr bond.
3.3. Mechanical milling-induced hydration of the milled amorphous silica product: hydrogen speciation and quantification of 1H contents by 1H MAS NMR analysis
 | ||
| Fig. 3 (a) 1H MAS NMR spectra (at 14.1 T) for milled SiO2 with varying milling speeds using a milling duration of 5 h (the number of protons in the unit sample weight estimated from the spectral intensity).37 The peak at ∼1.3 ppm (#) is due to organic contamination such as finger grease.67 (b) 1H MAS NMR spectra (at 14.1 T) obtained for milled SiO2 with varying milling durations at 800 rpm (the number of protons in the unit sample weight estimated from the spectral intensity). | ||
The impact of the milling duration on the hydrogen environment of the milled amorphous silica product at a constant milling speed of 800 rpm is illustrated in Fig. 3b. The NMR results confirm an overall increase in the hydrogen content in the milled amorphous silica product with increasing milling duration and milling speed; this is because milling-induced comminution and a larger surface area tend to promote the hydration and hydroxylation of milled amorphous silica product surfaces. These results also indicate that, under rapid frictional shear and a relatively long milling duration (of 30 min under 800 rpm), frictional heating during such intense milling can also dehydrate and/or dehydroxylate the phase during amorphization.
3.4. Evolution of network structures of the milled amorphous silica product under frictional shear: insights from 29Si MAS NMR analysis
 | ||
| Fig. 4 (a) 29Si MAS NMR spectra for milled SiO2 with varying milling speeds with a milling duration of 5 h and 7 nm amorphous silica, the used recycle delay is 10 s. The 29Si spectrum of 7 nm amorphous silica dehydrated at 1473 K was from the previous study.37 (b) 29Si MAS NMR spectra for milled SiO2 with varying milling speeds with a milling duration of 5 h, the used recycle delay is 4200 s. (c) 29Si MAS NMR spectra for milled SiO2 with varying milling durations at 800 rpm, the used recycle delay is 3600 s (7200 s for the milled SiO2 for 180 min). (d) 29Si MAS NMR spectra for milled SiO2 at 800 rpm with milling durations of 10 and 180 min. (e) 1H–29Si heteronuclear correlation (HetCor) NMR spectra and projections in 1H and 29Si dimensions for milled SiO2 with varying milling speeds using a milling duration of 5 h. | ||
We also collected the 29Si NMR spectra for 17O-enriched crystalline SiO2 (cristobalite) without milling and those with milling durations of 15 and 60 min under 800 rpm (Fig. S7†). The spectral results are compared to those of 17O-enriched amorphous SiO2 (Fig. S6–S8†). Using 17O-enriched a-SiO2 and 17O-enriched c-SiO2 (cristobalite) as starting materials, the 29Si MAS NMR spectra for the milled amorphous phase are rather similar, both showing an increase in peak intensity around ∼−90 to ∼−105 ppm (see Fig. S6–S8†). We note that with a short NMR recycle delay of 10 s used in the current 29Si NMR spectral acquisition, the fraction of the crystalline peak is underestimated (see the section of Amorphous structures at varying milling speeds: 29Si NMR results with a short recycle delay time above for further discussion). Nevertheless, the results confirm that the amorphous phases with different starting materials (i.e., cristobalite and a-SiO2) undergo similar amorphization paths, involving network depolymerization, hydroxylation, and the formation of extrinsic bonds resulting from interactions with the milling media.
 | (1) |
The estimated Q4(mZr) fractions are as follows: Q4(0Zr), ∼0.69; Q4(1Zr), ∼0.27; Q4(2Zr), ∼0.04; and Q4(3Zr), ∼0.003. Because the chemical shifts of these peaks are higher than those for Q4(0Zr) (i.e., less negative peak position), the formation of Q4(mZr) species accounts for the observed peak shift in the 29Si MAS NMR spectra for the milled SiO2 (see ESI-9 and Fig. S14b†). Second, an increase in network depolymerization due to hydroxylation of the milled amorphous silica product and, thus, the formation of Q2 with germinal silanol [(Si–O)2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(OH)2] and Q3 with single silanol [(Si–O)3
Si(OH)2] and Q3 with single silanol [(Si–O)3![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH]37 may partially account for the residual intensity, as the spectral patterns may not be fully explained by the estimated Q4(mZr) fractions alone (see ESI-9 and Fig. S14c†). Lastly, because the 29Si NMR chemical shift increases with decreasing Si–O–Si bond angle (see ref. 37 and the references therein), the current positive shift indicates a deformation-induced decrease in the Si–O–Si bond angles, as also implied by the FT-IR results (see ESI-10 and Fig. S15†).
Si–OH]37 may partially account for the residual intensity, as the spectral patterns may not be fully explained by the estimated Q4(mZr) fractions alone (see ESI-9 and Fig. S14c†). Lastly, because the 29Si NMR chemical shift increases with decreasing Si–O–Si bond angle (see ref. 37 and the references therein), the current positive shift indicates a deformation-induced decrease in the Si–O–Si bond angles, as also implied by the FT-IR results (see ESI-10 and Fig. S15†).
To further identify the relative contributions of both Zr and H to Si speciation (i.e., Q2(2OH), Q3(1OH), Q4, and Q4(mZr) species), the fractions of Qn species in the amorphous phase were estimated by deconvoluting the 29Si MAS NMR spectra collected under longer NMR recycle delays (see ESI-11 and ESI-12† for the effects of NMR relaxation time on quantification; ESI-13† for the effects of 10 and 4200 s recycle delays on the 29Si MAS NMR spectra). Here, we first constrained the Q4 peak position to ∼−110 ppm based on the NMR signal of pure SiO2 glass only with Q4 species (Fig. 4a). The relative Q4(mZr) proportions were then estimated based on the Si–O–Zr fraction from 17O NMR (eqn (1)). The Q4(2Zr) and Q4(1Zr) peak positions were constrained to ∼−92 and ∼−101 ppm (ESI-9 and Fig. S14b†), with the relative intensities set to match the Q4(2Zr), Q4(1Zr), and Q4 [i.e., Q4(0Zr)] ratios of approximately 4%, 27%, and 69%, respectively. Additionally, the residual spectral intensities of the experimental 29Si MAS NMR signals have been attributed to the combination of Q2 and Q3 species at ∼−92 and ∼−101 ppm, respectively, accounting for the hydroxylation-induced changes in the Si species (see ESI-9 and Fig. S14c†). The fractions of Si species resulting from SiO2 being subjected to frictional shear at 800 rpm were as follows: Q2, 10%; Q3, 24%; Q4, 45%; Q4(1Zr), 18%; and Q4(2Zr), 3%. Although the Q4 peak position was fixed at approximately −110 ppm, the potential positive peak shift in Q4 could contribute to increase the fractions of Q4 species, particularly the Q4(1Zr) and Q4(2Zr) species. Additionally, the fraction of Si–O–Zr bonds may be slightly underestimated because the Si–O–H bonds overlapped with the Si–O–Si bonds in the 17O MAS NMR spectra (Fig. 2). Therefore, the current simulation results represent a conservative estimation of the fractions of Q4(0Zr), Q4(1Zr), and Q4(2Zr) species in SiO2 (see ESI-9† for further details and uncertainties).
Furthermore, although the effects of Si–O–Si bond angle reduction during deformation have not been explicitly considered, the systematic peak shift in the Q4 species (toward a more positive frequency) can potentially provide quantitative insight into deformation-induced topological contraction (see Fig. S19† for milling-driven 29Si peak shift). Regardless of this uncertainty, the current 29Si results clearly highlight diverse deformation processes during milling, such as milling-induced chemical interactions, hydroxylation, and potential topological contraction (i.e., bond-angle change) in SiO2 networks during amorphization.
3.5. Quantification of the degree of amorphization in SiO2 under mechanical shear
 | ||
| Fig. 5 (a) 29Si MAS NMR spectra with varying NMR recycle delays (dark red) for milled SiO2 at 800 rpm with varying milling durations (dark blue) from 10 to 180 min. (b) 29Si MAS NMR spectra with varying NMR recycle delays (dark red) of 10, 300, and 3600 s for 10 to 120 min and 7200 s for 180 min for milled SiO2 at 800 rpm with varying milling durations (dark blue) from 10 to 180 min (right: normalized spectra). | ||
In Fig. 5b, the milling-driven evolution of 29Si MAS NMR spectra for SiO2 under distinct NMR recycle delays (10, 300, and 3600 s) is shown, which were used to determine the optimal NMR conditions for quantification of the NMR spectral features. The normalized spectra (Fig. 5b, right) show that the spectral intensities within the higher frequency range of −80 to −100 ppm are more prominent under the shorter recycle delay of 10 s. This is because the deformation-induced changes in the species with shorter T1 relaxation times, such as Q2, Q3, Q4(1Zr), and Q4(2Zr), are more pronounced under the shorter NMR recycle delay; the electron spin resonance spectra (see ESI-12 and Fig. S17†) reveal an increase in the concentration of radical species (i.e., unpaired electron spins) with increasing milling duration,71 which reduces the T1 relaxation times of the milled SiO2. Our ESR spectra reveal the peroxy radicals, which originates from the increased contact of SiO2 particles with the air (or oxidation) during milling. Furthermore, the reduced oxidation may preserve the E′ center and/or non-bridging oxygen hole center (NBOHC) (ESI-12† for further details).72 This increase also indicates that the NMR relaxation delay time for the acquisition of the quantitative spectrum can be shortened (ESI-12†). Taking these factors into consideration, it is concluded that an NMR recycle delay of ≥3600 s provides the fully quantitative 29Si MAS NMR spectra, enabling robust estimation of the Qn and/or Q4(mZr) species in milled SiO2 (see ESI-17† for further details).
 | ||
| Fig. 6 (a) Variation in the fractions of crystalline SiO2 and the milled amorphous silica product with varying milling speeds with a milling duration of 5 h, the used recycle delay is 4200 s. The solid lines show the shifted exponential function to model the fractions of c-SiO2 and milled amorphous silica product upon increase in the milling speeds, revealing the threshold milling speed of ∼185 rpm. The blue box indicates that the mechanical amorphization could not be achieved at milling speeds below ∼185 rpm. (b) Variation in the fractions of crystalline SiO2 and the milled amorphous silica product with varying milling durations at 800 rpm, the used recycle delay is 3600 s for 10 to 120 min and 7200 s for 180 min. The solid lines were fitted by a single exponential function. (c) Enlarged variation in the fractions for 10 to 60 min. | ||
Fig. 6b and c illustrate the relationship between c-SiO2 and milled amorphous silica product fractions and milling duration at 800 rpm. The fraction of the milled amorphous silica product sharply increased with increasing milling duration up to approximately 30 min. The fractions of the milled amorphous silica product were clearly saturated for milling durations above ∼30 min. The relationship between milled amorphous silica product fraction and milling duration can also be explained using a single exponential function: A(t) = A0[1 − exp(−t/D1)], where A(t) is the amorphous fraction for various milling durations and A0 is the saturated amorphous fraction. The mechanical amorphization time, D1, is defined as the milling duration required for the amorphization of ∼63% of c-SiO2. Under the current milling conditions, D1 was estimated to be ∼7 min. Although the extent of ball milling-induced mechanical amorphization had remained unknown, the current experimental results provide the first quantitative information on the fractions of crystalline and amorphous components in SiO2 milled under varying speeds and durations. The extensive 29Si NMR results, in conjunction with the inputs from the 17O and 1H results, enable us to establish the first predictive models for mechanical amorphization during intense milling.
3.6. Implications and applications
Our current results provide guidelines for controlling the structural disorder and fractions of crystalline–amorphous phases by tuning the milling speed and duration, and can be practically useful as novel mechanical milling-based synthesis protocols. Here, we discuss the implications further and the practical aspects of the current experimental findings and conceptual advances.4. Conclusions
In the current study, we investigated extensively the extent of network amorphization of prototypical SiO2 during intense milling and revealed the substantial degree of hydration, hydroxylation, and chemical interactions with the milling media at varying milling speeds and durations via multinuclear (1H, 17O, and 29Si) NMR spectroscopy, together with XRD, TEM-EDS, and FT-IR. The current results constitute a full-spectrum analysis of the mechanical amorphization-driven structural evolution and interactions during milling, which has not been previously reported. Notably, as the milling speed and duration increase, the crystallinity of SiO2 gradually decreased and it became amorphous. During milling-driven mechanical amorphization, the high-resolution 17O NMR revealed the formation of Si–O–Zr bonds through the interaction of SiO2 with ZrO2 milling media. The 1H NMR results also confirmed the hydration of the amorphous SiO2 complex through the formation of Si–O–H species. The fully relaxed 29Si MAS NMR spectra were used to quantify the fractions of c-SiO2 and amorphous SiO2 phase consisting of a hydroxyl group and Si–O–Zr at varying milling speeds and durations. The quantification of the fraction of the milled amorphous silica product revealed a threshold milling speed and duration. These findings help to explain the atomistic structural changes resulting from applying a ball-milling process to diverse materials with comparatively high hardness. Furthermore, our findings regarding deformation-induced chemical interactions and hydration also provide insight into structural changes in silicate rocks and melts during natural frictional processes (e.g., fault slip). The amorphous product obtained via mechanical amorphization of c-SiO2 has potential as an anode material for batteries and other energy storage materials. Future studies on comprehensive investigation of the formation of extrinsic and intrinsic chemical bonds for complex materials are needed. Our experimental findings provide guidelines for novel mechanical deformation-based synthesis routes for amorphous materials.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Jin Jung Kweon: data curation, formal analysis, investigation, validation, visualization, writing – original draft, review & editing; Hoon Khim: data curation, formal analysis, investigation, visualization; Yong-Hyun Kim: writing – review & editing; Sung Keun Lee: conceptualization, formal analysis, methodology, funding acquisition, supervision, writing – original draft, review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT to S. K. Lee. (2020R1A3B2079815). We are grateful to Prof. Seung Mo Oh and Dr Jae Gil Lee of the School of Chemical and Biological Engineering, Seoul National University for their discussion on synthesizing amorphous anode materials in July 2013. We thank Dr Jeongjae Lee for suggestions and editing of the earlier version of the manuscript.References
- A.-M. El-Sayed, Y. Wimmer, W. Goes, T. Grasser, V. V. Afanas'ev and A. L. Shluger, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 92, 014107 CrossRef.
- K. Möller and T. Bein, Chem. Mater., 2016, 29, 371–388 CrossRef.
- F. F. E. Kohle, J. A. Hinckley, S. Li, N. Dhawan, W. P. Katt, J. A. Erstling, U. Werner-Zwanziger, J. Zwanziger, R. A. Cerione and U. B. Wiesner, Adv. Mater., 2019, 31, 1806993 CrossRef PubMed.
- A. A. Nayl, A. I. Abd-Elhamid, A. A. Aly and S. Brase, RSC Adv., 2022, 12, 13706–13726 RSC.
- S. K. Lee, R. Han, E. J. Kim, G. Y. Jeong, H. Khim and T. Hirose, Nat. Geosci., 2017, 10, 436–441 CrossRef CAS.
- V. Samae, P. Cordier, S. Demouchy, C. Bollinger, J. Gasc, S. Koizumi, A. Mussi, D. Schryvers and H. Idrissi, Nature, 2021, 591, 82–86 CrossRef CAS PubMed.
- N. Watanabe, H. Abe, A. Okamoto, K. Nakamura and T. Komai, Sci. Rep., 2021, 11, 5340 CrossRef CAS PubMed.
- Y. Wang, K. Xie, X. Guo, W. Zhou, G. Song and S. Cheng, New J. Chem., 2016, 40, 8202–8205 RSC.
- O. Verho, F. Gao, E. V. Johnston, W. Wan, A. Nagendiran, H. Zheng, J.-E. Bäckvall and X. Zou, APL Mater., 2014, 2, 113316 CrossRef.
- V. G. Chandrashekhar, T. Senthamarai, R. G. Kadam, O. Malina, J. Kašlík, R. Zbořil, M. B. Gawande, R. V. Jagadeesh and M. Beller, Nat. Catal., 2021, 5, 20–29 CrossRef.
- C. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, Adv. Mater., 2004, 16, 1959–1966 CrossRef.
- K. K. Qian and R. H. Bogner, J. Pharm. Sci., 2012, 101, 444–463 CrossRef CAS PubMed.
- S.-G. Kang, W. Jeong, J. Paeng, H. Kim, E. Lee, G.-S. Park, S. Han, H. Nam Han and I.-S. Choi, Mater. Today, 2023, 66, 62–71 CrossRef CAS.
- X. Hou, M. Zhang, J. Wang, S. Hu, X. Liu and Z. Shao, J. Alloys Compd., 2015, 639, 27–35 CrossRef CAS.
- V. Zarei, M. Mirzaasadi, A. Davarpanah, A. Nasiri, M. Valizadeh and M. J. S. Hosseini, Processes, 2021, 9, 334 CrossRef CAS.
- H. N. Kim, J. W. Kim, B.-D. So, Y. Keehm, B. H. Lee and J. C. Kim, Geosci. J., 2022, 26, 703–713 CrossRef.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- K. Y. Baek, W. Lee, J. Lee, J. Kim, H. Ahn, J. I. Kim, J. Kim, H. Lim, J. Shin, Y. J. Ko, H. D. Lee, R. H. Friend, T. W. Lee, J. Lee, K. Kang and T. Lee, Nat. Commun., 2022, 13, 4263 CrossRef CAS PubMed.
- G. Gorrasi and A. Sorrentino, Green Chem., 2015, 17, 2610–2625 RSC.
- F. Delogu, G. Gorrasi and A. Sorrentino, Prog. Mater. Sci., 2017, 86, 75–126 CrossRef CAS.
- S. F. H. Lambregts, L. M. de Kort, F. Winkelmann, M. Felderhoff, P. Ngene, E. R. H. van Eck and A. P. M. Kentgens, J. Phys. Chem. C, 2024, 128, 12186–12193 CrossRef CAS PubMed.
- C. H. Chen, E. Gaillard, F. Mentink-Vigier, K. Chen, Z. Gan, P. Gaveau, B. Rebiere, R. Berthelot, P. Florian, C. Bonhomme, M. E. Smith, T. X. Metro, B. Alonso and D. Laurencin, Inorg. Chem., 2020, 59, 13050–13066 CrossRef CAS PubMed.
- C. H. Chen, F. Mentink-Vigier, J. Trebosc, I. Goldberga, P. Gaveau, E. Thomassot, D. Iuga, M. E. Smith, K. Chen, Z. Gan, N. Fabregue, T. X. Metro, B. Alonso and D. Laurencin, Chem.–Eur. J., 2021, 27, 12574–12588 CrossRef CAS PubMed.
- A. F. Fuentes and L. Takacs, J. Mater. Sci., 2012, 48, 598–611 CrossRef.
- S. Hwang, S. Gratz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC.
- T. Yamamoto, S. Ashida, N. Inubuse, S. Shimizu, Y. Miura, T. Mizutani and K.-i. Saitow, J. Mater. Chem. A, 2024, 12, 30906–30918 RSC.
- W.-S. Chang, C.-M. Park, J.-H. Kim, Y.-U. Kim, G. Jeong and H.-J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 RSC.
- D. Choi and K.-L. Choy, Electrochim. Acta, 2016, 218, 47–53 CrossRef CAS.
- D. Choi and K. Choy, Dalton Trans., 2017, 46, 14226–14233 RSC.
- H. Kim, J. E. Lee, S.-M. Jo and S. Wooh, ACS Sustain. Chem. Eng., 2022, 10, 9679–9686 CrossRef CAS.
- J. J. Kweon, H. Khim and S. K. Lee, Korean Journal of Mineralogy and Petrology, 2023, 36, 95–106 Search PubMed.
- P. F. M. de Oliveira, R. M. Torresi, F. Emmerling and P. H. C. Camargo, J. Mater. Chem. A, 2020, 8, 16114–16141 RSC.
- A. P. Yuda, P. Y. E. Koraag, F. Iskandar, H. S. Wasisto and A. Sumboja, J. Mater. Chem. A, 2021, 9, 18906–18926 RSC.
- G. Di Toro, R. Han, T. Hirose, N. De Paola, S. Nielsen, K. Mizoguchi, F. Ferri, M. Cocco and T. Shimamoto, Nature, 2011, 471, 494–498 CrossRef CAS PubMed.
- T. Watanabe, T. Isobe and M. Senna, J. Solid State Chem., 1997, 130, 284–289 CrossRef CAS.
- H. N. Kim and S. K. Lee, Geochim. Cosmochim. Acta, 2013, 120, 39–64 CrossRef CAS.
- J. Lee and S. K. Lee, Acta Mater., 2022, 241, 118413 CrossRef CAS.
- A. C. Lee and S. K. Lee, Geochim. Cosmochim. Acta, 2024, 370, 199–218 CrossRef CAS.
- W. Yang, Z. Wang, J. Huang and Y. Jiang, J. Phys. Chem. C, 2021, 125, 10179–10197 CrossRef CAS.
- J. J. Kweon, H.-I. Kim, S.-h. Lee, J. Kim and S. K. Lee, Acta Mater., 2022, 226, 117657 CrossRef CAS.
- N. Lopatik, A. De, S. Paasch, A. Schneemann and E. Brunner, Phys. Chem. Chem. Phys., 2023, 25, 30237–30245 RSC.
- S. M. Chemtob, G. R. Rossman and J. F. Stebbins, Am. Mineral., 2012, 97, 203–211 CrossRef CAS.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS.
- J. F. Stebbins and X. Xue, Rev. Mineral. Geochem., 2014, 78, 605–653 CrossRef CAS.
- S. K. Lee and S. Ryu, J. Phys. Chem. Lett., 2018, 9, 150–156 CrossRef CAS PubMed.
- S. K. Lee, K. Y. Mun, Y. H. Kim, J. Lhee, T. Okuchi and J. F. Lin, J. Phys. Chem. Lett., 2020, 11, 2917–2924 CrossRef CAS PubMed.
- S. K. Lee, J. L. Mosenfelder, S. Y. Park, A. C. Lee and P. D. Asimow, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21938–21944 CrossRef CAS PubMed.
- D. M. Pickup, G. Mountjoy, G. W. Wallidge, R. J. Newport and M. E. Smith, Phys. Chem. Chem. Phys., 1999, 1, 2527–2533 RSC.
- S. K. Lee and E. J. Kim, J. Phys. Chem. C, 2014, 119, 748–756 CrossRef.
- M. Avramovska, D. Freude, W. Schwieger, T. Fey, J. Kärger and J. Haase, J. Phys. Chem. C, 2023, 127, 19833–19840 CrossRef CAS.
- W. J. Malfait and X. Xue, Geochim. Cosmochim. Acta, 2010, 74, 719–737 CrossRef CAS.
- A. Kaiser, M. Lobert and R. Telle, J. Eur. Ceram. Soc., 2008, 28, 2199–2211 CrossRef CAS.
- M. Andrianainarivelo, R. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Mater. Chem., 1996, 6, 1665–1671 RSC.
- F. Del Monte, W. Larsen and J. D. Mackenzie, J. Am. Ceram. Soc., 2000, 83, 1506–1512 CrossRef CAS.
- R. A. B. Devine, J. Non-Cryst. Solids, 1993, 152, 50–58 CrossRef CAS.
- C. J. Brinker, R. K. Brow, D. R. Tallant and R. J. Kirkpatrick, J. Non-Cryst. Solids, 1990, 120, 26–33 CrossRef CAS.
- S. W. Lee and R. A. Condrate, J. Mater. Sci., 1988, 23, 2951–2959 CrossRef CAS.
- J. Anderson, C. Fergusson, I. Rodriguezramos and A. Guerreroruiz, J. Catal., 2000, 192, 344–354 CrossRef CAS.
- J. B. Miller and E. I. Ko, J. Catal., 1996, 159, 58–68 CrossRef CAS.
- E. R. H. van Eck, M. E. Smith and S. C. Kohn, Solid State Nucl. Magn. Reson., 1999, 15, 181–188 CrossRef CAS.
- S. K. Lee, J. Phys. Chem. B, 2004, 108, 5889–5900 CrossRef CAS.
- D. R. Spearing, I. Farnan and J. F. Stebbins, Phys. Chem. Miner., 1992, 19, 307–321 CrossRef CAS.
- X. Xue, J. F. Stebbins and M. Kanzaki, Am. Mineral., 1994, 79, 31–42 CAS.
- Y.-S. Kwon, K. B. Gerasimov and S.-K. Yoon, J. Alloys Compd., 2002, 346, 276–281 CrossRef CAS.
- L. Takacs and J. S. McHenry, J. Mater. Sci., 2006, 41, 5246–5249 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- J.-H. Choy, J.-B. Yoon, H. Jung and J.-H. Park, J. Porous Mater., 2004, 11, 123–129 CrossRef CAS.
- O. B. Lapina, D. F. Khabibulin and V. V. Terskikh, Solid State Nucl. Magn. Reson., 2011, 39, 47–57 CrossRef CAS PubMed.
- S. K. Lee and J. F. Stebbins, Am. Mineral., 1999, 84, 937–945 CrossRef CAS.
- K. Gobindlal, Z. Zujovic, P. Yadav, J. Sperry and C. C. Weber, J. Phys. Chem. C, 2021, 125, 20877–20886 CrossRef CAS.
- M. Hasegawa, T. Ogata and M. Sato, Powder Technol., 1995, 85, 269–274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta03462k |
| This journal is © The Royal Society of Chemistry 2025 |